

Abstract
In this study we investigated adaptive mechanisms associated with aromatase inhibitor (AI) resistance in breast cancer cells and show that sensitivity to AIs can be extended through dual inhibition of Estrogen Receptor (ER) and Human Epidermal Receptor-2 (Her-2) signaling. We utilized human ER-positive breast cancer cells stably transfected with the aromatase gene (MCF-7Ca). These cells grow as tumors in nude mice and are inhibited by AIs. Despite continued treatment, tumors eventually become insensitive to AI letrozole. The cells isolated from these Long-Term Letrozole Treated tumors (LTLT-Ca) were found to have decreased ERα levels. Our results suggest that LTLT-Ca cells survive estrogen deprivation by activation of Her-2/Mitogen Activated Protein Kinase (MAPK) pathway. Here, we demonstrate that trastuzumab (antibody against Her-2; IC50=0.4mg/ml) was very effective in restoring the ERα levels and sensitivity of LTLT-Ca cells to endocrine therapy by down-regulation of Her-2/MAPK pathway and upregulation of ERα. In contrast, trastuzumab was ineffective in the parental hormone responsive MCF-7Ca cells (IC50=4.28mg/ml) and xenografts. By blocking Her-2, trastuzumab also up-regulates ERα, aromatase expression and hyper-sensitized MCF-7Ca cells to E2. We show that trastuzumab is beneficial in hormone refractory cells and xenografts by restoring ER, implicating Her-2 as a negative regulator of ERα. In xenograft studies the combination of trastuzumab plus letrozole is equally effective in inhibiting growth of MCF-7Ca tumors as letrozole alone. However, upon the acquisition of resistance and increased Her-2 expression the combination of letrozole plus trastuzumab provided superior benefit over letrozole or trastuzumab alone.
Keywords: aromatase inhibitors, estrogen, trastuzumab, ERα, Her-2, breast cancer
Introduction
Aromatase inhibitors (AIs) such as letrozole and anastrozole that reduce estrogen production have now been shown to be more effective than antiestrogen (AE) tamoxifen in estrogen receptor positive (ER+) breast cancer patients and have few side effects. Nevertheless, not all patients respond and resistance to treatment may eventually occur in others. To investigate the mechanisms involved in the loss of sensitivity of the tumors to AIs, we developed a cell line isolated from tumors of human estrogen receptor (ER) positive breast cancer cells (MCF-7) stably transfected with aromatase (MCF-7Ca) grown in mice treated with letrozole for an extended period of time (1-3). We have previously reported that during treatment with letrozole, MCF-7Ca xenografts up-regulate Her-2 and proteins in the downstream MAPK signaling pathway (1). These cells also exhibited lower expression of ERα and apparent “estradiol independent” growth. We have also shown that signaling pathways such as the Her-2/MAPK are key regulators of the growth of letrozole refractory cells (1, 4). Similarly, several other investigators have reported the importance of members of Epidermal Growth Factor Receptor (EGFR) family (Her-2/EGFR) in resistance to endocrine therapy (5-8). We and others have suggested that the combination of EGFR/Her-2 tyrosine kinase inhibitors or trastuzumab (monoclonal humanized antibody against Her-2, Herceptin®) in combination with AIs or AEs may delay acquisition of resistance (4, 9-13).
In this study we investigated the effects of trastuzumab on the growth of letrozole refractory breast cancer cells. Upon examination, MCF-7Ca derived tumors treated with letrozole, upregulated Her-2 four weeks into treatment despite continued responsiveness to letrozole. Moreover, the level of Her-2 protein was found to be six fold higher in letrozole refractory tumors than the control tumors (1). However, when Her-2 was inhibited ERα levels were restored. This suggests that Her-2 is a negative regulator of ERα. This observation led to our hypothesis that inhibition of both the Her-2 and estrogen signaling pathways is required to prolong the responsiveness of the tumors to endocrine therapies (14, 15).
Thus, the combination of an AI plus Her-2 inhibitor could provide a substantial benefit in tumor growth inhibition when used after the acquisition of letrozole resistance.
Materials and Methods
Materials
Dulbecco's Modified Eagle Medium (DMEM), Modified Improved Minimum Essential medium (IMEM), penicillin/streptomycin solution (10,000IU each), 0.25% trypsin-1 mM EDTA solution, Dulbecco's phosphate-buffered saline (DPBS), and geneticin (G418) were obtained from Invitrogen (Carlsbad, CA). Androstenedione (Δ4A), tamoxifen, and Matrigel were obtained from Sigma Chemical Company (St. Louis, MO). Antibodies against Her-2, p-Her-2 were purchased from Upstate (now Millipore, Billerica, MA) antibodies against p-MAPK, MAPK, p-Elk-1, and p-p90RSK were purchased from Cell Signaling Technology, (Beverly, MA). Antibodies against ERα, and aromatase (CYP 19) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Radioactive ligands for ER binding assay and aromatase assay, 3H-E2 (40 Ci/mmole) and 3H-Δ4A (23.5 Ci/mmole) were purchased from Perkin Elmer (Boston, MA).
MCF-7 human breast cancer cells stably transfected with the human aromatase gene (MCF-7Ca) were provided by Dr. S. Chen (City of Hope, Duarte, CA). Letrozole was provided by Dr. D. Evans (Novartis Pharma, Basel, Switzerland). The pure antiestrogen fulvestrant and anastrozole was supplied by Dr. E. Anderson (AstraZeneca Pharmaceuticals, Macclesfield, U.K.).
Cell culture
MCF-7Ca cells were routinely cultured in DMEM supplemented with 5% FBS, 1% penicillin/streptomycin, 700μg/mL G418. LTLT-Ca cells were developed from MCF-7Ca cells as described earlier from tumors of mice treated with letrozole for 56 weeks (1). The cells were maintained in steroid-depleted medium, which consisted of phenol red-free IMEM supplemented with 5% dextran-coated charcoal-treated serum, 1% penicillin/streptomycin, and 750 μg/ml G418 and 1μM of letrozole. Cell proliferation assays performed using the MTT assay as described earlier (4). The results were expressed as a percentage of the cell number in the Δ4A-treated control wells. IC50 values for inhibitors were calculated from the linear regression line of the plot of percentage inhibition versus log inhibitor concentration.
Tumor Growth in Ovariectomized Female Athymic Nude Mice
All animal studies were performed according to the guidelines and approval of the Animal Care Committee of the University Of Maryland, Baltimore. Female ovariectomized BALB/c athymic nude mice 4-6 weeks of age were obtained from the National Cancer Institute-Frederick Cancer Research and Development Center (Frederick, MD). The mice were housed in a pathogen-free environment under controlled conditions of light and humidity and received food and water ad libitum.
The tumor xenografts of MCF-7Ca cells were grown in the mice as previously described (1, 16, 17). Each mouse received subcutaneous inoculations in one site per flank with 100μL of cell suspension containing ~ 2.5×107 cells. The mice were injected daily with Δ4A (100μg/day). Weekly tumor measurements and treatments began when the tumors reached ~ 300 mm3. Mice were assigned to groups for treatment so that there was no statistically significant difference in tumor volume among the groups at the beginning of treatment. Letrozole and Δ4A for injection were prepared in 0.3% HydroxyPropylCellulose (HPC). Trastuzumab for injection was prepared as 20 mg/ml stock solution in bacteriostatic water for injection and then diluted in 0.9% NaCl solution to obtain the required concentrations. Mice were then injected subcutaneously (sc) 5 times weekly with the indicated drugs: 100μg/mouse/day of Δ4A plus 10μg/mouse/day of letrozole or 10μg/mouse/day of letrozole plus trastuzumab or 100μg/mouse/day of Δ4A plus trastuzumab for indicated time. The doses of letrozole and Δ4A used are as previously determined and reported (1, 2). Mice in the trastuzumab group received 5mg/kg/week of the drug intra-peritoneally (ip) divided in two doses. Mice in the Δ4A and trastuzumab group were treated for 7 weeks after which they were sacrificed due to large tumor volumes; by decapitation and the blood was collected for analysis. The other groups (letrozole, trastuzumab plus letrozole, letrozole switched to trastuzumab plus letrozole and letrozole switched to trastuzumab) were sacrificed on week 28.
Western blotting
The protein extracts from tumor tissues were prepared by homogenizing the tissue in ice-cold DPBS containing protease inhibitors (18). Total 50μg of protein from each sample was analyzed by SDS-PAGE as described previously (4). Bands were quantitated by densitometry using Molecular Dynamics Software (ImageQuant®, Sunnyvale, CA). The densitometric values are corrected for loading control.
Competitive binding studies
Binding of trastuzumab to ER in MCF-7Ca cells was assessed by competitive binding assay as described before (3). MCF-7Ca cells were transferred to steroid depleted medium for 1 day before plating for the binding assay. 3H-E2 was used as a ligand and non-radio-labeled E2 was used to determine non-specific binding. Triamcinolone (1μM) was used to saturate glucocorticoid receptors.
3H2O release assay for aromatase activity measurement
The radiometric 3H2O release assay was performed as described previously (19). For pre-treatment studies, cells were treated with indicated agent for 24 hours before incubating with [1-β3H] Δ4A for 18 hours. For measuring aromatase activity in tumor samples, the tumors were homogenized in ice-cold DPBS. The resulting homogenate was used for aromatase activity assay. (19) The activity of the enzyme is corrected for protein concentration in the tumor homogenates and cells.
ERβ trans-activation assay
To measure activation of ERβ, an ELISA based ER trans-activation assay was performed as per manufacturer's guidelines (Panomics, Fremont, CA). Briefly, the nuclear-lysates of cells or tumors were generated as described by the manufacturer. Activated ER, from nuclear extracts was allowed to bind to an ER consensus binding site (ER Probe) on a biotinylated oligonucleotide. These oligonucleotides were then immobilized on a streptavidin coated 96-well plate. The ER bound to the oligonucleotide, is then detected by an antibody directed against ER. An additional horseradish peroxidase (HRP)-conjugated secondary antibody provided colorimetric readout quantified by reading absorbance at 450 nm.
RNA Extraction and Reverse Transcription (RT)
RNA was extracted and purified using the RNeasy Mini Kit (Qiagen, Valencia, CA) as per manufacturer's protocol. Total RNA concentration and purity were determined from 260 nm and 280 nm absorbances. RNA was diluted with water to 0.08 μg/μl and reverse transcribed as described by Kazi et al (20).
PCR
Analysis of ERα, CYP-19 and pS2 mRNA expression was done by conventional PCR. Each 30 μl reaction consisted of 3 μl RT, 3 μl 10X buffer, 2.4 μl dNTP mix, 3 μl 5 μM primer mix, 0.15 μl Taq DNA polymerase (Qiagen), and 18.45 μl molecular biology grade water. The following primers were used for the PCR analysis:
Human pS2 - forward 5'-ACCATGGAGAACAAGGTGAT-3' and reverse 5'-AAATTCACACTCCTCTTCTG-3' (21)
Human ERα - forward. 5'-GATCCTTCTAGACCCTTCAGTG-3' and reverse 5'-TCTTCCAGAGACTTCAAGGTGCT.
Human CYP-19 - (forward 5'-GAATATTGGAAGGATGCACAGACT-3' and reverse 5'-GGGTAAAGATCATTTCCAGCATGT-3') (19, 22)
Human 18s ribosomal RNA - (forward 5'-CAACTTTCGATGGTAGTCGC-3' and reverse 5'-CGCTATTGGAGCTGGAATTAC-3') (20)
ChIP Assay
For in vitro ChIP assay, the treated cells were washed with DPBS and fixed with 1% formaldehyde/DPBS for 10 minutes at 37°C after which the cells were washed with ice-cold DPBS containing protease inhibitors. The cells were collected into 1ml DPBS and pelleted by centrifugation at 6000rpm for 5minutes at 4°C. The cell pellet was resuspended in nuclear lysis buffer (ChIP Kit, Upstate) and incubated on ice for 15 minutes.
For in vivo ChIP assay, tumor slices were immersed in 2% formaldehyde/DPBS mixture and incubated at room temperature for 15 minutes. Fixation was stopped by adding 1M glycin and incubating for 5 minutes at room temperature. The tissues were rinsed in ice-cold DPBS containing the protease inhibitor tablet and homogenized on ice in modified RIPA buffer. The tissue homogenate was centrifuged at 12000rpm for 5 minutes at 4°C; the nuclear pellet was resuspended in nuclear lysis buffer and incubated on ice for 15 minutes. Samples were sonicated on ice for 10 × 10 sec cycles, with 20 sec pauses between each cycle. The sonicated samples were centrifuged at 14000rpm for 10 minutes at 4°C. Sonicated samples were diluted 1:10 with dilution buffer (ChIP kit) before being immunocleared in a solution containing of Protein A or G Sepharose slurry (Amersham, Piscataway, NJ) in Tris/EDTA buffer, salmon sperm DNA (Invitrogen), and normal mouse or rabbit serum (Sigma) for 2 h at 4°C. Immunocleared supernatants incubated overnight at 4°C with anti-ERα antibody (Santa Cruz Biotechnology). Protein A or G Sepharose beads and salmon sperm DNA were then added and incubated for 1 h at 4°C. The beads were then washed sequentially with 1 ml each of wash buffers. The protein-DNA complexes were then eluted by twice incubating beads in elution buffer for 10 min at room temperature with vigorous mixing. To separate immunoprecipitated protein and DNA, the pooled elutes were incubated at 65°C overnight. The DNA was purified using the Qiaquick PCR purification kit (Qiagen). Alternatively, immunoprecipitated ERα on the beads was subjected to western immunoblotting. The boiled (denatured) protein samples were resolved by SDS-PAGE and membranes were probed for Histone H3 and RNA Polymerase II.
The yield of target region DNA in each sample after ChIP was analyzed by conventional PCR. The following primers were used for PCR analysis (34 cycles at 60°C annealing temperature):
Human pS2: forward 5'-GGCCATCTCTCACTATGAATC-3' and reverse 5'-GGCAGG CTCTGTTTGCTTAAA-3' (20).
Human CYP-19 Promoter I.3/II: forward 5'-CGGAGTCAACGGATTTGGTCGTAT-3' and reverse 5'-AGCCTTCTCCATGGTGGTGAAGAC-3'. (23)
The promoter that was analyzed was I.3/II, which is the main aromatase promoter utilized in breast cancer cells lines such as MCF-7 (24) and thus measures the effect of trastuzumab on endogenous aromatase in MCF-7 cells. The MCF-7Ca cells contain human placental aromatase cDNA (placental CYP-19 uses promoter I.1 and I.2) (25).
Statistics
For in vivo studies, mixed-effects models were used. The tumor volumes were analyzed with S-PLUS (7.0, Insightful Corp.) to estimate and compare an exponential parameter (βi) controlling the growth rate for each treatment groups. The original values for tumor volumes were log transformed. In the process of searching for a suitable model we observed that the piecewise (knot at 15 weeks) model fits the data reasonably well (on groups switched after week 15 to another drug). The spline model with a single knot at time = week-15 weeks was used to accommodate the non-linearity with a piece-wise linear model.
All p values less than 0.05 were considered statistically significant. The graphs are represented as mean±standard error of the mean (SEM).
Results
Trastuzumab inhibits growth of LTLT-Ca cells and tumors and down-regulates MAPK activation
Trastuzumab was found to inhibit growth of LTLT-Ca cells in a dose dependent manner with an IC50 of 400μg/ml (2.75μM) compared to 4.28mg/ml (29.41μM) in parental MCF-7Ca cells (Figure 1A). In LTLT-Ca cells trastuzumab (100μg/ml) also decreased the activation of proteins of the MAPK pathway along with Her-2 in a time dependent manner (Figure 1B). This down-regulation was initiated as early as 15 minutes and was sustained until at least 72 hours. In addition, ERα was nearly restored to the level of the parental MCF-7Ca cells.
Figure 1A. Effect of trastuzumab on proliferation of MCF-7Ca and LTLT-Ca cells in vitro.

