

SUMMARY
Human salivary α-amylase (HSAmy) has three distinct functions relevant to oral health: 1) hydrolysis of starch; 2) binding to hydroxyapatite; and 3) binding to bacteria (e.g. viridans streptococci). Although the active site of HSAmy for starch hydrolysis is well characterized, the regions responsible for the bacterial binding are yet to be defined. Since HSAmy possesses several secondary saccharide-binding sites in which aromatic residues are prominently located, we hypothesized that one or more of the secondary saccharide binding sites harboring the aromatic residues may play an important role in bacterial binding. To test this hypothesis, the aromatic residues at five secondary binding sites were mutated to alanine to generate six mutants representing either single (W203A, Y276A and W284A), double (Y276A/W284A and W316A/W388A) or multiple (HSAmy-ar; W134A/W203A/Y276A/W284A/W316A/W388A) mutations. The crystal structure of HSAmy-ar was determined at a resolution of 1.5 Å as an acarbose complex and compared with the existing wild type acarbose complex. The wild type and the mutant enzymes were characterized for their abilities to exhibit enzyme activity, starch binding, hydroxyapatite and bacterial binding activities. Our results clearly showed that 1) mutation of aromatic residues does not alter the overall conformation of the molecule; 2) the single or double mutants showed either moderate or minimal changes in both starch and bacterial binding activities activity whereas the HSAmy-ar showed significant reduction in these activities; 3) the starch hydrolytic activity was reduced 10-fold in HSAmy-ar; 4) oligosaccharide hydrolytic activity was reduced in all the mutants but the action pattern was similar to that of the wild type enzyme; and 5) the hydroxyaptite binding was unaffected in HSAmy-ar. These results clearly show that the aromatic residues at the secondary saccharide binding sites in HSAmy play a critical role in bacterial binding and starch hydrolytic functions of HSAmy.
Keywords: α-amylase, site directed mutagenesis, enzyme mechanism, hydrolysis, bacterial binding, enamel binding, aromatic residues
INTRODUCTION
Alpha-amylases (α-1,4-glucan-4-glucanohydrolases, EC 3.2.1.1), widespread in all three domains of life, are ubiquitous enzymes belonging to the glycoside hydrolase family 13 1 that catalyze the hydrolysis of starch and related polysaccharides containing α-1,4-glucosidic bonds. Human salivary α-amylase (HSAmy), like other salivary proteins, is an important enzyme in the oral cavity carrying out several functions 2 and exists in several isoforms in salivary secretions. The structure of HSAmy, consisting of a single polypeptide chain of 496 amino acids 3, can be divided into three domains: domain A (residues 1-99, 170-404), domain B (residues 100-169) and domain C (residues 405-496). The molecular structure adapted by HSAmy is similar to the other mammalian α-amylases including the human pancreatic α-amylase with which it shares over 97% sequence identity 3,4.
Apart from the well-documented and studied starch hydrolytic property of HSAmy, other functions of HSAmy include the ability to bind to oral bacteria and enamel 5. Previous studies have shown that HSAmy binds to numerically prominent species of oral streptococci such as Streptococcus gordonii, S. crista and S. mitis 6. The α-amylase binding bacterial species constitute a substantial proportion of the total cultivable flora from the human teeth 5,7 and appear to colonize the mouth of animals that possess amylase activity. Such a selective binding ability may have an evolutionary advantage over bacteria that do not possess HSAmy binding property such as S. sanguis, S. oralis, S. mutans, or Actinomyces viscosus 8,9,10,11. Saliva-bacterium interactions are thought to be of key importance in the establishment of dental plaque/biofilm, which is responsible for oral diseases including caries and periodontal disease 12.
HSAmy binds to S. gordonii near the cell division sites on the surfaces of actively dividing cells 13. The binding of S. gordonii G9B to HSAmy is calcium independent, saturable and leads to a functionally irreversible complex 9. Two S. gordonii proteins, AbpA (20 kDa) and AbpB (80 kDa), mediate binding of HSAmy to the bacteria 13,14,15,16. The gene encoding the 20 kDa AbpA has been cloned and studied extensively 16,17 and its role in biofilm formation has been reported 18. A recent study provided further support for the role of AbpA in human saliva-supported biofilm formation by S. gordonii 17. While significant progress has been made in studies regarding bacterial proteins that bind to HSAmy, very little is known about the molecular features of HSAmy that are essential for the interaction.
Earlier biochemical and structural studies have commented on the similarity between bacterial and substrate binding sites and showed that bacteria-bound HSAmy has been shown to exhibit significant hydrolytic activity 19. Studies targeting residues at or in the vicinity of the active site using site directed mutagenesis have shown that mutation of three histidine residues in HSAmy did not affect the respective mutant’s (H52A, H299A and H305A) binding to S. gordonii but affected their hydrolytic activities 20. Recent structural studies showed the presence of multiple secondary oligosaccharide binding sites in HSAmy (Fig. 1) 21. The observation of secondary saccharide binding studies in HSAmy underscores the similarity noted between bacterial binding and substrate binding sites 19. The prominent feature at these secondary saccharide binding sites is the presence of aromatic residues (Tyr and Trp), which is consistent with many sugar-binding proteins that utilize stacking interaction provided by them for saccharide binding 22. The importance of aromatic residues in substrate binding and enzymatic activity of HSAmy was highlighted recently when an aromatic residue at the active site, W58, was mutated to Ala 23. The mutant W58A showed a drastic loss in hydrolytic activity. Since bacteria bound HSAmy retains hydrolytic activity 19, the utilization of active site aromatic residue for bacterial binding would negatively impact on hydrolytic activity. Hence bacterial binding at the active site would be inhibitory for the survival of bacteria. Therefore, we hypothesized that S. gordonii may utilize one or more of the secondary saccharide binding sites in HSAmy 21. We reasoned that the conspicuous location of aromatic residues and the potential stacking interactions at these sites might play a significant role in bacterial binding as well as in starch binding activities. In order to test these hypotheses, we used site directed mutagenesis to replace tryptophan and tyrosine residues with alanine residues at the four secondary sites observed in HSAmy 21. We generated single, double and multiple mutants of HSAmy which were expressed in a baculovirus expression system and the results of the characterization of the purified mutant enzymes for their enzyme activity, bacterial, starch, and hydroxyapatite (HA) binding abilities are reported here.
Figure 1.

Secondary saccharide binding sites in HSAmy. The six aromatic residues at sites 2, 3,4 and 5 were mutated to Ala. Site 2 and site 3 have two aromatic residues each whereas sites 4 and 5 have one each. The sites 2 and 3 are located on opposite surface locations in domain A. While the residues Y276 and W284 occur on the surface as part of domain A and residues W316 and W388 occur in the interface between domains A and C.
RESULTS
Design of the mutants of HSAmy
The hypothesis that aromatic residues are involved in starch and/or bacterial binding of HSAmy was tested using the mutants listed in Table 1. Of the four secondary sites present in HSAmy, residues at sites 2, 3, 4 and 5 (Fig. 1), which bound at least two saccharide units, were mutated either singly or in combination. The mutant DNA was derived from the parent cDNA for HSAmy by mutating one site at a time (mutants 1, through 3) or 2 sites (mutants 4 and 5) or all aromatic residues (mutant 6; HSAmy-ar). When necessary, subsequent mutations were introduced by using the single or double mutant DNAs. We designed HSAmy-ar to test whether the mutational effect on starch and bacterial binding, if any, is additive compared to the single or double mutations. In this regard, we have reported that single mutations at W316 and W388 also did not affect bacterial binding 24. Our earlier studies have shown that mutation of W58 at the active site completely abolished the enzyme activity 23. Therefore, the aromatic residues at the active site, W58, W59, Y62 and Y151 were not mutated. The mutant proteins of HSAmy were expressed using the Bac-To-Bac Baculovirus Expression system described in Material and Methods and purified using procedures already established in our laboratory 21,23. HSAmy mutant proteins were obtained at greater than 99% purity with a yield of 3-5 mg/L of the cell culture.
Table 1.
Mutagenic primers used in the construction of HSAmy mutants.
| Mutation | Sequence | Primer |
|---|---|---|
| W134A | 5’ - CCCATATTCTGGAGCGGATTTTAATGATGG - 3’ | Forward |
| W134A | 5’ - CCATCATTAAAATCCGCTCCAGAATATGGG- 3’ | Reverse |
| W203A | 5’ - CAAGCACATGGCGCCTGGAGAC- 3’ | Forward |
| W203A | 5’ - GTCTCCAGGCGCCATGTGCTTG - 3’ | Reverse |
| Y276A | 5’ - GAGAGAAGATGTCTGCGTTAAAGAACTGGG - 3’ | Forward |
| Y276A | 5’ - CCCAGTTCTTTAACGCAGACATCTTCTCTC - 3’ | Reverse |
| W284A | 5’ - GGGAGAAGGTGCGGGTTTCATGC- 3’ | Forward |
| W284A | 5’ - GCATGAAACCCGCACCTTCTCCC- 3’ | Reverse |
|
| ||
| Mutants Generated | ||
| Mutant 1: W203A | ||
| Mutant 2: Y276A | ||
| Mutant 3: W284A | ||
| Mutant 4: Y276A/W284A | ||
| Mutant 5: W316A/W388A | ||
| Mutant 6 (HSAmy-Ar): W134A/W203A/Y276A/W284A/W316A/W388A | ||
Secondary structure content of HSAmy and HSAmy-ar
To ensure that the mutations did not alter the overall conformation of HSAmy-ar, we measured the structural characteristics of the wild type and the mutant proteins using circular dichroism spectroscopy. As can be seen in Fig. 2, both HSAmy and HSAmy-ar showed characteristics of a protein having ordered secondary structures suggesting that the overall conformation of HSAmy is still retained in HSAmy-ar. Notably, the wild type structure has an unusual minimum at 227 nm, while the HSAmy-ar spectrum resembles a typical protein spectrum. The unusual negative minimum at 227 nm observed in the wild type enzyme HSAmy could be attributed to arise from interactions involving aromatic residues 25,26. The lack of this minimum in HSAmy-ar is consistent with the absence of aromatic residues due to mutation. The estimation of the secondary structure content from the spectra (Table 2), were analyzed using the program suite CD-PRO 27. While the values estimated by the individual programs were inconsistent among each other, comparison of the average values with those derived from the X-ray structure (PDB Code 1SMD) (α-helix 19% and β-sheet 21%) suggests that no gross conformational changes have occurred in the structure of HSAmy-ar. However, to further ascertain that the structure did not change, we undertook the crystal structure analysis of HSAmy-ar.
