

Abstract
Protective ventilation strategies have been universally embraced because of reduced mortality. We tested the hypothesis that tidal volume (VT) in an in vivo model of mechanical ventilation would modulate bactericidal function of alveolar macrophages (AMs). Adult New Zealand White rabbits were mechanically ventilated for 4 h with a VT of 6 ml/kg (low) or a VT of 12 ml/kg (traditional), with each group receiving 3 cm H2O positive end-expiratory pressure with and without intratracheal lipopolysaccharide (LPS) instillation (20 mg/kg). AMs were isolated from bronchoalveolar lavage fluid taken from the whole left lung and used for bacterial killing assays. There were no significant differences in steady-state levels of nitrite or AM phagocytosis and killing of Klebsiella pneumoniae, although these values trended to be slightly higher in the traditional VT group. However, bronchoalveolar lavage fluid protein concentrations were significantly increased in traditional VT groups receiving LPS compared with animals ventilated with a low VT (1,407.8 ± 121.4 versus 934.7 ± 118.2; P < 0.001). Lung wet:dry weight ratio in the traditional VT group was increased when compared with the low VT group without LPS (7.3 ± 0.4 versus 6.1 ± 0.3, respectively; P < 0.05). Additionally, IL-8 expression was significantly greater under conditions of LPS treatment and mechanical ventilation at VT of 12 ml/kg. These results suggest that the traditional ventilator approach (12 ml/kg VT) in a model of in vivo mechanical ventilation results in lung pathology without affecting AM antibacterial function.
Keywords: acute lung injury, acute respiratory distress syndrome, alveolar macrophages, lipopolysaccharide, protective ventilation
The basic purpose of mechanical ventilation is to support patients whose respiratory systems have failed until adequate function returns. Mechanical ventilation can also relieve respiratory distress in patients for whom the work of breathing has increased. However, traditional approaches to mechanical ventilation have been demonstrated to contribute to lung injury via upregulation of local pulmonary inflammatory responses and worsened increased alveolar capillary permeability (1–5). The release of inflammatory mediators can increase lung inflammation and contribute to injury to other vital organ systems, thus contributing to or sustaining the multiple organ dysfunction syndrome (4, 6). Mechanical ventilation also induces stress by overstretching small airways and alveoli due to increased volumes in some regions of the lung, the so-called “volu-trauma.” This continuous cycle of opening and closing of alveolar groups in other regions also causes severe wall stress and surfactant depletion, leading to recruitment-derecruitment injury (7, 8). Thus, lung injury, or worsened lung injury during mechanical ventilation, is a matter of major concern.
Animal studies show that ventilation using large tidal volumes (Vt) and low positive end-expiratory pressure causes the disruption of pulmonary epithelium and endothelium, lung inflammation, atelectasis, hypoxemia, and the release of inflammatory mediators (4, 9–11). However, a large prospective study showed that mortality was reduced from 40 to 31% in patients with acute respiratory distress syndrome (ARDS) with the use of a ventilatory strategy that used low Vt and limited plateau pressure to < 30 cm H2O (12). Although it is not known why ventilation at lower Vt results in an improved outcome, possibilities include a reduction in injurious lung stretch (13, 14), less baro-trauma (15), and diminished inflammatory cytokine production (4, 16, 17).
Likewise, persons subjected to mechanical ventilation are susceptible to bacterial infections (18). Because intubation and mechanical ventilation alter first-line patient defenses, they greatly increase the risk for nosocomial bacterial pneumonia. Lipopolysaccharide (LPS), a component of the cell wall of gram-negative bacteria, is the causative agent of endotoxin shock. Severe lung injury caused by endotoxin is challenging to control or treat and often causes death. It is a major complication in the control of infections in many patients. Exposure of the lower respiratory tract to LPS by intratracheal installation of LPS is a common model of acute lung inflammation and ARDS (19–22). LPS activates alveolar macrophages (AMs) and causes neutrophils to infiltrate and damage the lungs (23). AMs are thought to be an important clearance vehicle due to their ability to phagocytize, transport, and digest inhaled particles. AMs also play a central and functional role in a variety of acute lung injury models via secretion of oxygen and nitrogen-derived free radicals and proinflammatory cytokines, such as TNF-α, IL-6, and IL-8 (24–26). Thus, it is imperative to understand the pathogenic mechanisms involved, especially as they relate to the AM role in ventilator–lung interactions. Innate immunity involving AMs is of major importance in the early response of the lung to air–space pathogens; thus, a reduction in this population with traditional ventilation strategies may increase susceptibility to infection. In fact, one study showed that the AM population and the early innate immune response of the lung were profoundly affected by mechanical ventilation (27). It is possible that mechanical ventilation initiates or enhances an inflammatory response, with release of proinflammatory cytokines, which may better activate AMs. However, O'Reilly and colleagues (28) showed that oxidative stress produced by increased levels of reactive oxygen nitrogen species impaired AM antibacterial function.
The objective of this study was to compare bactericidal function of AMs at low (6 ml/kg) and traditional (12 ml/kg) Vt in in vivo models of mechanical ventilation. To better mimic the in vivo situation, we repeated these measurements after infusion of LPS, which resulted in significant levels of acute lung injury. Our results indicate that animals treated with LPS and ventilated at traditional Vt exhibit the greatest degree of lung injury; however, the AM seems to be a noncontributory participant in provoking injury.
