

Abstract
We investigated the binding of a naturally occurring antibiotic, actinonin, to the Ni2+ reconstituted recombinant form of E. coli peptide deformylase (PDFEc) via isothermal titration microcalorimetry. The binding data conformed to both exothermic and endothermic phases with magnitudes of ΔG°, ΔH°, and TΔS° being equal to −12, −2.7, and 9.3, and −8.7, 3.9, and 12.6 kcal/mol, respectively. Evidently, although both phases are dominated by favorable entropic changes, the exothermic phase has about 6.7 kcal/mol enthalpic advantage over the endothermic phase. We observed that the removal of bound Ni2+ from PDFEc abolished the exothermic phase without affecting the endothermic phase, but it was regained upon addition of Zn2+. In conjunction with metal analysis data, we propose that the recombinant form of PDFEc is expressed in two stable conformational states that yield markedly distinct ITC profiles (i.e., exothermic versus endothermic) upon interaction with actinonin. The existence of two conformational states of PDFEc is further supported by the observation of two distinct and independent transitions during the thermal unfolding of the enzyme. In addition, the thermodynamic data reveals that the formation of the PDFEc-actinonin complex results in the transfer of one H+ from the enzyme phase to the bulk solvent at pH 6.3. Both exothermic and endothermic phases produce highly negative ΔCp° values, but there is no apparent enthalpy-entropy compensation effect upon formation of the PDFEc-actinonin complex. In view of the known structural features of the enzyme, arguments are presented that the alternative conformational states of PDFEc are modulated by the metal ligation at the enzyme site.
There has been a growing realization that the increased resistance to antibiotics in pathogenic microbes is being manifested in serious problems for infected patients worldwide (1). Due to the resistance to antibiotics, many drugs that were once mainstay therapeutics have become ineffective and are no longer utilized. To fill the void left by obsolete/resistant antibiotics, there has been a widespread push to develop antibiotics that elicit their effects via new molecular targets. One of the dominating themes in the de novo design of antibiotics is the identification of compounds which selectively inhibit enzymes present in microbes but not in humans (2; 3). Peptide deformylase (PDF) is one such enzyme (4), and thus has become a target for the design of new antibiotics.
In prokaryotic organisms as well as in the organelles of eukaryotes, protein synthesis is initiated by a formylated methionine residue (FMet) (5-7). Activation of nascent proteins generally requires the removal of this residue from their N-termini by the actions of two enzymes, namely peptide deformylase and methionine aminopeptidase (8). The removal of the formyl group from FMet proteins is facilitated by the action of PDF (Scheme 1).
Scheme 1.

Reaction catalyzed by peptide deformylase.
Whereas PDF is ubiquitous in prokaryotes, the genomes of eukaryotes may or may not contain the def gene that encodes the enzyme. Eukaryotes such as A. thaliana that contain one or more def genes have been shown to express active PDF isozymes (9), and in recent years an isoform of PDF was discovered to be present in the human mitochondrion (10-12). However, the latter PDF has been shown to be weakly active in vitro due to a specific active site mutation (10; 11), and consequently (although evidence suggests that the enzyme may have a role in malignant cell types (13)), human PDF has been postulated to be non-functional in normal cells (11). Consistent with this premise is that proteins within the human mitochondrion retain their N-terminal formyl groups (14; 15). Since these proteins are able to function with FMet intact, this supports a minimal functional role of the mitochondrial PDF in its resident milieu. On the other hand, the retention of the N-terminal formyl group in nascent proteins is lethal to prokaryotes and results in the termination of cellular function (16). These features lead to the suggestion that the inhibition of PDF has the potential to eliminate, or at least suppress, the growth of microbial pathogens. Hence, it is not surprising that peptide deformylase falls in the category of a high-priority antibiotic target.
Due to its biomedical relevance, coupled with the ease of cloning, expression, and purification of the recombinant form of PDF from E. coli (PDFEc), there has been a growing interest in the detailed mechanistic and structural-functional studies of this enzyme. Several structures of PDF have been determined via NMR (17) and X-ray crystallographic (18; 19) methods in the absence and presence of inhibitors. These studies revealed that PDFEc is a monomeric enzyme comprised of approximately 40% alpha-helix (Figure 1) (20). The largely hydrophobic active site pocket of the enzyme contains a bound metal ion, which is tetrahedrally coordinated by His132, His136, Cys90 and a water molecule in the ligand-free state. The latter is replaced by the metal binding groups of putative inhibitors. Due to its Lewis acid characteristics, the active site metal ion of the enzyme deprotonates the bound water molecule and thus facilitates the nucleophilic attack to the formyl moiety of the substrate during catalysis (19).