Viability of cells was measured by MTT assay after 6 day treatment with trastuzumab as described in Materials and Methods. The treatment with trastuzumab is significantly more effective in reducing cell viability of LTLT-Ca cells compared to MCF-7Ca cells at all doses. At each dose of trastuzumab tested (0.1mg/ml through 10mg/ml) in LTLT-Ca cells growth was inhibited significantly better (*p-value <0.0001) than in MCF-7Ca cells.
Figure 1B. Effect of trastuzumab treatment at various time points on the expression of protein in the Her-2/MAPK pathway, ERα and aromatase in LTLT-Ca cells.

Expression of proteins was examined using western imunoblotting as described in materials and methods. Blot shows Her-2, p-Her-2 at 185 kDa, p-MAPK, MAPK at 42-44 kDa, phospho-p90RSK at 90 kDa, phospho-Elk-1 at 41 kDa, , ERα at 66 kDa, aromatase at 55 kDa and β-actin at 45 kDa. The blots show a single representative of three independent experiments.
To study the effect of trastuzumab in vivo, LTLT-Ca cells were inoculated sc into ovariectomized nude mice. The following day the mice were divided in two groups. One group received letrozole (n = 5) and the other group received vehicle only (Control, n = 25). When the tumors of the mice in the control group reached ~300mm3, this group was regrouped into 5 (n= 5) and injected with vehicle or 2, 5, 10, and 20 mg/kg/week of trastuzumab ip (divided in two doses). As shown in Figure 1C, the tumors in all of the trastuzumab treated groups regressed over the course of the experiment. The dose of 5mg/kg/week trastuzumab was selected for future investigations. As shown previously (1), treatment with letrozole did not result in a statistically significant difference in tumor growth than observed in controls (p = 0.91).
Figure 1C. Effect of trastuzumab treatment at various doses on the growth of LTLT-Ca xenografts.

