

Summary
S100A4 (metastasin) is a member of the S100 family of calcium-binding proteins that is directly involved in tumorgenesis. Until recently, the only structural information available was the solution NMR structure of the inactive, calcium-free form of the protein. Here we report the crystal structure of human S100A4 in the active, calcium-bound state at 2.03 Å resolution that was solved by molecular replacement in the space group P65 with two molecules in the asymmetric unit from perfectly merohedrally twinned crystals. The Ca2+-bound S100A4 structure reveals a large conformational change in the three-dimensional structure of the dimeric S100A4 protein upon calcium binding. This calcium-dependent conformational change opens up a hydrophobic binding pocket that is capable of binding to target proteins such as annexin A2, the p53 tumor suppressor protein, and myosin IIA. The structure of the active form of S100A4 provides insight into its interactions with its binding partners and a better understanding on its role in metastasis.
Keywords: S100, metastasis, merohedral twinning, angiogenesis, calcium-binding
Introduction
The family of S100 proteins belongs to the largest subgroup of EF-hand calcium-binding proteins and has been described as having either intracellular or extracellular function, or both.1 Currently 21 S100 family members have been identified in humans and are expressed in a cell-specific manner suggesting that each S100 protein has a different function and role.2; 3; 4 Some of these functions include cell differentiation, cell growth, cell-cycle regulation and the assembly/disassembly of cytoskeletal proteins.5; 6 Another feature of S100 proteins is their association with different stages and types of cancers. S100A4, S100A6, S100A7 and S100B have all been shown to be over-expressed in cancer cells.7 Although the exact role of S100 proteins in the development of cancer is unclear, it appears that the calcium-dependent conformational change of S100 proteins is important for forming protein interactions with targets involved in cancer progression.8
S100 proteins, named based on the observation that they are soluble in 100% saturated ammonium sulfate, are low molecular weight (10–12 kDa), acidic proteins that normally exist as symmetric homodimers stabilized by noncovalent interactions.9 Each S100 monomer is composed of two EF-hand calcium-binding domains (helix-loop-helix motif).10 The N-terminal EF-hand (also known as the S100 hand, pseudo EF-hand or half EF-hand) encompasses 14 amino acids and coordinates calcium weakly via backbone carbonyl oxygen atoms, while the C-terminal EF-hand (also known as the canonical EF-hand) is composed of 12 amino acid residues and coordinates calcium with a higher affinity (Kd = 2.6 µM) via side-chain carboxylate oxygens.11 Although most S100 proteins bind calcium, there are a few exceptions, including S100A10, which does not bind calcium due to deletions in the calcium coordinating residues in its EF-hand loops.12 For most S100 proteins that are capable of binding calcium, the apo state is known as the inactive, closed conformation and the calcium-bound state is known as the active, open conformation. Upon calcium binding, a large conformational change results in the exposure of a hydrophobic binding pocket in each monomer, which is capable of binding to intracellular or extracellular proteins.13; 14; 15 Several S100 proteins have been shown to form complexes with intracellular and extracellular proteins, including S100A10/annexin A2, S100A11/annexin A1, S100B/p53 and S100B/CapZ.16; 17; 18; 19 Human S100A4, also known as metastasin, has been shown to form complexes with several intracellular and extracellular targets in a calcium-dependent manner, which may be related to S100A4’s role in tumor progression.11 Some of S100A4’s targets include the p53 tumor suppressor protein, the heavy chain of nonmuscle myosin–II, liprin β-1 and annexin A2.20; 21; 22; 23
Human S100A4 (metastasin) is the best-characterized member of the S100 protein family in terms of its role in cancer and metastasis formation and has been recently shown to contribute to tumor-induced angiogenesis.23; 24 Over-expression of S100A4 has been observed in several metastatic cancers, including breast, pancreatic, prostate, gallbladder and lung cancer to name a few.25; 26; 27; 28; 29 High levels of S100A4 protein have been shown to be present in human carcinoma cells, primary colorectal carcinomas, gastric adenocarcinoma and invasive pancreatic carcinomas.26; 30; 31; 32 In addition, S100A4 has been immunocytochemically detected in the carcinoma cells of human breast cancers.33; 34 The metastatic phenotype in in vitro and in vivo models of mammary carcinogenesis has been strongly associated with up-regulated levels of S100A4.35 Previous studies have shown the development of the metastasis phenotype in nonmetastatic rat mammary epithelial cells when transfected with rodent or human S100A4 genes.36; 37 Furthermore, S100A4 has been shown to be secreted by cancer cells and is detectable in the serum of cancer patients indicating that S100A4 may serve as a prognostic marker in human cancers.11; 38 Determining the three-dimensional structure of Ca2+-bound S100A4 is essential, since it is the calcium-bound form of S100A4 that generates a biological effect.
Until now, the only structural information published on S100A4 has been that of the inactive, calcium-free form determined by solution NMR.39 In order to understand the calcium-dependent activation of S100A4 and its interactions with target proteins we have determined the active, Ca2+-bound structure of S100A4. Since then, two other coordinate sets of Ca2+-bound S100A4 have been deposited into the Protein Data Bank by two different groups without accompanying publications. Both deposited coordinate sets are from trigonal spacegroups (P3221 and P32, PDB codes 3C1V and 2Q91, respectively). In this paper, we present a structural analysis of Ca2+-bound S100A4 determined using perfectly merohedrally twinned crystals in the hexagonal spacegroup P65 at 2.03 Å resolution.
Results and Discussion
Detection of merohedral twinning
The x-ray diffraction data were processed in spacegroups P65 and P6522, which resulted in Rmerge values of 7.5% and 7.8%, respectively (Table. 1). Since the data merged well in both spacegroups without any significant increase in Rmerge or the number of rejected reflections, both spacegroups were initially considered. Unlike in the case of crystals of the integral membrane protein bacteriorhodopsin40, in the case of S100A4 the higher-symmetry spacegroup could not be excluded based on packing density, because the P65 cell contains two molecules in the asymmetric unit, and the P6522 cell contains one molecule. A Patterson self-rotation plot was calculated using the program MOLREP from the CCP441 suite (Collaborative Computational Project, Number 4, 1994) in the lower-symmetry spacegroup (P65) that revealed the presence of a two-fold axis along the crystallographic c axis (ψ = 90°, φ = 90°, κ = 180°), which is expected for a crystallographic six-fold axis (Figure 1a). However, the self-rotation plot revealed additional two-fold peaks occurring every 30° in the a/b plane, which is usually an indication of a P6x22 space group. Based on the results of the Patterson self-rotation plot, the Ca2+-bound S100A4 structure was initially treated as spacegroup P6522 with one monomer in the asymmetric unit. After several rounds of model building and refinement at 2.03 Å resolution with cycles of energy minimization and torsion-angle simulated annealing using the maximum-likelihood target function in CNS v.1.142, the R and Rfree values stalled at 36% and 41%, respectively. This lead us to consider that the Ca2+-bound S100A4 crystals might be merohedrally twinned and belong to spacegroup P65. There were several indications that the Ca2+-bound S100A4 structure might be merohedrally twinned. First, the Rmerge value for the higher symmetry Laue group 6/mmm (Rmerge of 7.8%) is only slightly higher than for the lower symmetry Laue group 6/m (Rmerge of 7.5%). Second, merohedral twinning is commonly reported for crystals in hexagonal spacegroups, especially when there is an unusually long c axis (c = 176.2 Å). In addition, we were able to find molecular replacement solutions in both spacegroups, P65 (two monomers per asymmetric unit) and P6522 (one monomer per asymmetric unit).
Table 1.