Figure 1.
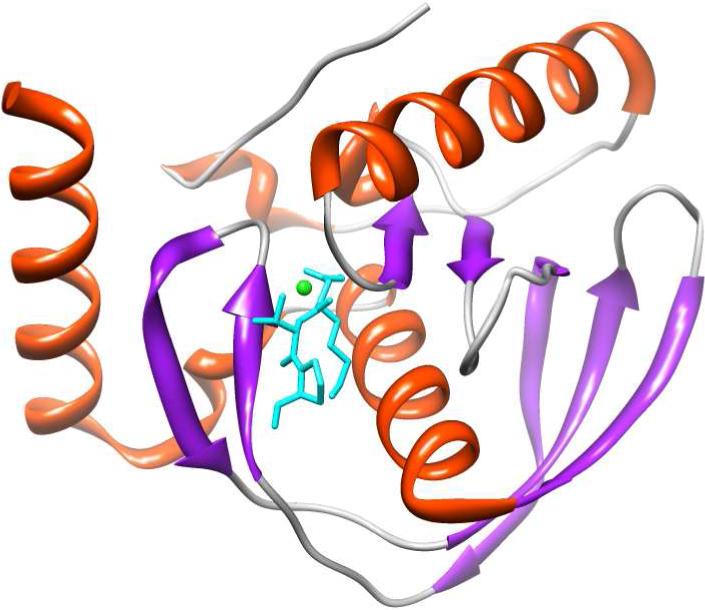
Structural depiction of E. coli PDF in complex with actinonin (PDB ID: 1G2A) as determined by Clements et al. (20). Actinonin is shown in cyan and the bound Ni2+ is depicted in green. The structural depiction of PDF was produced using the UCSF Chimera modeling package (21) from the Resource for Biocomputing, Visualization, and Informatics (University of California, San Francisco; supported by NIH P41 RR-01081).
Although PDFEc was discovered in 1968 (21; 22), the enzyme could not be purified in a fully-active form for about thirty years (23). The first highly purified form of PDFEc was shown to contain a bound Zn2+, and the enzyme was proposed to be a Zn2+ metalloprotease due to its similarities with thermolysin and matrix metalloproteinases (17; 24; 25). However, the purified Zn2+-containing enzyme had a much lower activity than that of the crude extract, causing uncertainty of the physiological metal ion. Subsequently, it was discovered that Fe2+ served as the physiological metal ion during the enzyme catalyzed deformylation reaction (26). The erroneous assignment of the weakly-active Zn2+ as the catalytic metal ion in PDFEc was due to the tight binding affinity of Zn2+ as compared to the fully-active Fe2+, which resulted in a significant fraction of PDFEc in complex with the former metal when expressed in rich media. Moreover, since the Fe2+ form of PDFEc undergoes slow inactivation under aerobic conditions (due to the Fe2+-assisted oxidation of the essential Cys90 residue (27)), recourse was made to prepare Ni2+ or Co2+ substituted PDFs for a variety of structural-functional studies as they retain high activity (28; 29).
The design and screening of nearly all PDF inhibitors has been accomplished using Ni2+ or Co2+-substituted enzyme, and the majority of such inhibitors contain functional groups that bind directly to the catalytic metal ion (30). One highly potent and selective inhibitor of PDF has been the naturally occurring antibiotic actinonin, which has been known to interact with the enzyme-bound metal ions (via its hydroxamate functional group) (31). Biophysical characterizations of the PDF-actinonin complex revealed that the binding to Ni2+-substituted S. aureus, H. influenzae and E. coli enzymes exhibited a two-step binding process that resulted in an effective Ki in the picomolar range (32). As a consequence of the tight binding affinity of actinonin for PDF isozymes, it has been successfully utilized as a lead compound in the further design of selective PDF inhibitors for use as potential antibiotics as well as cancer therapeutics (13; 33).
Recently, we became interested in ascertaining the mechanistic and structural-functional features of PDFEc from the point of view of designing novel inhibitors of the enzyme. In this endeavor, we discovered that the unfolding pathway of the enzyme was suddenly reversed following formation of an intermediary species (34). An extension of these studies provided evidence that the Ni2+-substituted PDFEc existed in at least two stable conformational states. To determine the impact of this finding on the binding of ligands to PDFEc and deciphering the thermodynamic basis of their interactions, isothermal titration calorimetric studies for the binding of actinonin to the enzyme were undertaken. Such studies have been sparse in the case of PDFEC.‡ As will be elaborated in the subsequent sections, this study has shed light on the alternative conformational states of the enzyme and the changes in their microenvironments upon binding of actinonin to the enzyme.
