

Abstract
Although there is a strong correlation between CAG repeat length and age at onset (AO) of motor symptoms, individual Huntington disease (HD) patients may differ dramatically in onset age and disease manifestations despite similar CAG repeat lengths. This has led to a search for genetic factors that influence AO. In order to identify such a genetic modifier, we analysed polymorphisms in the PGC-1alpha gene. Recent data indicate inhibition of PGC-1alpha function by mutant Htt supporting a link between transcriptional deregulation and mitochondrial dysfunction in HD. In > 400 HD patients, a polymorphism located within intron 2, a potential recombination hot spot, explains a small, but statistically significant, amount of the variability in AO. Our data suggest that PGC-1alpha has modifying effects on the pathogenic process in HD.
Findings
Huntington Disease (HD) is an autosomal-dominant disorder due to lesions in the striatum that cause involuntary choreiform movements and progressive behavioral and cognitive impairment. The underlying mutation is an expansion of an unstable CAG repeat in the HD gene resulting in an expanded polyglutamine tract in huntingtin protein (Htt) [1]. HD shows highly variable clinical expression, as exemplified by the wide variation of AO. The strong inverse relationship between AO and number of CAG repeats is well-defined. Yet, there is substantial variation in AO that is not explained by the HD repeat [2-6].
To date, several genetic modifiers of HD have been described in independent studies. All of these modifiers relate to various mechanisms implicated in HD pathology such as excitotoxicity, dopamine toxicity, metabolic impairment, transcription deregulation, protein misfolding and oxidative stress [5,7-11]. Additionally, genomewide linkage scans revealed potential loci that may contain genes that modify AO [12-14].
Increasing evidence implicates mitochondrial dysfunction and metabolic impairment in HD pathology [for review see [15]]. In particular, PGC-1alpha (peroxisome proliferator-activated receptor [PPAR]-g coactivator 1a) as a key transcriptional co-regulator is an important mediator in protecting neurons against oxidative damage and seems to be involved in HD pathogenesis. PGC-1alpha induces the transcription of cellular programs regulating mitochondrial respiration, oxidative stress defense and adaptive thermogenesis [16]. Recent data indicate inhibition of PGC-1alpha function by mutant Htt supporting a link between transcriptional deregulation and mitochondrial dysfunction in HD [17-19]. Altered PGC-1alpha function may, therefore, contribute to HD pathogenesis.
A total of 15 single nucleotide polymorphisms (SNPs) in the peroxisome proliferators-activated receptor γ coactivator 1 α (PPARGC1A) gene (rs2970865, rs2970866, rs4383605, rs2946386, rs2970869, rs17576121, rs2970870, rs7695542, rs2970873, rs2946385, rs12374310, rs7665116, rs2970855, rs2970848, rs8192678) were selected for genotyping in a German HD cohort of more than 400 unrelated patients recruited from the Huntington Center NRW in Bochum. Clinical assessment and determination of the motor AO was performed exclusively by experienced neurologists of the Center. The expanded CAG repeats ranged from 40 to 66 trinucleotide units and AO ranged from 16 to 76 years of age, with a mean of 45 years. HD CAG repeat sizes were determined by polymerase chain reaction using an assay counting the perfectely repeated (CAG)n units. Informed consent was obtained from all patients and controls. The studies were performed in a manner that fully complies with the Code of Ethics of the World Medical Association (Declaration of Helsinki) and was approved by the relevant university review board.
The polymorphisms were selected from NCBI SNP database due to their potential functional relevance, e.g. polymorphisms in the 5'-UTR were included due to their potential to influence gene expression, and their relative frequency. The genotype distributions of all the chosen polymorphisms were consistent with Hardy-Weinberg equilibrium (HWE).
Controlling for the effect of CAG repeat length on AO revealed an R2 value of 0.729 indicating that nearly 73% of the variation in AO could be explained by the mutation itself (Table 1). In addition to the number of the expanded CAG repeats, the modifying effects of the polymorphisms in PGC-1alpha on the AO were examined.
Table 1.