MATERIALS AND METHODS
All experimental work conformed to the National Institutes of Health guidelines and was approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Animal Model
New Zealand white rabbits weighing 2.5–3.0 kg were used. Rabbits were anesthetized with intravenous ketamine (10 mg/kg) administered via a marginal ear vein. Anesthesia was maintained with ketamine (10 mg/kg/h) and xylazine (3 mg/kg/h). Intermittent neuromuscular blockade was maintained with intravenous administration of pancuronium bromide (0.5 mg/kg/h). A 3.5-mm endotracheal tube was placed via tracheostomy. Ventilation was initiated with Vt at 6 ml/kg (a physiologic tidal volume) or 12 ml/kg, positive end-expiratory pressure at 3 cm H2O, with a respiratory rate to maintain a PaCO2 values between 35–45 mm Hg and FiO2 = 0.4 for 4 h. Mean arterial pressure (mm Hg), heart rate (beats per minute), and esophageal temperature (°C) were measured throughout the study protocol and recorded at 30-min intervals. In one group of rabbits, lung injury was induced by a single intratracheal instillation of LPS (20 mg/kg) (Escherichia coli, serotype 055:B5; Sigma Chemical, St. Louis, MO), which was administered ∼ 15 min after the initiation of mechanical ventilation.
Isolation of AMs
Resident AMs were obtained by bronchoalveolar lavage (BAL) of the left lung with five aliquots of 30 ml 0.9% NaCl. Cells in the BAL fluid (BALF) were pelleted (800 × g, 10 min, 4°C), washed in ammonium chloride lysing buffer to lyse red blood cells, and resuspended in Dulbecco's modified Eagle's medium (DMEM). Cell numbers were counted with a hemacytometer, and the number of different cells was assessed via cytospin preparations stained with Diff-Quik (Dade AG, Dudingen, Switzerland).
Bacterial Killing Assay
We used Klebsiella pneumoniae (American Type Culture Collection 43816, type 2). Bacterial stocks were thawed, inoculated into broth, and grown to log phase. All bacteria were washed free of growth media before infection. Internalization and intracellular killing of K. pneumoniae was monitored using a modification of the assay by Bidani and colleagues (29). In brief, 5 × 105 AMs were suspended in DMEM and infected with K. pneumoniae in a concentration ratio of 100:1 to 200:1 bacteria per cell on a shaker platform at 37°C for 1 h. The cell suspensions were chilled on ice and centrifuged (100 × g, 7 min, 4°C). The supernatants were collected, and cells were washed twice in DMEM and lysed. Quantitative bacterial culture was performed on the supernatants and cell lysates to obtain CFU in the extracellular and intracellular cell environments, respectively. CFUs were determined by enumeration after serial dilution and inoculation onto agar plates. K. pneumoniae was grown in BBL brain heart infusion broth and nutrient agar (DIFCO; Becton Dickinson) and incubated in room air at 37°C for 18–24 h. Phagocytosis was quantified as the difference in the CFUs between paired studies with and without cells. Intracellular killing of the phagocytosed bacteria was calculated as the difference in the CFUs of phagocytosed bacteria and that of cell lysate in paired studies.
Phagocytosis Assay
K. pneumoniae was labeled with 5- (and 6-) Carboxyfluorescein succinymidyl ester (Molecular Probes, Eugene, OR) as described (30) and stored at −20°C. AMs (5 × 105) were incubated with fluorescent bacteria in 0.5 ml serum-free DMEM on a shaker at 37°C for 1 h, pelleted by centrifugation at 180 × g, and washed in ice-cold PBS with 0.02% EDTA to stop phagocytosis and remove extracellular bacteria. Cytospin preparations were made from cells, fixed with 3% formaldehyde, permeabilized with 0.1% Triton X-100 in PBS, and stained with AlexaFluor 594 phalloidin (Molecular Probes) diluted 1:40 in 1% BSA. Sections were mounted in VectaShield containing 4,6-diamidino-2-phenylindole (Vector Laboratories, Burlingame, CA). Imaging was performed on a Leica DMIRBE inverted epifluorescence/Nomarski microscope outfitted with Leica TCS NT Laser Confocal optics (Leica Inc., Exton, PA). This method allowed us to distinguish intracellular from outer-membrane–adhered pathogens.
Lung Injury Indices
The right lower lobe was used for determination of lung wet-to-dry weight ratio. Lung segments were weighed before and after drying in an oven at 120°C for 2 wk to calculate the wet-to-dry weight ratios. Nitric oxide (NO) concentration in the BAL was quantified by measuring nitrite 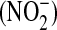 , a stable oxidation product of NO, as previously described (30). Nitrate
, a stable oxidation product of NO, as previously described (30). Nitrate 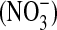 was converted to
was converted to 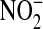 with Escherichia coli reductase, and 100 μl of sample was incubated with 25 μl of freshly prepared 2,3-diaminonaphthalene (0.05 mg/ml in 0.62 M HCl) and incubated for 10 min. The reaction was stopped by the addition of 25 μl of 5.6 N NaOH, and the signal was measured using a fluorescent plate reader with excitation at 360 nm and emission at 450 nm.
with Escherichia coli reductase, and 100 μl of sample was incubated with 25 μl of freshly prepared 2,3-diaminonaphthalene (0.05 mg/ml in 0.62 M HCl) and incubated for 10 min. The reaction was stopped by the addition of 25 μl of 5.6 N NaOH, and the signal was measured using a fluorescent plate reader with excitation at 360 nm and emission at 450 nm. 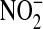 concentrations were determined using a NaNO2 standard. Total protein concentration in the BAL was measured using the bicinchoninic acid (BCA) assay (Pierce Biotechnology, Inc., Rockford, IL).
concentrations were determined using a NaNO2 standard. Total protein concentration in the BAL was measured using the bicinchoninic acid (BCA) assay (Pierce Biotechnology, Inc., Rockford, IL).