Data collection and crystallographic refinement statistics for Ca2+-bound S100A4.
| Data Collection | |
| Spacegroup | P65 |
| Unit-cell parameters | |
| a = b (Å) | 47.1 |
| c (Å) | 176.2 |
| Matthews coefficient (Å3 Da−1) | 2.57 |
| No. of molecules in the ASU | 2 |
| Solvent content (%) | 52.1 |
| Resolution (Å) | 50.0 - 2.03 (2.07 - 2.03) |
| Total observations | 187,833 |
| Unique reflections | 14,094 |
| Completeness (%) | 99.2 (98.6) |
| Rmerge#(%) | 7.5 (47.5) |
| Average I/σ(I) | 39.7 (2.9) |
| Mosaicity (°) | 0.47 |
| Refinement | |
| Twin Fraction | 0.5 |
| Resolution range (Å) | 50.0 - 2.03 |
| Number of reflections | |
| Working set [(Fo > 4σ (Fo)/all data] | 11,289/13,399 |
| Test set [(Fo > 4σ (Fo)/all data] | 594/700 |
| Rwork (%) [(Fo > 4σ (Fo)/all data]a | 23.6/25.5 |
| Rfree (%) [(Fo > 4σ (Fo)/all data] b | 28.5/33.1 |
| Average B factor (Å2) | 31.3 |
| Ramachandran plotc | |
| Most favorable regions (%) | 81.5% |
| Allowed regions (%) | 18.5% |
| Disallowed regions (%) | 0.0% |
| Model details | |
| No. of protein atoms | 1,412 |
| No. of water molecules | 51 |
| No. of calcium ions | 4 |
Values in parentheses are for the highest resolution shell.
Rmerge = Σhkl Σj|Ij(hkl)- <I(hkl)>|/ΣhklΣj<I(hkl)>, where Ij(hkl) and <I(hkl)> are the intensity of measurement j and the mean intensity for the reflection with indices hkl, respectively.
Rwork = Σ|Fobs-Fcalc|/(Σ|Fobs|).
The Rfree is the R-factor based on 5% of the data excluded from refinement.
Determined by PROCHECK.71
The final model contains amino acids 2–90 for molecule A and molecule B (out of 101 each).
Figure 1. Analysis of merohedral twinning for data reduced in spacegroup P65.



(a) Self-rotation function for κ = 180°. In addition to the crystallographic two-fold peak at ψ = 90°, φ = 90°, the plot shows two strong two-fold peaks every 30° in the a/b plane, which is usually an indication of a P622 spacegroup. The resolution limits are 25-2.03 Å and the Patterson vector cutoff radius is 30.0 Å. (b) Results from the twinning server (Twin Detector: Padilla-Yeates Algorithm) suggests that the crystal is not twinned.43 (c) The plot from the twinning analysis program TRUNCATE from CCP441 also suggests that the crystal is not twinned. The expected values of (I2)/(I)2 are 2.0 for non-twined data and 1.5 for perfectly twinned data.
To confirm the presence of merohedral twinning, the Ca2+-bound S100A4 data set was tested for twinning using the Twin Detector: Padilla-Yeates Algorithm (http://nihserver.mbi.ucla.edu/pystats/) (Figure 1b) and the Merohedral Crystal Twinning Server (http://nihserver.mbi.ucla.edu/Twinning).43; 44 To our surprise, the perfect merohedral twinning test from the Merohedral Crystal Twinning Server revealed that the calculated values of (I2)/(I)2 for acentric reflections were closer to 2.0 (no twinning) than to 1.5 (perfect twinning), indicating that the crystals were not twinned.44 Also, no strong evidence of merohedral twinning was found using the twinning analysis program TRUNCATE from CCP441 (Figure 1c). However, the partial merohedral twinning test from the Merohedral Crystal Twinning Server44 did estimate a twin fraction of 0.46 (SHELXL commands “TWIN 1 0 0 −1 −1 0 0 0 −1” and “BASF 0.461516”), which contradicted the results from the perfect merohedral twinning test and the twinning analysis program TRUNCATE. Failure to detect merohedral twinning by the majority of the twinning servers and analysis programs indicated above may be due to the close alignment of the non-crystallographic symmetry (NCS) two-fold axis with that of the twinning operator. A similar instance was reported by Eisenberg and coworkers in 2003; they also were unable to detect merohedral twinning using the conventional twinning servers despite having the typical warning signs of twinning and high R factors after several rounds of refinement.45 They were only able to obtain reasonable R factors after including a twinning operator and twinning fraction during refinement and concluded that merohedral twinning was not detected because it was masked by NCS. Based on these results, refinement in SHELXL-97 was attempted using a twin fraction of 0.46 and the twinning operator k, h –l as suggested by the partial merohedral twinning test.46 After the first round of refinement in SHELXL-97, the twinning fraction refined to very close 0.5, so no detwinning was attempted. Manual model-building and several rounds of refinement were performed in SHELXL-97, which led to the final R values of 23.6% (Fo > 4σ (Fo)) / 25.5% (no cutoff) and Rfree values of 28.1% (Fo > 4σ (Fo)) / 33.1% (no cutoff). The R and Rfree dropped dramatically after the introduction of twinning during refinement, which strongly supported our initial suspicions of merohedral twinning. The final model of Ca2+-bound S100A4 exhibits good stereochemistry and consists of 176 amino acids (dimer in the asymmetric unit), 51 water molecules and four calcium ions. To further investigate the issue of the alignment of the non-crystallographic symmetry (NCS) two-fold axis with that of the twinning operator, we utilized the Xtriage program, a tool for analyzing structure factor data to identify the presence of twinning using the NZ test, the L test (Padilla Yeates) and Britton and H analyses.47 The following interpretation was offered by the Xtriage program: “As the data does not appear to be twinned, one can interpret this as origination from a 2-fold NCS axis parallel to the putative twin axis or that the spacegroup is too low. It might be useful to refine a putative twin fraction during refinement and/or re-assess your data processing.” These analyses suggest that we have a case of masked merohedral twinning similar to that reported by Eisenberg and coworkers in 2003.45
Overall structure of active, Ca2+-bound S100A4
The crystal structure of active, Ca2+-bound S100A4 was solved by molecular replacement using the structure of the most closely related S100 protein, active Ca2+-bound S100A6 (PDB code: 1K96).13 The Ca2+-bound crystal structure of S100A4 exhibits the same overall topology seen in other S100 proteins and like other S100 proteins crystallized as a tight homodimer (Figure 2). Each monomer consists of four α-helices (H1, H2, H3 and H4) connected by short loops (L1, L2 (hinge) and L3) (Figure 3). The first helix (H1) is formed by residues Leu5-Ser20, the second (H2) by residues Lys31-Glu41, the third (H3) by residues Phe55-Leu62 and the fourth (H4) by residues Phe72-Glu88. The loop regions L1, L2 and L3 connect the pairs of helices H1-H2, H2-H3 and H3-H4, respectively. In S100A4, the C-terminal loop is long and very basic and makes it unique in comparison to other S100 proteins; however, in the Ca2+-bound S100A4 crystal structure the C-terminal loop (Phe89-Lys101) was disordered in the electron density maps. The hinge region (L2) links helices H2 and H3 and shares low sequence homology to other S100 proteins and has been shown to be involved in target binding. Each S100A4 monomer consists of two helix-loop-helix calcium-binding domains known as EF-hands (EF-hand I and EF-hand II) which are brought into close proximity in the structure. The N-terminal EF-hand (EF-hand I), which is also known as the pseudo EF-hand, is formed by H1-L2-H2 and is comprised of 14 residues (Ser20-Glu33). The calcium ion in the pseudo EF-hand (Kd > 50 µM) is primarily coordinated by backbone carbonyl oxygen atoms.48; 49 The C-terminal EF-hand (EF-hand II), which is also known as the canonical EF-hand, is formed by H3-L3-H4 and is comprised of 12 residues (Asp63-Glu74). The calcium ion in the canonical EF-hand is coordinated with a higher affinity (Kd = 2.6 µM) primarily through side chain oxygen atoms.48; 49 In the pseudo EF-hand, the calcium ion is coordinated in a pentagonal bipyramidal coordination sphere formed by the backbone carbonyl oxygen atoms of Ser20, Glu23, Asp25, Lys28, the bidentate carboxylate group of Glu33 and a water molecule (Figure 4a). In the canonical EF-hand, the calcium ion is coordinated in a hexagonal coordination sphere formed by the monodentate carboxylate groups of Asp63 and Asp67, the side-chain carbonyl of Asn65 and the bidentate carboxylate group of Glu74 and the backbone carbonyl group of Glu69 (Figure 4b).
Figure 2. Ribbon diagram of the Ca2+-bound S100A4 homodimer.

Monomer A is shown in blue, monomer B is shown in green and the calcium ions are represented as red spheres. The N termini and C termini are labeled as Nt and Ct, respectively. The figure was prepared using the programs Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
Figure 3. Ribbon representation of the Ca2+-bound S100A4 monomer.