Figure 2.

CD spectra of HSAmy and HSAmy-ar proteins. The double minima of the spectrum are characteristic of proteins with significant secondary structures such as α-helix and β-sheet.
Table 2.
Secondary structure content analysis of CD spectra of HSAmy and HSAmy-ar (values in parentheses).
| Method | α-helix | β-sheet | Turns | other | Sum |
|---|---|---|---|---|---|
| CDNN | 0.24 (0.21) | 0.24 (0.27) | 0.19 (0.20) | 0.40 (0.43) | 1.07 (1.11) |
| SELCON | 0.14 (0.13) | 0.16 (0.29) | 0.30 (0.20) | 0.36 (.028) | 0.96 (0.90) |
| CONTIN LL | 0.28 (0.16) | 0.26 (0.33) | 0.20 (0.21) | 0.26 (0.30) | 1.00 (1.00) |
| CDSSTR | 0.24 (0.09) | 0.27 (0.35) | 0.20 (025) | 0.28 (0.31) | 1.00 (1.00) |
| Average | 0.23 (0.15) | 0.23 (0.31) | 0.22 (0.21) | 0.33 (0.33) | 1.00 (1.00) |
| Stdev | 0.05 (0.04) | 0.05 (0.03) | 0.04 (0.02) | 0.06 (0.06) | 0.04 (0.07) |
| X-ray | 0.19 | 0.21 | 0.21 | 0.39 | 1.00 |
Overall structure of HSAmy-ar
An important feature of the structure of HSAmy-ar is that in spite of the number of mutations, it exhibits an overall fold very similar to the native molecule ((Fig. 3); r.m.s deviation, 0.18 Å). In support of the CD studies that showed retention of conformation as outlined above, we note that some of the salient features of the α-amylase structures such as the chloride binding site, the calcium binding site, the mobile loop containing the His305 residue are all well-defined in the mutant as well as the complex HSAmy-ar/acarbose structures. Analysis of the difference and weighted density maps clearly showed the absence of density corresponding to the side chain at the mutated sites (Fig. 4). The data collection and refinement parameters are given in Table 3.
Figure 3.

Superposition of the HSAmy (green) and HSAmy-ar structures (magenta). No significant gross conformational deviations are present between the two structures.
Figure 4.

Drawing of the 2Fo-Fc difference density maps contoured around the mutated residues of HSAmy-ar showing A134, A203, A276, A284, A316 and A388. The density contours are at 1.0 σ. The absence of density for the aromatic moiety of the Trp side chain is evident in all mutants. The final refined coordinates of the corresponding protein residues are overlaid and shown as thick black lines.
Table 3.
Summary of diffraction data collection parameters and structure refinement statistics
| Parameters | HSAmy-ar/acarbose |
|---|---|
| Space group | P212121 |
| Cell dimensions: a,b,c (Å) | 52.74×73.72×134.10 |
| Resolution range (Å) | 30.9 – 1.5 |
| Total / unique .no. of reflections | 84462/83812 |
| Completeness (%): overall / last shell | 99.4/99.2 |
| Mean I/σI: overall/last shell | 40.9/3.6 |
| Multiplicity | 6.3 |
| Rmerge (%) (overall / last shell) | 10.2/36.8 |
| Number of protein/ solvent/ other | 3934/444/47 |
| Number of reflections used | 83812 |
| <B> factor (Å2): protein/solvent/other | 33.1/37.1/37.3 |
| R-/Rfree (%) | 0.179/0.196 |
| r.m.s deviations: bonds (Å) / angles (°) | 0.011/1.329 |
Structure of HSAmy-ar/acarbose complex
Examination of the structure of the acarbose complex showed that the saccharide binding in HSAmy-ar/acarbose was restricted to the active site only. Unlike the wild type/acarbose complex, wherein acarbose was modified into a hexasaccharide (PDB code 1MFV), only a pseudotetrasaccharide (glucose-acarvosine-glucose) was fully occupied in the complex HSAmy-ar/acarbose (Fig. 5a). The four sugar rings of the bound ligand occupy subsites -2, -1, +1 and +2 28 in a manner nearly identical to the corresponding tetrasaccharide component in the wild type enzyme (Fig. 5b) 21. The two subsites, subsites -4, and -3, corresponding to the non-reducing end observed in 1MFV, are not clearly defined in the maps. The presence of the modified tetrasaccharide and not acarbose at the active site of HSAmy-ar/acarbose complex suggests that HSAmy-ar appears to possess the enzyme activity in the crystal, a property that has been noted in several α-amylase crystals 21,29,30,31,32,33,34,35. The saccharide units at subsites -4 and -3, if any, are not clearly defined the electron density maps, probably due to positional disorder the sugar units are not clearly defined in the electron density maps. This positional disorder is even present, to some extent, at subite -2 since atoms of the sugar unit at this site have high thermal parameters (<B> > 43 Å2) compared to the other atoms of subsites -1, +1 and +2 (<B> of 24 Å2).
Figure 5.

a) The density for the bound oligosaccharide, contoured at 1 σ is shown. Note that there are only four carbohydrate moieties that could be fitted. The identity of the residues were determined using the method described earlier 21. The final refined coordinates of the corresponding oligosaccharide residues are overlaid and are shown as thick black lines. b) Superposition of the pseudotetrasaccharide observed in the HSAmy-ar/acarbose structure with that of the pseudohexasaccharide observed in the recombinant HSAmy complexed with acarbose (PDB Code 1MFV). The two corresponding tetrasaccharides fit well at the active sites.
The structural analyses also reveal that the inhibitor binding at subsites -2, −1, +1 and +2 has little impact on the interactions and orientation of the catalytic groups in the active site. Interestingly, the complex HSAmy-ar/acarbose does not display secondary sugar-binding site on its surface centered on the residues W203, Y276A/W284 and W316/W388 21. In a separate structural study 24, we observed that the structures of complexes, W316A/acarbose (PDB Code 3BLK) or W388/acarbose (PDB Code 3BLP), were also devoid of any bound acarbose molecule at site 3 containing residues W316 and W388 24. Clearly, mutation of aromatic residues at these sites has resulted in the inability of these sites to bind a saccharide moiety as observed in the wild type enzyme.
Hydrolytic activity and raw starch binding ability of HSAmy enzymes
The specific activity of HSAmy mutants for the starch hydrolysis was measured and compared against the same activity for wild type (Fig. 6a). For HSAmy-ar, the specific activity decreased by 87% (a 10-fold reduction) compared to HSAmy indicating that the absence of saccharide binding ability at the secondary binding sites in HSAmy-ar has resulted in a significant reduction in the enzyme activity. The specific activity of the enzyme is also affected by about 2-fold when the aromatic residues at position 203 is mutated. Among the single or double mutants studied, W203A exhibits the least activity against starch hydrolysis. Although the site harboring the aromatic residue W284 has been observed in mammalian α-amylases as the second saccharide-binding site 36, mutations at this site did not alter the specific activity of the enzyme to any significant extent. Double mutations at site 3 harboring residues W316 and W388, (mutant W316A/W388A) also did not affect activity. Based on the specific activity results, it appears that a single mutation does not reduce the activity of the wild type enzyme to the extent of HSAmy-ar although the mutant W203A has affected the activity to a larger extent than any of the mutants studied. To achieve a reduction equivalent to HSAmy-ar, a synergistic effect between residue W203 (site 4) and others might be necessary.
Figure 6.

Binding abilities of HSAmy and HSAmy mutants. a) The specific activity for starch hydrolysis for the various mutants compared to HSAmy. b) Comparison of starch binding. Higher value for HSAmy indicates better binding to starch; c) Bacterial binding abilities of HSAmy mutants. About 10 7 bacterial cells were incubated with increasing amounts of [125I] labeled enzymes until saturation was achieved. HSAmy (
 ), W203A (
), W203A (
 ), Y276A (
), Y276A (
 ), W284A (
), W284A (
 ), Y276A/W284A (
), Y276A/W284A (
 ), W316A/W388A (
), W316A/W388A (
 ) and HSAmy-ar (
) and HSAmy-ar (
 ); d) AbpA binding. The efficiency of AbpA binding was calculated from the residual specific activities of the enzymes in the supernatant with and without added AbpA.
); d) AbpA binding. The efficiency of AbpA binding was calculated from the residual specific activities of the enzymes in the supernatant with and without added AbpA.
The reduction in specific activity of HSAmy-ar towards starch hydrolysis is also reflected in the ability of HSAmy-ar to bind to raw starch as shown in Fig. 6b. The starch binding ability rb 37 of HSAmy was 91% whereas the value of rb for HSAmy-ar was only 13%. In agreement with the starch hydrolyzing activity, HSAmy-ar shows the least starch binding ability. However, the starch binding ability of W203A is not significantly affected and its rb value (80%) is comparable to the rb value of the wild type enzyme. Among the mutants studied, mutant W284A and the double mutant Y276A/W284A, both possess an rb value of only 60% suggesting that W284 is a significant residue in the starch binding activity exhibited by HSAmy. Interestingly, greater effect is seen in the double mutant W316A/W388A, suggesting the possibility that site 3 residues may contribute, at least partially, to the starch binding ability of HSAmy. Taken together, the higher reduction in starch binding ability of HSAmy-ar suggests that a synergistic effect involving multiple residues might be involved in starch binding by HSAmy.