Measurement of TNF-α
A 100-mg section of the upper right lung lobe was homogenized in Hanks' balanced salt solution. Total protein concentration was measured using the BCA protein assay. TNF-α expression was measured using a sandwich ELISA. Each well of a 96-well plate was coated with purified polyclonal goat anti-rabbit TNF-α (1:250 dilution; BD Biosciences, Mountain View, CA). Protein from homogenized lungs (1 mg/ml) was added into antibody-coated wells. Wells were washed, and biotinylated monoclonal mouse anti-rabbit TNF-α (1:5,000 dilution; Amersham Biosciences, Piscataway, NJ) was added. After extensive washing, avidin-HRP was added to detect the “antibody-antigen-antibody sandwich.” Absorbance was read at 450 nm, and results are expressed as percentage of change.
Immunohistology
Paraffin-embedded lung tissue sections 5-μm in thickness were stained for IL-8 as previously described (31). In preparation for immunohistochemistry, sections were dewaxed and rehydrated through graded alcohols to a final wash in Tris-buffered saline (TBS). Sections were treated with 0.3% hydrogen peroxide for 10 min to block endogenous peroxidase activity and washed with TBS containing 0.1% Triton X-100 and TBS. Nonspecific protein binding was blocked with 5% normal donkey serum for 2 h at room temperature. Sections were incubated with a goat polyclonal IL-8 antibody (1:40 dilution) (Santa Cruz Biotechnology, Inc., Santa Cruz, CA) overnight at 4°C. Control sections were treated in parallel but incubated with normal goat serum (as a negative control) instead of the primary antibody. All sections were incubated in a moist chamber. Sections were incubated with biotin-conjugated donkey anti-goat IgG (1:200 dilution) (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA) for 30 min. After 30 min of incubation in an avidin-biotin complex, the reaction product was visualized by 3,3′-diaminobenzidine (DAKO Corp., Carpinteria, CA). Finally, sections were dehydrated and cleared in ethanol and xylene and mounted in Permount (Fisher Scientific, Pittsburgh, PA). Representative photomicrographs were acquired via a Nikon Diaphot microscope with bright-field optics and a Nikon Coolpix 950 digital color camera (Nikon, Tokyo, Japan).
ELISA Measurement of IL-8
Concentrations of IL-8 in the BALF were measured using ELISA. Total protein concentration of BALF was measured using the BCA protein assay. Each well of a 96-well plate was coated with 1 ml diluted BALF fluid, which is 1 mg/ml. Blocking buffer (PBS containing 1% BSA and 0.02% azide) was added to block nonspecific protein binding. These wells were incubated with goat anti-human IL-8 antibody (Santa Cruz Biotechnology, Santa Cruz, CA). The concentrations of IL-8 were visualized with a horseradish peroxidase-conjugated secondary antibody followed by the addition of 3,3′,5,5′-tetramethylbenzidine peroxidase substrate. The acid stop solution was added after incubated for 15 min at room temperature, and the expression was evaluated by the reading at 450 nm.
Statistical Analysis
The data are presented as means ± SEM. Data were analyzed by one-way ANOVA followed by Tukey-Kramer's multiple group comparison of the means for parametric data and by the Kruskal-Wallis ANOVA and Dunn's multiple group comparison of the means for nonparametric data. A P value of ⩽ 0.05 was considered statistically significant.
RESULTS
Indices of Lung Injury
The ratio between PaO2 and FiO2 was used as a measure of lung injury. A decrease in this ratio to < 300 signified the onset of lung injury (Figure 1). Lung wet-to-dry weight ratios, BALF protein, and steady-state levels of nitrite were used to express indices of inflammatory-mediated lung injury. Lung wet-to-dry weight ratio in the traditional Vt group was increased when compared with the low Vt group without LPS (7.3 ± 0.4 versus 6.1 ± 0.3, respectively; P < 0.05) (Figure 2A). LPS administration significantly increased lung wet-to-dry weight ratios in the low Vt and traditional Vt groups compared with untreated lungs (9.1 ± 0.9 versus 6.1 ± 0.3 and 10.1 ± 0.9 versus 7.3 ± 0.4, respectively; P < 0.05) (Figure 2A). Protein concentration was significantly greater in the LPS-treated low Vt and traditional Vt groups (934.7 ± 118.2 versus 227.1 ± 45.4 and 1,407.8 ± 121.4 versus 199.8 ± 28.8, respectively; P < 0.001) (Figure 2B). There were no differences in protein concentration between low and traditional Vt groups without LPS; however, there was a significant increase in the LPS-treated traditional Vt group compared with the LPS-treated low Vt group (1,407.8 ± 121.4 versus 934.7 ± 118.2; P < 0.05) (Figure 2B). Steady-state levels of nitrate were significantly increased in the traditional Vt group after LPS treatment when compared with the untreated group (13.5 ± 2.5 versus 0.9 ± 0.3; P < 0.001) (Figure 2C). There was also a significant difference in steady-state levels of nitrate between LPS-treated traditional and low Vt groups (13.5 ± 2.5 versus 6.6 ± 1.1; P < 0.05) (Figure 2C).
Figure 1.

PaO2/FiO2 values for mechanically ventilated rabbits. Mean arterial pressure (mm Hg) was measured and recorded at 30-min intervals. Data from each treatment group represents the average of five experiments ± SEM. Vt 6 = tidal volume 6 ml/kg; Vt 12 = tidal volume 12 ml/kg. Solid triangles, Vt 6; open triangles, Vt 12; solid squares, Vt 6 + LPS; open squares, Vt 12 + LPS.
Figure 2.



Lung wet-to-dry weight ratios (A), BALF protein (B), and nitric oxide (C) content from ventilated rabbits untreated or treated with LPS. Data are mean values ± SEM. For each condition, n = 5. *P < 0.05; ***P < 0.001.