Helix H1 is shown in blue, helix H2 is shown in dark pink, helix H3 is shown in green, helix H4 is shown in light pink, loop L2 is shown in red, loop L2 (hinge) is shown in orange and loop L3 is shown in cyan. The calcium ions are represented as gold spheres, the N terminus (Nt) is shown in yellow and the C terminus (Ct) is shown in purple. The figure was prepared using the programs Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
Figure 4. Coordination of the calcium ions in the EF-hand I and EF-hand II loops in the Ca2+-bound S100A4 structure.


Calcium ions are represented as red spheres, water molecules as yellow spheres and the coordination between the calcium ion and oxygen atoms is indicated by cyan dotted lines. (a) Calcium coordination in the EF-hand I loop. The coordination distances between the calcium ion and the coordinating oxygen atoms (Ca-O) are as follows: Ca-O Ser20 2.45 Å, Ca-O Glu23 2.54 Å, Ca-O Asp25 2.22 Å, Ca-O Lys28 2.64 Å, Ca-OE1 Glu33 2.77 Å, Ca-OE2 Glu33 2.37 Å and Ca-O HOH124 2.36 Å. (b) Calcium coordination in the EF-hand II loop. The coordination distances between the calcium ion and the coordinating oxygen atoms (Ca-O) are as follows: Ca-OD1 Asp63 2.31 Å, Ca-OD1 Asn65 2.15 Å, Ca-OD1 Asp67 2.75 Å, Ca-O Glu69 2.27 Å, Ca-OE1 Glu74 2.26 Å and Ca-OE2 Glu74 2.61 Å. The figure was prepared in the molecular graphics programs Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
The overall size of the Ca2+-bound S100A4 dimer is approximately 54 × 41 × 31 Å3 and the total buried surface area between monomers A and B is 3,065 Å 2. Similar to the apo-S100A4 structure and other S100 structures, the Ca2+-bound S100A4 homodimer is held together by the X-type four-helix bundle formed between helices H1 and H4 of monomer A and helices H1’ and H4’ of monomer B. Yeast two-hybrid assays revealed that residues Phe72, Tyr75, Phe78 and Leu79 from helix H4 are essential for dimerization in vivo.50 Mutation studies on S100A4 revealed that side chains Phe72, Tyr75, Phe78 and Leu79 from helix H4 (or helix H4’) form noncovalent interactions with the hydrophobic side chains Met12, Val13 and Phe16 from helix H1’ (or helix H1) to form the dimerization interface.50 In addition to the residues that form the X-type four helix bundle, residues Leu34, Leu37 and Leu38 of helix H2, Leu62 of helix H3, Leu29 of loop L1, Leu42 and Leu46 of loop L2 and Val70 of loop L3 contribute to monomer integrity. A sequence alignment of S100A4 with five other S100 proteins (S100A1, S100A6, S100A10, S100A11 and S100B) reveals similar residues involved in dimerization (Figure 5). In helix H4, the phenylalanine residue at position 72 in S100A4 is strictly conserved in the other S100 sequences, while residues Tyr75, Phe78 and Leu79 in S100A4 have aromatic or aliphatic substitutions throughout the sequences. In helix H1, the phenylalanine residue at position 16 in S100A4 is strictly conserved in the other S100 sequences, while residues Met12 and Val13 have aliphatic substitutions throughout the S100 sequences. The amino acid residues important for S100 dimerization appears to be either strictly conserved or have homologous substitutions, which may be the reason why heterodimerization is possible between different members of the S100 family.
Figure 5. Sequence alignment of S100A4 and S100 family members S100A1, S100A6, S100A10, S100A11 and S100B.

The secondary structural elements for S100A4 are indicated above the alignment. Helices H1, H2, H3 and H4 and loop regions L1, L2 (hinge) and L3 are labeled. The N terminus and C terminus are labeled as Nt and Ct, respectively. Residues denoted as white letters on red background are strictly conserved. Residues boxed in blue and denoted in red are homologous while residues denoted in black are non-homologous in this group. Highlighted in green are residues that coordinate the calcium ions in EF-hand I (pseudo EF hand) and EF-hand II. Highlighted in yellow are residues that form hydrophobic interactions with the annexin A2 peptide in complex with S100A10 and the proposed residues that may form hydrophobic interactions with annexin A2 in complex with S100A4. The sequence alignment was created using the program CLUSTALW65; 66 and the figure was prepared with the program ESPript.67
Most members of the S100 protein family contain one or more cysteine residues and are capable of forming disulfide-linked homotetramers under oxidizing conditions.51 S100B and S100A10 have been shown to form disulfide-linked dimers via surface-exposed cysteine residues (Cys61 and Cys70, respectively) in the EF-hand II loop.51; 52 S100A4 contains four cysteine residues, one near the N-terminus (Cys3) and three in helix H4 (Cys76, Cys81 and Cys86). Due to the presence of four cysteine residues, Ca2+-bound S100A4 was crystallized under reducing conditions (in the presence of 10 mM dithiothreitol) to prevent the formation of disulfide linked S100A4 dimers. In the Ca2+-bound S100A4 structure, Cys76 and Cys81 only move by 1.41 Å and 0.83 Å, respectively (Cys3 is not visible in the electron density maps). The lack of conformational change is probably because Cys76 is located at the S100A4 dimer interface, while Cys81 is exposed to the solvent in both the calcium-free and calcium-bound forms. On the other hand, Cys86 undergoes the most dramatic conformational change in response to the addition of calcium by moving 2.98 Å. Cys86 is one of the residues comprising the hydrophobic target-binding site and is buried in the calcium-free (inactive) form, while it is exposed in the calcium-bound (active) form such that it could possibly interact with other S100A4 dimers under oxidizing conditions or different protein targets under reducing conditions. Therefore, the Ca2+-bound S100A4 structure reported here most likely represents the S100A4 dimer present in the reducing environment of the cytosol.
Comparison with the inactive apo S100A4 structure
A comparison of the inactive apo-S100A4 structure and the active Ca2+-bound S100A4 structure highlights the major conformational change S100A4 undergoes in the presence of calcium (Figure 6a).39 A structural comparison of the apo-S100A4 and Ca2+-bound S100A4 structures reveals dramatic differences in the overall shape and size of the dimers. The inactive apo-S100A4 dimer structure is more compact (43 × 40 × 35 Å3) in comparison to active Ca2+-bound S100A4 dimer structure (54 × 41 × 31 Å3). The root mean square distance (r.m.s.d.) of the Cα atoms between apo-S100A4 and Ca2+-bound S100A4 is 1.67 Å, illustrating significant conformational changes between the apo and calcium-bound states (Figure 6b). The most noticeable differences occurs in helices H2 and H3 and in the loop regions L1 (EF-hand I), L2 (hinge) and L3 (EF-hand II) (Figures 7 (a) and 7 (b)). A superimposition of the apo and Ca2+-bound S100A4 structures reveals a 70° reorientation of helix H3 such that helix H3 is now nearly perpendicular to helix H4 in the presence of calcium. This dramatic reorientation of helix H3 opens up the target binding region formed by helix H4 and loop L2 (hinge). In addition, helix H3 is one turn shorter in the Ca2+-bound structure than in the apo structure, which extends the length of the L2 loop (hinge) by two amino acids. The regions of the Ca2+-bound structure that change the least in comparison to apo structure are helices H1 and H4. This area of the structure changes the least since the hydrophobic residues from helices H1 and H4 hold the dimer together via the X-type four-helix bundle.
Figure 6. Comparison of apo (inactive) and calcium-bound (active) S100A4 structures.


(a) Ribbon representation of the apo S100A4 NMR structure (blue) superimposed on the Ca2+-bound S100A4 (pink) crystal structure. Calcium ions are represented as blue spheres in the Ca2+-bound S100A4 crystal structure. (b) The Ca2+-bound S100A4 dimer colored from blue to red according to its r.m.s.d. from apo S100A4, with regions with the largest deviations depicted in red. Helices H1, H2, H3 and H4 and loop regions L1, L2 (hinge) and L3 are labeled. Superposition of the apo and Ca2+-bound structures was performed with the program Coot68 (Cα r.m.s.d. 1.67 Å) and the figure was prepared in the molecular graphics programs Swiss-Pdb Viewer69, Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
Figure 7. Overlay of EF-hands I and II from monomer A of S100A4 in the apo (pink) and Ca2+-bound (blue) states.