Bacterial binding ability of HSAmy enzymes
The saturability of binding of HSAmy and HSAmy mutants to S. gordonii G9B was analyzed using a method previously described 9 and the results are provided in Fig. 6c. Clearly, the three single mutants (Table 1) although showing some reduction in bacterial binding, did not exhibit a reduction as high as HSAmy-ar (Fig. 6c). Perturbation of all of the aromatic residues in the secondary saccharide binding sites (HSAmy-ar) has resulted in at least 2-3 log unit reduction in the ability of HSAmy-ar to bind S. gordonii compared to HSAmy. Among the single and double mutants tested for bacterial binding activity, the double mutant Y276A/W284A showed an intermediate reduction in activity compared to HSAmy-ar. As with starch binding ability noted above, the secondary saccharide binding site 2, harboring residue W284 is again involved in a second function of HSAmy, namely, the bacterial binding.
Previous studies have shown the identities of bacterial protein(s) responsible for this interaction between S. gordonii and HSAmy 16,17. In this study, we also cloned, expressed and purified AbpA (Fig. 7; see Methods) to analyze the efficiency of binding between HSAmy mutants and AbpA. We noted that when equimolar mixtures of AbpA and HSAmy are incubated in ice, the complex HSAmy:AbpA precipitates (data not shown). Using this property, we determined the residual enzyme activity in the supernatant of the equimolar mixture of HSAmy enzymes and AbpA. The efficiency of binding to AbpA for HSAmy was 70% compared to 9% for HSAmy-ar (Fig. 6d). Both W203A and the site 2 mutants (Y276A, W284A, and the double mutant Y276A/W284A) showed a moderate reduction in efficiency but the site 3 mutant W316A/W388A, showed no reduction in efficiency compared to HSAmy suggesting that the site 2 and site 4 residues and not the site 3 residues are involved in the binding to AbpA. Our results from both bacterial binding and AbpA binding are in agreement with each other with respect to the fact that aromatic residues are involved in such a binding interaction. In addition, our results also extend our knowledge with respect to the identity of the site and residues involved in the binding. A common feature that is highlighted by our results is the fact that the secondary binding sites 2 and 4 are likely to play a major role in bacterial binding as well.
Figure 7.

Purification profile for AbpA using Ni2+ affinity column. The left Y-axis shows the UV absorbance at 280 nm and the right Y-axis shows the imidazole gradient. The protein AbpA elutes at about 100 mM imidazole concentration indicated by the rectangular box. Inset shows the purified protein electrophoresed on a 10% SDS-PAGE gel. The solid arrow represents the 20 kDa band corresponding to AbpA.
AbpA binding using SPR spectroscopy
Binding of both HSAmy and HSAmy-ar (wild-type or mutant) to immobilized AbpA was observed as an increase in resonance units (RU). As shown in Fig. 8, incremental concentrations of the mutant (1.25 - 10 μM) resulted in a dose-dependent increase in binding of HSAmy-ar. Analysis of the binding data (BIAevaluation3.2 software) yielded a KD (equilibrium dissociation constant) value of 14.9 μM. The other apparent binding parameters are given in Table 4. These values indicate a weak affinity of the mutant HSAmy-ar coupled to faster dissociation and slower association rate. In sharp contrast, the value of KD for HSAmy (0.0113 μM), which is over 1300-fold higher compared to the KD for HSAmy-ar, suggests a very tight binding to AbpA.
Figure 8.
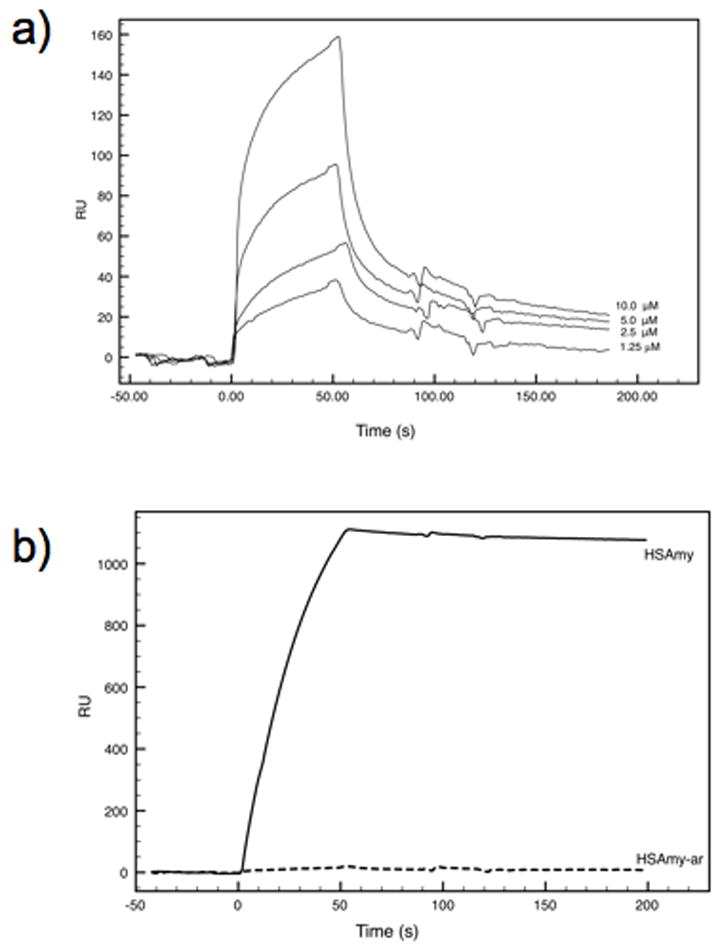
Binding of HSAmy and HSAmy-ar to AbpA. a) A set of sensorgrams depicting binding of the mutant HSAmy-ar, supplied at concentrations ranging from 1.25 μM to 10 μM. The curve at 20 μM (not shown) was identical to that obtained with 10 μM. The curves were adjusted for the effect of buffers and salts on resonance signals using the uncoated (blank) surface in flow cell 1 as a reference. b) Sensorgrams comparing the binding of HSAmy (thick line) and mutant HSAmy-ar (dashed line) to AbpA. Both proteins were supplied in the mobile phase at 1 μM concentration.
Table 4.
Kinetic parameters for the binding of AbpA to HSAmy enzymes
| Parameter | HSAmy-ar | HSAmy |
|---|---|---|
| kon (M-1s-1) | 1134 | 27006 |
| koff (s-1) | 0.017 | 3.07 ×10-4 |
| KA (M-1) | 6.6 × 104 | 8.79 × 107 |
| KD (M) | 1.49 × 10-5 | 1.13 × 10-8 |
| Requilibrium (RU) | 20 | 1419 |
HA binding ability
The abilities of HSAmy and its mutants to bind to HA crystallites were compared. The HA binding ability of HSAmy-ar was comparable to the binding ability of HSAmy at all concentrations of labeled enzymes tested (Fig. 9). Since the HA binding ability of HSAmy-ar did not change, the HA binding abilities of the other mutants generated here were not tested as the mutations in them are a subset of the mutations in HSAmy-ar. Thus, the aromatic residues in HSAmy do not affect the conformation of the HA binding sites in the mutants. The absence of reduction in HA binding by the aromatic residue mutation is expected since the mutation at the secondary saccharide binding sites did not involve any acidic residues, which are considered to contribute to the HA binding 38,39,40. Studies in our laboratory in this regard have shown that when several acidic residues are mutated, a significant reduction in HA binding is observed (unpublished data).
Figure 9.

Comparison of HA binding between labeled HSAmy and HSAmy-ar. HSAmy and HSAmy-ar enzymes bound to HA crystallites equally well.
Oligosaccharide hydrolysis
Since the starch hydrolyzing activity differences might arise as a result of longer substrate, we compared the abilities of the mutants to hydrolyze oligosaccharides, maltoheptapentaoside and maltopheptaoside and analyzed the product distribution with HPLC (Fig. 10a and b). The pentasaccharide is of the right length to fill the active site subsites whereas the heptasaccharide is longer. The results as depicted in Fig.10a clearly show that all mutant enzymes possess the ability to hydrolyze the pentasaccharide substrate and generate an action pattern similar to HSAmy. Of notable interest are the hydrolytic abilities of W203A, W284A and HSAmy-ar. These three mutants generated less of the products, especially W203A and HSAmy-ar, which also required higher enzyme concentration (600 nM vs. 60 nM for HSAmy and other mutants) and time (5 min vs. 2 min for HSAmy and other mutants). Similar results were obtained for the heptasaccharide substrate as well.
Figure 10.
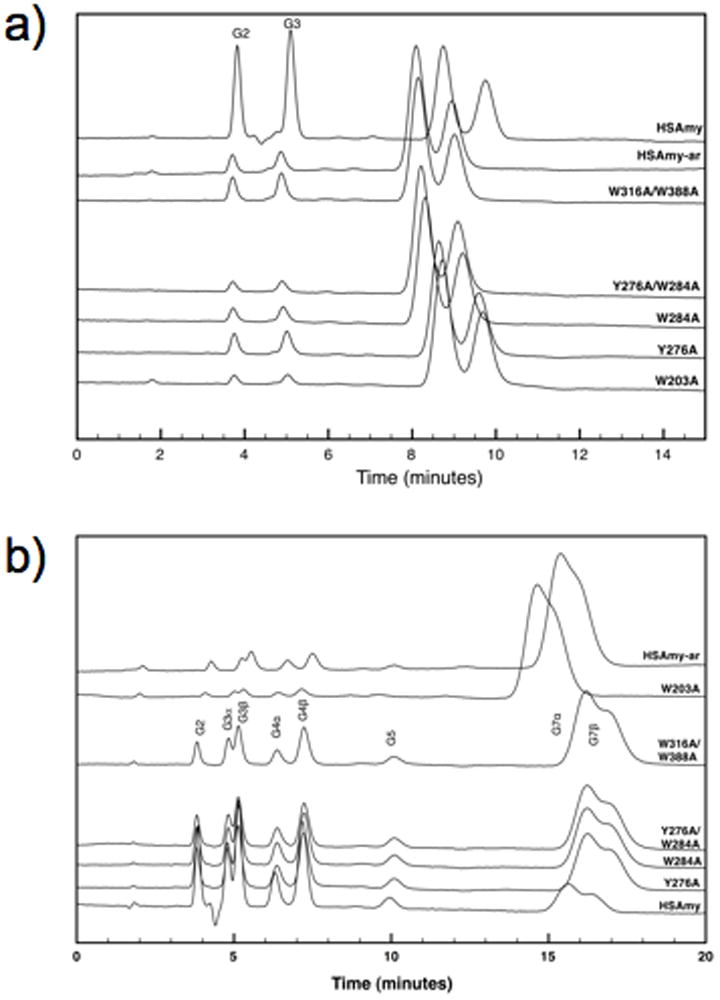
Hydrolytic action profiles of the HSAmy mutants for G5 (a) and G7 (b). The action pattern is identical for the mutants albeit differences in the amount of products generated.