Phagocytosis and Intracellular Killing of K. pneumoniae
Phagocytosis and intracellular killing of K. pneumoniae was observed by use of quantitative bacterial culture as previously described (5). There were no significant differences in phagocytosis and killing between low and traditional Vt groups (Figures 3A and 3B). Phagocytosis was significantly increased only in the LPS-treated low Vt group (P < 0.001) (Figure 3A); however, killing was significantly increased in the low and high Vt LPS-treated groups (P < 0.01 and P < 0.001) (Figure 3B). LPS treatment resulted in increased percentage of killing in low and traditional Vt groups when compared with untreated controls (73.5% versus 33.2% and 70.8% versus 43.7%, respectively). Treatment with LPS resulted in an influx of neutrophils into the alveolar space, thereby increasing the BALF neutrophil cell count. We assessed intracellular bacterial killing in AM and neutrophil cell populations isolated from BALF of LPS-treated animals. There was no significant difference in percentage killing between these two cell types (Figure 4).
Figure 3.


Effects of mechanical ventilation on phagocytosis (A) and intracellular killing (B) of K. pneumoniae by AMs over 1 h. Data are mean values ± SEM. Data represent the average of five experiments. **P < 0.01; ***P < 0.001. Solid bars, no LPS; shaded bars, 20 mg/kg LPS.
Figure 4.

Bacterial killing in AMs (solid bars) and neutrophils (shaded bars) isolated from LPS-treated mechanically ventilated rabbits. AMs and neutrophils were isolated from the BALF, and cell populations were separated using Ficoll-Paque PLUS (Amersham Biosciences) according to the manufacturer's protocol. Bacterial phagocytosis and killing were assessed in each population.
Immunofluorescent detection of bacterial phagocytosis corroborated our findings in the quantitative bacterial culture assays. Cells were infected with fluorescein-labeled Klebsiella in the presence or absence of 10 μM cytochalasin D. LPS treatment increased phagocytosis of fluorescein-labeled bacteria compared with controls (Figures 5A and 5B), whereas cytochalasin D treatment decreased phagocytosis (data not shown).
Figure 5.

Immunofluorescent analysis of phagocytosis of Klebsiella in cells isolated from mechanically ventilated rabbits. Cells were infected with fluorescein-labeled K. pneumoniae in the presence or absence of 10 μM cytochalasin D for 1 h at 37°C with gentle shaking. Original magnification: ×100. Cells are from rabbits ventilated at 6 ml/kg (A, B) or 12 ml/kg (C, D). B and D represent LPS treatment.
ELISA for TNF-α
TNF-α is one of several inflammatory cytokines that have been shown to be crucial to antibacterial host defense in gram-negative infection. There were no significant differences between ventilatory strategies with regard to TNF-α expression. However, significantly higher TNF-α levels were seen after intratracheal instillation of LPS (P < 0.001) (Figure 6). In both Vt groups, ventilation after intratracheal LPS was associated with TNF-α levels five times greater than those seen in the untreated control groups.
Figure 6.

TNF-α levels in lung tissue of mechanically ventilated rabbits. For each condition, n = 5. ***P < 0.001.
IL-8 Immunohistochemistry
Lungs were analyzed by immunohistochemistry for expression of IL-8. IL-8 staining of bronchoalveolar segments (Figure 7) was noticeably increased in animals receiving LPS. Staining was greatest in animals ventilated with traditional Vt when compared with low Vt.
Figure 7.

Immunohistochemistry of IL-8 in bronchopulmonary segments of mechanically ventilated rabbits. Paraffin-embedded lung sections from rabbits ventilated at 6 ml/kg (A, B) or 12 ml/kg (C, D) were stained for IL-8. Lungs were untreated (A, C) or treated (B, D) with LPS. Original magnification: ×20.
IL-8 ELISA
Lung BALF was analyzed for IL-8 expression after exposing the animals to 6 h of the experimental conditions described. IL-8 expression was significantly increased under stimulation of LPS treatment but further increased when exposed to Vt of 12 ml/kg compared with 6 ml/kg (1,200 ± 130 versus 980 ± 20, respectively; P < 0.05) (Figure 8). Both increases in Vt and stimulation with LPS increased IL-8 expression.
Figure 8.

ELISA of IL-8 in the BALF of mechanically ventilated rabbits. BALF collected from rabbits ventilated under the conditions described. Data are means ± SEM (n = 5). *P < 0.05.
DISCUSSION
The major goal of this study was to determine whether “protective” (6 ml/kg Vt) and “injurious” (12 ml/kg Vt) ventilation strategies would differ in their impact on bactericidal function of AMs. Studies show that ventilation with tidal volumes lower than those used conventionally reduces the relative risk of mortality (12, 14, 32). However, the mechanisms associated with the protective effect are not fully understood; thus, information gained shedding light on contributing variables or noncontributors are important to the critical care physician. Potential mechanisms include decreased production of proinflammatory cytokines (1, 3, 4, 12), decreased disruption of the alveolar-capillary barrier (11, 33, 34), and reduction in injurious lung stretch (13, 14). The results of our study show that animals treated with LPS and ventilated with traditional Vt displayed the greatest degree of lung injury regarding increased wet-to-dry weight ratios, BALF protein, and steady-state levels of nitrite. Likewise, other studies show that LPS treatment causes an increase in lung permeability (35–38), BALF protein (4, 17, 36, 37, 39), and NO concentration (35, 36, 39). In our study, although the wet-to-dry weight ratios were greatest in the “injurious” Vt group receiving LPS, they were not significantly greater than the “protective” group treated with LPS, probably due to the variability, the low number of animals studied, and the short duration of ventilation. A study by Peng and colleagues (40) showed that inducible nitric oxide synthase gene expression and activity are significantly upregulated by high Vt ventilation, are accompanied by nitrotyrosine deposition (mainly in capillary endothelial cells), and correlate with increased lung capillary permeability. However, the animals used in this study were not treated with LPS. Because NO (the product of inducible nitric oxide synthase induction) is involved in host defense, mechanical ventilation may stimulate or decrease innate immunity.
