

Abstract
Following a pathologic insult, the adult mammalian heart undergoes hypertrophic growth and remodeling of the extracellular matrix. While a small sub-population of cardiomyocytes can re-enter the cell cycle following cardiac injury, the myocardium is largely thought to be incapable of significant regeneration. Periostin, an extracellular matrix protein, has recently been proposed to induce re-entry of differentiated cardiomyocytes back into the cell cycle and promote meaningful repair following myocardial infarction. Here, we show that while periostin is induced in the heart following injury, it does not stimulate DNA synthesis, mitosis or cytokinesis of cardiomyocytes in vitro or in vivo. Mice lacking the gene encoding periostin and mice with inducible overexpression of full-length periostin were analyzed at baseline and after myocardial infarction. There was no difference in heart size or a change in cardiomyocyte number in either periostin transgenic or gene-targeted mice at baseline. Quantification of proliferating myocytes in the peri-infarct area showed no difference between periostin overexpressing and null mice compared with strain-matched controls. In support of these observations, neither overexpression of periostin in cell culture, via an adenoviral vector, nor stimulation with recombinant protein induced DNA synthesis, mitosis or cytokinesis. Periostin is a regulator of cardiac remodeling and hypertrophy and may be a reasonable pharmacological target to mitigate heart failure, but manipulation of this protein appears to have no obvious effect on myocardial regeneration.
Keywords: Fibroblast, remodeling, periostin, cardiomyocyte proliferation, myocardial infarction
Introduction
Myocardial infarction (MI) leads to an irreversible loss of cardiomyocytes with expansion and remodeling of the extracellular matrix (ECM) and scar formation. This “fibrous scar” lacks contractile capacity and results in adverse changes to chamber geometry, global systolic dysfunction and poor ventricular compliance. If adult cardiomyocytes could proliferate and repopulate the injured area, cardiac function may be rescued, although the ability of cardiomyocytes to proliferate in a physiological meaningful manner remains an area of ongoing controversy.1-3 Recent reports have proposed that a population of post-mitotic cardiomyocytes, if exposed to the proper environment, may be capable of proliferation.3-6
Although a targeted therapy to drive cardiomyocyte proliferation has not been reported, one may be possible given that detectable, albeit extremely small increases in cardiomyocyte cell cycle re-entry can occur after myocardial injury. 3, 5, 7, 8 The number of cardiomyocytes that enter the cell cycle at baseline, evident by markers for DNA synthesis, occurs at a very low frequency of approximately 0.008%.8 The trigger that drives this small percentage of cells to proliferate has not yet been determined. A recent publication has proposed that the ECM protein periostin leads to the initiation of DNA synthesis, mitosis and ultimately cytokinesis.9 These results have sparked controversy among those interested in the function of periostin since most other reports have only observed an effect on the ECM and collagen fibrillogenesis.10, 11
Periostin is a 90-kDa secreted protein that contains four fasciclin domains12, 13 and is similar to the insect protein fasciclin I, which is involved in neuronal cell-cell adhesion.12 Periostin is detectable in the developing heart but is not present in the adult ventricular myocardium at baseline.14-19 It is re-expressed exclusively by fibroblasts or cells that adopt a fibroblast-like phenotype following an injurious event such as MI or pressure overload stimulation.20-23 Using transgenic and gene-deleted mice, periostin was shown to affect cardiac hypertrophy and ventricular remodeling.15 Periostin null (Pn−/−) mice are prone to rupture following MI, but if they survive the first 10 days of this insult, their cardiac morbidity is improved compared to wild-type mice.15 These data support the hypothesis that periostin orchestrates cellular remodeling and collagen deposition in the heart. Periostin also functions in a similar fashion when secreted by neoplastic cells.24-26 An increased concentration of periostin correlates with enhanced cellular migration and adhesion and is proportional to the cancer cells ability to invade and metastasize.24-28
Although the exact role of periostin has not been defined, the contention that it mediates physiological meaningful myocyte proliferation in the heart following MI is currently a matter of great interest considering the therapeutic ramifications. Here we used transgenic mice to determine if the presence of periostin leads to cardiomyocyte proliferation, or if its absence in gene-targeted mice antithetically reduces this index. We also examined a model of cellular proliferation in cultured neonatal cardiomyocytes, which have a finite capacity for proliferation. In both models we determined that periostin had no effect on any aspect of cell cycle re-entry in cardiomyocytes.
Materials and Methods
Animals
All mouse protocols were approved by the Institutional Animal Care and Use Committee of Cincinnati Children's Hospital Medical Center. Pn−/− and transgenic overexpressors (PntTA) were described previously.15
Western Blot Analysis
Western blot analysis confirmed overexpression of periostin protein in PntTA mice and its absence in Pn−/− mice. A rabbit polyclonal antibody to periostin was used.24 Western blotting procedures were described previously.29
Myocardial Infarction and BrdU labeling
MI was performed by permanent ligation of the left coronary artery as previously described.30 Due to the small size of the Pn−/− mice, body weight-matched 10−12 week-old Pn−/− mice (23.4 ± 0.6g) were used for MI while 8 week-old C57BL/6 mice (24.1 ± 0.5g) were used as controls. Because the growth of the PntTA mice is not affected, 8−10 week old PntTA mice and FVB littermates were used for MI. Seven days post MI, mice received intraperitoneal (i.p.) injections with 5-bromodeoxyuridine (BrdU) labeling reagent (1 ml/100g) (Zymed). Four hours after injection organs were harvested, fixed in buffered 10% formalin, embedded in paraffin, and cut into 6 μm sections. Duodenum was harvested and fixed as a positive BrdU labeling control.