LTLT-Ca xenografts were grown in female OVX nude mice as described in Materials and Methods. The mice in the control and letrozole treated group exhibited similar rate of tumor growth. The difference in the exponential parameter governing growth was -0.027 (p = 0.71) over first eight weeks and 0.001 (p value = 0.97) over 19 weeks. Four doses of trastuzumab were tested. All of the tested doses caused a marked regression of LTLT-Ca xenografts. The growth rate of tumors of mice treated with trastuzumab (5mg/kg/week) was significantly different from mice in the control (p =0.0008) and letrozole treated (p = 0.0002) mice. The difference in the exponential parameter governing growth between control and trastuzumab was 0.24 (p = 0.0008). The difference in the exponential parameter governing growth between letrozole and trastuzumab was 0.27 (p value = 0.0002).
Trastuzumab restores sensitivity of LTLT-Ca cells to AEs and AIs
As shown in our earlier studies, LTLT-Ca cells exhibit cross-resistance to growth inhibitory effects of tamoxifen, fulvestrant, exemestane, and anastrozole (1). However, the combination of trastuzumab (100μg/mL) with AEs or AIs produced synergistic growth inhibition. As shown in Figure 1D, the combination of trastuzumab with AE/AI at 1μM each produced synergistic growth inhibition. The combination was statistically better than either drug alone (p<0.0001) or control (p<0.0001), whereas single drug treatment with AE or AI was not statistically different from control. As shown in Figure 2A, proliferation of LTLT-Ca cells was not inhibited with letrozole (10-12M - 10-5M), whereas co-treatment with trastuzumab (100μg/mL) and letrozole inhibited the growth of LTLT-Ca cells in a dose dependent manner. However, in MCF-7Ca cells, the combination of letrozole plus trastuzumab was not significantly different from letrozole at 10-9M - 10-5M, but different from letrozole alone at 10-12M - 10-10M (Figure 2B). Although the IC50 value of letrozole in the presence of trastuzumab was 2-log lower than that in the absence of trastuzumab, it should be noted that this effect was not seen in vivo.
Figure 1D. Effect of combination of trastuzumab with AEs tamoxifen, fulvestrant and AIs exemestane, and anastrozole in LTLT-Ca cells.

Viability of cells was measured by MTT assay after 6-day treatment with AEs and AIs in presence or absence of 100μg/ml of trastuzumab as described in Materials and Methods. The GLM method was applied to estimate and assess differences among groups' means. The cell viability was found to be significantly lower in treatment groups treated with the combination of trastuzumab plus AI or AE versus control (*p<0.0001) or trastuzumab alone (*p<0.0001) or the endocrine agent alone (*p<0.0001).
Figure 2A. Effect of combination of letrozole and trastuzumab in LTLT-Ca cells.
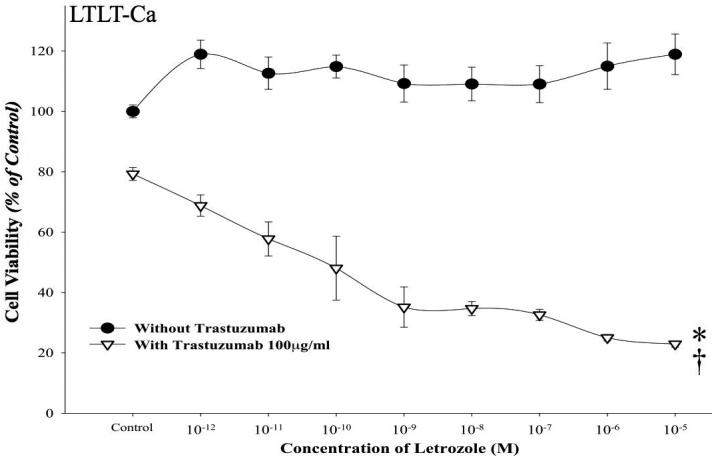
Viability of cells was measured by MTT assay after 6-day treatment with letrozole (10-12M-10-5M) alone or in presence of trastuzumab (100μg/ml) as described in Materials and Methods. The treatment with the combination of letrozole plus trastuzumab was significantly better than single drug treatment or control, *p < 0.01 (10-12M-10-9M), †p < 0.0001 (10-8M-10-5M).
Figure 2B. Effect of combination of letrozole and trastuzumab in MCF-7Ca cells.
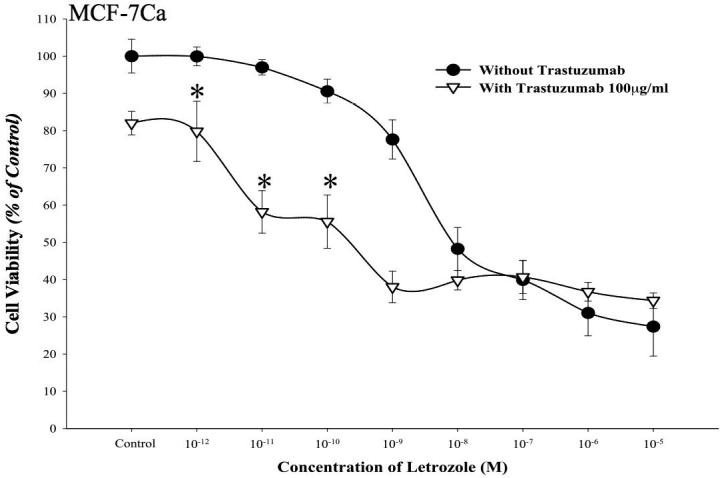
Viability of cells was measured by MTT assay after 6-day treatment with letrozole (10-12M-10-5M) alone or in presence of trastuzumab (100μg/ml) as described in Materials and Methods. Combination of letrozole plus trastuzumab was significantly better than single drug treatment or control, *p < 0.0001 (10-12M-10-9M), †p < 0.00001 (10-8M-10-5M).
The effect of combining trastuzumab plus letrozole was also examined in letrozole refractory LTLT-Ca xenografts (data not shown). The tumors of mice receiving trastuzumab plus letrozole regressed significantly in volume. The combination was significantly better than trastuzumab alone (p<0.0001), letrozole alone (p=0.0001) and control (p<0.0001), although trastuzumab alone was significantly better than control (p<0.0001). This data suggests that combining letrozole plus trastuzumab was significantly more effective than single agent in letrozole refractory LTLT-Ca xenografts (26).
Trastuzumab up-regulated ERα and E2 mediated transcription in LTLT-Ca cells
Treatment with trastuzumab up-regulated ERα in LTLT-Ca cells (Figure 1B). The maximum increase of ERα was seen at 24 hours and was found to be 0.83 fold compared to basal level of ERα in MCF-7Ca cells. A 10 fold higher level of ERα was found in LTLT-Ca cells compared to basal levels (0.83 compared to 0.08). In addition, trastuzumab pre-treatment followed by E2 treatment for 1-hour increased ER mediated transcription in a time dependent manner (Figure 2C). A 72-hour pre-treatment of LTLT-Ca cells with trastuzumab followed by 1 hour of E2 treatment induced ERα transcriptional activation to the same extent as in parental MCF-7Ca cells stimulated with E2 alone.
Figure 2C. Effect of trastuzumab treatment on trans-activation of ERαin LTLT-Ca cells.