Calcium ions are represented as blue spheres in the Ca2+-bound S100A4 crystal structure and helices H1, H2, H3 and H4 and loop regions L1, L2 (hinge) and L3 are labeled. (a) Superimposition of the N-terminal EF-hand (EF-hand I) and (b) the C-terminal EF-hand (EF-hand II) in the apo and Ca2+-bound states illustrating how these two regions change conformation upon calcium binding. Superposition of the apo and Ca2+-bound structures was performed with the program Coot68 and the figure was prepared in the molecular graphics programs Pymol [http://pymol.sourceforge.net/] and POVRAY [http://www.povray.org/].
Upon calcium binding, the amino acid residues in both the pseudo EF-hand (loop L1 or EF-hand I) and canonical EF-hand (loop L2 or EF-hand II) undergo changes to accommodate the calcium ions. In the pseudo EF-hand the coordination of the calcium ion is mediated primarily by main chain carbonyls and the conformational change is less dramatic than in the canonical EF-hand where the calcium ion is coordinated primarily by side chain carboxylates. In the pseudo EF-hand the only major change occurs in residue Glu33 which moves by ~3.8 Å to coordinate the calcium ion via its bidentate carboxylate group. In contrast, all the residues involved in the calcium coordination in the canonical EF-hand undergo major conformational changes with residues Asp63 and Asn65 undergoing the most dramatic changes by moving ~7.7 Å and ~8.1 Å, respectively.
Interestingly, previous researchers have measured the calcium affinity of S100 proteins by measuring changes in tyrosine fluorescence intensity.53; 54 Typically, the calcium-induced conformational change resulted in a significant increase in tyrosine fluorescence, which was correlated with the exposure of the hydrophobic binding sites in the S100 dimer.53 There are two tyrosine residues (Tyr19 and Tyr75) in human S100A4 located in helices H1 and H4, respectively. Only slight conformational changes are observed in Tyr19 and Tyr75 in the presence of calcium, which only move by 0.64 Å and 0.67 Å, respectively in comparison to the calcium-free form of S100A4. Therefore, using tyrosine fluorescence to estimate the calcium affinity of S100A4 is not a reliable method, since the tyrosine residues are located in the helices that form the S100A4 dimer interface and not in the hydrophobic binding sites that are exposed upon calcium activation.
Comparison with other S100 structures
The overall structure of Ca2+-bound S100A4 displays the same topology as the other members of the S100 family. The r.m.s.d. values of the Cα atoms between Ca2+-bound S100A4 and the Ca2+-bound structures of S100A1, S100A6 and S100B and the apo form of S100A10 are 1.57 Å, 1.08 Å, 1.57 Å and 1.22 Å, respectively (PDB codes: 1ZFS, 1K96, 1UWO and 1A4P respectively).13; 15; 51; 55 S100A10 is one of the few members of the S100 family that is incapable of binding calcium due to mutations and deletions in its EF-hands; however, S100A10 is still capable of binding to protein targets in the absence of calcium and is therefore constitutively active.12; 51 The EF-hand I loop of S100A10 is three amino acids shorter than the EF-hand I loop in S100A4 and in a significantly different conformation. Surprisingly, the conformation of the EF-hand II loop in S100A10 is similar to that of S100A4 despite three major substitutions in the corresponding calcium-coordinating residues (Asn65→Cys61, Glu69→Lys65 and Glu74→Ser70). The major reason why S100A10 is in a permanently active conformation is likely due to the similarity of conformations of the EF-hand II loops that lead to the exposure of hydrophobic target binding sites formed by helix H4 and loop L2 (hinge) in both the S100A10 and Ca2+-bound S100A4 structures. The reason why apo S100A10 has a similar overall EF-hand II conformation as in Ca2+-bound S100A4 could be due to conserved acidic residues Asp59 and Asp63 and the formation of a salt bridge between Asp63 and Lys65.
Based on the r.m.s.d. values reported above, the Ca2+-bound S100A4 structure is most similar to the apo form of S100A10 and the Ca2+-bound structure of S100A6. However, the r.m.s.d. values indicate there are significant differences between the Ca2+-bound S100A4 structure and the Ca2+-bound structures of S100A1 and S100B. Superimposition of the S100 structures shows that helices H1 and H2 are in a similar conformation among the S100 structures (Figure 8). However, there are significant differences in the orientation of helices H3 and H4 in the Ca2+-bound S100A4 structure in comparison to the Ca2+-bound structures of S100A1 and S100B. The differences in the positions of helices H3 and H4 are likely due to the variations in the amino acid sequence in the C-terminal loop of helix H4 and the loop L2 region (hinge) preceding helix H3. The lack of sequence homology in loop L2 and the C-terminal loop presumed to determine which specific protein target will interact with specific members of the S100 family.49
Figure 8. Superposed Cα ribbon representations of five S100 proteins with bound calcium: S100A4 in blue, the S100A10 in red, S100A1 in yellow, S100A6 in mint and S100B in pink.

Helices H3 and H4 are labeled to illustrate the differences between the orientations of these helices among the S100 family members. Bound calcium ions were removed for clarity. Superposition of the S100 protein structures was performed in the molecular graphics programs Swiss-Pdb Viewer69 and the figure was rendered with POVRAY [http://www.povray.org/].
Implications for target binding to annexin A2 and other targets
S100A4 is the best characterized of all the members of the S100 family in its role in cancer; however, its exact mechanism of action in the development of cancer is unknown.24 The interactions between S100A4 and its protein targets are dimerization-dependent and calcium-dependent and may reveal clues on the metastasis-promoting properties of S100A4.11 To date, both intracellular and extracellular targets of S100A4 have been identified: the p53 tumor suppressor protein, F-actin, tropomyosin, S100A1 and annexin A2 to name a few.20; 23; 50; 56; 57 Most of the S100A4 protein targets that have been identified thus far are found intracellularly. However, accumulating evidence has shown that S100A4 plays an extracellular role as well.11 Although the mechanism of secretion are unclear it is known that S100A4 is secreted by tumor cells and it has been recently discovered that S100A4 binds to the endothelial plasminogen co-receptor, annexin A2, another protein that is secreted by a poorly understood mechanisms.23 Several annexins have been shown to bind to members of the S100 family to form heterotetramer complexes including: annexin A1 and S100A11, annexin A11 and S100A6 and annexin A2 and S100A10.16; 17; 58 Annexins are a family of cytosolic proteins that bind to negatively charged phospholipids in a Ca2+-dependent manner and have been implicated in several membrane fusion processes.59 The annexin A2/S100A10 heterotetramer complex is the best characterized of the annexin/S100 complexes discovered thus far and has been shown to be the only complex that is calcium-independent due to mutations in the EF-hands of S100A10 that render it incapable of binding calcium, yet arrest it in the active conformation.12; 16 Previously, only the annexin A2/S100A10 heterotetramer complex was shown to activate tissue plasminogen activator (t-PA)-mediated plasmin formation.60 However, a report was recently published that provided evidence that S100A4 in complex with annexin A2 accelerates t-PA-mediated plasminogen activation on the surface of endothelial cells.23
Immunoprecipitation and NMR studies demonstrated direct binding of S100A4 and annexin A2, and the binding site was found to be located in the first thirteen residues of the N-terminal region of annexin A2.23 The report proposed that the angiogenic and metastasis properties of S100A4 are induced when annexin A2 is translocated to the surface of endothelial cells while S100A4 is secreted by tumor cells. The report proposed that formation of the S100A4/annexin A2 heterotetramer complex increases the t-PA-mediated conversion of plasminogen to plasmin, activating matrix metalloproteinases (MMPs), which in turn induce extracellular matrix remodeling leading to angiogenesis.23
The Ca2+-dependent activation of dimeric S100A4 exposes two symmetrically positioned hydrophobic target binding sites, each of which is thought to bind specifically to the N-terminal domain of annexin A2. Electrostatic surface representations of the apo and Ca2+-bound S100A4 structures reveal that several of the hydrophobic patches buried in the apo structure are exposed in the Ca2+-bound structure (Figures 9 (a) and 9 (b)). The electrostatic distribution is most dramatically affected near helices H4 and H4’ and loops L2 and L2’ upon calcium-binding. In the Ca2+-bound structure the major reorientation of helix H3 unveils hydrophobic residues from helices H4 and H4’ (residues Leu79, Ile82, Cys86 and Phe89) and loops L2 and L2’ (residues Leu42, Phe45 and Leu46), which likely participate in target binding to annexin A2. Sequence alignment of S100A4 and S100A10 reveals high sequence similarity in the annexin A2 target binding region (Figure 5). The crystal structure of S100A10 and the annexin A2 N-terminal peptide (first 13 amino acids) revealed that each annexin A2 peptide was bound to a pocket formed by helix H4 and loop L2 of one monomer and the N-terminal portion of helix H1’ from the second monomer.51 The crystal structure of the S100A10/annexin A2 peptide complex showed that the hydrophobic residues lining the target binding region in S100A10 are Phe38 and Phe41 of the loop L2 region and Leu78, Cys82 and Tyr85 of helix H4. Based on a sequence alignment and structural comparison of S100A4 and the S100A10/annexin A2 complex structure, we propose that hydrophobic residues Leu42 and Phe45 of the loop L2 region and Ile82, Cys86 and Phe89 of helix H4 from the Ca2+-bound S100A4 structure form the target binding region for annexin A2 (Figures 10 (a) and (b)). We propose that similar residues involved in the complex between S100A10 and the annexin A2 N-terminal peptide may also be involved in the S100A4 and annexin A2 N-terminal peptide interaction; however, a crystal structure of S100A4 in complex with full-length annexin A2 complex would provide more information on additional interactions.