Variability in AO attributable to the CAG repeat length was assessed by linear regression using the logarithmically transformed AO as the dependent variable and SNP genotypes as independent variables.
| Model | R2 | ΔR2 | % additionally explained variance | P value | |
| HD CAG 40–66 (n = 401) | 0.729 | - | < 0.0005 | ||
| PGC-1alpha Polymorphis ms | |||||
| rs2970865 | promoter region | - | - | - | - |
| rs2970866 | promoter region | - | - | - | - |
| rs4383605 | promoter region | - | - | - | - |
| rs2946386 | promoter region | - | - | - | - |
| rs2970869 | promoter region | - | - | - | - |
| rs17576121 | promoter region | - | - | - | - |
| rs2970870 | promoter region | - | - | - | - |
| rs7695542 | promoter region | - | - | - | - |
| rs2970873 | intron1 | - | - | - | - |
| rs2946385 | intron2 | - | - | - | - |
| rs12374310 | intron2 | - | - | - | - |
| rs7665116 | intron2 | ||||
| 0,1,2 | 0.732 | 0.003 | 1.1 | 0.025 | |
| 0,1,1 | 0.733 | 0.004 | 1.5 | 0.012 | |
| rs2970855 | intron5 | - | - | - | - |
| rs2970848 | intron7 | 0.731 | - | - | 0.054 |
| rs8192678 | exon 8 | - | - | - | - |
R2 illustrates the relative improvement of the regression model when the genotypes are considered in addition to the CAG repeats; ΔR2 values quantify these differences. (-) indicates no increase in R2.
Here, we saw evidence of association of the rs7665116 SNP. The R2 statistic rose modestly but significantly (from 0.729 to 0.732, p = 0.025 in the additive model, TT vs TC vs CC, and from 0.729 to 0.733, p = 0.012, in the dominant model, TT vs TC+CC) when rs7665116 genotypes were added to the regression model (Table 1). The mean AO in patients homozygous for the wildtype allele T is 45.08 years of age, while the mean AO for patients homozygous for the C allele is 47.3 years of age. SNP rs2970848 in intron 7 shows a trend towards association, for all other polymoprhism no impact on the R2 statistic could be observed.
Figure 1 shows the HapMap r2 values among these SNPs in the HD cohort and reflects the previously reported rough subdivision into 2 main haplotype blocks [20]. The first one includes the polymorphisms in the promoter region and intron 1 (rs2970865, rs2970866, rs4383605, rs2946386, rs2970869, rs17576121, rs2970870, rs7695542, rs2970873), whereas the second includes the SNPs located 3' downstream of rs7665116 in intron 2. The two haplotype blocks are separated by a region of high recombination frequency. These results comply with the HapMap database.
Figure 1.
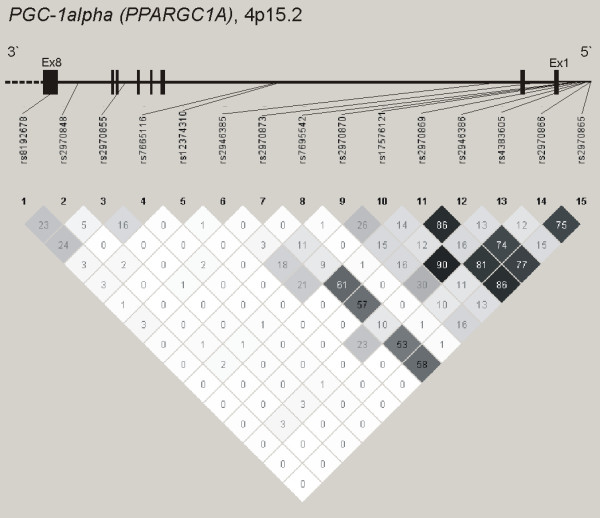
Schematic representation of the PGC-1alpha gene and linkage disequilibrium (r2) in the analysed HD cohort. Black boxes and a horizontal line represent exons and introns respectively. The numbers in the cells denote the r2 between the two SNPs corresponding to the cell. Cell shading indicates strength of r2 as shown by the number.
The rs7665116 T > C polymorphism is located within intron 2, a potential recombination hot spot. Sequence alignments of multiple species show that this SNP is located at the beginning of a 233 bp highly conserved sequence. Yet, the rs7665116 polymorphism itself is not conserved to any significant extent across species. No other SNPs are described in this conserved sequence.
In silico-analysis of rs7665116 using MatInspector [21] revealed loss of potential binding sites for cAMP-responsive element binding proteins (V$CHOP.01) in case of the C allele as compared to the wildtype T allele. On the other hand, in case of the C allele a new binding site is generated for v-Myb (V$VMYB.02) and X-box binding protein RFX1 (V$RFX1.01). Yet, it can only be speculated that the conserved region around rs7665116 represents a regulatory region controlling constitutive functions of PGC-1alpha. Functional studies are needed to assess whether PGC-1alpha is a true modifier gene and to identify the causal genetic variations contributing in the pathogenesis of HD in this region.