Effective host defense against lung bacterial infection is primarily dependent on the rapid clearance of the organism from the respiratory tract. Early clearance is mediated by neutrophils and macrophages, which must be recruited and activated at the site of infection (41). Functional impairment or reduction in this population of cells increases host susceptibility to lung infection (42, 43). Several studies have shown that injurious ventilation results in a marked reduction in immune functioning (44–49). In our study, there were no significant differences in bacterial phagocytosis and killing between the low and traditional Vt groups, although both parameters were slightly increased in the traditional Vt group. In fact, although LPS treatment significantly increased phagocytosis in the low Vt group and killing in low and high Vt groups, there was no difference between low and traditional Vt. Other studies involving ex vivo phagocytosis after LPS treatment show significant increases in AM uptake of degraded lipids (50) and apoptotic neutrophils (51). In our study, it is possible that the increased phagocytosis due to LPS treatment is a consequence of ventilation alone because previous studies reported that LPS had no effect on phagocytosis by AM (52, 53). Moreover, Brackenbury and colleagues (54) recently demonstrated that “low-stretch” mechanical ventilation alone did not significantly alter the pulmonary response to a bacterial challenge within a normal host over a 4 h time course.
Evidence suggests that mechanical ventilation, especially of injured lungs, causes the release of inflammatory mediators, which may pass into the circulation (4, 55, 56). Leukocyte emigration and AM-derived cytokines may contribute to lung microvascular injury associated with ARDS. Pugin and colleagues (26), using an in vitro model of mechanical ventilation of cell monolayers, identified the lung macrophage as the likely source for the secretion of proinflammatory mediators (e.g., TNF-α, IL-8, IL-6, and MMP-9) in response to mechanical ventilation. We measured TNF-α and IL-8 levels because of published data suggesting an association between mechanical stretch and sepsis (4, 26). TNF-α plays an important role in innate immunity. It is believed to enhance the release of chemokines and cytokines, increase lung vascular permeability, and modulate the eventual recruitment and activation of neutrophils (57, 58). IL-8, a potent neutrophil chemoattractant, was chosen as a relevant mediator because a sign of ventilator-induced lung injury is the lung recruitment of neutrophils and neutrophil-mediated tissue injury (59, 60). In our study, TNF-α and IL-8 expression measured in lung BALF and tissues, respectively, were significantly increased in animals treated with LPS, and increases in animals mechanically ventilated with “traditional” Vt were observed. It is possible that conventional or higher-volume mechanical ventilation does not cause a significant release of proinflammatory cytokines. Numerous studies involving proinflammatory cytokine production after high-volume mechanical ventilation have been conducted. Our results, like the results of these studies, are contradictory but continue to add to the knowledge base of this complex subject matter. Some investigators found increased concentrations of proinflammatory cytokines in lungs subjected to moderate to high-volume ventilation (1, 4, 26, 56, 61, 62), whereas others saw no effect on TNF-α production (17, 26, 62, 63). Clinical studies show that the lower tidal volume ventilation strategy is associated with a greater decrease in plasma cytokine levels and morbidity and mortality in patients with acute lung injury and ARDS (3, 12, 16, 32).
There are a few limitations associated with the present study. In our rabbit model of acute lung injury, LPS may already be a maximum stimulus for the AMs; therefore, the addition of mechanical ventilation may not result in any additional effect on bactericidal activity. Because the administration of LPS alone does not completely mimic the systemic or pulmonary effects of bacteremia or endotoxemia, use of another model of acute lung injury that is not dependent on LPS, such as cecal ligation and puncture (64), may show differences in AM bactericidal activity. Previously, Cardozo and colleagues (65) demonstrated increased phagocytosis by AMs 12 h after intravenous injection of LPS. No change was observed at 4 h. However, they used intravenous instead of intratracheal LPS and used a different animal species (rats). Because the rabbits in our study were ventilated for only 4 h, it may be possible that any effect of ventilation on phagocytosis did not manifest. However, intratracheal delivery of LPS generally upregulates pulmonary inflammatory responses in a more timely manner.
In conclusion, in this animal model of LPS-induced lung injury, animals ventilated with traditional Vt exhibited the greatest degree of lung injury. Although the addition of LPS enhanced injury between the tidal volumes prospectively tested, the AM seemed to be a noncontributory participant in provoking injury. Future investigations should be encouraged to reconfirm this observation and to elucidate additional possible mechanisms by which lower tidal volumes lead to reduced patient mortality.
Acknowledgments
The authors thank Albert Tousson at the High Resolution Imaging Facility located at the University of Alabama at Birmingham for his excellent assistance in helping in the imaging of cells and lung tissue sections. The authors also thank Dr. Y- C.T. Huang of Duke University Medical Center for kindly providing the IL-8 antibody methodology.
N.G.H. and Y.L. contributed equally to this work as first authors; S.M. and J.D.L. contributed equally as senior authors.
This work was supported by National Institutes of Health Grants HL31197, HL51173 (S.M.), and HL067982 (J.L.). N.G.H. was supported by Lung Training Grant.
Originally Published in Press as DOI: 10.1165/rcmb.2005-0463OC on February 10, 2006
Conflict of Interest Statement: None of the authors has a financial relationship with a commercial entity that has an interest in the subject of this manuscript.