Figure 9. Electrostatic surface potential representations of (a) apo S100A4 and (b) Ca2+-bound S100A4.


Red, blue and white areas indicate negatively charged, positively charged and hydrophobic regions, respectively. Helices H4 and H4’ and loop regions L2 and L2’ are labeled. The electrostatic surface representation of the Ca2+-bound S100A4 structure highlights the exposure of two symmetrically hydrophobic target binding sites formed by helix H4 and loop L2, and by helix H4’ and loop L2’, which are buried in the apo S100A4 structure. The figure was prepared in the CCP4 molecular graphics program.70
Figure 10. Ca2+-bound S100A4 structure modeled in complex with the N-terminal domain of annexin A2, based on a superimposition of the S100A10/N-terminal annexin A2 peptide complex structure on the Ca2+-bound S100A4 structure.51.

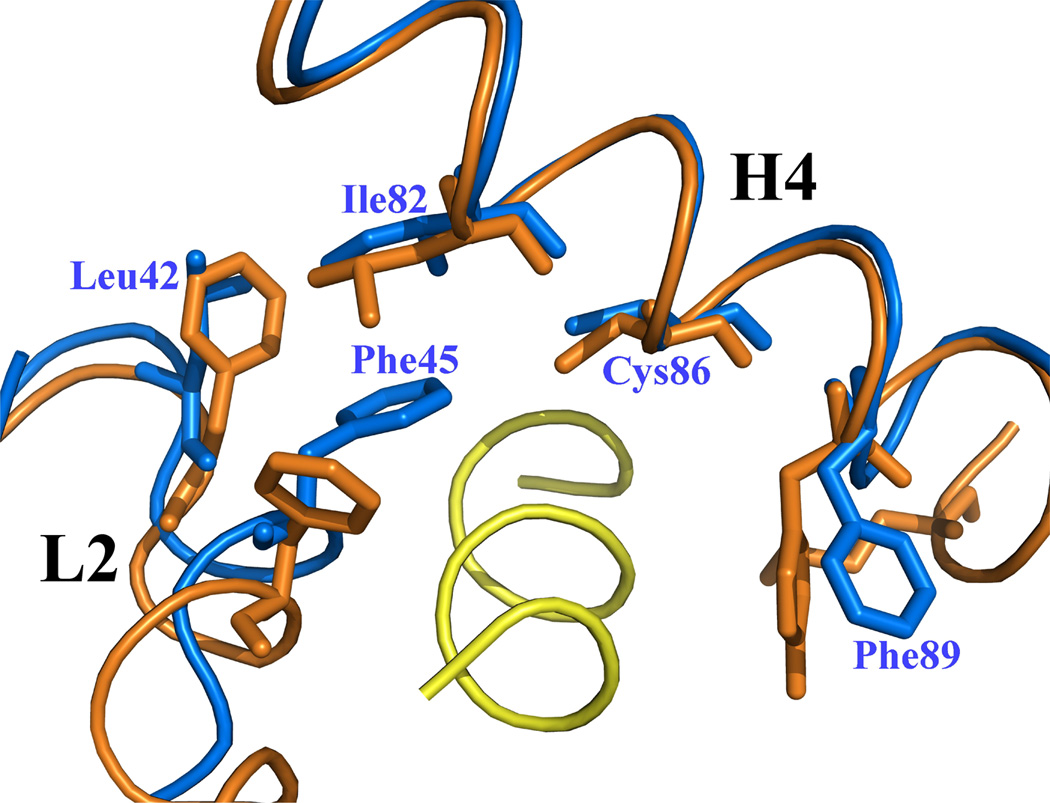
Helices H4 and H4’ and loop regions L2 and L2’ are labeled. (a) Ribbon representation of Ca2+-bound S100A4 (blue) homodimer with two annexin A2 peptides (yellow) bound to the target-binding regions formed by helix H4 and loop L2, and by helix H4’ and loop L2’. Calcium ions are represented as red spheres. (b) A close-up view on the overlay of the target-binding regions of Ca2+-bound S100A4 (blue) and S100A10 (orange) with the N-terminal annexin A2 peptide (yellow). The residues (orange) forming hydrophobic interactions in S100A10 with the N-terminal annexin A2 peptide are shown as sticks. Corresponding residues Leu42, Phe45, Ile82, Cys86 and Phe89 in S100A4 that may form hydrophobic interactions with the N-terminal annexin A2 peptide are labeled and shown as sticks. Superposition of the Ca2+-bound S100A4 and S100A10/N-terminal annexin A2 peptide complex structures was performed with the program Coot68 and the figure was prepared in the program Pymol [http://pymol.sourceforge.net/].
Conclusions
We have solved the crystal structure of the active, Ca2+-bound S100A4 (metastasin) from perfectly merohedrally twinned crystals. The structure of active S100A4 reveals a dramatic conformational change that opens up two symmetrically positioned target binding sites that are buried in the apo S100A4 structure. Based on the structural information we have gained from Ca2+-bound S100A4 we propose a model of calcium induced target binding to annexin A2 and how this interaction may contribute to S100A4’s role in metastasis and angiogenesis. Although conventional twinning servers and analysis programs did not detect twinning, refinement of the Ca2+-bound S100A4 structure was only successful after including a twinning operator and a twin fraction of 0.5. We believe that conventional twinning servers failed to detect twinning because NCS masked the twinning operator, a phenomenon that has been previously reported.45
Materials and Methods
Expression and purification
The pET23a-human S100A4 expression construct was kindly provided by Dr. David J. Weber, University of Maryland School of Medicine, Maryland, USA and Dr. Anne R. Bresnick, Albert Einstein College of Medicine, New York, USA. The recombinant plasmid pET23a-S100A4 was expressed in BL21 (DE3) Escherichia coli cells (Novagen, San Diego, CA, USA). Cells grown from an overnight pre-culture incubated at 37 °C were grown in 1 l Luria-Bertani (LB) broth medium containing 50 µg/ml ampicillin at 37 °C. At an optical density of 0.8 at 600 nm, the cells were induced with 1 mM isopropyl β-D-thiogalactopyranoside (IPTG) at 37 °C. The cells were harvested 4 h after induction by centrifugation at 6000g for 15 min at 4 °C, then frozen and stored at −20 °C until lysis.