The ERα trans-activation assay was performed as described in Materials and Methods. The treatment with trastuzumab increased ERα activation in a time dependent manner (* p<0.001).
In vitro ChIP assay and RT-PCR analysis were performed to examine the effect of trastuzumab alone or in combination with E2 on ERα mediated transcription. When stimulated with trastuzumab MCF-7Ca and LTLT-Ca cells exhibit increased transcriptional activation as evidenced by increase in the expression of pS2 mRNA, a known ERα responsive gene. This transcriptional activation was found to be dose dependent (Figure 2D). In addition to pS2, the transcription of ERα and CYP-19 gene was found to be upregulated in a dose dependent manner. In vitro ChIP assay was performed to examine recruitment of ERα to the promoter region of pS2 and aromatase gene. The western blotting for histone H3 confirm recruitment of ERα to the DNA and RNA polymerase II expression confirms transcriptional activation (Figure 3A). As shown in Figure 3B, E2 treatment in MCF-7Ca cells induces recruitment of ERα to the pS2 promoter. Similarly, trastuzumab induced association of ERα to the pS2 promoter region. However, trastuzumab plus E2 were not able to increase this association further. In LTLT-Ca cells, the basal level of promoter activity was not changed with E2 or trastuzumab treatment. However, the combination of E2 plus trastuzumab increased the recruitment of ERα to the pS2 promoter by 1.9-fold compared to the control. These results confirm that trastuzumab induces ERα mediated transcription of downstream genes such as pS2. In addition, ERα was also recruited to the aromatase I.3/II promoter after treatment with trastuzumab, E2 and the combination in LTLT-Ca cells. In MCF-7Ca cells trastuzumab and E2 increase recruitment of ERα to the aromatase I.3/II promoter, however the combination did not increase this further. The MCF-7Ca cells may require different treatment period with trastuzumab to exhibit similar effect. These results suggest that trastuzumab activated the aromatase transcription in ERα dependent manner in LTLT-Ca cells.
Figure 2D. Effect of trastuzumab at various doses on the mRNA expression of ERα, pS2 and Aromatase in MCF-7Ca and LTLT-Ca cells.
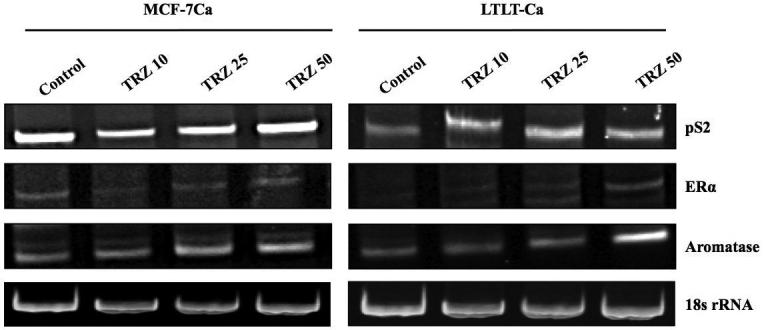
Expression of mRNA was examined using RT-PCR as described in materials and methods. Blot shows ERα at 419bp, pS2 at 245bp and aromatase (CYP-19) 293bp and 18s ribosomal RNA (rRNA) at 283bp.
Figure 3A. Effect of trastuzumab and E2 alone or in combination on the ERα mediated transcriptional activation in MCF-7Ca and LTLT-Ca cells.

The in vitro ChIP assay was performed as described in Materials and Methods. E2-Induced Recruitment of ERα to the DNA and transcriptional activation in MCF-7Ca and LTLT-Ca cells was measured by western blotting. The blot shows Histone H3 at 15kDa and RNA polymerase II at 300kDa. Left panel shows MCF-7Ca and right panel shows LTLT-Ca cells. For both panels lane 1-E2W control, lane 2 E2-10nM, lane 3-trastuzumab- 100μg/ml and lane 4-trastuzumab plus E2.
Figure 3B. Effect of trastuzumab and E2 alone or in combination on the ERα mediated transcriptional activation in MCF-7Ca and LTLT-Ca cells.

The in vitro ChIP assay was performed as described earlier. E2-Induced Recruitment of ERα to the Aromatase I.3/II and pS2 promoter in MCF-7Ca and LTLT-Ca cells was examined by PCR. The blot shows aromatase product at 317bp and pS2 at 415bp. Input indicates samples before immunoprecipitation. Left panel shows MCF-7Ca and right panel shows LTLT-Ca cells. For both panels lane 1-E2W control, lane 2 E2-10nM, lane 3-trastuzumab- 100μg/ml and lane 4-trastuzumab plus E2.
Trastuzumab amplified mitogenic effects of estradiol in vitro and in vivo
E2 stimulation of MCF-7Ca cells resulted in a typical biphasic dose response curve where maximum stimulation occurs at a concentration of approximately 10-9M. (Figure 3C). In contrast, as reported in our earlier studies, proliferation of LTLT-Ca cells is not stimulated by E2. (1, 27) However, when pre-treated with trastuzumab (100μg/mL) for 72 hours, LTLT-Ca cells exhibited a marked stimulation of proliferation at concentrations of 10-12M to 10-7M when compared to E2 alone (p<0.0001). In addition, when MCF-7Ca cells were pretreated with trastuzumab, E2 stimulated proliferation was increased at concentrations of 10-11M to 10-10M (p = 0.02 and 0.03 respectively). The results suggest that inhibition of Her-2 restores ERα and E2 sensitivity and that Her-2 is a negative regulator of ER.
Figure 3C. Effect of estradiol on proliferation of MCF-7Ca and LTLT-Ca cells in presence or absence of trastuzumab pre-treatment.

Viability of cells was measured by MTT assay after 6-day treatment with E2 (10-12M-10-5M) alone or in presence of trastuzumab (100μg/ml). When pre-treated with trastuzumab (100μg/mL), LTLT-Ca cells exhibit a significantly marked stimulation of proliferation in response to E2 at concentrations of 10-12M through 10-7M when compared to E2 alone (p <0.0001). When MCF-7Ca cells were pretreated with trastuzumab, E2 stimulated proliferation was increased at concentrations 10-11M through 10-10M (p = 0.02 and 0.03 respectively).
Pre-treatment of MCF-7Ca, MCF-7 and LTLT-Ca cells with trastuzumab for 24 hours also increased aromatase activity in a dose dependent manner (Figure 3D). At the same time, aromatase protein expression was upregulated by trastuzumab in a dose dependent manner along with ERα (Figure 4A). Trastuzumab caused an increase in the aromatase activity of MCF-7, suggesting that trastuzumab influences endogenous aromatase. To investigate whether suppressing Her-2 resulted in upregulation of ERα and aromatase was limited to LTLT-Ca cells, we carried out the same experiment with ER negative and Her-2 positive SKBr-3 cells. The results were similar confirming that inhibition of Her-2 results in increase aromatase activity (Figure 3D inset) and ERα upregulation (data not shown). However, this effect was not seen in ER and Her-2 negative MDA-MB-231 cells (data not shown). Trastuzumab did not bind competitively to ERα (Figure 4B) suggesting that the stimulatory effects were ERα dependent, not due to intrinsic estrogenic properties of trastuzumab. In addition, when MCF-7Ca cells were co-treated with increasing doses of trastuzumab for 24 hour followed by 1nM of E2, for 1-hour, transcriptional activity of ERα was found to be upregulated in dose dependent manner. This increase was found to be higher than E2 (1nM) treatment alone. (Figure 4C).
Figure 3D. Effect of trastuzumab treatment at various doses on aromatase activity of MCF-7, MCF-7Ca and LTLT-Ca cells.
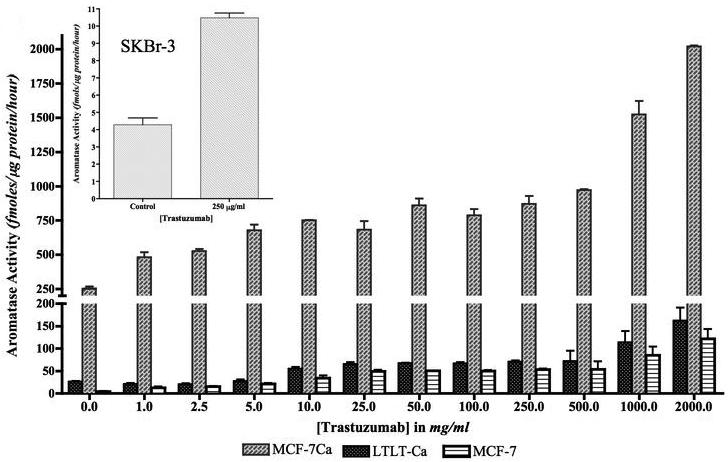
MCF-7, MCF-7Ca and LTLT-Ca cells were treated with trastuzumab at varying doses for 24 hours and then incubated with 3H-androstenedione for 18 hours. The activity of the enzyme after treatment is corrected for total protein amount after treatment. Inset: Effect of trastuzumab treatment at 250μg/ml on aromatase activity of SKBr-3 cells: SKBr-3 cells were treated with trastuzumab at 250μg/ml for 24 hours and then incubated with 3H-androstenedione for 18 hours. The activity of the enzyme after treatment is corrected for total protein amount after treatment.
Figure 4A. Effect of trastuzumab treatment at various concentrations on protein expression of ERα, Her-2 and CYP-19 in MCF-7Ca cells.

Expression of proteins was examined using western imunoblotting as described in materials and methods. Blot shows ERα at 66 kDa, CYP-19 at 55 kDa, Her-2 at 185 kDa and β-actin at 45 kDa. The blots show a single representative of three independent experiments. The blots were stripped and reprobed for β-actin to verify equal loading.
Figure 4B. Effect of trastuzumab treatment on binding affinity of estradiol to the ERα in MCF-7Ca cells.