DISCUSSION
Circular dichroism spectroscopy of HSAmy and HSAmy-ar
While the CD spectra in the far-ultraviolet region is very well known to reflect the secondary structure of proteins, the tertiary structure sometimes influences the spectrum due to the interaction among aromatic residues. Examples in which the side chain of aromatic residues, notably Trp, influence the peptide CD of some proteins have been reported in the literature 25. It has also been shown that the exciton coupling between Trp47 and Trp74 side chains of dihydrofolate reductase cause a change in the far UV region in the W74L mutant compared to the wild type 26. The CD spectrum of HSAmy suggests that the aromatic residues are responsible for the change in the far UV region compared to a typical protein spectrum. When some of these residues are mutated, the CD spectrum of the mutant HSAmy-ar reverts back to exhibit a typical protein spectrum. The structure of HSAmy shows that the aromatic residues W134, W213, Y276, W284, W316 and W388 are in the vicinity of other aromatic residues. In addition, the active site contains residues W58 and W59, which are juxtaposed next to each other. If these interactions have contributed to the CD spectrum, then it is entirely possible for the mutant HSAmy-ar would exhibit an altered spectrum compared to HSAmy. It is also conceivable that the estimation of secondary structure content in Table 2 using algorithms based on well-behaved proteins structures will be somewhat skewed due to the unusual contributions from the aromatic residues.
Structures of HSAmy enzymes and starch hydrolytic activity
In the HSAmy-ar complex, clear density is present only for 4 residues (1.0 σ) in the 2Fo-Fc map at subsites -2, -1, +1, and +2 (Fig. 5a) corresponding to a pseudotetrasaccharide unit at the active site. That this tetrasaccharide is not acarbose is clearly evident from the electron density map since acarbose (acarvosine-glucose-glucose) has two glucose units at the reducing end unlike the present structure where in the pseuodtetrasaccharide is glucose-acarvosine-glucose. Since the electron density maps clearly showed a sugar moiety attached to the HMC moiety at the non-reducing end, the bound tetrasaccharide could only be generated after the initial hydrolysis of acarbose and subsequence modification of the trisaccharide product. One of the characteristics of α-amylases soaked with acarbose is their ability to rearrange the pseuodtetrasaccharide acarbose 21,29,31,32,34,41. However, in most α-amylases, including the crystal structures of W316A/acarbose (PDB Code: 3BLK) and W388/acarbose (PDB Code: 3BLP) 24, a minimum of five sugar units is observed whereas in the structure of HSAmy-ar, only a tetrasaccharide is observed. Although in the HSAmy-ar, additional sugar units are absent, the observation of a modified acarbose bodes well with the ability of HSAmy-ar to carry out catalytic reaction in the crystal.
The weak binding at subsites -3 and -4 in HSAmy-ar/acarbose complex could be due to fact that the saccharide binding itself could be affected at these subsites due to flexible binding pocket. In this regard, it is of interest to note that HSAmy-ar exhibits differences in the <B> values, especially for residues in the B domain (residues 100 through 167) compared to wild type HSAmy. In HSAmy-ar complex structure, some of the ordered structures (helices and strands) in the B domain are lost, as calculated by the program PyMol 42. For example, the region S132 through F136 is not classified as a helical turn whereas it is a helix in the wild type. Also, two regions H101-G104 and L165-D167, which contain the calcium binding residues H101 and D167, are no longer classified as beta strands in the mutant structure. Our observation that HSAmy-ar has significantly lower hydrolytic activity in solution suggests that saccharide binding at all the secondary binding sites, in addition to the active site, might be necessary for the enzyme to exhibit optimal hydrolytic activity. This is augmented by the observation that single or double mutants (Table 1) did not affect the hydrolytic activity of the enzyme to any significant extent.
Aromatic residues and starch binding
To our knowledge, this is the first report on the starch binding and bacterial binding ability of HSAmy mediated by the aromatic residues. Starch adsorption in many α-amylases is usually dependent upon the existence of a separate domain, which has repeat regions 43. The functions of such domains in barley α-amylase have been studied using recombinant proteins 44,45. However, in HSAmy, no such domain exists. Our results show that removal of aromatic residues acting as platforms for sugar binding at the secondary saccharide binding sites leads to weaker starch binding. Since HSAmy has a high affinity toward raw starch, a starch binding capacity of only 13% for HSAmy-ar suggests that the mutated residues are of crucial importance for starch binding. A higher reduction observed in the mutants W284A, Y276A/W284A and W316A/W388A, but not in the mutant W203A, suggests that the region around W284 and to some extent around W316 and W388, might contribute to starch binding. The involvement of multiple surface sites, distal to the active site, in starch binding has been proposed in barley a-amylase. These sites might play a significant role in localizing the enzyme on starch granule to enhance processivity 46. Interestingly, the site 3 region (W316 and W388) is the interface between domains A and C in HSAmy. In this regard, it has been demonstrated that many of the glycoside hydrolases, including some amylases contain a carbohydrate-binding module (CBM), which is usually present contiguous to a carbohydrate-active enzyme 47. The major purpose of CBMs is to position the substrate near the active site and to improve hydrolysis. CBMs have been classified into 52 families based on their sequence similarities, according to the CAZy database (http://www.cazy.org). While some of these families have starch-binding domains (SBDs; families CBM20, CBM21, CBM25, CBM26, CBM34, CBM41, and CBM45), HSAmy does not possess a SBD. However, among the putative SBD sequences recognized in various plant and animal proteins, two tryptophans (or at least two aromatic residues) are conserved in their sequences 48,49,50. Our results on the aromatic residues reported here suggest a potential role of domain C, which contains two tryptophans W316 and W388, in HSAmy. While the sites 2 and 3 are not akin to the “ sugar tongs” previously demonstrated in barley α-amylase 46,51,52, our results raise the possibility that distal sites in HSAmy might also be important in starch binding, and to some extent in enzyme activity.
Aromatic residues and bacterial binding
We designed HSAmy mutants to determine whether S. gordonii bound to the active site of HSAmy or to one of the secondary saccharide binding sites. In all the mutants, the active site is left intact and therefore, if bacterial binding occurs at this site, all the mutants should show comparable binding to that exhibited by the wild type HSAmy. However, we observed that, as with the starch binding above, there is disparity in the ability of bacteria to bind to the single and double mutants. Conspicuously, we observed that the mutant Y276A/W284A exhibited a reduction in bacterial binding that was in excess of the other mutants studied but less than what was observed for HSAmy-ar. In this regard, our results on AbpA binding add some insight into the bacterial binding by HSAmy. Our results show that the ability of AbpA to bind HSAmy is impaired when the aromatic residues are absent. This conclusion is also supported by our results using Biacore, which clearly showed a very tight binding between HSAmy and AbpA. However, the dissociation of the HSAmy/AbpA complex does not occur even after 600 s. Even using condition such as 0.1% Triton-X could not break the binding between HSAmy and AbpA. Therefore, the relative binding abilities of the mutant enzymes could be not be reliably compared with HSAmy since the initial chip was no longer usable. Interestingly, the sensogram for one of the mutants (Y276A/W284A) was similar to the HSAmy and also formed a tighter complex that could not be dissociated easily. These technical difficulties prevented us from carrying out the determination of KD values for the mutants. Irrespective of this setback, it is clear from the data in Table 4 that the aromatic residues located at the secondary binding sites play a significant role in contributing to the affinity at equilibrium as well as the association/dissociation kinetics of the binding of HSAmy to AbpA.
Local architecture and interactions around aromatic residues
The W203 residue is spatially closer to the subsite +2 where a Tyr151 is present. Analysis of the structure of HSAmy-ar/acarbose complex revealed that there was no significant perturbation of the structure around this region. The residue W203 is also sequentially closer to H201, which binds to an essential calcium ion. Comparison and analysis of the calcium-binding site in the two structures revealed no significant changes. Thus, the starch or bacterial binding at this site might involve nearby residues. In the immediate vicinity of W203, the residue K140 makes a hydrogen bond with the bound substrate. In the structure of HSAmy-ar/acarbose complex, this hydrogen bond is lacking. Alternate conformation was also noted for another residue, S163, at the opposite end of the active site near subsite -2. Because of this minimal structural deviations exhibited by HSAmy and HSAmy-ar, the cause for loss in enzyme activity exhibited by HSAmy-ar, is unclear at this time. One possible explanation might be that HSAmy-ar is more flexible compared to the wild type. The flexibility in the mutant is enzyme is illustrated by the alternate conformation adopted by the residues that occur at the edges of the active site such as K140 and S163 and by the higher <B> values for domain B in HSAmy-ar. Oligosaccharide hydrolytic pattern and amount of product generated (Fig. 9) suggests that even single mutation of the aromatic residues such as W203A or W284A, can impair the enzyme activity to considerable extent. These two aromatic residues W203 and W284 are located at the either end of the active site pocket. The small structural changes noted above due K140 and S163 residues might affect the way in which the substrates, even smaller oligosaccharides, are guided into the active site. However, once the substrate is properly bound at the active site, hydrolysis can occur. In this regard, one or more of the secondary binding sites involving aromatic residues could be assisting to hold the starch molecule or guide the oligosaccharide into the active site for efficient hydrolysis. Interestingly, two aromatic residues W278/W279, have been proposed as starch granule-binding site in barley α-amylase 44. Based on our results, the low activity exhibited by HSAmy-ar could arise as a result of slow initial substrate-binding step.