References
- 1.Chiumello D, Pristine G, Slutsky AS. Mechanical ventilation affects local and systemic cytokines in an animal model of acute respiratory distress syndrome. Am J Respir Crit Care Med 1999;160:109–116. [DOI] [PubMed] [Google Scholar]
- 2.Haitsma JJ, Uhlig S, Goggel R, Verbrugge SJ, Lachmann U, Lachmann B. Ventilator-induced lung injury leads to loss of alveolar and systemic compartmentalization of tumor necrosis factor-alpha. Intensive Care Med 2000;26:1515–1522. [DOI] [PubMed] [Google Scholar]
- 3.Ranieri VM, Suter PM, Tortorella C, De TR, Dayer JM, Brienza A, Bruno F, Slutsky AS. Effect of mechanical ventilation on inflammatory mediators in patients with acute respiratory distress syndrome: a randomized controlled trial. JAMA 1999;282:54–61. [DOI] [PubMed] [Google Scholar]
- 4.Tremblay L, Valenza F, Ribeiro SP, Li J, Slutsky AS. Injurious ventilatory strategies increase cytokines and c-fos m-RNA expression in an isolated rat lung model. J Clin Invest 1997;99:944–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wrigge H, Zinserling J, Stuber F, von Spiegel T, Hering R, Wetegrove S, Hoeft A, Putensen C. Effects of mechanical ventilation on release of cytokines into systemic circulation in patients with normal pulmonary function. Anesthesiology 2000;93:1413–1417. [DOI] [PubMed] [Google Scholar]
- 6.Slutsky AS, Tremblay LN. Multiple system organ failure: is mechanical ventilation a contributing factor? Am J Respir Crit Care Med 1998;157:1721–1725. [DOI] [PubMed] [Google Scholar]
- 7.Marini JJ. New options for the ventilatory management of acute lung injury. New Horiz 1993;1:489–503. [PubMed] [Google Scholar]
- 8.Mead J, Takishima T, Leith D. Stress distribution in lungs: a model of pulmonary elasticity. J Appl Physiol 1970;28:596–608. [DOI] [PubMed] [Google Scholar]
- 9.Parker JC, Hernandez LA, Peevy KJ. Mechanisms of ventilator-induced lung injury. Crit Care Med 1993;21:131–143. [DOI] [PubMed] [Google Scholar]
- 10.Tsuno K, Miura K, Takeya M, Kolobow T, Morioka T. Histopathologic pulmonary changes from mechanical ventilation at high peak airway pressures. Am Rev Respir Dis 1991;143:1115–1120. [DOI] [PubMed] [Google Scholar]
- 11.Webb HH, Tierney DF. Experimental pulmonary edema due to intermittent positive pressure ventilation with high inflation pressures: protection by positive end-expiratory pressure. Am Rev Respir Dis 1974;110:556–565. [DOI] [PubMed] [Google Scholar]
- 12.Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. The Acute Respiratory Distress Syndrome Network. N Engl J Med 2000;342:1301–1308. [DOI] [PubMed] [Google Scholar]
- 13.Hickling KG, Henderson SJ, Jackson R. Low mortality associated with low volume pressure limited ventilation with permissive hypercapnia in severe adult respiratory distress syndrome. Intensive Care Med 1990;16:372–377. [DOI] [PubMed] [Google Scholar]
- 14.Hickling KG, Walsh J, Henderson S, Jackson R. Low mortality rate in adult respiratory distress syndrome using low-volume, pressure-limited ventilation with permissive hypercapnia: a prospective study. Crit Care Med 1994;22:1568–1578. [DOI] [PubMed] [Google Scholar]
- 15.Dos Santos CC, Slutsky AS. Invited review: mechanisms of ventilator-induced lung injury: a perspective. J Appl Physiol 2000;89:1645–1655. [DOI] [PubMed] [Google Scholar]
- 16.Parsons PE, Eisner MD, Thompson BT, Matthay MA, Ancukiewicz M, Bernard GR, Wheeler AP. Lower tidal volume ventilation and plasma cytokine markers of inflammation in patients with acute lung injury. Crit Care Med 2005;33:1–6. [DOI] [PubMed] [Google Scholar]
- 17.Ricard JD, Dreyfuss D, Saumon G. Production of inflammatory cytokines in ventilator-induced lung injury: a reappraisal. Am J Respir Crit Care Med 2001;163:1176–1180. [DOI] [PubMed] [Google Scholar]
- 18.Chastre J, Fagon JY. Ventilator-associated pneumonia. Am J Respir Crit Care Med 2002;165:867–903. [DOI] [PubMed] [Google Scholar]
- 19.Finsnes F, Skjonsberg OH, Lyberg T, Christensen G. Endothelin-1 production is associated with eosinophilic rather than neutrophilic airway inflammation. Eur Respir J 2000;15:743–750. [DOI] [PubMed] [Google Scholar]
- 20.Schmal H, Shanley TP, Jones ML, Friedl HP, Ward PA. Role for macrophage inflammatory protein-2 in lipopolysaccharide-induced lung injury in rats. J Immunol 1996;156:1963–1972. [PubMed] [Google Scholar]
- 21.Su X, Song Y, Jiang J, Bai C. The role of aquaporin-1 (AQP1) expression in a murine model of lipopolysaccharide-induced acute lung injury. Respir Physiol Neurobiol 2004;142:1–11. [DOI] [PubMed] [Google Scholar]
- 22.van Helden HP, Kuijpers WC, Langerwerf PE, Langen RC, Haagsman HP, Bruijnzeel PL. Efficacy of Curosurf in a rat model of acute respiratory distress syndrome. Eur Respir J 1998;12:533–539. [DOI] [PubMed] [Google Scholar]
- 23.Blackwell TS, Lancaster LH, Blackwell TR, Venkatakrishnan A, Christman JW. Chemotactic gradients predict neutrophilic alveolitis in endotoxin-treated rats. Am J Respir Crit Care Med 1999;159:1644–1652. [DOI] [PubMed] [Google Scholar]