Immunohistochemistry
Sections, 6 μm thick, were deparaffinized and incubated with primary antibody to BrdU (Sigma). Slides were stained with wheat germ agglutinin (WGA) -fluorescein-4-isothiocyanate (FITC) (Sigma) to distinguish the sarcolemma membrane. Nuclei were stained with TO-PRO-3 (Molecular Probes). Fluorescent secondary antibody was used (Molecular Probes) and slides were examined with confocal microscopy. To determine the number of cells positive for phosphorylated histone H3 (Cell Signaling), immunohistochemistry was performed following antigen retrieval with sodium citrate. These slides were also stained with WGA-FITC and TO-PRO-3. The peri-infarct area of four animals, >100,000 cells, were counted per cohort for both BrdU and phosphorylated histone H3. Data were presented as percent of positive cardiomyocyte nuclei versus total cardiomyocyte nuclei.
Histology and Cell Surface Area Measurements
Hearts were harvested and fixed in 10% buffered formalin, embedded in paraffin and cut into 6 μm sections. Sections were stained for fibrosis with Masson's trichrome or with WGA for the sarcolemma membrane. Measurement of myocyte cross sectional area was counted from both the apex and the center region of the left ventricle, in a minimum of 3 separate hearts from each cohort; >2000 cardiomyocytes were measured. Cross sectional area was calculated using Image J software.
Recombinant Adenovirus and in vitro Proliferation Experiments
A periostin recombinant adenovirus, AdPn, was generated using the full-length mouse periostin, subcloned into the shuttle vector for the Adeasy system (Invitrogen). All in vitro experiments were performed in primary neonatal rat cardiomyocytes and fibroblasts, which were isolated from 1- to 2- day old neonates and differentially plated as previously described.31 The cardiomyocytes were initially plated in 2% fetal bovine serum (FBS) in M199 media on gelatin coated chamber slides and infected/stimulated on day 2, after which the serum was reduced for experimentation of proliferation rates. For adenoviral infection, cardiomyocytes and fibroblasts were incubated for 2-hr with AdPn and AdGFP. The positive control cells were grown in media supplemented with 15% FBS, all other conditions were performed in 0.2% FBS media. Similar methods and conditions were used for the neonatal cardiac fibroblasts. To determine the effect of periostin, cells were infected with AdPn or stimulated daily with recombinant human periostin, a C-terminal truncated version of the full-length protein (BioVendor, Czech Republic). The source of recombinant periostin is the same as used by Kühn and colleagues.9 AdGFP and vehicle controls were also used under identical conditions. Infection efficiency was examined with AdGFP infection. Forty eight hours following infection, greater than 95% of the cells were GFP positive. A similar dosage of AdPn infection was used to achieve greater than 95% infectivity. To assess proliferation, cells were labeled with BrdU for the final 24 hours of experimentation. To detect DNA synthesis, immunohistochemistry for BrdU was performed, along with cell-type specific markers for cardiomyocytes; α-actinin (Sigma) and troponin (Cell Signaling), while fibroblasts were detected with vimentin (Sigma) antibody. Further immunohistochemistry was performed with antibodies to phosphorylated histone H3 and aurora B kinase (Abcam) to detect mitosis and cytokinesis, respectively. Fluorophoreconjugated secondary antibodies were provided by Invitrogen. Proliferating cells in culture were quantified (N>1000) and data presented as percent of positive nuclei versus total nuclei. Each experiment was repeated in quadruplicate.
Statistics
All data are expressed as mean ± SEM. Statistical significance was determined with a paired Student's t-test between 2 groups or one-way ANOVA where appropriate. Probability values of <0.05 were considered statistically significant.
Results
Periostin Does Not Induce Neonatal Cardiomyocyte or Fibroblast Proliferation in vitro
A previous report concluded that fibroblasts produce periostin to promote neighboring cardiomyocytes to re-enter the cell cycle in the heart.9 To examine this potential phenomenon in greater detail, in vitro experiments were first performed with isolated neonatal cardiomyocytes since they possess some proliferative capacity at baseline, suggesting they would be permissive to cell cycle re-entry. Cardiomyocytes were pulse-labeled with BrdU to detect DNA synthesis. As a positive control, 15% FBS was used, which induced a 2.1- fold increase in BrdU positive cardiomyocyte nuclei after 48 hrs compared with 0.2% FBS cultured myocytes. However, overexpression of periostin by infection with a recombinant adenovirus, AdPn, did not increase DNA synthesis rates compared with AdGFP infection (Figure 1A,B). AdPn treatment for 3 days also did not increase DNA synthesis (data not shown). Treatment with recombinant periostin also did not induce a significant change in DNA synthesis (Figure 1A,B). As neonatal cardiomyocyte preparations are rarely free of fibroblasts, myocytes were identified by expression of α-actinin and/or troponin. We further characterized cardiomyocyte proliferation by utilizing specific immunohistochemical markers of mitosis and cytokinesis. The percent of nuclei positive for phosphorylated histone H3, a marker for mitosis, was not significantly different between the AdPn infected cells or with recombinant periostin compared with their respective controls (Figure 1C,D). The percent of positive cells treated with recombinant periostin and vehicle were lower than the negative control, possibly due to the pH of the buffer required to reconstitute the protein. Cytokinesis, the final event of proliferation, was detected by aurora B kinase staining. Once again, neither infecting with AdPn or treatment with exogenous periostin significantly increased the percentage of cardiomyocytes undergoing cytokinesis, although serum stimulation was readily effective (Figure 1E,F). To rule out post translational processing as a requirement for bioactivity we also collected conditioned media of myocytes and fibroblasts infected with AdPn to incubate with freshly prepared neonatal myocytes for 48 hrs but, once again, no effect on DNA synthesis or other indexes of proliferation was observed (data not shown). As a control, AdPn infection of cardiomyocytes, or conditioned media from AdPn infected cardiomyocytes both showed expression of full-length periostin by Western blotting (Figure 1G).