Biological significance of the role of aromatic residues
Three different functions, such as starch hydrolysis, bacterial binding and hydroxyapatite binding, have been attributed to HSAmy in the oral cavity. Since it has been shown previously that bacteria bound HSAmy still retains its hydrolytic activity, starch binding and bacterial binding (AbpA) could be independent of each other if two different sites are utilized for the two activities/functions. Our results have clearly demonstrated the independence of these two binding interactions. An interesting outcome our study is that both starch binding as well as AbpA binding to HSAmy might use multiple sites. Our results suggest that, both starch binding and AbpA might be assisted by at least two secondary binding sites. The retention of hydroxyapatite binding ability in HSAmy-ar suggests that this third function might utilize yet another site for binding. Mutation of acidic residues located on the surface of HSAmy has indicated that these surface acidic residues are involved in HA binding (unpublished data). However, the hydroxyapatite binding ability of HSAmy is unaffected the secondary site aromatic residues. Overall, it is tempting to suggest that the three functions of HSAmy, viz. starch hydrolysis, enamel binding and bacterial binding are independent of each other with regard to their interaction domains in HSAmy. Although as a component of the acquired enamel pellicle, HSAmy renders some protection against bacterial insult, the likely independence of its three functions might render a negative impact on the oral health since cariogenic bacteria could obtain nutrients and sustain in the oral cavity.
CONCLUSIONS
We show that the surface aromatic residues, located at the secondary saccharide binding sites in HSAmy, play a critical role in starch as well as bacterial binding activities. In addition, at least two of these aromatic residues, W203 and W284, might play a role in the hydrolytic activity as well as bacterial binding activity. Finally, the surface aromatic residues might provide a synergistic effect in the functions exhibited by HSAmy.
MATERIALS AND METHODS
General procedures
All buffer reagents and other chemicals were obtained from Sigma Chemical Co. All oligonucleotides used in this study were synthetic products purchased from Integrated DNA Technologies. All nucleotide sequencing was performed at the Molecular Resource Facility at University of Medicine and Dentistry of New Jersey, NJ.
Bacterial strain, media and plasmids
The variant of HSAmy was generated using the Bac-To-Bac Baculovirus Expression System using procedures outlined previously 53,54. The forward primers and the corresponding reverse oligonucleotide primers used to create the mutant reported here are provided in Table 1. All primers were used in vector pFASTBAC1 (Invitrogen) into which HSAmy gene was cloned 54. We used a sequential approach to mutate one site at a time in order to introduce the multiple mutations at the various positions. After each mutation was performed, the mutated sites were confirmed by nucleotide sequencing using appropriate primers. The plasmid pFASTBAC1 with mutant HSAmy was transformed into MAX EFFICIENCY DH10BAC™(Invitrogen) cells that contained baculovirus genomic DNA (bacmid) as well as a helper plasmid. Transformed cells were plated on Kanamycin, Gentamycin, Tetracycline, Bluo-gal and IPTG-containing plates. A single white colony was cultured overnight and the high molecular recombinant bacmid DNA was isolated and transfected into Sf9 cells using CELLFECTIN Reagent ™ (Invitrogen). After 72 h of incubation at 28 °C in SF900II serum-free medium (Invitrogen), recombinant baculovirus was harvested from the medium. Viral stocks were amplified by re-infection into suspension culture of Sf9 cells at 28 °C with continuous shaking at a speed of 140 rpm.
Protein expression and purification
The mutant protein was isolated from Sf9 cell culture grown in 1 L of the medium by following protocol previously established for native HSAmy 54. Briefly, cell debris was removed from a three to five-day post-infected medium, adjusted to pH 8.0 with 5N NaOH. After centrifugation, the supernatant was further clarified by passing through a 0.45 μM filter (Corning Inc.) and the low molecular weight proteins were removed by ultrafiltration using a 10 kDa cut-off spiral cartridge (Prep/SCALE® TFF Cartridge, Millipore). The medium was lyophilized and resuspended in 100 mM Tris.HCl, pH. 8.0. Following extensive dialysis against a buffer (5 mM Tris.HCl, pH 8.0) containing 2 mM CaCl2, and centrifugation, the supernatant was applied to a 3 × 13 cm DEAE-52 cellulose column (Whatman). Proteins were eluted from the column as previously described 54. Fractions containing recombinant protein were pooled based on SDS-PAGE 55 and western blotting and dialyzed extensively against cold deionized water using Spectra/Por 2 (MWCO of 12-14,000 Da; Spectrum Medical Industries, Inc.) and lyophilized. At this stage, the enriched proteins were subjected to a BioGel P60 size exclusion chromatography following a procedure described previously 9. After pooling the fractions containing the desired mutant enzymes based on western blotting, enzymes with greater than 99% purity were obtained at approximately 5 mg/L of the culture medium. The mass and purity of the mutant enzyme were confirmed by mass spectral analysis using Perspective Biosystems, a DE Pro MALDI-TOF instrument equipped with a laser at 337 nm and operated with a positive or negative detection with 6 kV acceleration potential.
Cloning and expression of AbpA
The abpA gene was amplified from the plasmid pCR2KB-7 provided by Dr. Scannapieco, University of Buffalo using the following primers: (5’-GCGCGCCATATGAAAAAAGTTTTATTGTCAAGCGTG-3’) forward (5’-GCAGGGTACCttaGTGGTGGTGGTGGTGGTGTTTAACTGCTGAAG-3’) reverse. The forward primer introduced an NdeI restriction site (underlined) while the reverse primer contained a KpnI restriction site (underlined) and a stop codon (lower case). The PCR product (620 bp) was digested with NdeI and KpnI and was ligated into the NdeI-KpnI sites of plasmid pET29b (Novagen). The insert of the resulting plasmid (designated pRC4) was subjected to DNA sequence analysis. Plasmid pRC4 carried a gene that encoded amino acids 1 to 195 of abpA fused directly to a hexahistidine metal-binding C-terminal tail to facilitate purification.
Expression and purification of AbpA
The protein AbpA was expressed using a procedure similar to the one previously established for the expression of a dispersin B 56. Briefly, a 2-liter Erlenmeyer flask containing 500 mL of Luria-Bertani broth supplemented with 30 μg of Kanamycin per mL was inoculated with 5 mL of an overnight culture of E. coli strain Rosetta™ (DE3) (Novagen) 57 transformed with the plasmid pCR4. The flask was incubated at 37 °C with agitation (200 rpm) until the optical density of the culture at 600 nm reached 0.6 (ca. 3 h). IPTG was added to a final concentration of 0.1 mM, and the flask was incubated for an additional 5 h with agitation. The cells were harvested by centrifugation for 15 min at 6000 × g.
The cell pellet was resuspended in 20 mL of lysis buffer (20 mM Tris-HCl, pH 8.0, 500 mM NaCl, 1 mM phenylmethylsulfonyl fluoride and 0.1% Nonidet P-40). The cell suspension was then sonicated on ice for 30 s (x5 with 2 min intervals) at 30% capacity with a 30% duty cycle by using a Branson model 450 sonicator equipped with a microprobe. The cell debris was pelleted by centrifugation (15,000 × g for 20 min; 4 °C), and the supernatant was filtered using 0.45 μ syringe filter (Corning) and purified by ÄKTA® FPLC system controlled by UNICORN™ software version 5.1 (GE Healthcare). Briefly, 2 mL of supernatant was loaded onto a sample loop, which was injected into a 1 mL HisTrap® Chelating HP column (GE Healthcare) at a flow rate of 1.0 mL/min. The column was activated with NiSO4 (100 mM) and equilibrated with buffer A (5 mM Tris.HCl , pH 8.0, 500 mM NaCl, containing 5 mM imidazole) prior to sample injection. Unbound proteins were eluted with 5 CV of buffer A. The bound proteins were further eluted with imidazole. Fractions (1 mL) of the eluate were collected and assayed for the presence of the protein by SDS-PAGE and Coomassie Blue staining 58. Fractions containing pure protein were pooled and dialyzed overnight against water (2 × 4L changes at 4 °C) by using a 10-kDa-cutoff dialysis membrane (Spectra/Por), and lyophillized. The purity and molecular mass of AbpA protein was determined by mass spectral analysis. The yield of AbpA obtained was 15 mg/L.
Enzyme activity assays
Dinitrosalicylic acid assay was used for measuring the starch-hydrolyzing activity of HSAmy and mutants at 25 °C for 3 min in 20 mM phosphate buffer (pH 6.9) containing 6 mM NaCl using 1% soluble starch as substrate 59. The concentrations of the HSAmy and the mutants were determined by a protein assay kit (BioRad) using BSA (Sigma) as a standard. All experiments were done in triplicate and the average value is reported.
Binding ability of HSAmy enzymes on raw starch
The enzymes HSAmy and mutants (0.1 mg/mL) were prepared in 20 mM phosphate buffer (pH 6.9) containing 6 mM NaCl. Raw corn starch (10 mg) was added to an aliquot of the stock solution (200 μL). The reaction mixture was shaken gently at room temperature for 10 min and centrifuged at 5,000 rpm for 5 min. An aliquot of the supernatant was used to measure the starch hydrolyzing activity (B) and compared with the activity of the original enzyme solution ithout starch (A). The starch binding activity (rb) of the enzymes was defined by the following equation:
Preparation of 125I labeled α-amylase and mutant proteins
Purified recombinant α-amylase (25 μg) was iodinated by the chloramine-T method in 100 μL of PBS 60. lodinated and not iodinated proteins were separated from free iodine by gel filtration on columns (10 mL disposable pipettes) of Bio-Gel P6DG (Bio-Rad) equilibrated in PBS. Fractions of 1 mL were monitored for radioactivity with a gamma counter (model 5500; Beckman). Void volume materials were used for the subsequent studies.