While this manuscript was under review, an article by Weydt et al. was published in this journal showing a modifying effect of PGC-1alpha haploblock 2 variations upon AO in an Italian cohort of 447 unrelated HD patients [22]. Our independent confirmation of their findings in a German cohort strengthens the conclusion that the PGC-1alpha gene appears to have modifying effects on the pathogenic process in HD and that it may be a therapeutically useful target for development of a treatment. Yet, it will be necessary to delineate of the precise basis for the PGC-1alpha modifier effect in order to effectively undertake a search for chemical compounds that delay HD onset.
Abbreviations
PGC-1alpha: peroxisome proliferator-activated receptor gamma co-activator
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
ETF carried out the molecular genetic studies and helped to design the study and draft the manuscript. CS and JA had ascertained the clinical status of the patients. SW interpreted the data and reviewed the manuscript. LA designed the study including statistical analysis and drafted the manuscript.
Contributor Information
Elahe Taherzadeh-Fard, Email: elahe.taherzadeh@yahoo.com.
Carsten Saft, Email: carsten.saft@rub.de.
Jürgen Andrich, Email: juergen.andrich@rub.de.
Stefan Wieczorek, Email: Stefan.Wieczorek@ruhr-uni-bochum.de.
Larissa Arning, Email: larissa.arning@rub.de.
References
- The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. Cell. 1993;72:971–983. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P, MacDonald ME, Gusella JF, Harper PS, Shaw DJ. Relationship between trinucleotide repeat expansion and phenotypic variation in Huntington's disease. Nat Genet. 1993;4:393–397. doi: 10.1038/ng0893-393. [DOI] [PubMed] [Google Scholar]
- Andrew SE, Goldberg YP, Kremer B, Telenius H, Theilmann J, Adam S, Starr E, Squitieri F, Lin B, Kalchman MA, et al. The relationship between trinucleotide (CAG) repeat length and clinical features of Huntington's disease. Nat Genet. 1993;4:398–403. doi: 10.1038/ng0893-398. [DOI] [PubMed] [Google Scholar]
- Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M, Folstein S, Ross C, Franz M, Abbott M, et al. Trinucleotide repeat length instability and age of onset in Huntington's disease. Nat Genet. 1993;4:387–392. doi: 10.1038/ng0893-387. [DOI] [PubMed] [Google Scholar]
- Kehoe P, Krawczak M, Harper PS, Owen MJ, Jones AL. Age of onset in Huntington disease: sex specific influence of apolipoprotein E genotype and normal CAG repeat length. J Med Genet. 1999;36:108–111. [PMC free article] [PubMed] [Google Scholar]
- The U.S.-Venezuela Collaborative Research Project and Wexler NS Venezuelan kindreds reveal that genetic and environmental factors modulate Huntington's disease age of onset. Proc Natl Acad Sci USA. 2004;101:3498–3503. doi: 10.1073/pnas.0308679101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger S, Bauer P, Tomiuk J, Laccone F, Didonato S, Gellera C, Soliveri P, Lange HW, Weirich-Schwaiger H, Wenning GK, et al. The S18Y polymorphism in the UCHL1 gene is a genetic modifier in Huntington's disease. Neurogenetics. 2006;7:27–30. doi: 10.1007/s10048-005-0023-z. [DOI] [PubMed] [Google Scholar]
- Arning L, Saft C, Wieczorek S, Andrich J, Kraus PH, Epplen JT. NR2A and NR2B receptor gene variations modify age at onset in Huntington disease in a sex-specific manner. Hum Genet. 2007;122:175–82. doi: 10.1007/s00439-007-0393-4. [DOI] [PubMed] [Google Scholar]