Figure 1.

Neonatal cardiomyocyte proliferation is not altered by periostin. A. Immunocytochemistry detection of BrdU incorporation in neonatal cardiomyocytes versus total cardiomyocyte nuclei. Positive control was 15% FBS and the negative control (-) was no stimulation. B. Image of representative BrdU positive cardiomyocyte nuclei. Red is troponin (myocyte), blue is nuclei, and green is BrdU (arrow). C and D, Immunocytochemical quantification and representative image of cardiomyocyte nuclei positive for phosphorylated histone H3. Red is troponin (myocyte), blue is nuclei, and green is phosphorylated histone H3 (arrow). E and F, Immunocytochemical quantification and representative image of cardiomyocyte nuclei positive for aurora B kinase staining compared to total cardiomyocyte nuclei. Red is troponin (myocyte), blue is nuclei, and green is aurora B kinase (arrow). G, Western blot for periostin protein from AdPn infected cardiomyocytes versus AdGFP infection, or concentrated conditioned media 48 hrs after AdPn or AdGFP infection. The positive control (+Con) is protein extract from 2 day-old neonatal feet. *P<0.05 positive vs. negative control. Abbreviations: rPn, recombinant periostin; Pn, periostin; veh, vehicle.
To further delineate the role of periostin in inducing cell cycle re-entry, we repeated the above experiments with neonatal cardiac fibroblasts. AdPn and recombinant periostin did not stimulate DNA synthesis in fibroblasts, while the positive control of 15% FBS was efficacious (Figure 2A,B). Phosphorylated histone H3 staining revealed a 3-fold increase in the percent of cells undergoing mitosis in the 15% FBS group, while AdPn and recombinant periostin did not significantly alter this index of cellular proliferation (Figure 2C,D). Furthermore, AdPn and recombinant periostin had no effect on cellular mitosis, as detected by aurora B kinase immunohistochemistry (Figure 2E,F). In the myocardium, fibroblasts and cardiomyocytes co-exist, and are immersed in the same milieu of growth factors and proteins, hence additional co-culture experiments were performed. Neonatal rat cardiac fibroblasts were infected with AdPn and were subsequently cultured with neonatal rat cardiomyocytes. Markers of cell cycle re-entry, (BrdU, phosphorylated histone H3 and aurora B kinase) were examined and revealed no significant change in the quantity of cells proliferating (data not shown). Collectively, these data indicate that periostin cannot induce cell cycle re-entry in two different cell types that are normally permissive to some level of proliferation.
Figure 2.

Neonatal fibroblast proliferation is not altered by periostin. A, Immunocytochemistry detection of BrdU incorporation in primary fibroblasts versus total fibroblast nuclei. The positive control was 15% FBS and the negative control (-) was no stimulation. B, Image of a representative BrdU positive fibrolast nuclei. Red is troponin to show contaminating myocytes, blue is nuclei, and green is BrdU (arrows). C and D, Immunocytochemical quantification and representative image of fibroblast nuclei positive for phosphorylated histone H3. Red is vimentin (fibroblast), blue is nuclei, and green is phosphorylated histone H3 (arrow). E and F, Immunocytochemical quantification and representative image of fibroblast nuclei positive for aurora B kinase staining compared to total fibroblast nuclei. Red is vimentin (fibroblast), blue is nuclei, and green is aurora B kinase (arrow). *P<0.05 positive vs. negative control. Abbreviations: rPn, recombinant periostin; Pn, periostin; veh, vehicle.
Periostin Null and Overexpressing Mice Do Not Have Altered Myocyte Numbers in the Heart at Baseline
We also reasoned that if periostin directly regulates myocyte proliferation it was most likely to do so throughout development when these cells are readily dividing. Indeed, a large number of genetically modified mouse models with altered cell cycle regulatory proteins have been created that display significant changes in total myocyte cell numbers due to a developmental effect of these proteins. 32-35 However, baseline characterization of the Pn−/− and periostin overexpressing transgenic (PntTA) mice at 6 weeks of age showed no difference in total cell number in the heart. At 6 weeks of age periostin is not detectable in the heart of the wildtype or Pn−/− mice (data not shown) but is abundantly present in the hearts of the PntTA mice (Figure 3A). Importantly, the heart weight to body weight ratio was not altered in the Pn−/− animals (4.3 ±0.5 mg/g) versus age-matched controls (4.2 ± 0.4 mg/g) (Figure 3B) or PntTA transgenic animals (5.1 ± 0.2 mg/g) versus appropriate controls (4.8 ± 0.3 mg/g). PntTA mice at 6 months of age also showed no significant difference in heart size normalized to body weight (5.4 ±0.2 mg/g) when compared to controls (5.3 ± 0.3, Figure 3C). To calculate total myocyte number in the heart the cross sectional area of myocytes measured from ventricular histological sections was analyzed at 6 weeks of age and determined to be similar between the Pn−/−, PntTA, and their controls (Figure 3D,E). Cell number and myocyte fraction was assessed by immunohistochemistry and once again no differences were noted between any of the groups (data not shown). Additionally, the degree of fibrosis was not significantly different in the hearts of 6-week-old, PntTA mice and age-matched controls as assessed by Masson's trichrome staining of histological sections (data not shown).