Cell pellets from 6 l of culture were resuspended in 150 ml of lysis buffer [50 mM imidazole-HCl pH 7.4, 2 mM EGTA, 150 mM NaCl, 10 mM MgCl2, 1 mM dithiothreitol and 1 mM phenylmethylsulfonyl fluoride (PMSF), containing a protease inhibitor cocktail [20 µg/ml aprotinin, 0.5 mM benzamidine and 10 µM trans-epoxysuccinyl-L-leucylamido(4-guanidino)butane (E-64)]. Lysozyme was added to a final concentration of 100 µg/ml and the resuspended cells were lysed using a French Press. The lysate was centrifuged at 40,000g for 1 h at 4 °C to remove cell debris and denatured protein. The supernatant was dialyzed against buffer A [10 mM imidazole-HCl (pH 7.4), 10 mM NaCl, 1 mM EGTA, 1 mM NaN3 and 1 mM PMSF].
Human S100A4 was purified using ion-exchange chromatography and all purification steps were performed at 4 °C. The supernatant was first applied to a 200 ml bed volume DEAE Sepharose column (Amersham Pharmacia, Piscataway, NJ, USA) preequilibrated with buffer A. Unbound protein was removed by washing the column with 200 ml of buffer A and the S100A4 protein was eluted in buffer B (10 mM imidazole-HCl (pH 7.4), 50 mM NaCl, 1 mM EGTA, 1 mM NaN3 and 1 mM PMSF). Fractions containing S100A4 were pooled and dialyzed against buffer C (50 mM MES-NaOH pH 5.6 and 1 mM dithiothreitol) and applied to a 200 ml bed volume MonoS cation-exchange column (Amersham Pharmacia, Piscataway, NJ, USA) pre-equilibrated with buffer C. The column was washed with 200 ml of buffer C to remove any unbound protein and S100A4 was eluted in 50 mM MES-NaOH pH 5.6, 350 mM NaCl and 1 mM dithiothreitol using a 50–500 mM NaCl step gradient composed of 50 mM steps. Fractions containing S100A4 were pooled and dialyzed against 50 mM MES-NaOH pH 5.6, 250 mM NaCl, 100 mM CaCl2 and 10 mM dithiothreitol in order to obtain Ca2+-bound crystals of human S100A4. Prior to crystallization S100A4 was concentrated to 20 mg/ml via ultrafiltration using a 10 kDa cutoff ultrafiltration membrane (Millipore Corporation, Bedford, MA, USA). The purity of the sample during each purification step was confirmed using 15% (w/v) SDS-PAGE stained with Coomassie Brilliant Blue.
Crystallization and data collection
Crystallization screens were set up using the hanging-drop vapor-diffusion method61 by mixing 2 µl of protein solution with 2 µl of precipitant solution over wells containing 1 ml of precipitant solution in EasyXtal Tool plates from Nextal Biotechnologies (Montreal, Quebec, Canada). Crystallization conditions were obtained in 20% (w/v) PEG 3350 and 100 mM Tris-HCl pH 8.5 at 22 °C. This condition was optimized to improve the quality of the crystals by using isopropanol as an additive and using various protein concentrations. The best crystals grew in three days at 22 °C by mixing 1.5 µl of protein solution (20 mg/ml) with 2 µl of precipitant solution (20% (w/v) PEG 3350, 10% (v/v) isopropanol and 100 mM Tris-HCl pH 8.5); the maximum dimensions of the crystals were 70 × 70 × 90 µm.
Prior to data collection, Ca2+-bound S100A4 crystals were soaked for a few seconds in the crystallization solution containing 30% (v/v) glycerol as a cryo-protectant before being flash-cooled in liquid nitrogen. X-ray diffraction data were collected on beamline 9-1 at the Stanford Synchrotron Radiation Laboratory (SSRL) using remote robotic data collection at a wavelength of 0.979 Å. The detector was an Area Detector Systems Corporation (ADSC) Q315 CCD detector. A total of 180 frames of 1° oscillation range and an exposure time of 30 s per frame were collected from a single crystal. The images were processed to 2.03 Å in spacegroups P65 and P6522 with the program HKL2000.62 Data-collection statistics are summarized in Table 1.
Structure determination and refinement
Molecular replacement was carried using PHASER63 using the Ca2+-bound S100A6 structure (PDB code: 1K96, 46% sequence identity) as the search model.13 The calcium ions were removed from the search model prior to molecular replacement.
Due to the presence of strong merohedral twinning, SHELX-97 was used for crystallographic refinement (TWIN and BASF options).46 In order to avoid bias due to the presence of the twinning operator the Rfree test reflections were selected to include all reflection (twin pairs) from several thin resolution shells. During each step of the refinement, twenty conjugate-gradient cycles were performed and after several rounds of refinement and manual model-building using the program Coot64, water molecules were added using SHELX-97 (SHELXWAT option). Final refinement statistics for Ca2+-bound S100A4 are listed in Table 1.
Detection of merohedral twinning
Merohedral twinning analysis was attempted using the Merohedral Twin Detector: Padilla-Yeates Algorithm available at http://nihserver.mbi.ucla.edu/pystats/ and the Merohedral Crystal Twinning Server available at http://nihserver.mbi.ucla.edu/Twinning.43; 44 Twinning analysis was also performed using the program TRUNCATE from CCP4 (Collaborative Computational Project, Number 4, 1994).41 Cumulative intensity distribution plots and the second moment of I were examined in TRUNCATE. The program Xtriage was used to analyze the structure factor data for the presence of twinning.47 The twinning fraction was refined with the program SHELXL-97.46
Acknowledgements
We would like to thank David J. Weber from the University of Maryland, School of Medicine and Anne R. Bresnick from the Albert Einstein College of Medicine for `providing the pET23a-S100A4 expression construct. This work was supported by NIH `grant R01-GM067808 and a Chancellor’s Fellowship to H.L.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Protein Data Bank accession codes
The atomic coordinates and structure factors of active, Ca2+ bound S100A4 have been deposited in the RCSB Protein Data Bank under the accession code 3CGA.
References
- 1.Schafer BW, Heizmann CW. The S100 family of EF-hand calcium-binding proteins: functions and pathology. Trends Biochem. Sci. 1996;21:134–140. doi: 10.1016/s0968-0004(96)80167-8. [DOI] [PubMed] [Google Scholar]
- 2.Zimmer DB, Cornwall EH, Landar A, Song W. The S100 protein family: history, function, and expression. Brain Res. Bull. 1995;37:417–429. doi: 10.1016/0361-9230(95)00040-2. [DOI] [PubMed] [Google Scholar]
- 3.Heizmann CW, Fritz G, Schafer BW. S100 proteins: structure, functions and pathology. Front Biosci. 2002;7:1356–1368. doi: 10.2741/A846. [DOI] [PubMed] [Google Scholar]
- 4.Marenholz I, Heizmann CW, Fritz G. S100 proteins in mouse and man: from evolution to function and pathology (including an update of the nomenclature) Biochem. Biophys. Res. Commun. 2004;322:1111–1122. doi: 10.1016/j.bbrc.2004.07.096. [DOI] [PubMed] [Google Scholar]
- 5.Donato R. S100: a multigenic family of calcium-modulated proteins of the EF-hand type with intracellular and extracellular functional roles. Int. J. Biochem. Cell. Biol. 2001;33:637–668. doi: 10.1016/s1357-2725(01)00046-2. [DOI] [PubMed] [Google Scholar]
- 6.Donato R. Intracellular and extracellular roles of S100 proteins. Microsc. Res. Tech. 2003;60:540–551. doi: 10.1002/jemt.10296. [DOI] [PubMed] [Google Scholar]
- 7.Emberley ED, Murphy LC, Watson PH. S100 proteins and their influence on pro-survival pathways in cancer. Biochem. Cell. Biol. 2004;82:508–515. doi: 10.1139/o04-052. [DOI] [PubMed] [Google Scholar]