EXPERIMENTAL PROCEDURES
MATERIALS
Actinonin was purchased from Peptides International, Inc. (Louisville, KY) and Chelex® 100 was purchased from Bio-Rad (Hercules, CA). All other reagents were of analytical reagent grade.
METHODS
Full-length E. coli peptide deformylase (PDFEc) was purified in the Ni2+-substituted form as described previously (34; 36). The purified preparation of the enzyme exhibited a single band on SDS-PAGE. The presence of Ni2+ in the enzyme preparation was ascertained by the presence of an absorption peak at 355 nm due to the formation of Cys90-Ni2+ charge transfer complex (28; 36). The enzyme activity was measured via the A. proteolytica aminopeptidase-coupled assay utilizing FMet-Leu-p-nitroanilide as a substrate (37). The stock solution of PDFEc was stored in a 50 mM HEPES, pH 7.5 containing 5 mM NiCl2, and was subjected to dialysis in appropriate buffers suited for other studies. The protein concentration of the enzyme was determined via the Bradford assay (38) using BSA as a standard, and a correction factor of 0.56 X Bradford concentration was used for calculating the PDFEc concentration (23). Removal of Ni2+ from PDFEc was accomplished by exhaustive dialysis against 50 mM potassium phosphate, pH 7.0 (treated with Chelex® 100) and confirmed by the disappearance of the charge transfer peak. The dialyzed PDFEc was either used as such or treated with 10 mM ZnCl2 and passed through a 0.2 micron filter to prepare the Zn2+-substituted PDFEc. A stock solution of 5 mM actinonin was prepared in 50 mM potassium phosphate buffer, pH 7.0 and diluted to 300−350 μM in the experimental buffer immediately prior to titration.
Metal Analysis
The metal contents of PDFEc were determined via Inductively Coupled Plasma Emission (ICPE) spectroscopy at the University of Georgia Chemical Analysis Laboratory (Athens, GA). The enzyme samples (30−50 μM) were dialyzed against 50 mM potassium phosphate buffer, pH 7.0 with or without 1 mM NiCl2 using dialyzate buffer as a blank.
Isothermal Titration Microcalorimetry
A VP-ITC isothermal titration microcalorimeter (MicroCal, Inc. Northampton, MA) was employed for all calorimetric experiments utilizing 0.02% sodium azide as a reference and 50 mM potassium phosphate, pH 7.0 containing 1 mM NiCl2 as sample buffer unless otherwise noted. For comprehensive reviews of ITC function, data analysis and applications, refer to Wiseman et al. (39) and Leavitt and Freire (40). Before initiating the titration run, both enzyme and actinonin solutions were thoroughly degassed under vacuum. The sample cell was filled either with 1.8 ml (effective volume = 1.36 ml) of buffer (control) or 31.5 μM PDFEc. Injections of 6-μl each plus an initial injection of 3-μl were titrated into the sample cell for a total of 40−50 aliquots of actinonin. To ensure proper mixing, the reaction mixture was stirred at 310 rpm throughout the entire experiment. All ITC experiments were performed twice with the exception of the temperature-dependent measurements which were performed once at each temperature above or below 25 °C.
Raw experimental data were collected as the amount of heat produced per second following each injection of ligand into the sample cell. This is converted into the amount of heat produced per injection via integration of the area under individual peaks (performed automatically by the analysis software provided with the instrument). The observed heats per injection in ITC experiments are a function of the heats of binding as well as background signals that are mainly due to heats of dilution. As the heats of dilution determined in control experiments were of essentially the same magnitude as the heats obtained at the end of the titration (at saturation), we utilized the last few injections as the background. Specifically, the heats per injection of the final 5−6 injections were calculated and their average subtracted from all the injections. Final data are presented as the corrected amount of molar heat produced per injection versus the molar ratio of actinonin to PDFEc.
To analyze the data, either the single or double binding site models included in MicroCal Origin® 5.0 software were utilized with all parameters (viz., n, Ka and ΔH°) allowed to vary without constraints. The standard errors listed are those derived from the best fit of the experimental data to the binding model. From the data analysis, the stoichiometry (n), association constant (Ka) and the standard enthalpy changes (ΔH°) for the binding of actinonin to PDFEc are obtained. The standard free energy changes (ΔG°) for the binding process were calculated by the relationship, ΔG° = −RT ln Ka, and the standard entropy changes (ΔS°) were then calculated according to the standard thermodynamic equation, ΔG° = ΔH°-TΔS°.