Figure 3.

Baseline myocyte content is not different in the hearts of Pn−/− and Pn transgenic mice. A, Western blot for periostin (Pn) from hearts of 6 week-old FVB, tTA and PntTA mice. GAPDH is a loading control. B, Assessment of heart weight (HW) normalized to body weight (BW) in Pn−/− versus wild type (C57BL/6) mice at 6 weeks of age. C, HW/BW in PntTA and wildtype (FVB) at 6 weeks and 6 months of age. D and E, Assessment of myocyte cross sectional cell areas from left ventricular histological sections of the indicated genotypes (n=3 hearts each, with a minimum of 2000 cells counted in total).
Periostin Does Not Induce Cardiomyocyte Proliferation in vivo Following MI
Following myocardial injury there is abundant re-expression and accumulation of periostin in the interstitial space within and surrounding the injured area.15 Periostin protein is detected as early as 4 days after MI and peaks at 7 days, and remains detectable for up to 8 weeks.15, 16 To examine if periostin expression and accumulation in the ECM stimulates cardiomyocyte cell cycle re-entry in vivo, MI injury was induced in both the Pn−/− and PntTA transgenic mice, and their appropriate wild type controls. There was no difference in infarct size between the Pn−/− mice, the PntTA mice, and their respective controls in agreement with our previous results (data not shown).15 Histological analysis was performed 7 days after MI, when periostin expression is maximal (Figure 4A). Four hours prior to harvesting the heart, animals underwent an intraperitoneal injection of BrdU to label cardiomyocytes that were undergoing DNA synthesis. On examination of the peri-infarct area of control mice (C57BL/6), consistent with published data, there was a very small number, 0.035%, of BrdU positive cardiomyocyte nuclei. Pn−/− and PntTA transgenic mice did not show a significant change in cardiomyocyte DNA synthesis (Figure 4B). A representative immunohistochemical image from the infarct border zone is shown with cells outlined with WGA-FITC and BrdU labeling, which revealed nonmyocytes (asterisk) and a single labeled cardiomyocyte (arrow) as undergoing DNA synthesis (Figure 4B). To further support these results, immunohistochemical analysis was done to detect phosphorylated histone H3 staining as evidence of mitosis. Only 0.012% of myocyte nuclei were positive for phosphorylated histone H3 in the peri-infarct area. There was no significant increase in the percent of positive nuclei in either Pn−/− or PntTA mice (Figure 4C). A representative immunohistochemical image from the infarct border zone is shown with cells outlined with WGA-FITC and phosphorylated histone H3 labeling, which revealed a labeled non-myocyte (asterisk) and cardiomyocyte (arrow) (Figure 4C). A minimum of 4 mice was analyzed in each group. These results indicate that overexpression of periostin or its loss does not appreciably affect myocyte cell cycle activity in the peri-infarct region.
Figure 4.
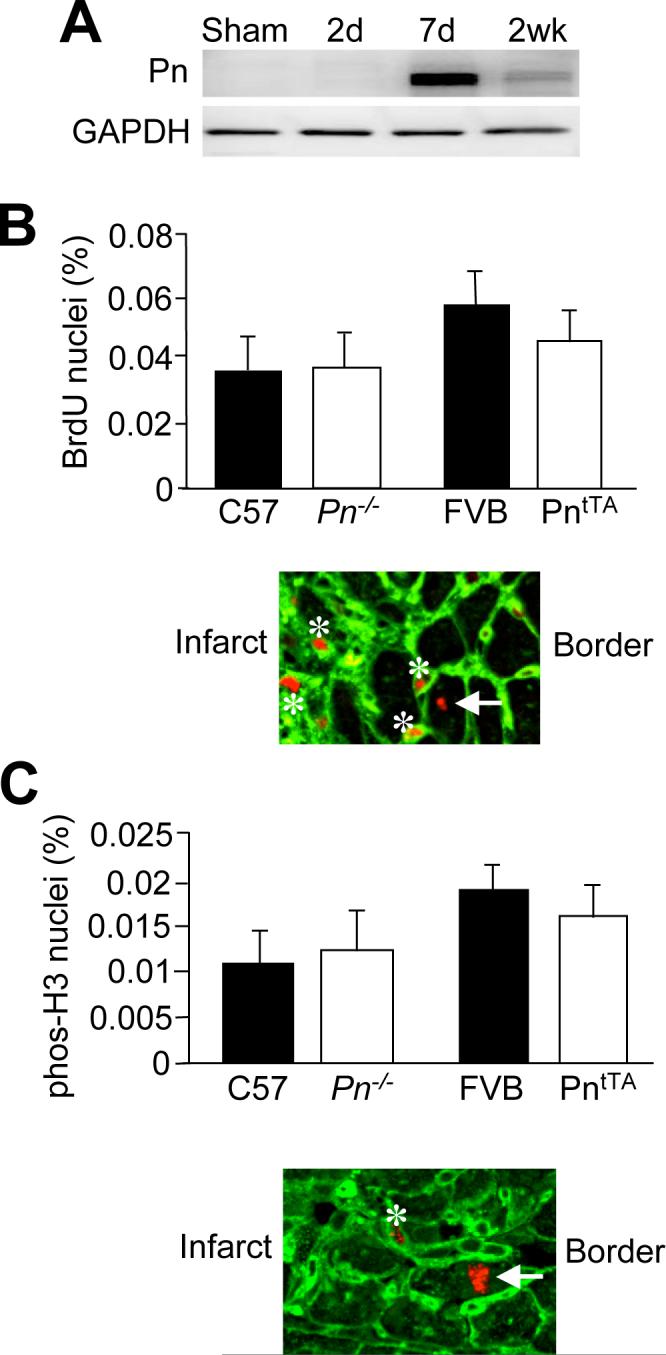
Assessment of cardiomyocyte proliferation 7 days after MI in Pn−/− and PntTA transgenic mice. A, Western blot analysis of periostin in the heart following MI. GAPDH is a loading control. B, Percent of BrdU positive cardiomyocyte nuclei when compared to total cardiomyocyte nuclei in the peri-infarct area of Pn−/−, PntTA mice and their appropriate controls. Below the graph a representative immunohistological image is shown with labeled non-myocytes (asterisk) and a labeled myocyte (arrow). C, Percent of phosphorylated histone H3 positive cardiomyocytes compared to total cardiomyocytes in the peri-infarct area of Pn−/−, PntTA mice and their appropriate controls. Below the graph a representative immunohistological image is shown with a labeled non-myocyte (asterisk) and a labeled myocyte (arrow). Experiments were performed on a minimum of 4 hearts per cohort. Greater then 100,000 cells were counted per experimental group. None of the groups were significantly different.
Discussion