- 8.Donato R. Functional roles of S100 proteins, calcium-binding proteins of the EF-hand type. Biochim. Biophys. Acta. 1999;1450:191–231. doi: 10.1016/s0167-4889(99)00058-0. [DOI] [PubMed] [Google Scholar]
- 9.Moore BW. A soluble protein characteristic of the nervous system. Biochem. Biophys. Res. Commun. 1965;19:739–744. doi: 10.1016/0006-291x(65)90320-7. [DOI] [PubMed] [Google Scholar]
- 10.Heizmann CW, Cox JA. New perspectives on S100 proteins: a multi-functional Ca(2+)-, Zn(2+)- and Cu(2+)-binding protein family. Biometals. 1998;11:383–397. doi: 10.1023/a:1009212521172. [DOI] [PubMed] [Google Scholar]
- 11.Garrett SC, Varney KM, Weber DJ, Bresnick AR. S100A4, a mediator of metastasis. J. Biol. Chem. 2006;281:677–680. doi: 10.1074/jbc.R500017200. [DOI] [PubMed] [Google Scholar]
- 12.Gerke V, Weber K. The regulatory chain in the p36-kd substrate complex of viral tyrosine-specific protein kinases is related in sequence to the S-100 protein of glial cells. EMBO J. 1985;4:2917–2920. doi: 10.1002/j.1460-2075.1985.tb04023.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otterbein LR, Kordowska J, Witte-Hoffmann C, Wang CL, Dominguez R. Crystal structures of S100A6 in the Ca(2+)-free and Ca(2+)-bound states: the calcium sensor mechanism of S100 proteins revealed at atomic resolution. Structure. 2002;10:557–567. doi: 10.1016/s0969-2126(02)00740-2. [DOI] [PubMed] [Google Scholar]
- 14.Drohat AC, Baldisseri DM, Rustandi RR, Weber DJ. Solution structure of calcium-bound rat S100B(betabeta) as determined by nuclear magnetic resonance spectroscopy. Biochemistry. 1998;37:2729–2740. doi: 10.1021/bi972635p. [DOI] [PubMed] [Google Scholar]
- 15.Wright NT, Varney KM, Ellis KC, Markowitz J, Gitti RK, Zimmer DB, et al. The three-dimensional solution structure of Ca(2+)-bound S100A1 as determined by NMR spectroscopy. J. Mol. Biol. 2005;353:410–426. doi: 10.1016/j.jmb.2005.08.027. [DOI] [PubMed] [Google Scholar]
- 16.Jost M, Gerke V. Mapping of a regulatory important site for protein kinase C phosphorylation in the N-terminal domain of annexin II. Biochim. Biophys. Acta. 1996;1313:283–289. doi: 10.1016/0167-4889(96)00101-2. [DOI] [PubMed] [Google Scholar]
- 17.Seemann J, Weber K, Gerke V. Structural requirements for annexin I-S100C complex-formation. Biochem. J. 1996;319(Pt 1):123–129. doi: 10.1042/bj3190123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wilder PT, Lin J, Bair CL, Charpentier TH, Yang D, Liriano M, et al. Recognition of the tumor suppressor protein p53 and other protein targets by the calcium-binding protein S100B. Biochim. Biophys. Acta. 2006;1763:1284–1297. doi: 10.1016/j.bbamcr.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 19.Garbuglia M, Verzini M, Rustandi RR, Osterloh D, Weber DJ, Gerke V, et al. Role of the C-terminal extension in the interaction of S100A1 with GFAP, tubulin, the S100A1- and S100B-inhibitory peptide, TRTK-12, and a peptide derived from p53, and the S100A1 inhibitory effect on GFAP polymerization. Biochem. Biophys. Res. Commun. 1999;254:36–41. doi: 10.1006/bbrc.1998.9881. [DOI] [PubMed] [Google Scholar]
- 20.Grigorian M, Andresen S, Tulchinsky E, Kriajevska M, Carlberg C, Kruse C, et al. Tumor suppressor p53 protein is a new target for the metastasis-associated Mts1/S100A4 protein: functional consequences of their interaction. J. Biol. Chem. 2001;276:22699–22708. doi: 10.1074/jbc.M010231200. [DOI] [PubMed] [Google Scholar]
- 21.Kriajevska MV, Cardenas MN, Grigorian MS, Ambartsumian NS, Georgiev GP, Lukanidin EM. Non-muscle myosin heavy chain as a possible target for protein encoded by metastasis-related mts-1 gene. J. Biol. Chem. 1994;269:19679–19682. [PubMed] [Google Scholar]
- 22.Kriajevska M, Fischer-Larsen M, Moertz E, Vorm O, Tulchinsky E, Grigorian M, et al. Liprin beta 1, a member of the family of LAR transmembrane tyrosine phosphatase-interacting proteins, is a new target for the metastasis-associated protein S100A4 (Mts1) J. Biol. Chem. 2002;277:5229–5235. doi: 10.1074/jbc.M110976200. [DOI] [PubMed] [Google Scholar]
- 23.Semov A, Moreno MJ, Onichtchenko A, Abulrob A, Ball M, Ekiel I, et al. Metastasis-associated protein S100A4 induces angiogenesis through interaction with Annexin II and accelerated plasmin formation. J. Biol. Chem. 2005;280:20833–20841. doi: 10.1074/jbc.M412653200. [DOI] [PubMed] [Google Scholar]
- 24.Barraclough R, Rudland PS. The S-100-related calcium-binding protein, p9Ka, and metastasis in rodent and human mammary cells. Eur. J. Cancer. 1994;30A:1570–1576. doi: 10.1016/0959-8049(94)00320-5. [DOI] [PubMed] [Google Scholar]
- 25.Pedersen KB, Nesland JM, Fodstad O, Maelandsmo GM. Expression of S100A4, E-cadherin, alpha- and beta-catenin in breast cancer biopsies. Br. J. Cancer. 2002;87:1281–1286. doi: 10.1038/sj.bjc.6600624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rosty C, Ueki T, Argani P, Jansen M, Yeo CJ, Cameron JL, et al. Overexpression of S100A4 in pancreatic ductal adenocarcinomas is associated with poor differentiation and DNA hypomethylation. Am. J. Pathol. 2002;160:45–50. doi: 10.1016/S0002-9440(10)64347-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gupta S, Hussain T, MacLennan GT, Fu P, Patel J, Mukhtar H. Differential expression of S100A2 and S100A4 during progression of human prostate adenocarcinoma. J. Clin. Oncol. 2003;21:106–112. doi: 10.1200/JCO.2003.03.024. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Ajiki T, Murao S, Kamigaki T, Maeda S, Ku Y, et al. Prognostic significance of S100A4 expression in gallbladder cancer. Int. J. Oncol. 2002;20:937–941. [PubMed] [Google Scholar]
- 29.Kimura K, Endo Y, Yonemura Y, Heizmann CW, Schafer BW, Watanabe Y, et al. Clinical significance of S100A4 and E-cadherin-related adhesion molecules in non-small cell lung cancer. Int. J. Oncol. 2000;16:1125–1131. doi: 10.3892/ijo.16.6.1125. [DOI] [PubMed] [Google Scholar]
- 30.Simpson PT, Shoker BS, Barraclough R, Halliwell N, Rudland PS, Sibson DR, et al. Examination of tumour histopathology and gene expression in a neu/S100A4 transgenic model of metastatic breast cancer. Int. J. Exp. Pathol. 2003;84:173–184. doi: 10.1046/j.1365-2613.2003.00350.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Takenaga K, Nakanishi H, Wada K, Suzuki M, Matsuzaki O, Matsuura A, et al. Increased expression of S100A4, a metastasis-associated gene, in human colorectal adenocarcinomas. Clin. Cancer Res. 1997;3:2309–2316. [PubMed] [Google Scholar]
- 32.El-Rifai W, Frierson HF, Jr, Harper JC, Powell SM, Knuutila S. Expression profiling of gastric adenocarcinoma using cDNA array. Int. J. Cancer. 2001;92:832–838. doi: 10.1002/ijc.1264. [DOI] [PubMed] [Google Scholar]
- 33.Platt-Higgins AM, Renshaw CA, West CR, Winstanley JH, De Silva Rudland S, Barraclough R, et al. Comparison of the metastasis-inducing protein S100A4 (p9ka) with other prognostic markers in human breast cancer. Int. J. Cancer. 2000;89:198–208. [PubMed] [Google Scholar]
- 34.Rudland PS, Platt-Higgins A, Renshaw C, West CR, Winstanley JH, Robertson L, et al. Prognostic significance of the metastasis-inducing protein S100A4 (p9Ka) in human breast cancer. Cancer Res. 2000;60:1595–1603. [PubMed] [Google Scholar]