The competitive binding study was performed as described in Materials and Methods. The difference between trastuzumab and control in the percent of radioactive estradiol bound to ERα was 18.053, which was not statistically significant (p = 0.4; using Kruskal-Wallis test and Dunn's Multiple comparison).
Figure 4C. Effect of trastuzumab treatment on at various doses on trans-activation of ERα in MCF-7Ca cells.

The ERα transactivation assay was performed to measure transcriptional activation of ERα in MCF-7Ca cells, as described in Materials and Methods. The treatment with trastuzumab increased ERα activation in a dose dependent manner. The differences between E2W control and trastuzumab treatment 100μg/ml, 200μg/ml, 400μg/ml and 1000μg/ml were -111.31 (†p = 0.06), - 156.23 (‡p<0.01), -279.91 (*p<0.001) and -305.45 (*p<0.001) and 0.04 for E2. When combined with letrozole 1μM, trastuzumab 1000μg/ml was did not stimulate ERα activation. Letrozole treatment was not significantly different from the E2W control. One Way ANOVA with post-hoc Tukey-Kramer test (* p<0.05).
The measurement of uterine weight is a useful bioassay that correlates with the levels of circulating estrogens in the body of the mice. The uteri of mice bearing LTLT-Ca xenografts treated with trastuzumab were removed and weighed at the completion of the experiment. Trastuzumab induced a statistically significant (p=0.001) dose dependent increase in uterine wet weight compared to controls (Figure 4D). Increased uterine weight suggests that co-treatment with trastuzumab acts to enhance ERα and effects of E2 on the uterus. Furthermore increased tumor aromatase activity suggests that trastuzumab increases E2 synthesis from Δ4A (data not shown).
Figure 4D. Effect of trastuzumab treatment at various doses on uterine wet weight of mice bearing LTLT-Ca xenografts.

The mice bearing LTLT-Ca xenografts were treated with trastuzumab at varying doses for 12 weeks after which the uteri were removed and weighed. The mice in control group had a mean uterine weight of 6.4 ± 2.2 mg that was the lowest uterine weight (exact 2-sided p=0.008 by the Wilcoxon test) and significantly lower than in mice treated with trastuzumab 2, 10 and 20 mg. The mean uterine wet weights ranged from 15.4 ± 2.2 mg; 20.4 ± 2.2 mg and 29.8 ± 2.2 mg respectively.
Trastuzumab is ineffective in inhibiting growth of MCF-7Ca tumors
MCF-7Ca xenografts were grown in female ovariectomized mice. When tumors reached measurable size ~300 mm3, mice were assigned to four groups, (i) control (n=5), (ii) trastuzumab - 5 mg/kg/week divided in two doses (n=5), (iii) letrozole - 10μg/day plus trastuzumab - 5 mg/kg/week divided in two doses (n=5) and (iv) letrozole - 10μg/day (n=30). The tumors were measured weekly and volume was calculated as described in Materials and Methods. Trastuzumab did not inhibit the growth of MCF-7Ca tumors (Figure 5A). The growth rate of control tumors (Δ4A-100μg/day) was (βi=0.17 ± 0.1) which was not statistically different from that of tumors in mice treated with trastuzumab (βi= 0.19 ± 0.14) over the first 7 weeks. The mice in both these groups (control and trastuzumab) were sacrificed at week 7 due to large tumor volume. The tumors and uteri of the treated mice were removed, weighed and stored at -80 °C for any additional analysis. The tumor weights (Figure 5B) of control (1.37 ± 0.57 g) and trastuzumab (2.68 ± 0.57 g,) were not statistically different; p=0.14. However, trastuzumab treatment alone was significantly less effective than other treatments; [vs. letrozole (p=0.0009) or vs. trastuzumab plus letrozole (p=0.0001)].
Figure 5A. Effect of trastuzumab alone or in combination with letrozole on the growth of MCF-7Ca xenografts.
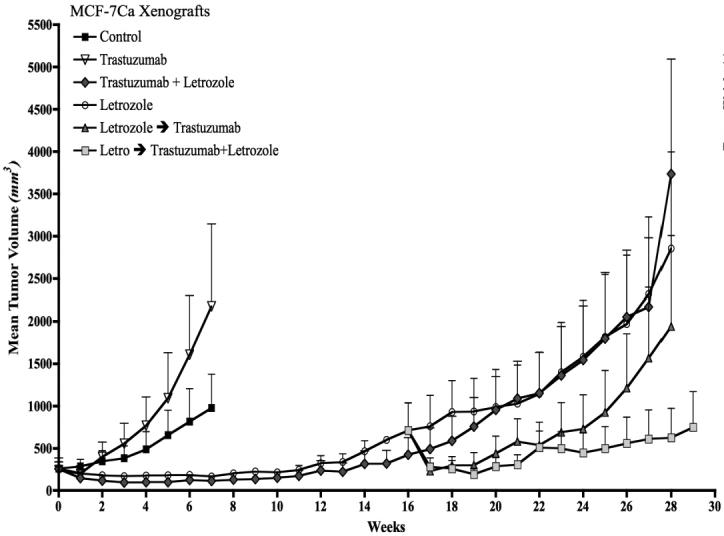
Trastuzumab (5mg/kg/week) did not inhibit the growth of MCF-7Ca tumors. The difference in the exponential parameter governing growth rate of control versus trastuzumab treatment was 0.02 ± 0.14, which was not statistically significant (p = 0.86). The difference in the exponential parameter governing growth rate of trastuzumab versus trastuzumab plus letrozole was 0.49; p = 0.0001. The difference in the exponential parameter governing growth rate of trastuzumab versus letrozole was 0.32, p = 0.0009.
The difference in the exponential parameter governing rate of letrozole versus letrozole switched to letrozole plus trastuzumab was 0.21 ± 0.08, p = 0.008. The difference in the exponential parameter governing tumor growth rate of letrozole plus trastuzumab versus letrozole switched to letrozole plus trastuzumab was 0.39 ± 0.09, p <0.0001. The difference in the exponential parameter governing rate of letrozole switched to trastuzumab versus letrozole switched to letrozole plus trastuzumab was 0.2 ± 0.08, p = 0.011, over weeks 15-28. When compared through week 29, the difference in the exponential parameter governing growth rate of letrozole versus letrozole switched to trastuzumab was 0.005 ± 0.08, p value = 0.97.
Figure 5B. Effect of trastuzumab on the tumor weight of the mice bearing MCF-7Ca xenografts.

The mean tumor weight of trastuzumab treated mice was 2.68 g ± 0.57, which was not significantly different from those of the Δ4A, treated mice (1.37 ± 0.57 g), p value = 0.14). The tumor weights of other groups are not compared due to difference in the time of termination.
As shown in Figure 5C, uteri in mice treated with trastuzumab weighed significantly more than those treated with Δ4A (p =0.008). The increase in uterine weight again suggests amplified growth stimulatory effects of E2 upon co-treatment with trastuzumab.
Figure 5C. Effect of trastuzumab on the uterine wet weight of mice bearing MCF-7Ca xenografts.