In vitro interaction of 125I -labeled HSAMY with bacteria
Streptococcus gordonii was cultured in TSB-Y and washed and resuspended in PBS with 0.1% lipid-free BSA. Cell density was calculated at A600. Experiments were performed in polypropylene microcentrifuge tubes precoated with 1.0% BSA (Sigma-Aldrich) to reduce the nonspecific interaction of the ligand with the tube walls. Reaction mixtures contained 0.1 mL of the bacterial suspension (Approximately 1 × 107), various amounts of 125I -labeled protein, and buffer added to a total volume of 200 μL. Triplicate samples were incubated at 27 °C for 30 min with continuous rotation (6 rpm) using mini lab roller (Labnet). The reaction was terminated by centrifugation and the bacterial cells were washed three times (1 mL) with PBS. Finally, the pellets were resuspended in 200 μL of PBS. The amount of 125I -labeled HSAmy in 10μL of the control samples (no bacteria, total cpm added) and in the bacterial suspension (bound cpm) was measured with a gamma counter (model 5500; Beckman). Negligible radioactivity (less than 0.01%) was recovered in the control microcentrifuge tubes from reaction mixtures, which did not contain bacteria.
Biacore assays
Surface plasmon resonance (SPR) experiments were performed on a Biacore X instrument equipped with research grade CM5 sensor chips. The AbpA surfaces were prepared at 25 °C by the amine-coupling method using reagents available from BIAcore (N-ethyl-N’-dimethylaminopropyl-carbodiimide [EDC], N-hydroxysuccinimide [NHS], and 1 M ethanolamine-HCl, pH 8.5). Flow cells were activated for 6 min with a solution containing 50 mM NHS and 0.2 mM EDC. AbpA was diluted to 1.5 μM in 10 mM sodium acetate (pH 5.0) and injected over the surface for 7 min. The remaining reactive groups were blocked with a 7 min injection of 1 M ethanolamine-HCl (pH 8.5). The immobilization of AbpA was achieved using a running buffer of NaPO4 (pH 6.9), 6 mM NaCl at a flow rate of 5 μL/min. The change in refractive index induced by the association of compounds interacting with the sensor surface is expressed in resonance units (RU). Typical immobilization levels of AbpA (~0.2 ng/mm2) yieled values between 2000 and 2100 RU.
Real-time binding analyses to AbpA were performed at room temperature, using HSAmy and HSAmy-ar. For binding experiments, the running buffer (20 mM, pH 6.9, containing 6 mM NaCl) was used at a flow-rate of 5 μL/min. A independent flow cell on the same sensor chip that was subjected to a “blank immobilization” (no ABP was added in the process of chip preparation) was used as control. Varying concentrations (1.25 - 20 mM) of HSAmy-ar was flushed over the experimental (AbpA-coated) and control (uncoated) flow cells. The amount of binding was observed as an increase in resonance units (RU) and maximal amplitudes were corrected for non-specific binding (control flow cell). Regeneration of the chip was achieved easily by short pulses (1 min) of 10 mM glycine pH 2.5 for experiments involving HSAmy-ar. However, for experiments using HSAmy regeneration was unsuccessful even after several regeneration cycles using 10 mM glycine pH 2.5, or 250 mM NaCl, or 2M NaCl, or with running buffer with increasing concentrations of TritonX 100 up to 1% or 20 mM glucose. Therefore, for the binding of the wild type, only a single concentration (1 μM) was used. The binding parameters – kon, koff, KA and KD – for the interaction were calculated using BIAevaluation 3.2 software. The curves were fitted according to a simple 1:1 interaction. The fitting residuals did not exhibit any significant systematic deviations and were minimal (-1 to 1).
In vitro binding of AbpA and HSAmy enzymes
Since AbpA has been suggested to bind to amylase 16, we tested the ability of HSAmy and its variants to bind to AbpA. Equimolar concentrations of HSAmy enzymes and AbpA in 20 mM phosphate buffer (pH 6.9) containing 6 mM NaCl were incubated in ice for 30 min. The mixture was centrifuged at 12,000 rpm for 5 min at 4 °C and an aliquot of the supernatant was assayed for enzyme activity using the dinitrosalicylic acid assay as described above. Control samples with out AbpA were treated similarly. All binding experiments were done in triplicate and repeated twice.
In vitro assay for HA binding
The ability of wild type HSAmy and HSAmy-ar to bind to HA crystallites as follows using iodinated enzymes. Known amounts (125 – 1000 × 103 cpm) of the wild type and mutant samples were equilibrated with HA crystallites (6 mg, with a surface area of approximately 0.24 m2/g of HA; Biorad) in a saturated solution (200 μL) at 37 °C, pH 6.5 using an ionic strength of 0.15 mol/L of imidazole for 1 hr in polycarbonate tubes precoated with 1 % BSA. The tubes were continuously rotated at 6 rpm using a mini lab roller (Labnet). After equilibration, the HA crystallites were separated from the supernatant solution by centrifugation (0.5 × g) for 5 minutes and washed (3x) with a saturated solution of HA (500 μl). Finally, the washed HA crystallites were resuspended in 200 μL of PBS and the amount of 125I -labeled HSAmy in 10μL of the control samples (no HA crystallites, total cpm added) and in the HA suspension (bound cpm) was measured with a gamma counter (model 5500; Beckman). Negligible radioactivity (less than 0.01%) was recovered in the control microcentrifuge tubes from reaction mixtures, which did not contain HA. All adsorption experiments were carried out in triplicate with similar results.
Hydrolysis of maltooligosaccharides
Assays measuring the products of oligosaccharide hydrolysis were carried out using a Varian HPLC (ProStar) system equipped with a single port manual injector and a refractive index detector (Model No. 350). The hydrolytic ability of the wild type and mutant enzymes was determined using the substrate maltopentaoside (G5) or maltoheptaoside (G7) by HPLC analysis at room temperature. The hydrolysates were analyzed using an analytical Dextropak column (100 × 8 mm) to which a Novapak C18 Guard Pak module was attached (Waters). Water was used as an eluent. Integration of the HPLC profiles was carried out using Varian Star software (Version 5.51). In a typical run, a total reaction volume (200 μL) consisted of either an enzyme concentration of 60 nM (600 nM for the mutants W203A and HSAmy-ar), oligosaccharide at 0.5 mM in water. The reaction mixture (20 μL) was injected in to the HPLC system after a specific interval of 2 min (5 min for W203A and HSAmy-ar). The amount of residual G5 or G7 was determined using the area under each peak and converting it in to molar concentration using values obtained previously for the standards.
Circular Dichroism Spectroscopy (CD)
CD spectra were recorded on an AVIV model 215 spectropolarimeter equipped with a five-position thermostated cell holder controlled by a Hewlett-Packard Peltier temperature controller. HSAmy and HSAmy-ar solutions of 0.12 mg/mL in 0.15 M NaCl, 0.01M sodium phosphate, pH 7.4, were transferred into quartz cells of path length 0.1 cm. Concentrations were determined using protein assay kit (Bio-Rad). Spectra were collected from 260 nm to approximately 200 nm with a step size of 0.5 nm and a bandwidth of 1.5 nm at room temperature and corrected for buffer baseline and measured in the same cells. These results were normalized to mean residue ellipticities ([θ]).
Structure determination of mutant and acarbose complexes
Crystals of HSAmy-ar were grown using conditions previously described 3,61 using a final protein concentration of 16 mg/mL. Diffraction quality crystals appeared over a period of one week. The protein complexes with acarbose were obtained by soaking the enzyme crystals with acarbose (1 mM final concentration) in 45% (+/-)-2-Methyl-2,4-pentanediol for 24 h and the soaked crystals were used for data collection. The crystals were mounted on loops (Hampton Research) and flash frozen to −170 °C in liquid nitrogen. Diffraction data were collected using synchrotron radiation at the CHESS beam line A-1 at liquid nitrogen temperatures and an ADSC Quantum 4 CCD detector. A total of 360 frames were collected with an exposure time of 10 -15 s and an oscillation interval of 0.5° to give a 99% complete data set to 1.6 Å. Intensity data were integrated, scaled and reduced to structure factor amplitudes using HKL2000 suite of programs 62. The unit cell parameters were found to be isomorphous with those of the wild type HSAmy 54.
Refinement of the mutant structure was carried out using Refmac5 in the CCP4 package 63 wherein cycles of rigid body, positional and thermal B factor refinements were carried out using the phase information from the native enzyme (PDB Code 1smd). A test set consisting of 5% of reflections was used to monitor the Rfree behavior. Manual model rebuilding was carried out using COOT 64. The complete polypeptide chain of HSAmy-ar was examined with Fo-Fc and 2Fo-Fc maps. During this process, the mutant structure clearly showed absence of side chain density for the mutated residues. The residue at positions 134, 203, 276 284, 316 and 388 were changed to Ala to reflect the mutation and for the remainder of the refinement these residues was treated as such.
At this stage, clear-cut continuous density corresponding to the oligosaccharide ligand was observed in the active site region at subsites −2, −1, +1 and +2. However, no oligosaccharide atoms were included in the refinement until the refinement of the protein reached convergence. The identity of the sugar moieties (either 5-hydroxymethylchonduritol or 4-amino-4,6-dideoxy-α-D-glucose or glucose) was deduced from the presence or absence of density for the hydroxyl group of the side chain at position C5 in the ring 21. The refinement was continued by the inclusion of the sugar atoms. Further examination of the density maps revealed no additional binding sites in the complex at sites harboring the mutated aromatic residues.