While adult cardiac myocytes are not thought to proliferate in vivo given their highly differentiated state, a finite population are proposed to re-enter the cell cycle following experimental MI. 8 Such myocytes are typically located in the peri-infarct area and are presumably responding to external stimuli, such as growth factors or changes in ECM composition. Our rates of endogenous cardiomyocyte cell cycle activity in the peri-infarct region of hearts after MI were comparable to previous accounts in the literature (0.005% − 0.04%)5, 8, 36, which given the extremely low value is unlikely to contribute to cardiac regeneration in a meaningful manner. We also employed an in vitro model in which neonatal cardiomyocytes were isolated from 1−2 day old rat pups. This model was selected because neonatal myocytes inherently possess some degree of cell cycle activity, suggesting they are physiologically primed to respond to proliferative signals if they occur. Indeed, serum stimulation promoted a greater than 2-fold activation of cell cycle activity in these myocytes. However, in neither context did periostin alter myocyte cell cycle activity or proliferation.
As a secreted ECM protein that is associated with areas of fibrosis, remodeling and inflammation, periostin can directly interact with other ECM proteins such as fibronectin, tenascin-C, collagen I, collagen V, and heparin.10, 11, 23 Periostin serves as a ligand for select integrins, such as α5β3, αvβ5, and α4β6, where it can affect the ability of cells (fibroblasts or cancer cells) to migrate and/or undergo a epithelial-mesenchymal transition (EMT) in select tissues.24, 25 MI or pressure overload stimulation to the adult heart induces abundant re-expression of periostin from resident fibroblasts located between myocytes within the heart parenchyma proper, where prior to such stimulation the only observed expression of periostin in the heart is within the collagen rich environment of the valves.14-19 Periostin expression is also induced at sites of vascular injury,37, 38 in the lung after injury/fibrosis,23, 39 in and around tumors,39 and at wound sites.40 While not unequivocal, these various studies suggest a common biologic response whereby periostin becomes re-expressed at injury or inflammatory sites within the adult organism, often associated with a need for ECM remodeling and cellular migration, not unlike many developmental processes associated with periostin expression.
The function of periostin upon re-expression in the injured myocardium is currently an area of controversy given disparate accounts in the literature. We originally reported that Pn−/− mice had an altered fibrotic response and were unable to form proper scars after MI injury, leading to greater rates of LV rupture and death.15 This result was independently confirmed the following year in separately generated Pn null mice.16 Conversely, we generated transgenic mice overexpressing full-length periostin in the heart, which were protected from wall rupture following MI injury.15 Abundant periostin overexpression in the heart did not induce fibrosis, although it did eventually promote a mild hypertrophic response in aged mice (32 weeks). Another interesting aspect of Pn−/− mice is that they are less susceptible to fibrotic heart disease associated with long-term pressure overload or MI, resulting in better ventricular performance.15 These results suggest that loss of periostin is protective to the heart long-term, provided the initial insult is survived.
An entirely novel function for periostin was recently reported whereby cardiomyocyte proliferation and the repair of injured myocardium was shown to be regulated by this protein.9 In addition to reporting increased proliferation, Kühn et al. showed that periostin expression prevented fibrosis and reduced cardiac hypertrophy after an injury event, results that are not consistent with other reports.15 A potential reason for these differences may be that Kühn et al. utilized a recombinant truncated form of periostin (amino acids 22−669 of 811) in ascertaining biological effects.9 Using this abridged protein, as well as a full-length protein, the authors reported increased proliferation rates in adult rat cardiac myocytes cultured for a total of 12 days.9 These later results may also be influenced by the known process of de-differentiation that occurs in adult myocytes cultured for this length of time. We failed to observe any induction of cell cycle activity in a model system that is known to be responsive to proliferative signals, neonatal myocytes in culture for 2 or 3 days.