The average weight of the atrophic uterus in ovariectomized mice is ~10 mg, the greater uterine weight of mice receiving Δ4A (22.42 ± 0.92 mg) indicates that aromatase in the tumors is producing enough estrogens to maintain the uterine weight similar to intact mice in diestrus. When mice were treated with trastuzumab, the uteri weighed significantly more (74.8 mg ± 0.92) than Δ4A treated mice (Wilcoxon rank sums test, 2-sided exact p-value=0.008). The uterus weights of other groups are not compared due to difference in the time of termination.
Combination of trastuzumab plus letrozole is more effective than either drug alone in letrozole refractory tumors
As shown in Figure 5A, combination of letrozole plus trastuzumab was effective in reducing the tumor growth rate (βi = -0.04 ± 0.04, over week 0-15; βi = 0.28 ± 0.06, over weeks 15-27). However, the combination was no more effective than letrozole as single agent (βi = -0.01 ± 0.02 over weeks 0-15; βi = 0.1 ± 0.08 over weeks 15-27).
Letrozole inhibited the growth of these tumors for a prolonged period (13 weeks). Nevertheless, tumors ultimately began to grow on continued treatment and had doubled in volume by week 15. At this time, the mice were sub-divided into three groups (i) trastuzumab - 5 mg/kg/week (two doses) (n=10), (ii) letrozole - 10μg/day plus trastuzumab - 5 mg/kg/week (n=10) and (iii) letrozole -10μg/day (n=10). The experiment was terminated on week 28. Tumors in mice switched to combination therapy following letrozole resistance responded better than combination treatment from week zero (p<0.0001). This suggests that upon letrozole resistance, the addition of trastuzumab to the treatment regimen is superior to combination therapy from the beginning. The growth rate of these tumors was significantly different from the tumors of mice that stayed on letrozole treatment, (p=0.008). Tumors treated with letrozole responded better when switched to letrozole plus trastuzumab than those kept on letrozole. Letrozole refractory tumors switched to trastuzumab alone responded to the treatment only for the first 3 weeks (weeks 16-19) but then began to re-grow at the rate similar to that of the letrozole treated mice (weeks 15-28); p = 0.97. Thus, upon resistance to letrozole, the addition of trastuzumab was more effective than the switch to trastuzumab suggesting that Her-2 inhibition extends sensitivity of letrozole.
Protein expression and activity of tumors treated with letrozole and trastuzumab alone or in combination
At 28 weeks (Figure 5A) tumors of mice bearing MCF-7Ca xenografts were analyzed for expression of Her-2, p-MAPK, MAPK, aromatase and ERα protein expression by western blotting (Figure 5D). Treatment with trastuzumab increased ERα and decreased Her-2 and p-MAPK protein expression. In contrast, letrozole reduced ERα and increased Her-2 and p-MAPK levels. The combination of letrozole plus trastuzumab did not affect ERα levels and caused moderate increases in Her-2 and p-MAPK. The tumors of mice switched from letrozole to trastuzumab showed upregulation of ER and down-regulation of Her-2 and p-MAPK, compared to letrozole. This would be consistent with stimulation of growth due to release from aromatase inhibition allowing estrogen production. In mice treated with letrozole and the addition of trastuzumab upon tumor volume doubling, ERα levels were moderately increased, p-MAPK was down-regulated and Her-2 was moderately reduced.
Figure 5D. Effect of trastuzumab and letrozole alone or in combination on protein expression of ERα, Her-2, MAPK and CYP-19 in MCF-7Ca xenografts.
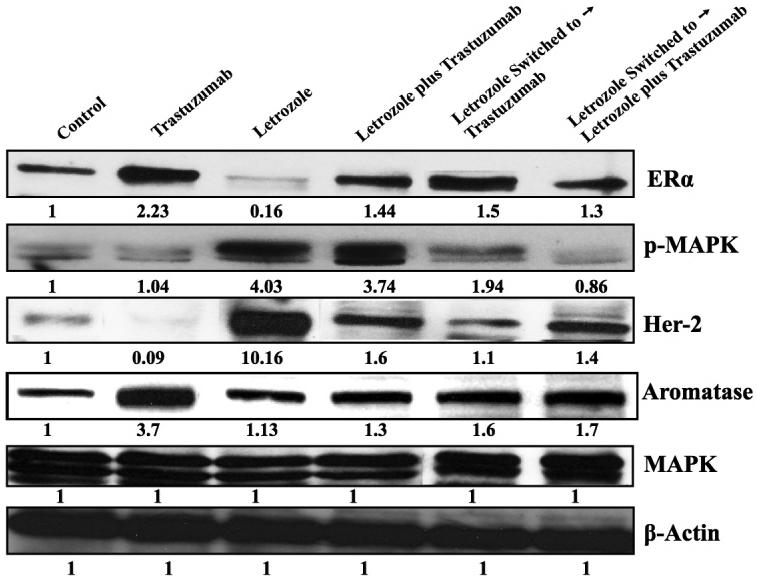
Expression of proteins was examined using western imunoblotting as described in materials and methods. Blot shows ERα at 66 kDa, Her-2 at 185 kDa, p-MAPK and MAPK at 42-44 kDa and CYP-19 at 55 kDa, β-actin at 45 kDa. The blots show a single representative of three independent experiments. The blots were stripped and reprobed for β-actin to verify equal loading.
As shown in Figure 6A, trastuzumab increased aromatase activity in the tumors, whereas letrozole inhibited aromatase activity. This increased activity was also accompanied by a 3.7-fold up-regulation of aromatase protein expression (Figure 5D). The aromatase activity in the tumor samples from letrozole, letrozole plus trastuzumab and letrozole switched to letrozole plus trastuzumab was found to be significantly lower than control (p<0.001) or, trastuzumab treatment (p<0.001). The aromatase activity of tumors from letrozole switched to trastuzumab alone treated mice was however not significantly different from control, but significantly higher than letrozole alone (p<0.01). However, aromatase protein expression in all these groups was not significantly different from control. The data suggests that letrozole maintains aromatase inhibition after prolonged treatment and switching to trastuzumab therapy removes the inhibitory effect of the AI on aromatase. Furthermore, tumors of mice treated with letrozole switched to letrozole plus trastuzumab show reduced aromatase activity, suggesting addition of trastuzumab to letrozole did not interfere directly with aromatase inhibition by letrozole.
Figure 6A. Effect of trastuzumab and letrozole alone or in combination on aromatase activity of MCF-7Ca xenografts.

The tumors of mice treated with letrozole, trastuzumab and the combinations were examined for aromatase activity as described in Materials and Methods. The activity of the enzyme after treatment is corrected for total protein amount in the tumors. Aromatase activity of MCF-7Ca xenografts treated with trastuzumab is significantly higher than control (*p<0.001). Aromatase activity of control is significantly higher compared to letrozole (p<0.001), letrozole plus trastuzumab (*p<0.001) and letrozole switched to letrozole plus trastuzumab is (*p<0.001). The aromatase activity of letrozole switched to trastuzumab was not significantly different from Δ4A treated control however significantly higher than letrozole (†p<0.01). One Way ANOVA, Tukey-Kramer multiple comparisons test.
Trastuzumab increases transcriptional activation of ERα in MCF-7Ca xenografts
The in vivo ChIP assay was used to measure the effect of trastuzumab on transcriptional activation of ERα in MCF-7Ca xenografts. Δ4A treated control and trastuzumab treated tumors exhibited active transcription of ERα, as evidenced by recruitment of histone H3 and RNA polymerase II into the transcriptional complex (Figure 6B). The treatment of MCF-7Ca tumors with trastuzumab increased recruitment of ERα to the pS2 and aromatase I.3/II promoter (Figure 6C). These findings support in vivo tumor volume data and suggest that trastuzumab treatment in a hormone sensitive breast cancer model is agonistic via its effects on Her-2 and hence the ER. As such, Her-2 inhibition without blockade of estrogen signaling in the clinical setting could be detrimental.
Figure 6B. Effect of trastuzumab and letrozole alone or in combination on the ERα mediated transcriptional activation in MCF-7Ca xenografts.