Solvent molecules were added using the arp/warp procedure 65 in the CCP4 package. The validity of the water molecules were assessed based on the following criteria: 1) the presence of a peak at least 3σ in the difference map with at least one hydrogen bond to a protein atom (N or O) or 2) if the located water molecule were part of a water chain connecting protein atoms, and 3) the water refined with a thermal factor less than 50 Å2. Manual fitting was interspaced between refinements as and when necessary. The programs PROCHECK 66, CCP4 and COOT were used for model analysis of the final refined structures (Table 2).
Protein Data Bank accession codes
The atomic coordinates and structure factors for HSAmy-ar/acarbose have been deposited with the RCSB PDB, with accession code 3DHP. Other structures used for comparison have the following PDB Codes: 1smd (HSAmy); 1mfv (HSAmy:acarbose); and 1mfu (Δ306-310HSAmy:acarbose).
Acknowledgments
We thank Dr. Norma Greenfield of the Circular dichroism facility at UMDNJ for help in obtaining the CD spectra and evaluating the secondary structural content using CD-PRO software suite. Part of this work is based upon research conducted at the Cornell High Energy Synchrotron Source (CHESS), which is supported by the National Science Foundation under award DMR 97-13424, using the Macromolecular Diffraction at CHESS (MacCHESS) facility, which is supported by award RR-01646 from the National Institutes of Health, through its National Center for Research Resources. We also thank Dr. Scannapieco for providing the clone for AbpA. This study was supported by the USPHS Grant DE12585 (NR).
Abbreviations
- AbpA
Amylase binding protein A
- CBM
carbohydrare binding module
- G5
maltopentasaccharide
- G7
maltoheptasaccharide
- HSAmy
human salivary α -amylase
- HSAmy-ar
human salivary α-amylase aromatic residue multiple mutant
- SBD
starch binding domain
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Literature cited
- 1.MacGregor EA, Janecek S, Svensson B. Relationship of sequence and structure to specificity in the alpha-amylase family of enzymes. Biochim Biophys Acta. 2001;1546:1–20. doi: 10.1016/s0167-4838(00)00302-2. [DOI] [PubMed] [Google Scholar]
- 2.Scannapieco FA, Torres GI, Levine MJ. Salivary amylase promotes adhesion of oral streptococci to hydroxyapatite. J Dent Res. 1995;74:1360–6. doi: 10.1177/00220345950740070701. [DOI] [PubMed] [Google Scholar]
- 3.Ramasubbu N, Paloth V, Luo Y, Brayer GD, Levine MJ. Structure of human salivary alpha-amylase at 1.6 A resolution: implications for its role in the oral cavity. Acta Crystallogr D Biol Crystallogr. 1996;52:435–46. doi: 10.1107/S0907444995014119. [DOI] [PubMed] [Google Scholar]
- 4.Brayer GD, Luo Y, Withers SG. The structure of human pancreatic alpha-amylase at 1.8 A resolution and comparisons with related enzymes. Protein Sci. 1995;4:1730–42. doi: 10.1002/pro.5560040908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Scannapieco FA, Solomon L, Wadenya RO. Emergence in human dental plaque and host distribution of amylase-binding streptococci. J Dent Res. 1994;73:1627–35. doi: 10.1177/00220345940730100701. [DOI] [PubMed] [Google Scholar]
- 6.Handley P, Coykendall A, Beighton D, Hardie JM, Whiley RA. Streptococcus crista sp. nov., a viridans streptococcus with tufted fibrils, isolated from the human oral cavity and throat. Int J Syst Bacteriol. 1991;41:543–7. doi: 10.1099/00207713-41-4-543. [DOI] [PubMed] [Google Scholar]
- 7.Tseng CC, Scannapieco FA, Levine MJ. Use of a replica-plate assay for the rapid assessment of salivary protein-bacteria interactions. Oral Microbiol Immunol. 1992;7:53–6. doi: 10.1111/j.1399-302x.1992.tb00021.x. [DOI] [PubMed] [Google Scholar]
- 8.Douglas CW. The binding of human salivary alpha-amylase by oral strains of streptococcal bacteria. Arch Oral Biol. 1983;28:567–73. doi: 10.1016/0003-9969(83)90003-1. [DOI] [PubMed] [Google Scholar]
- 9.Scannapieco FA, Bergey EJ, Reddy MS, Levine MJ. Characterization of salivary alpha-amylase binding to Streptococcus sanguis. Infect Immun. 1989;57:2853–63. doi: 10.1128/iai.57.9.2853-2863.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scannapieco FA, Torres G, Levine MJ. Salivary alpha-amylase: role in dental plaque and caries formation. Crit Rev Oral Biol Med. 1993;4:301–7. doi: 10.1177/10454411930040030701. [DOI] [PubMed] [Google Scholar]
- 11.Brown AE, Rogers JD, Haase EM, Zelasko PM, Scannapieco FA. Prevalence of the amylase-binding protein A gene (abpA) in oral streptococci. J Clin Microbiol. 1999;37:4081–5. doi: 10.1128/jcm.37.12.4081-4085.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scannapieco FA. Saliva-bacterium interactions in oral microbial ecology. Crit Rev Oral Biol Med. 1994;5:203–48. doi: 10.1177/10454411940050030201. [DOI] [PubMed] [Google Scholar]
- 13.Scannapieco FA, Haraszthy GG, Cho MI, Levine MJ. Characterization of an amylase-binding component of Streptococcus gordonii G9B. Infect Immun. 1992;60:4726–33. doi: 10.1128/iai.60.11.4726-4733.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Douglas CW. Characterization of the alpha-amylase receptor of Streptococcus gordonii NCTC 7868. J Dent Res. 1990;69:1746–52. doi: 10.1177/00220345900690110701. [DOI] [PubMed] [Google Scholar]
- 15.Gwynn JP, Douglas CW. Comparison of amylase-binding proteins in oral streptococci. FEMS Microbiol Lett. 1994;124:373–9. doi: 10.1111/j.1574-6968.1994.tb07311.x. [DOI] [PubMed] [Google Scholar]
- 16.Rogers JD, Haase EM, Brown AE, Douglas CW, Gwynn JP, Scannapieco FA. Identification and analysis of a gene (abpA) encoding a major amylase-binding protein in Streptococcus gordonii. Microbiology. 1998;144(Pt 5):1223–33. doi: 10.1099/00221287-144-5-1223. [DOI] [PubMed] [Google Scholar]
- 17.Rogers JD, Palmer RJ, Jr, Kolenbrander PE, Scannapieco FA. Role of Streptococcus gordonii amylase-binding protein A in adhesion to hydroxyapatite, starch metabolism, and biofilm formation. Infect Immun. 2001;69:7046–56. doi: 10.1128/IAI.69.11.7046-7056.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Loo CY, Corliss DA, Ganeshkumar N. Streptococcus gordonii biofilm formation: identification of genes that code for biofilm phenotypes. J Bacteriol. 2000;182:1374–82. doi: 10.1128/jb.182.5.1374-1382.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scannapieco FA, Bhandary K, Ramasubbu N, Levine MJ. Structural relationship between the enzymatic and streptococcal binding sites of human salivary alpha-amylase. Biochem Biophys Res Commun. 1990;173:1109–15. doi: 10.1016/s0006-291x(05)80900-3. [DOI] [PubMed] [Google Scholar]
- 20.Tseng CC, Miyamoto M, Ramalingam K, Hemavathy KC, Levine MJ, Ramasubbu N. The roles of histidine residues at the starch-binding site in streptococcal-binding activities of human salivary amylase. Arch Oral Biol. 1999;44:119–27. doi: 10.1016/s0003-9969(98)00112-5. [DOI] [PubMed] [Google Scholar]
- 21.Ramasubbu N, Ragunath C, Mishra PJ. Probing the role of a mobile loop in substrate binding and enzyme activity of human salivary amylase. J Mol Biol. 2003;325:1061–76. doi: 10.1016/s0022-2836(02)01326-8. [DOI] [PubMed] [Google Scholar]
- 22.Nagy T, Simpson P, Williamson MP, Hazlewood GP, Gilbert HJ, Orosz L. All three surface tryptophans in Type IIa cellulose binding domains play a pivotal role in binding both soluble and insoluble ligands. FEBS Lett. 1998;429:312–6. doi: 10.1016/s0014-5793(98)00625-5. [DOI] [PubMed] [Google Scholar]
- 23.Ramasubbu N, Ragunath C, Mishra PJ, Thomas LM, Gyemant G, Kandra L. Human salivary alpha-amylase Trp58 situated at subsite -2 is critical for enzyme activity. Eur J Biochem. 2004;271:2517–29. doi: 10.1111/j.1432-1033.2004.04182.x. [DOI] [PubMed] [Google Scholar]
- 24.Ragunath C, Manuel SG, Kasinathan C, Ramasubbu N. Structure-function relationships in human salivary alpha-amylase: role of aromatic residues in a secondary binding site. Biologia. 2008;63 doi: 10.1016/j.jmb.2008.09.089. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vuilleumier S, Sancho J, Loewenthal R, Fersht AR. Circular dichroism studies of barnase and its mutants: characterization of the contribution of aromatic side chains. Biochemistry. 1993;32:10303–13. doi: 10.1021/bi00090a005. [DOI] [PubMed] [Google Scholar]
- 26.Kuwajima K, Garvey EP, Finn BE, Matthews CR, Sugai S. Transient intermediates in the folding of dihydrofolate reductase as detected by far-ultraviolet circular dichroism spectroscopy. Biochemistry. 1991;30:7693–703. doi: 10.1021/bi00245a005. [DOI] [PubMed] [Google Scholar]
- 27.Sreerama N, Woody RW. Estimation of protein secondary structure from circular dichroism spectra: comparison of CONTIN, SELCON, and CDSSTR methods with an expanded reference set. Anal Biochem. 2000;287:252–60. doi: 10.1006/abio.2000.4880. [DOI] [PubMed] [Google Scholar]