More remarkably, Kühn et al. reported that application of the truncated form of periostin with a gelfoam patch onto infarcted rat hearts reduced fibrosis, reduced cardiac hypertrophy, increased myocyte proliferation, and improved cardiac function.9 However, overexpression of full-length periostin in the mouse heart by transgenesis (via the well-characterized α-myosin heavy chain promoter), which results in the proper accumulation of periostin protein in the ECM without detectable intracellular retention, did not increase myocyte number at baseline, nor did it augment indices associated with cardiac repair after MI injury.15 However, one potential difference with our study is that the Pn transgene was used in a constitutive manner so that periostin protein was always present, while Kühn et al. applied recombinant periostin acutely. We could not mimic this acute affect because the inducible Pn transgene takes 4−6 weeks to achieve reasonable expression due to the time it takes doxycycline to clear the mouse's system. Also to be considered, we did not observe differences in infarct size in any of the experimental or control groups, suggesting that the presence of the tetracycline transactivator was not a confounding variable (data not shown). Another potential difference is that we used the mouse while Kühn et al used the rat for infarct studies. Finally, our transgenic mice expressed only one isoform of periostin, of which there are many alternately spliced versions present in the heart15. Thus, it is possible that another spliced form of periostin could have cell cycle promoting activity that is akin to the truncated version of periostin employed by Kühn et al.9
While our data are negative concerning a role for periostin in regulating myocyte proliferation, we believe that they are credible given the nature of the model systems employed and our consistent use of positive and negative controls. First, we carefully assessed the possibility that lack of periostin or constitutive expression of periostin throughout embryonic and postnatal development would alter myocyte numbers in the heart, although no effect was observed. By comparison, transgene mediated overexpression of calmodulin in the developing mouse heart promoted a 73% increase in myocyte number by embryonic day 19 and a 40% increase in myocyte number in the adult heart.32 Overexpression of either insulin-like growth factor 1 in the heart or Bcl-2 increased myocyte number as measured by assessment of myocyte size normalized to heart weight and volume fractions.33, 34 Overexpression of telomerase in the heart by transgenesis also produced greater myocyte content in the heart with some hypertrophy.35 Second, neither Pn−/− mice nor periostin overexpressing transgenic mice showed any difference in stimulated cell cycle activity in myocytes within the peri-infarct zone of the adult mouse heart. Third, application of periostin by multiple methods was without effect on proliferation and cell cycle activity in cultured neonatal cardiomyocytes. Forth, Shimazaki, et al. also noted no difference in Ki67 positive cells in the infarct border zone of Pn−/− mice.16 In conclusion, while real differences in experimental conditions may have underlied an inability to detect proliferation and regeneration in our mice, we favor the alternate hypothesis that periostin mostly functions in collagen fibrillogenesis, ventricular remodeling, and in facilitating cellular movements in areas of injury or ongoing EMT.
Acknowledgements and source of funding
This work was supported by grants from the National Institutes of Health (J.D.M), the Fondation Leducq (Heart failure network grant to J.D.M), and the Howard Hughes Medical Institute (J.D.M.). A.L was supported by a physician scientist award pediatric center grant from the National Institutes of Health, Institute of Child Health and Human Development.
Footnotes
Disclosures: None
References
- 1.Rumyantsev PP. Interrelations of the proliferation and differentiation processes during cardiact myogenesis and regeneration. Int Rev Cytol. 1977;51:186–273. [PubMed] [Google Scholar]
- 2.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circ Res. 2002;90:1044–1054. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 3.Soonpaa MH, Field LJ. Survey of studies examining mammalian cardiomyocyte DNA synthesis. Circ Res. 1998;83:15–26. doi: 10.1161/01.res.83.1.15. [DOI] [PubMed] [Google Scholar]
- 4.Engel FB, Schebesta M, Duong MT, Lu G, Ren S, Madwed JB, Jiang H, Wang Y, Keating MT. p38 MAP kinase inhibition enables proliferation of adult mammalian cardiomyocytes. Genes Dev. 2005;19:1175–1187. doi: 10.1101/gad.1306705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P. Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med. 2001;344:1750–1757. doi: 10.1056/NEJM200106073442303. [DOI] [PubMed] [Google Scholar]