- 35.Barraclough R. Calcium-binding protein S100A4 in health and disease. Biochim. Biophys. Acta. 1998;1448:190–199. doi: 10.1016/s0167-4889(98)00143-8. [DOI] [PubMed] [Google Scholar]
- 36.Davies BR, Davies MP, Gibbs FE, Barraclough R, Rudland PS. Induction of the metastatic phenotype by transfection of a benign rat mammary epithelial cell line with the gene for p9Ka, a rat calcium-binding protein, but not with the oncogene EJ-ras-1. Oncogene. 1993;8:999–1008. [PubMed] [Google Scholar]
- 37.Lloyd BH, Platt-Higgins A, Rudland PS, Barraclough R. Human S100A4 (p9Ka) induces the metastatic phenotype upon benign tumour cells. Oncogene. 1998;17:465–473. doi: 10.1038/sj.onc.1201948. [DOI] [PubMed] [Google Scholar]
- 38.Ambartsumian N, Klingelhofer J, Grigorian M, Christensen C, Kriajevska M, Tulchinsky E, et al. The metastasis-associated Mts1(S100A4) protein could act as an angiogenic factor. Oncogene. 2001;20:4685–4695. doi: 10.1038/sj.onc.1204636. [DOI] [PubMed] [Google Scholar]
- 39.Vallely KM, Rustandi RR, Ellis KC, Varlamova O, Bresnick AR, Weber DJ. Solution structure of human Mts1 (S100A4) as determined by NMR spectroscopy. Biochemistry. 2002;41:12670–12680. doi: 10.1021/bi020365r. [DOI] [PubMed] [Google Scholar]
- 40.Luecke H, Richter HT, Lanyi JK. Proton transfer pathways in bacteriorhodopsin at 2.3 angstrom resolution. Science. 1998;280:1934–1937. doi: 10.1126/science.280.5371.1934. [DOI] [PubMed] [Google Scholar]
- 41.Collaborative Computational Project, N. The CCP4 suite: programs for protein crystallography. Acta Crystallogr. D Biol. Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 42.Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr. D Biol. Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 43.Padilla JE, Yeates TO. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallogr. D Biol. Crystallogr. 2003;59:1124–1130. doi: 10.1107/s0907444903007947. [DOI] [PubMed] [Google Scholar]
- 44.Yeates TO. Detecting and overcoming crystal twinning. Methods Enzymol. 1997;276:344–358. [PubMed] [Google Scholar]
- 45.Lee S, Sawaya MR, Eisenberg D. Structure of superoxide dismutase from Pyrobaculum aerophilum presents a challenging case in molecular replacement with multiple molecules, pseudo-symmetry and twinning. Acta Crystallogr. D Biol. Crystallogr. 2003;59:2191–2199. doi: 10.1107/s0907444903019942. [DOI] [PubMed] [Google Scholar]
- 46.Sheldrick GM, Schneider TR. SHELXL: high-resolution refinement. Methods Enzymol. 1997;277:319–343. [PubMed] [Google Scholar]
- 47.Zwart P, Grosse-Kunstleve R, Adams P. CCP4 Newslett. 2006;8 [Google Scholar]
- 48.Baudier J, Gerard D. Ions binding to S100 proteins. II. Conformational studies and calcium-induced conformational changes in S100 alpha alpha protein: the effect of acidic pH and calcium incubation on subunit exchange in S100a (alpha beta) protein. J. Biol. Chem. 1986;261:8204–8212. [PubMed] [Google Scholar]
- 49.Kligman D, Hilt DC. The S100 protein family. Trends Biochem. Sci. 1988;13:437–443. doi: 10.1016/0968-0004(88)90218-6. [DOI] [PubMed] [Google Scholar]
- 50.Tarabykina S, Scott DJ, Herzyk P, Hill TJ, Tame JR, Kriajevska M, et al. The dimerization interface of the metastasis-associated protein S100A4 (Mts1): in vivo and in vitro studies. J. Biol. Chem. 2001;276:24212–24222. doi: 10.1074/jbc.M009477200. [DOI] [PubMed] [Google Scholar]
- 51.Rety S, Sopkova J, Renouard M, Osterloh D, Gerke V, Tabaries S, et al. The crystal structure of a complex of p11 with the annexin II N-terminal peptide. Nat. Struct. Biol. 1999;6:89–95. doi: 10.1038/4965. [DOI] [PubMed] [Google Scholar]
- 52.Barger SW, Wolchok SR, Van Eldik LJ. Disulfide-linked S100 beta dimers and signal transduction. Biochim. Biophys. Acta. 1992;1160:105–112. doi: 10.1016/0167-4838(92)90043-d. [DOI] [PubMed] [Google Scholar]
- 53.Stradal TB, Troxler H, Heizmann CW, Gimona M. Mapping the zinc ligands of S100A2 by site-directed mutagenesis. J. Biol. Chem. 2000;275:13219–13227. doi: 10.1074/jbc.275.18.13219. [DOI] [PubMed] [Google Scholar]
- 54.Kordowska J, Stafford WF, Wang CL. Ca2+ and Zn2+ bind to different sites and induce different conformational changes in human calcyclin. Eur. J. Biochem. 1998;253:57–66. doi: 10.1046/j.1432-1327.1998.2530057.x. [DOI] [PubMed] [Google Scholar]
- 55.Smith SP, Shaw GS. A novel calcium-sensitive switch revealed by the structure of human S100B in the calcium-bound form. Structure. 1998;6:211–222. doi: 10.1016/s0969-2126(98)00022-7. [DOI] [PubMed] [Google Scholar]
- 56.Watanabe Y, Usada N, Minami H, Morita T, Tsugane S, Ishikawa R, et al. Calvasculin, as a factor affecting the microfilament assemblies in rat fibroblasts transfected by src gene. FEBS Lett. 1993;324:51–55. doi: 10.1016/0014-5793(93)81530-d. [DOI] [PubMed] [Google Scholar]
- 57.Takenaga K, Nakamura Y, Sakiyama S, Hasegawa Y, Sato K, Endo H. Binding of pEL98 protein, an S100-related calcium-binding protein, to nonmuscle tropomyosin. J. Cell. Biol. 1994;124:757–768. doi: 10.1083/jcb.124.5.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sudo T, Hidaka H. Regulation of calcyclin (S100A6) binding by alternative splicing in the N-terminal regulatory domain of annexin XI isoforms. J. Biol. Chem. 1998;273:6351–6357. doi: 10.1074/jbc.273.11.6351. [DOI] [PubMed] [Google Scholar]
- 59.Gerke V, Moss SE. Annexins: from structure to function. Physiol. Rev. 2002;82:331–371. doi: 10.1152/physrev.00030.2001. [DOI] [PubMed] [Google Scholar]
- 60.Kwon M, MacLeod TJ, Zhang Y, Waisman DM. S100A10, annexin A2, and annexin a2 heterotetramer as candidate plasminogen receptors. Front Biosci. 2005;10:300–325. doi: 10.2741/1529. [DOI] [PubMed] [Google Scholar]
- 61.McPherson A., Jr Crystallization of proteins from polyethylene glycol. J. Biol. Chem. 1976;251:6300–6303. [PubMed] [Google Scholar]
- 62.Otwinowski Z, Minor W. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 63.Storoni LC, McCoy AJ, Read RJ. Likelihood-enhanced fast rotation functions. Acta Crystallogr. D Biol. Crystallogr. 2004;60:432–438. doi: 10.1107/S0907444903028956. [DOI] [PubMed] [Google Scholar]
- 64.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. D Biol. Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 65.Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chenna R, Sugawara H, Koike T, Lopez R, Gibson TJ, Higgins DG, et al. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003;31:3497–3500. doi: 10.1093/nar/gkg500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gouet P, Courcelle E, Stuart DI, Metoz F. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics. 1999;15:305–308. doi: 10.1093/bioinformatics/15.4.305. [DOI] [PubMed] [Google Scholar]
- 68.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 69.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 70.Potterton L, McNicholas S, Krissinel E, Gruber J, Cowtan K, Emsley P, et al. Developments in the CCP4 molecular-graphics project. Acta Crystallogr. D Biol. Crystallogr. 2004;60:2288–2294. doi: 10.1107/S0907444904023716. [DOI] [PubMed] [Google Scholar]
- 71.Morris AL, MacArthur MW, Hutchinson EG, Thornton JM. Stereochemical quality of protein structure coordinates. Proteins. 1992;12:345–364. doi: 10.1002/prot.340120407. [DOI] [PubMed] [Google Scholar]