- 28.Davies GJ, Wilson KS, Henrissat B. Nomenclature for sugar-binding subsites in glycosyl hydrolases. Biochem J. 1997;321(Pt 2):557–9. doi: 10.1042/bj3210557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qian M, Haser R, Buisson G, Duee E, Payan F. The active center of a mammalian alpha-amylase. Structure of the complex of a pancreatic alpha-amylase with a carbohydrate inhibitor refined to 2.2-A resolution. Biochemistry. 1994;33:6284–94. doi: 10.1021/bi00186a031. [DOI] [PubMed] [Google Scholar]
- 30.Qian M, Spinelli S, Driguez H, Payan F. Structure of a pancreatic alpha-amylase bound to a substrate analogue at 2.03 A resolution. Protein Sci. 1997;6:2285–96. doi: 10.1002/pro.5560061102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brzozowski AM, Davies GJ. Structure of the Aspergillus oryzae alpha-amylase complexed with the inhibitor acarbose at 2.0 A resolution. Biochemistry. 1997;36:10837–45. doi: 10.1021/bi970539i. [DOI] [PubMed] [Google Scholar]
- 32.Brayer GD, Sidhu G, Maurus R, Rydberg EH, Braun C, Wang Y, Nguyen NT, Overall CM, Withers SG. Subsite mapping of the human pancreatic alpha-amylase active site through structural, kinetic, and mutagenesis techniques. Biochemistry. 2000;39:4778–91. doi: 10.1021/bi9921182. [DOI] [PubMed] [Google Scholar]
- 33.Qian M, Nahoum V, Bonicel J, Bischoff H, Henrissat B, Payan F. Enzyme-catalyzed condensation reaction in a mammalian alpha-amylase. High-resolution structural analysis of an enzyme-inhibitor complex. Biochemistry. 2001;40:7700–9. doi: 10.1021/bi0102050. [DOI] [PubMed] [Google Scholar]
- 34.Aghajari N, Roth M, Haser R. Crystallographic evidence of a transglycosylation reaction: ternary complexes of a psychrophilic alpha-amylase. Biochemistry. 2002;41:4273–80. doi: 10.1021/bi0160516. [DOI] [PubMed] [Google Scholar]
- 35.Remenyik J, Ragunath C, Ramasubbu N, Gyemant G, Liptak A, Kandra L. Introducing transglycosylation activity into human salivary alpha-amylase (HSA) Org Lett. 2003;5:4895–8. doi: 10.1021/ol035999k. [DOI] [PubMed] [Google Scholar]
- 36.Qian M, Haser R, Payan F. Carbohydrate binding sites in a pancreatic alpha-amylase-substrate complex, derived from X-ray structure analysis at 2.1 A resolution. Protein Sci. 1995;4:747–55. doi: 10.1002/pro.5560040414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iefuji H, Chino M, Kato M, Iimura Y. Raw-starch-digesting and thermostable alpha-amylase from the yeast Cryptococcus sp. S-2: purification, characterization, cloning and sequencing. Biochem J. 1996;318(Pt 3):989–96. doi: 10.1042/bj3180989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Raj PA, Johnsson M, Levine MJ, Nancollas GH. Salivary statherin. Dependence on sequence, charge, hydrogen bonding potency, and helical conformation for adsorption to hydroxyapatite and inhibition of mineralization. J Biol Chem. 1992;267:5968–76. [PubMed] [Google Scholar]
- 39.Johnsson M, Levine MJ, Nancollas GH. Hydroxyapatite binding domains in salivary proteins. Crit Rev Oral Biol Med. 1993;4:371–8. doi: 10.1177/10454411930040031601. [DOI] [PubMed] [Google Scholar]
- 40.Ramasubbu N, Thomas LM, Bhandary KK, Levine MJ. Structural characteristics of human salivary statherin: a model for boundary lubrication at the enamel surface. Crit Rev Oral Biol Med. 1993;4:363–70. doi: 10.1177/10454411930040031501. [DOI] [PubMed] [Google Scholar]
- 41.Gilles C, Astier JP, Marchis-Mouren G, Cambillau C, Payan F. Crystal structure of pig pancreatic alpha-amylase isoenzyme II, in complex with the carbohydrate inhibitor acarbose. Eur J Biochem. 1996;238:561–9. doi: 10.1111/j.1432-1033.1996.0561z.x. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. The PyMOl User’s Manual. Delano Scientific; San Carlos, CA: 2002. [Google Scholar]
- 43.Sumitani J, Tottori T, Kawaguchi T, Arai M. New type of starch-binding domain: the direct repeat motif in the C- terminal region of Bacillus sp. no. 195 alpha-amylase contributes to starch binding and raw starch degrading. Biochem J. 2000;350(Pt 2):477–84. [PMC free article] [PubMed] [Google Scholar]
- 44.Tibbot BK, Wong DW, Robertson GH. A functional raw starch-binding domain of barley alpha-amylase expressed in Escherichia coli. J Protein Chem. 2000;19:663–9. doi: 10.1023/a:1007148202270. [DOI] [PubMed] [Google Scholar]
- 45.Sogaard M, Kadziola A, Haser R, Svensson B. Site-directed mutagenesis of histidine 93, aspartic acid 180, glutamic acid 205, histidine 290, and aspartic acid 291 at the active site and tryptophan 279 at the raw starch binding site in barley alpha-amylase 1. J Biol Chem. 1993;268:22480–4. [PubMed] [Google Scholar]
- 46.Robert X, Haser R, Mori H, Svensson B, Aghajari N. Oligosaccharide binding to barley alpha-amylase 1. J Biol Chem. 2005;280:32968–78. doi: 10.1074/jbc.M505515200. [DOI] [PubMed] [Google Scholar]
- 47.Boraston AB, Bolam DN, Gilbert HJ, Davies GJ. Carbohydrate-binding modules: fine-tuning polysaccharide recognition. Biochem J. 2004;382:769–81. doi: 10.1042/BJ20040892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Machovic M, Janecek S. The evolution of putative starch-binding domains. FEBS Lett. 2006;580:6349–56. doi: 10.1016/j.febslet.2006.10.041. [DOI] [PubMed] [Google Scholar]
- 49.Machovic M, Janecek S. Starch-binding domains in the post-genome era. Cell Mol Life Sci. 2006;63:2710–24. doi: 10.1007/s00018-006-6246-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Machovic M, Svensson B, MacGregor EA, Janecek S. A new clan of CBM families based on bioinformatics of starch-binding domains from families CBM20 and CBM21. Febs J. 2005;272:5497–513. doi: 10.1111/j.1742-4658.2005.04942.x. [DOI] [PubMed] [Google Scholar]
- 51.Robert X, Haser R, Gottschalk TE, Ratajczak F, Driguez H, Svensson B, Aghajari N. The structure of barley alpha-amylase isozyme 1 reveals a novel role of domain C in substrate recognition and binding: a pair of sugar tongs. Structure. 2003;11:973–84. doi: 10.1016/s0969-2126(03)00151-5. [DOI] [PubMed] [Google Scholar]
- 52.Bozonnet S, Jensen MT, Nielsen MM, Aghajari N, Jensen MH, Kramhoft B, Willemoes M, Tranier S, Haser R, Svensson B. The ‘pair of sugar tongs’ site on the non-catalytic domain C of barley alpha-amylase participates in substrate binding and activity. Febs J. 2007;274:5055–67. doi: 10.1111/j.1742-4658.2007.06024.x. [DOI] [PubMed] [Google Scholar]
- 53.Mishra PJ, Ragunath C, Ramasubbu N. The mechanism of salivary amylase hydrolysis: role of residues at subsite S2’. Biochem Biophys Res Commun. 2002;292:468–73. doi: 10.1006/bbrc.2002.6682. [DOI] [PubMed] [Google Scholar]
- 54.Ragunath C, Sundar K, Ramasubbu N. Expression, characterization, and biochemical properties of recombinant human salivary amylase. Protein Expr Purif. 2002;24:202–11. doi: 10.1006/prep.2001.1559. [DOI] [PubMed] [Google Scholar]
- 55.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–5. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 56.Ramasubbu N, Thomas LM, Ragunath C, Kaplan JB. Structural analysis of dispersin B, a biofilm-releasing glycoside hydrolase from the periodontopathogen Actinobacillus actinomycetemcomitans. J Mol Biol. 2005;349:475–86. doi: 10.1016/j.jmb.2005.03.082. [DOI] [PubMed] [Google Scholar]
- 57.Dubendorff JW, Studier FW. Creation of a T7 autogene. Cloning and expression of the gene for bacteriophage T7 RNA polymerase under control of its cognate promoter. J Mol Biol. 1991;219:61–8. doi: 10.1016/0022-2836(91)90857-3. [DOI] [PubMed] [Google Scholar]
- 58.Sambrook J, Fritsch E, Maniatis T. Molecular cloning A laborartory manual. 2. Cold Spring Harbor Laboratory Press; Cold Spring Harbor: 1989. [Google Scholar]
- 59.Bernfeld P. Amylase, a and b. Methods Enzymol. 1955;1:49–150. [Google Scholar]
- 60.Greenwood FC, Hunter WM, Glover JS. The Preparation of I-131-Labelled Human Growth Hormone of High Specific Radioactivity. Biochem J. 1963;89:114–23. doi: 10.1042/bj0890114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ramasubbu N, Bhandary KK, Scannapieco FA, Levine MJ. Crystallization and preliminary X-ray diffraction studies of human salivary alpha-amylase. Proteins. 1991;11:230–2. doi: 10.1002/prot.340110308. [DOI] [PubMed] [Google Scholar]
- 62.Otwinowski Z, Minor W. Processing of X-ray Crystallographic Data in Oscillation Mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 63.CCP4. The CCP4 Suite: Programs for Protein Crystallography. Acta Cryst. 1994;D50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 64.Emsley P, Cowtan K. Coot: modelbuilding tools for molecular graphics. Acta Cryst D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 65.Lamzin VS, Wilson KS. Automated refinement of protein models. Acta Cryst. 1993;D49:129–149. doi: 10.1107/S0907444992008886. [DOI] [PubMed] [Google Scholar]
- 66.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]