- 6.Anversa P, Nadal-Ginard B. Myocyte renewal and ventricular remodelling. Nature. 2002;415:240–243. doi: 10.1038/415240a. [DOI] [PubMed] [Google Scholar]
- 7.Nakajima H, Nakajima HO, Tsai SC, Field LJ. Expression of mutant p193 and p53 permits cardiomyocyte cell cycle reentry after myocardial infarction in transgenic mice. Circ Res. 2004;94:1606–1614. doi: 10.1161/01.RES.0000132279.99249.f4. [DOI] [PubMed] [Google Scholar]
- 8.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. Am J Physiol. 1997;272:H220–226. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 9.Kuhn B, del Monte F, Hajjar RJ, Chang YS, Lebeche D, Arab S, Keating MT. Periostin induces proliferation of differentiated cardiomyocytes and promotes cardiac repair. Nat Med. 2007;13:962–969. doi: 10.1038/nm1619. [DOI] [PubMed] [Google Scholar]
- 10.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, Trusk T, Potts JD, Goodwin RL, Davis J, Hoffman S, Wen X, Sugi Y, Kern CB, Mjaatvedt CH, Turner DK, Oka T, Conway SJ, Molkentin JD, Forgacs G, Markwald RR. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101:695–711. doi: 10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kii I, Amizuka N, Minqi L, Kitajima S, Saga Y, Kudo A. Periostin is an extracellular matrix protein required for eruption of incisors in mice. Biochem Biophys Res Commun. 2006;342:766–772. doi: 10.1016/j.bbrc.2006.02.016. [DOI] [PubMed] [Google Scholar]
- 12.Zinn K, McAllister L, Goodman CS. Sequence analysis and neuronal expression of fasciclin I in grasshopper and Drosophila. Cell. 1988;53:577–587. doi: 10.1016/0092-8674(88)90574-0. [DOI] [PubMed] [Google Scholar]
- 13.Horiuchi K, Amizuka N, Takeshita S, Takamatsu H, Katsuura M, Ozawa H, Toyama Y, Bonewald LF, Kudo A. Identification and characterization of a novel protein, periostin, with restricted expression to periosteum and periodontal ligament and increased expression by transforming growth factor beta. J Bone Miner Res. 1999;14:1239–1249. doi: 10.1359/jbmr.1999.14.7.1239. [DOI] [PubMed] [Google Scholar]
- 14.Snider P, Hinton RB, Moreno-Rodriguez RA, Wang J, Rogers R, Lindsley A, Li F, Ingram DA, Menick D, Field L, Firulli AB, Molkentin JD, Markwald R, Conway SJ. Periostin is required for maturation and extracellular matrix stabilization of noncardiomyocyte lineages of the heart. Circ Res. 2008;102:752–760. doi: 10.1161/CIRCRESAHA.107.159517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, Lorts A, Brunskill EW, Dorn GW, 2nd, Conway SJ, Aronow BJ, Robbins J, Molkentin JD. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shimazaki M, Nakamura K, Kii I, Kashima T, Amizuka N, Li M, Saito M, Fukuda K, Nishiyama T, Kitajima S, Saga Y, Fukayama M, Sata M, Kudo A. Periostin is essential for cardiac healing after acute myocardial infarction. J Exp Med. 2008;205:295–303. doi: 10.1084/jem.20071297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Johnatty SE, Dyck JR, Michael LH, Olson EN, Abdellatif M. Identification of genes regulated during mechanical load-induced cardiac hypertrophy. J Mol Cell Cardiol. 2000;32:805–815. doi: 10.1006/jmcc.2000.1122. [DOI] [PubMed] [Google Scholar]
- 18.Stanton LW, Garrard LJ, Damm D, Garrick BL, Lam A, Kapoun AM, Zheng Q, Protter AA, Schreiner GF, White RT. Altered patterns of gene expression in response to myocardial infarction. Circ Res. 2000;86:939–945. doi: 10.1161/01.res.86.9.939. [DOI] [PubMed] [Google Scholar]
- 19.Wang D, Oparil S, Feng JA, Li P, Perry G, Chen LB, Dai M, John SW, Chen YF. Effects of pressure overload on extracellular matrix expression in the heart of the atrial natriuretic peptide-null mouse. Hypertension. 2003;42:88–95. doi: 10.1161/01.HYP.0000074905.22908.A6. [DOI] [PubMed] [Google Scholar]
- 20.Kruzynska-Frejtag A, Machnicki M, Rogers R, Markwald RR, Conway SJ. Periostin (an osteoblast-specific factor) is expressed within the embryonic mouse heart during valve formation. Mech Dev. 2001;103:183–188. doi: 10.1016/s0925-4773(01)00356-2. [DOI] [PubMed] [Google Scholar]
- 21.Wilde J, Yokozeki M, Terai K, Kudo A, Moriyama K. The divergent expression of periostin mRNA in the periodontal ligament during experimental tooth movement. Cell Tissue Res. 2003;312:345–351. doi: 10.1007/s00441-002-0664-2. [DOI] [PubMed] [Google Scholar]
- 22.Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, Kawashima K, Kaneda Y, Ogihara T, Sugimura K. Periostin as a novel factor responsible for ventricular dilation. Circulation. 2004;110:1806–1813. doi: 10.1161/01.CIR.0000142607.33398.54. [DOI] [PubMed] [Google Scholar]
- 23.Takayama G, Arima K, Kanaji T, Toda S, Tanaka H, Shoji S, McKenzie AN, Nagai H, Hotokebuchi T, Izuhara K. Periostin: a novel component of subepithelial fibrosis of bronchial asthma downstream of IL-4 and IL-13 signals. J Allergy Clin Immunol. 2006;118:98–104. doi: 10.1016/j.jaci.2006.02.046. [DOI] [PubMed] [Google Scholar]