- 24.Fujii Y, Goldberg P, Hussain SN. Contribution of macrophages to pulmonary nitric oxide production in septic shock. Am J Respir Crit Care Med 1998;157:1645–1651. [DOI] [PubMed] [Google Scholar]
- 25.Lentsch AB, Czermak BJ, Bless NM, Van RN, Ward PA. Essential role of alveolar macrophages in intrapulmonary activation of NF-kappaB. Am J Respir Cell Mol Biol 1999;20:692–698. [DOI] [PubMed] [Google Scholar]
- 26.Pugin J, Dunn I, Jolliet P, Tassaux D, Magnenat JL, Nicod LP, Chevrolet JC. Activation of human macrophages by mechanical ventilation in vitro. Am J Physiol 1998;275:L1040–L1050. [DOI] [PubMed] [Google Scholar]
- 27.Whitehead TC, Zhang H, Mullen B, Slutsky AS. Effect of mechanical ventilation on cytokine response to intratracheal lipopolysaccharide. Anesthesiology 2004;101:52–58. [DOI] [PubMed] [Google Scholar]
- 28.O'Reilly PJ, Hickman-Davis JM, Davis IC, Matalon S. Hyperoxia impairs antibacterial function of macrophages through effects on actin. Am J Respir Cell Mol Biol 2003;28:443–450. [DOI] [PubMed] [Google Scholar]
- 29.Bidani A, Reisner BS, Haque AK, Wen J, Helmer RE, Tuazon DM, Heming TA. Bactericidal activity of alveolar macrophages is suppressed by V-ATPase inhibition. Lung 2000;178:91–104. [DOI] [PubMed] [Google Scholar]
- 30.Hickman-Davis JM, O'Reilly P, Davis IC, Peti-Peterdi J, Davis G, Young KR, Devlin RB, Matalon S. Killing of Klebsiella pneumoniae by human alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2002;282:L944–L956. [DOI] [PubMed] [Google Scholar]
- 31.Kotani M, Kotani T, Li Z, Silbajoris R, Piantadosi CA, Huang YC. Reduced inspiratory flow attenuates IL-8 release and MAPK activation of lung overstretch. Eur Respir J 2004;24:238–246. [DOI] [PubMed] [Google Scholar]
- 32.Amato MB, Barbas CS, Medeiros DM, Magaldi RB, Schettino GP, Lorenzi-Filho G, Kairalla RA, Deheinzelin D, Munoz C, Oliveira R, et al. Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N Engl J Med 1998;338:347–354. [DOI] [PubMed] [Google Scholar]
- 33.Dreyfuss D, Soler P, Basset G, Saumon G. High inflation pressure pulmonary edema: respective effects of high airway pressure, high tidal volume, and positive end-expiratory pressure. Am Rev Respir Dis 1988;137:1159–1164. [DOI] [PubMed] [Google Scholar]
- 34.Parker JC, Ivey CL. Isoproterenol attenuates high vascular pressure-induced permeability increases in isolated rat lungs. J Appl Physiol 1997;83:1962–1967. [DOI] [PubMed] [Google Scholar]
- 35.Lang JD, Figueroa M, Sanders KD, Aslan M, Liu Y, Chumley P, Freeman BA. Hypercapnia via reduced rate and tidal volume contributes to lipopolysaccharide-induced lung injury. Am J Respir Crit Care Med 2005;171:147–157. [DOI] [PubMed] [Google Scholar]
- 36.Liaudet L, Pacher P, Mabley JG, Virag L, Soriano FG, Hasko G, Szabo C. Activation of poly(ADP-Ribose) polymerase-1 is a central mechanism of lipopolysaccharide-induced acute lung inflammation. Am J Respir Crit Care Med 2002;165:372–377. [DOI] [PubMed] [Google Scholar]
- 37.Sato K, Kadiiska MB, Ghio AJ, Corbett J, Fann YC, Holland SM, Thurman RG, Mason RP. In vivo lipid-derived free radical formation by NADPH oxidase in acute lung injury induced by lipopolysaccharide: a model for ARDS. FASEB J 2002;16:1713–1720. [DOI] [PubMed] [Google Scholar]
- 38.Yamamoto T, Kajikawa O, Martin TR, Sharar SR, Harlan JM, Winn RK. The role of leukocyte emigration and IL-8 on the development of lipopolysaccharide-induced lung injury in rabbits. J Immunol 1998;161:5704–5709. [PubMed] [Google Scholar]
- 39.Liaudet L, Mabley JG, Pacher P, Virag L, Soriano FG, Marton A, Hasko G, Deitch EA, Szabo C. Inosine exerts a broad range of antiinflammatory effects in a murine model of acute lung injury. Ann Surg 2002;235:568–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Peng X, Abdulnour RE, Sammani S, Ma SF, Han EJ, Hasan EJ, Tuder R, Garcia JG, Hassoun PM. Inducible nitric oxide synthase contributes to ventilator-induced lung injury. Am J Respir Crit Care Med 2005;172:470–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Toews GB, Gross GN, Pierce AK. The relationship of inoculum size to lung bacterial clearance and phagocytic cell response in mice. Am Rev Respir Dis 1979;120:559–566. [DOI] [PubMed] [Google Scholar]
- 42.Imanaka H, Shimaoka M, Matsuura N, Nishimura M, Ohta N, Kiyono H. Ventilator-induced lung injury is associated with neutrophil infiltration, macrophage activation, and TGF-beta 1 mRNA upregulation in rat lungs. Anesth Analg 2001;92:428–436. [DOI] [PubMed] [Google Scholar]
- 43.Nelson S, Summer WR. Innate immunity, cytokines, and pulmonary host defense. Infect Dis Clin North Am 1998;12:555–567. (vii.). [DOI] [PubMed] [Google Scholar]
- 44.Baleeiro CE, Wilcoxen SE, Morris SB, Standiford TJ, Paine R III. Sublethal hyperoxia impairs pulmonary innate immunity. J Immunol 2003;171:955–963. [DOI] [PubMed] [Google Scholar]