The in vivo ChIP assay was performed as described in Materials and Methods. E2-Induced Recruitment of ERα to the DNA in MCF-7Ca xenografts. The blot shows Histone H3 at 15kDa and RNA polymerase II at 300kDa. ChIP analysis was done using ERα antibody and Input indicates samples before immunoprecipitation.
Figure 6C. Effect of trastuzumab and letrozole alone or in combination on the ERα mediated transcriptional activation in MCF-7Ca xenografts.
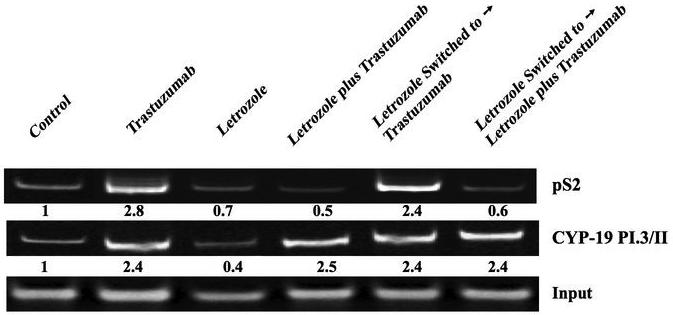
The in vivo ChIP assay was performed as described earlier. Treatment induced Recruitment of ERα to the Aromatase I.3/II and pS2 promoter in MCF-7Ca xenografts was examined by PCR. Input indicates samples before immunoprecipitation. The blot shows aromatase product at 317bp and pS2 at 415bp.
Discussions
Although AI letrozole is more effective and tumor growth is suppressed over an extended period compared to tamoxifen, eventually tumors become resistant to AI therapy (1). Previous results indicate that tumor growth is maintained by activating Her-2/MAPK signaling pathways in letrozole resistant LTLT-Ca cells and letrozole refractory tumors. In addition ER expression is decreased and cells become insensitive to E2, suggesting a hormone refractory phenotype. Our previous studies also indicated that the combination of letrozole with MAPK inhibitor PD98059 up-regulated ERα expression consistent with the reports that hyperactivation of MAPK induces loss of ERα in breast cancer cells (1, 28). Her-2 is overexpressed in about 25-30% of breast cancers and correlates with poor disease free survival and overall survival (29, 30). With the development of trastuzumab, effective targeted treatment of Her-2 overexpressing cancers has been possible. Treatment with trastuzumab also resulted in upregulation of ERα. We have shown that loss of ERα expression induced by MAPK hyper-activation is reversible as observed in findings of others (31). Our studies show that trastuzumab by blocking Her-2 can reverse the suppression of ERα. To prevent development of letrozole resistance and restore hormone sensitivity of resistant cells, we utilized trastuzumab to inhibit the Her-2/MAPK pathway in the present study.
The combination of trastuzumab with letrozole reversed resistance to the AI and restored responsiveness of the tumors. However, this effect was limited to letrozole refractory and Her-2 overexpressing cells and tumors. Trastuzumab alone was only effective for a brief period of 4 weeks in letrozole refractory tumors. In the tumors with low Her-2 levels, the letrozole plus trastuzumab combination was no more effective than letrozole single agent. However, in Her-2 overexpressing tumors, trastuzumab plus letrozole was far superior in inhibiting tumor growth compared to either single agent (Figure. 5A). These results suggest that trastuzumab restores sensitivity of LTLT-Ca cells to letrozole by inhibiting Her-2 and up-regulation of ERα. As such, blockade of the ERα pathway remains an essential component of tumor and cell growth inhibition. Up-regulation of intratumoral aromatase was also seen with trastuzumab treatment, would make the cells more responsive to AIs.
In our current study we have shown that inhibition of Her-2 with trastuzumab results in activation of ERα mediated signaling pathway. Based on this data we hypothesize that an inverse and compensatory relationship exists between Her-2 and ERα and inhibition of one pathway leads to activation of the other. Several reports have suggested that translocation of ERα to the membrane may be responsible for the crosstalk with EGFR family members in endocrine resistant phenotype (32-34) whereas a few reports have also suggested that EGFR family transmembrane receptors such as Her-2 can translocate to the nucleus and act as transcription factors (35-38).
Studies performed in trastuzumab resistant variants of breast cancer cell lines have indicated upregulation of TGFα and VEGF (39, 40). Interestingly, both VEGF and TGFα are known to be estrogen responsive genes (41). These data are consistent with our results and suggest that Her-2 suppresses ERα, as resistance to trastuzumab is associated with up-regulation of ERα responsive genes (39, 40). Our studies demonstrate that trastuzumab alone was not an effective treatment strategy in this model and the data suggest that it is likely to be due to up-regulation of intra-tumoral aromatase and ERα. To distinguish whether up-regulation of ERα was a result of increased protein synthesis or reduced degradation, cells were treated with trastuzumab in presence or absence of cyclohexamide (translational inhibitor). Western immunoblotting analysis revealed that trastuzumab treatment increased ERα to a greater extent in absence of cyclohexamide, suggesting trastuzumab modulates protein synthesis. However, even in the presence of cyclohexamide ERα protein was upregulated by trastuzumab treatment, suggesting trastuzumab treatment affects protein degradation as well (data not shown). In addition, RT-PCR analysis revealed that ERα mRNA expression is increased with trastuzumab treatment in a dose dependent manner (Figure. 2D).
In agreement with our findings, clinical studies have shown that trastuzumab as single agent has promising effects in first line treatment of about 40% breast cancer patients (42). However, the median duration of response is only about 8 months. Furthermore, intrinsic or de novo resistance to trastuzumab in Her-2 overexpressing breast cancers has been reported. As the use of trastuzumab is now extended to adjuvant treatment of breast cancer, it is important to assess the molecular effects of trastuzumab on Her-2 overexpressing breast cancer cells and determine appropriate strategies for optimal treatment. Our studies clearly demonstrate the importance of inhibiting both estrogen signaling by AI letrozole and the growth factor receptor Her-2 pathway in overcoming resistance to therapy. However, if used for treatment of hormone responsive tumors, trastuzumab may amplify estrogen signaling and stimulate tumor growth. The results indicate that inhibition of Her-2 via trastuzumab can restore the responsiveness of tumors to letrozole and trastuzumab, thus extending the use of AIs and delay the need for chemotherapy.
Acknowledgements
This work was supported by grant CA-62483 to Dr. Brodie from the National Cancer Institute, National Institute of Health. We would like to thank Dr. Armina Kazi for her help with the RT-PCR and ChIP assays.
Abbreviations used
- ER
Estrogen Receptor
- Δ4A
Androstenedione
- E2
Estradiol
- Her-2
Human Epidermal Growth factor Receptor- 2
- MAPK
Mitogen Activated Protein Kinase
- AIs
Aromatase Inhibitors
- AEs
Antiestrogens
- TRZ
Trastuzumab
- Let
Letrozole
Footnotes
Note: Novartis Pharma (Basel, Switzerland) supplied the letrozole used in this study. This work was also presented in part at the Centennial AACR Meeting, Los Angeles, CA, April 2007, 29th San Antonio Breast Cancer Symposium, San Antonio, TX, December 2006 and XIIIth International Aromatase Meeting, Baltimore, MD, September 2006.
References
- 1.Jelovac D, Sabnis G, Long BJ, et al. Activation of mitogen-activated protein kinase in xenografts and cells during prolonged treatment with aromatase inhibitor letrozole. Cancer Res. 2005;65:5380–9. doi: 10.1158/0008-5472.CAN-04-4502. [DOI] [PubMed] [Google Scholar]
- 2.Long BJ, Jelovac D, Handratta V, et al. Therapeutic strategies using the aromatase inhibitor letrozole and tamoxifen in a breast cancer model. J Natl Cancer Inst. 2004;96:456–65. doi: 10.1093/jnci/djh076. [DOI] [PubMed] [Google Scholar]
- 3.Long BJ, Jelovac D, Thiantanawat A, Brodie AM. The effect of second-line antiestrogen therapy on breast tumor growth after first-line treatment with the aromatase inhibitor letrozole: long-term studies using the intratumoral aromatase postmenopausal breast cancer model. Clin Cancer Res. 2002;8:2378–88. [PubMed] [Google Scholar]
- 4.Sabnis GJ, Jelovac D, Long B, Brodie A. The role of growth factor receptor pathways in human breast cancer cells adapted to long-term estrogen deprivation. Cancer Res. 2005;65:3903–10. doi: 10.1158/0008-5472.CAN-04-4092. [DOI] [PubMed] [Google Scholar]
- 5.Shim WS, Conaway M, Masamura S, et al. Estradiol hypersensitivity and mitogen-activated protein kinase expression in long-term estrogen deprived human breast cancer cells in vivo. Endocrinology. 2000;141:396–405. doi: 10.1210/endo.141.1.7270. [DOI] [PubMed] [Google Scholar]
- 6.Yue W, Wang JP, Conaway MR, Li Y, Santen RJ. Adaptive hypersensitivity following long-term estrogen deprivation: involvement of multiple signal pathways. J Steroid Biochem Mol Biol. 2003;86:265–74. doi: 10.1016/s0960-0760(03)00366-2. [DOI] [PubMed] [Google Scholar]