- Andresen JM, Gayan J, Cherny SS, Brocklebank D, Alkorta-Aranburu G, Addis EA, Cardon LR, Housman DE, Wexler NS. Replication of twelve association studies for Huntington's disease residual age of onset in large Venezuelan kindreds. J Med Genet. 2007;44:44–50. doi: 10.1136/jmg.2006.045153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arning L, Monté D, Hansen W, Wieczorek S, Jagiello P, Akkad DA, Andrich J, Kraus PH, Saft C, Epplen JT. ASK1 and MAP2K6 as modifiers of age at onset in Huntington's disease. J Mol Med. 2008;86:485–490. doi: 10.1007/s00109-007-0299-6. [DOI] [PubMed] [Google Scholar]
- Metzger S, Rong J, Nguyen HP, Cape A, Tomiuk J, Soehn AS, Propping P, Freudenberg-Hua Y, Freudenberg J, Tong L, Li SH, Li XJ, Riess O. Huntingtin-associated protein-1 is a modifier of the age-at-onset of Huntington's disease. Hum Mol Genet. 2008;17:1137–1146. doi: 10.1093/hmg/ddn003. [DOI] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Almqvist EW, Brinkman RR, Durr A, Dode C, Morrison PJ, Suchowersky O, Ross CA, Margolis RL, et al. A genome scan for modifiers of age at onset in Huntington disease: The HD MAPS study. Am J Hum Genet. 2003;73:682–687. doi: 10.1086/378133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JL, Hayden MR, Warby SC, Durr A, Morrison PJ, Nance M, Ross CA, Margolis RL, Rosenblatt A, Squitieri F, et al. Genome-wide significance for a modifier of age at neurological onset in Huntington's disease at 6q23-24: the HD MAPS study. BMC Med Genet. 2006;7:71. doi: 10.1186/1471-2350-7-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayán J, Brocklebank D, Andresen JM, Alkorta-Aranburu G, US-Venezuela Collaborative Research Group. Zameel Cader M, Roberts SA, Cherny SS, Wexler NS, Cardon LR, Housman DE. Genomewide linkage scan reveals novel loci modifying age of onset of Huntington's disease in the Venezuelan HD kindreds. Genet Epidemiol. 2008;5:445–453. doi: 10.1002/gepi.20317. [DOI] [PubMed] [Google Scholar]
- Gil JM, Rego AC. Mechanisms of neurodegeneration in Huntington's disease. Eur J Neurosci. 2008;27:2803–2820. doi: 10.1111/j.1460-9568.2008.06310.x. [DOI] [PubMed] [Google Scholar]
- Puigserver P, Spiegelman BM. Peroxisome proliferatoractivated receptor-g coactivator 1a (PGC-1a): transcriptional coactivator and metabolic regulator. Endocr Rev. 2003;24:78–90. doi: 10.1210/er.2002-0012. [DOI] [PubMed] [Google Scholar]
- Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59–69. doi: 10.1016/j.cell.2006.09.015. [DOI] [PubMed] [Google Scholar]
- St-Pierre J, Drori S, Uldry M, Silvaggi JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, Simon DK, Bachoo R, Spiegelman BM. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- Weydt P, Pineda VV, Torrence AE, Libby RT, Satterfield TF, Lazarowski ER, Gilbert ML, Morton GJ, Bammler TK, Strand AD, Cui L, Beyer RP, Easley CN, Smith AC, Krainc D, Luquet S, Sweet IR, Schwartz MW, La Spada AR. Thermoregulatory and metabolic defects in Huntington's disease transgenic mice implicate PGC-1alpha in Huntington's disease neurodegeneration. Cell Metab. 2006;4:349–62. doi: 10.1016/j.cmet.2006.10.004. [DOI] [PubMed] [Google Scholar]
- Iglseder B, Oberkofler H, Felder TK, Klein K, Paulweber B, Krempler F, Tregouet DA, Patsch W. Associations of PPARGC1A haplotypes with plaque score but not with intima-media thickness of carotid arteries in middle-aged subjects. Stroke. 2006;37:2260–2265. doi: 10.1161/01.STR.0000238584.57864.d4. [DOI] [PubMed] [Google Scholar]
- Werner T. Computer-assisted analysis of transcription control regions. Matinspector and other programs. Methods Mol Biol. 2000;132:337–349. doi: 10.1385/1-59259-192-2:337. [DOI] [PubMed] [Google Scholar]
- Weydt P, Soyal S, Gellera C, DiDonato S, Weidinger C, Oberkofler H, Landwehrmeyer B, Patsch W. The gene coding for PGC-1alpha modifies age at onset in Huntington's Disease. Molecular Neurodegeneration. 2009;4:3. doi: 10.1186/1750-1326-4-3. [DOI] [PMC free article] [PubMed] [Google Scholar]