- 45.Nahum A, Hoyt J, Schmitz L, Moody J, Shapiro R, Marini JJ. Effect of mechanical ventilation strategy on dissemination of intratracheally instilled Escherichia coli in dogs. Crit Care Med 1997;25:1733–1743. [DOI] [PubMed] [Google Scholar]
- 46.Savel RH, Yao EC, Gropper MA. Protective effects of low tidal volume ventilation in a rabbit model of Pseudomonas aeruginosa-induced acute lung injury. Crit Care Med 2001;29:392–398. [DOI] [PubMed] [Google Scholar]
- 47.Tateda K, Deng JC, Moore TA, Newstead MW, Paine R III, Kobayashi N, Yamaguchi K, Standiford TJ. Hyperoxia mediates acute lung injury and increased lethality in murine Legionella pneumonia: the role of apoptosis. J Immunol 2003;170:4209–4216. [DOI] [PubMed] [Google Scholar]
- 48.Verbrugge SJC, Sorm V, van't Veen A, Mouton J, Gommers D, Lachmann B. Lung overinflation without positive end-expiratory pressure promotes bacteremia after experimental Klebsiella pneumoniae inoculation. Intensive Care Med. 1998;24:172–177. [DOI] [PubMed] [Google Scholar]
- 49.Vreugdenhil HA, Heijnen CJ, Plotz FB, Zijlstra J, Jansen NJ, Haitsma JJ, Lachmann B, van Vught AJ. Mechanical ventilation of healthy rats suppresses peripheral immune function. Eur Respir J 2004;23:122–128. [DOI] [PubMed] [Google Scholar]
- 50.Quintero OA, Wright JR. Clearance of surfactant lipids by neutrophils and macrophages isolated from the acutely inflamed lung. Am J Physiol Lung Cell Mol Physiol 2002;282:L330–L339. [DOI] [PubMed] [Google Scholar]
- 51.Reidy MF, Wright JR. Surfactant protein A enhances apoptotic cell uptake and TGF-beta1 release by inflammatory alveolar macrophages. Am J Physiol Lung Cell Mol Physiol 2003;285:L854–L861. [DOI] [PubMed] [Google Scholar]
- 52.Frevert CW, Warner AE, Weller E, Brain JD. The effect of endotoxin on in vivo rat alveolar macrophage phagocytosis. Exp Lung Res 1998;24:745–758. [DOI] [PubMed] [Google Scholar]
- 53.Wizemann TM, Laskin DL. Enhanced phagocytosis, chemotaxis, and production of reactive oxygen intermediates by interstitial lung macrophages following acute endotoxemia. Am J Respir Cell Mol Biol 1994;11:358–365. [DOI] [PubMed] [Google Scholar]
- 54.Brackenbury AM, McCaig LA, Yao LJ, Veldhuizen RA, Lewis JF. Host response to intratracheally instilled bacteria in ventilated and nonventilated rats. Crit Care Med 2004;32:2502–2507. [DOI] [PubMed] [Google Scholar]
- 55.Ranieri VM, Giunta F, Suter PM, Slutsky AS. Mechanical ventilation as a mediator of multisystem organ failure in acute respiratory distress syndrome. JAMA 2000;284:43–44. [DOI] [PubMed] [Google Scholar]
- 56.von Bethmann AN, Brasch F, Nusing R, Vogt K, Volk HD, Muller KM, Wendel A, Uhlig S. Hyperventilation induces release of cytokines from perfused mouse lung. Am J Respir Crit Care Med 1998;157:263–272. [DOI] [PubMed] [Google Scholar]
- 57.Roach DR, Bean AG, Demangel C, France MP, Briscoe H, Britton WJ. TNF regulates chemokine induction essential for cell recruitment, granuloma formation, and clearance of mycobacterial infection. J Immunol 2002;168:4620–4627. [DOI] [PubMed] [Google Scholar]
- 58.Strieter RM, Kunkel SL. Acute lung injury: the role of cytokines in the elicitation of neutrophils. J Investig Med 1994;42:640–651. [PubMed] [Google Scholar]
- 59.Baggiolini M, Dewald B, Moser B. Interleukin-8 and related chemotactic cytokines: CXC and CC chemokines. Adv Immunol 1994;55:97–179. [PubMed] [Google Scholar]
- 60.Imai Y, Kawano T, Miyasaka K, Takata M, Imai T, Okuyama K. Inflammatory chemical mediators during conventional ventilation and during high frequency oscillatory ventilation. Am J Respir Crit Care Med 1994;150:1550–1554. [DOI] [PubMed] [Google Scholar]
- 61.Takata M, Abe J, Tanaka H, Kitano Y, Doi S, Kohsaka T, Miyasaka K. Intraalveolar expression of tumor necrosis factor-alpha gene during conventional and high-frequency ventilation. Am J Respir Crit Care Med 1997;156:272–279. [DOI] [PubMed] [Google Scholar]
- 62.Vlahakis NE, Schroeder MA, Limper AH, Hubmayr RD. Stretch induces cytokine release by alveolar epithelial cells in vitro. Am J Physiol 1999;277:L167–L173. [DOI] [PubMed] [Google Scholar]
- 63.Verbrugge SJ, Uhlig S, Neggers SJ, Martin C, Held HD, Haitsma JJ, Lachmann B. Different ventilation strategies affect lung function but do not increase tumor necrosis factor-alpha and prostacyclin production in lavaged rat lungs in vivo. Anesthesiology 1999;91:1834–1843. [DOI] [PubMed] [Google Scholar]
- 64.Chinnaiyan AM, Huber-Lang M, Kumar-Sinha C, Barrette TR, Shankar-Sinha S, Sarma VJ, Padgaonkar VA, Ward PA. Molecular signatures of sepsis: multiorgan gene expression profiles of systemic inflammation. Am J Pathol 2001;159:1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Cardozo C, Edelman J, Lesser M. Lipopolysaccharide-induced stimulation of alveolar macrophage opsonin-independent phagocytosis. J Surg Res 1992;53:170–174. [DOI] [PubMed] [Google Scholar]