- 7.Martin LA, Farmer I, Johnston SR, et al. Enhanced estrogen receptor (ER) alpha, ERBB2, and MAPK signal transduction pathways operate during the adaptation of MCF-7 cells to long term estrogen deprivation. J Biol Chem. 2003;278:30458–68. doi: 10.1074/jbc.M305226200. [DOI] [PubMed] [Google Scholar]
- 8.Shin I, Miller T, Arteaga CL. ErbB receptor signaling and therapeutic resistance to aromatase inhibitors. Clin Cancer Res. 2006;12:1008s–12s. doi: 10.1158/1078-0432.CCR-05-2352. [DOI] [PubMed] [Google Scholar]
- 9.Shou J, Massarweh S, Osborne CK, et al. Mechanisms of tamoxifen resistance: increased estrogen receptor-HER2/neu cross-talk in ER/HER2-positive breast cancer. J Natl Cancer Inst. 2004;96:926–35. doi: 10.1093/jnci/djh166. [DOI] [PubMed] [Google Scholar]
- 10.Johnston SR. Clinical efforts to combine endocrine agents with targeted therapies against epidermal growth factor receptor/human epidermal growth factor receptor 2 and mammalian target of rapamycin in breast cancer. Clin Cancer Res. 2006;12:1061s–68s. doi: 10.1158/1078-0432.CCR-05-2125. [DOI] [PubMed] [Google Scholar]
- 11.Johnston SR. Combinations of endocrine and biological agents: present status of therapeutic and presurgical investigations. Clin Cancer Res. 2005;11:889s–99s. [PubMed] [Google Scholar]
- 12.Johnston SR. Clinical trials of intracellular signal transductions inhibitors for breast cancer--a strategy to overcome endocrine resistance. Endocr Relat Cancer. 2005;12(Suppl 1):S145–57. doi: 10.1677/erc.1.00992. [DOI] [PubMed] [Google Scholar]
- 13.Kurokawa H, Arteaga CL. Inhibition of erbB receptor (HER) tyrosine kinases as a strategy to abrogate antiestrogen resistance in human breast cancer. Clin Cancer Res. 2001;7:4436s–42s. discussion 11s-12s. [PubMed] [Google Scholar]
- 14.Brodie A, Jelovac D, Macedo L, et al. Therapeutic observations in MCF-7 aromatase xenografts. Clin Cancer Res. 2005;11:884s–8s. [PubMed] [Google Scholar]
- 15.Brodie A, Jelovac D, Sabnis G, et al. Model systems: mechanisms involved in the loss of sensitivity to letrozole. J Steroid Biochem Mol Biol. 2005;95:41–8. doi: 10.1016/j.jsbmb.2005.04.026. [DOI] [PubMed] [Google Scholar]
- 16.Yue W, Brodie A. MCF-7 human breast carcinomas in nude mice as a model for evaluating aromatase inhibitors. J Steroid Biochem Mol Biol. 1993;44:671–3. doi: 10.1016/0960-0760(93)90278-5. [DOI] [PubMed] [Google Scholar]
- 17.Yue W, Zhou D, Chen S, Brodie A. A new nude mouse model for postmenopausal breast cancer using MCF-7 cells transfected with the human aromatase gene. Cancer Res. 1994;54:5092–5. [PubMed] [Google Scholar]
- 18.Sabnis G, Goloubeva O, Jelovac D, Schayowitz A, Brodie A. Inhibition of the Phosphatidylinositol 3-Kinase/Akt Pathway Improves Response of Long-term Estrogen-Deprived Breast Cancer Xenografts to Antiestrogens. Clin Cancer Res. 2007;13:2751–7. doi: 10.1158/1078-0432.CCR-06-2466. [DOI] [PubMed] [Google Scholar]
- 19.Long BJ, Tilghman SL, Yue W, et al. The steroidal antiestrogen ICI 182,780 is an inhibitor of cellular aromatase activity. J Steroid Biochem Mol Biol. 1998;67:293–304. doi: 10.1016/s0960-0760(98)00122-8. [DOI] [PubMed] [Google Scholar]
- 20.Kazi AA, Jones JM, Koos RD. Chromatin immunoprecipitation analysis of gene expression in the rat uterus in vivo: estrogen-induced recruitment of both estrogen receptor alpha and hypoxia-inducible factor 1 to the vascular endothelial growth factor promoter. Mol Endocrinology. 2005;19:2006–19. doi: 10.1210/me.2004-0388. [DOI] [PubMed] [Google Scholar]
- 21.Saegusa M, Hashimura M, Hara A, Okayasu I. Up-regulation of pS2 expression during the development of adenocarcinomas but not squamous cell carcinomas of the uterine cervix, independently of expression of c-jun or oestrogen and progesterone receptors. J Pathol. 2000;190:554–63. doi: 10.1002/(SICI)1096-9896(200004)190:5<554::AID-PATH557>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- 22.Nakamura J, Lu Q, Aberdeen G, Albrecht E, Brodie A. The effect of estrogen on aromatase and vascular endothelial growth factor messenger ribonucleic acid in the normal nonhuman primate mammary gland. J Clin Endocrinol Metab. 1999;84:1432–7. doi: 10.1210/jcem.84.4.5641. [DOI] [PubMed] [Google Scholar]
- 23.Deb S, Zhou J, Amin SA, et al. A novel role of sodium butyrate in the regulation of cancer-associated aromatase promoters I.3 and II by disrupting a transcriptional complex in breast adipose fibroblasts. J Biol Chem. 2006;281:2585–97. doi: 10.1074/jbc.M508498200. [DOI] [PubMed] [Google Scholar]
- 24.Zhou C, Zhou D, Esteban J, et al. Aromatase gene expression and its exon I usage in human breast tumors. Detection of aromatase messenger RNA by reverse transcription-polymerase chain reaction. J Steroid Biochem Mol Biol. 1996;59:163–71. doi: 10.1016/s0960-0760(96)00100-8. [DOI] [PubMed] [Google Scholar]
- 25.Sebastian S, Bulun SE. A highly complex organization of the regulatory region of the human CYP19 (aromatase) gene revealed by the Human Genome Project. J Clin Endocrinol Metab. 2001;86:4600–2. doi: 10.1210/jcem.86.10.7947. [DOI] [PubMed] [Google Scholar]
- 26.Brodie A, Sabnis G, Macedo L. Xenograft models for aromatase inhibitor studies. J Steroid Biochem Mol Biol. 2007;106:119–24. doi: 10.1016/j.jsbmb.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Sabnis GJ, Schayowitz A, Goloubeva O, Brodie AMH. Trastuzumab increases sensitivity of hormone dependent and hormone refractory breast cancer cells to endocrine agents. AACR Meeting Abstracts. 2007 Abstract 991. [Google Scholar]
- 28.Oh AS, Lorant LA, Holloway JN, et al. Hyperactivation of MAPK induces loss of ERalpha expression in breast cancer cells. Mol Endocrinology. 2001;15:1344–59. doi: 10.1210/mend.15.8.0678. [DOI] [PubMed] [Google Scholar]
- 29.Piccart M, Lohrisch C, Di Leo A, Larsimont D. The predictive value of HER2 in breast cancer. Oncology. 2001;61(Suppl 2):73–82. doi: 10.1159/000055405. [DOI] [PubMed] [Google Scholar]
- 30.Slamon DJ, Clark GM, Wong SG, et al. Human breast cancer: correlation of relapse and survival with amplification of the HER-2/neu oncogene. Science. 1987;235:177–82. doi: 10.1126/science.3798106. [DOI] [PubMed] [Google Scholar]
- 31.Creighton CJ, Hilger AM, Murthy S, et al. Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors. Cancer Res. 2006;66:3903–11. doi: 10.1158/0008-5472.CAN-05-4363. [DOI] [PubMed] [Google Scholar]
- 32.Levin ER, Pietras RJ. Estrogen receptors outside the nucleus in breast cancer. Breast Cancer Res Treat. 2007 doi: 10.1007/s10549-007-9618-4. [DOI] [PubMed] [Google Scholar]
- 33.Santen RJ, Song RX, McPherson R, et al. The role of mitogen-activated protein (MAP) kinase in breast cancer. J Steroid Biochem Mol Biol. 2002;80:239–56. doi: 10.1016/s0960-0760(01)00189-3. [DOI] [PubMed] [Google Scholar]
- 34.Song RX, Fan P, Yue W, Chen Y, Santen RJ. Role of receptor complexes in the extranuclear actions of estrogen receptor alpha in breast cancer. Endocr Relat Cancer. 2006;13(Suppl 1):S3–13. doi: 10.1677/erc.1.01322. [DOI] [PubMed] [Google Scholar]
- 35.Hsu SC, Hung MC. Characterization of a novel tripartite nuclear localization sequence in the EGFR family. J Biol Chem. 2007;282:10432–40. doi: 10.1074/jbc.M610014200. [DOI] [PubMed] [Google Scholar]
- 36.Lo HW, Hsu SC, Hung MC. EGFR signaling pathway in breast cancers: from traditional signal transduction to direct nuclear translocalization. Breast Cancer Res Treat. 2006;95:211–8. doi: 10.1007/s10549-005-9011-0. [DOI] [PubMed] [Google Scholar]
- 37.Lo HW, Hung MC. Nuclear EGFR signalling network in cancers: linking EGFR pathway to cell cycle progression, nitric oxide pathway and patient survival. Br J Cancer. 2007;96(Suppl):R16–20. [PubMed] [Google Scholar]
- 38.Xie Y, Hung MC. Nuclear localization of p185neu tyrosine kinase and its association with transcriptional transactivation. Biochem Biophys Res Commun. 1994;203:1589–98. doi: 10.1006/bbrc.1994.2368. [DOI] [PubMed] [Google Scholar]
- 39.du Manoir JM, Francia G, Man S, et al. Strategies for delaying or treating in vivo acquired resistance to trastuzumab in human breast cancer xenografts. Clin Cancer Res. 2006;12:904–16. doi: 10.1158/1078-0432.CCR-05-1109. [DOI] [PubMed] [Google Scholar]
- 40.Valabrega G, Montemurro F, Aglietta M. Trastuzumab: mechanism of action, resistance and future perspectives in HER2-overexpressing breast cancer. Ann Oncol. 2007;18:977–84. doi: 10.1093/annonc/mdl475. [DOI] [PubMed] [Google Scholar]
- 41.Osborne CK, Shou J, Massarweh S, Schiff R. Crosstalk between estrogen receptor and growth factor receptor pathways as a cause for endocrine therapy resistance in breast cancer. Clin Cancer Res. 2005;11:865s–70s. [PubMed] [Google Scholar]
- 42.Cardoso F, Piccart MJ, Durbecq V, Di Leo A. Resistance to trastuzumab: a necessary evil or a temporary challenge? Clin Breast Cancer. 2002;3:247–57. doi: 10.3816/CBC.2002.n.028. discussion 58-9. [DOI] [PubMed] [Google Scholar]