- 24.Gillan L, Matei D, Fishman DA, Gerbin CS, Karlan BY, Chang DD. Periostin secreted by epithelial ovarian carcinoma is a ligand for alpha(V)beta(3) and alpha(V)beta(5) integrins and promotes cell motility. Cancer Res. 2002;62:5358–5364. [PubMed] [Google Scholar]
- 25.Baril P, Gangeswaran R, Mahon PC, Caulee K, Kocher HM, Harada T, Zhu M, Kalthoff H, Crnogorac-Jurcevic T, Lemoine NR. Periostin promotes invasiveness and resistance of pancreatic cancer cells to hypoxia-induced cell death: role of the beta4 integrin and the PI3k pathway. Oncogene. 2007;26:2082–2094. doi: 10.1038/sj.onc.1210009. [DOI] [PubMed] [Google Scholar]
- 26.Tai IT, Dai M, Chen LB. Periostin induction in tumor cell line explants and inhibition of in vitro cell growth by anti-periostin antibodies. Carcinogenesis. 2005;26:908–915. doi: 10.1093/carcin/bgi034. [DOI] [PubMed] [Google Scholar]
- 27.Yan W, Shao R. Transduction of a mesenchyme-specific gene periostin into 293T cells induces cell invasive activity through epithelial-mesenchymal transformation. J Biol Chem. 2006;281:19700–19708. doi: 10.1074/jbc.M601856200. [DOI] [PubMed] [Google Scholar]
- 28.Bao S, Ouyang G, Bai X, Huang Z, Ma C, Liu M, Shao R, Anderson RM, Rich JN, Wang XF. Periostin potently promotes metastatic growth of colon cancer by augmenting cell survival via the Akt/PKB pathway. Cancer Cell. 2004;5:329–339. doi: 10.1016/s1535-6108(04)00081-9. [DOI] [PubMed] [Google Scholar]
- 29.Kaiser RA, Liang Q, Bueno O, Huang Y, Lackey T, Klevitsky R, Hewett TE, Molkentin JD. Genetic inhibition or activation of JNK1/2 protects the myocardium from ischemia-reperfusion-induced cell death in vivo. J Biol Chem. 2005;280:32602–32608. doi: 10.1074/jbc.M500684200. [DOI] [PubMed] [Google Scholar]
- 30.Wilkins BJ, Dai YS, Bueno OF, Parsons SA, Xu J, Plank DM, Jones F, Kimball TR, Molkentin JD. Calcineurin/NFAT coupling participates in pathological, but not physiological, cardiac hypertrophy. Circ Res. 2004;94:110–118. doi: 10.1161/01.RES.0000109415.17511.18. [DOI] [PubMed] [Google Scholar]
- 31.Oka T, Dai YS, Molkentin JD. Regulation of calcineurin through transcriptional induction of the calcineurin A beta promoter in vitro and in vivo. Mol Cell Biol. 2005;25:6649–6659. doi: 10.1128/MCB.25.15.6649-6659.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Colomer JM, Terasawa M, Means AR. Targeted expression of calmodulin increases ventricular cardiomyocyte proliferation and deoxyribonucleic acid synthesis during mouse development. Endocrinology. 2004;145:1356–1366. doi: 10.1210/en.2003-1119. [DOI] [PubMed] [Google Scholar]
- 33.Limana F, Urbanek K, Chimenti S, Quaini F, Leri A, Kajstura J, Nadal-Ginard B, Izumo S, Anversa P. bcl-2 overexpression promotes myocyte proliferation. Proc Natl Acad Sci U S A. 2002;99:6257–6262. doi: 10.1073/pnas.092672899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Reiss K, Cheng W, Ferber A, Kajstura J, Li P, Li B, Olivetti G, Homcy CJ, Baserga R, Anversa P. Overexpression of insulin-like growth factor-1 in the heart is coupled with myocyte proliferation in transgenic mice. Proc Natl Acad Sci U S A. 1996;93:8630–8635. doi: 10.1073/pnas.93.16.8630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Oh H, Taffet GE, Youker KA, Entman ML, Overbeek PA, Michael LH, Schneider MD. Telomerase reverse transcriptase promotes cardiac muscle cell proliferation, hypertrophy, and survival. Proc Natl Acad Sci U S A. 2001;98:10308–10313. doi: 10.1073/pnas.191169098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kajstura J, Leri A, Finato N, Di Loreto C, Beltrami CA, Anversa P. Myocyte proliferation in end-stage cardiac failure in humans. Proc Natl Acad Sci U S A. 1998;95:8801–8805. doi: 10.1073/pnas.95.15.8801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li G, Oparil S, Sanders JM, Zhang L, Dai M, Chen LB, Conway SJ, McNamara CA, Sarembock IJ. Phosphatidylinositol-3-kinase signaling mediates vascular smooth muscle cell expression of periostin in vivo and in vitro. Atherosclerosis. 2006;188:292–300. doi: 10.1016/j.atherosclerosis.2005.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen YF, Feng JA, Li P, Xing D, Ambalavanan N, Oparil S. Atrial natriuretic peptide-dependent modulation of hypoxia-induced pulmonary vascular remodeling. Life Sci. 2006;79:1357–1365. doi: 10.1016/j.lfs.2006.03.051. [DOI] [PubMed] [Google Scholar]
- 39.Hayashi N, Yoshimoto T, Izuhara K, Matsui K, Tanaka T, Nakanishi K. T helper 1 cells stimulated with ovalbumin and IL-18 induce airway hyperresponsiveness and lung fibrosis by IFN-gamma and IL-13 production. Proc Natl Acad Sci U S A. 2007;104:14765–14770. doi: 10.1073/pnas.0706378104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Roy S, Patel D, Khanna S, Gordillo GM, Biswas S, Friedman A, Sen CK. Transcriptome-wide analysis of blood vessels laser captured from human skin and chronic wound-edge tissue. Proc Natl Acad Sci U S A. 2007;104:14472–14477. doi: 10.1073/pnas.0706793104. [DOI] [PMC free article] [PubMed] [Google Scholar]