The temperature-dependent experiments were conducted in the standard 50 mM phosphate buffer, pH 7.0 containing 1 mM NiCl2. The following buffers containing 1 mM NiCl2 were used to determine proton uptake or release upon binding of actinonin to PDFEc: 50 mM potassium phosphate (ΔH° ion = 1.22 kcal mol−1) or 100 mM Tris (ΔH° ion = 11.51 kcal mol−1) plus 50 mM potassium phosphate at pH 6.3 and 7.0. The ionization enthalpies (ΔH° ion) of the above buffer species were taken from Morin & Freire (41).
Circular Dichroism
Thermodynamic analyses of PDFEc unfolding as a function of temperature were performed in triplicate on a Jasco J-815 spectropolarimeter (Tokyo, Japan) equipped with a peltier temperature control. Using a 1-mm path length quartz cuvette, PDFEc samples (35 μM in 5 mM HEPES, pH 7.5 containing 5 mM NiCl2) were ramped at a heating rate of 2 °C per minute from 30 °C to 95 °C and the ellipticity at 223 nm was monitored as a function of temperature with the midpoint of the cooperative (sigmoidal) transitions taken as the transition temperature (Tm). To test for interconversion between the two apparent conformations of PDF, samples were first heated at 60 °C for 10 min, cooled on ice and centrifuged to remove precipitate prior to scanning.
RESULTS
The purification protocol for the recombinant form of E. coli PDF consistently yielded large quantities of active enzyme which exhibited a single band on the SDS-PAGE at approximately 22 kD (as reported previously for the recombinant PDFEc preparations; (23)), attesting to an apparent homogeneity of the enzyme preparation. The latter feature implied that the purified PDFEc predominated in a single population state. However, due to its differential avidity for the metal cofactors, the homogenous preparation of the enzyme can have different types of metal bound forms, and such forms may exhibit differential kinetic and thermodynamic properties (36). To determine the relative distribution of metal ions, we subjected the purified preparation of the Ni2+ reconstituted form of PDFEc to metal analysis via Inductively Coupled Plasma Emission (ICPE) spectroscopy (see Materials and Methods). Such analysis revealed that our enzyme preparation contained a nearly equal amount (i.e., mole fraction) of both Ni2+ and Zn2+. This was despite the fact that all buffers used in the preparation of PDFEc were made using high purity reagents and water that had been treated with a chelating resin to remove the trace of metal contaminants (particularly Zn2+, since it has the highest binding affinity for the enzyme) from our samples. Apparently, Zn2+ contamination of our enzyme came from growing the E. coli cells in rich media, which presumably contained ample Zn2+. Thus, it could easily compete with the physiological Fe2+ in vivo prior to the replacement of the latter by Ni2+ during purification. This finding has been consistent with the previous reports that the Zn2+ contents of PDFEc can vary from 0.1 to 1.2 (mol/mol) (23; 36).
It is established that Zn2+ and Ni2+-containing PDFEc is inhibited by actinonin with similar IC50 values in the nanomolar range (31). We became interested whether or not the above metal-bound forms of the enzyme exhibited the same thermodynamic profiles for the binding with actinonin. Consequently, we performed a preliminary ITC experiment to determine if there was a measurable heat signal upon binding of actinonin to PDFEc, and whether the total heat release/absorbed per injection was kinetically controlled. The latter was important since actinonin has been known to function as a slow-binding inhibitor for PDF with the overall binding process conforming to a two-step mechanism (32). Our preliminary experiment revealed that the heat released upon injection of 6-μl of 300 μM actinonin into 42 μM PDFEc returned to the baseline and remained stable up to 4 min (the time interval set for the subsequent injections). Apparently, the amount of heat released upon titration of PDFEc by actinonin was not kinetically controlled during our experimental conditions. We further noted that in our standard phosphate buffer, a minimal PDFEc concentration of approximately 30 μM generated reliable heat signals when titrated with 300−350 μM actinonin; a range of inhibitor concentrations deemed reasonable to reach saturating conditions based on the previously reported Ki values (31; 32).
Figure 2 shows the titration of 31.5 μM PDF with 40, 6-μL aliquots (plus an initial injection of 3-μL) of 350 μM actinonin in 50 mM phosphate, pH 7.0 containing 1 mM NiCl2 at 25 °C. The top panel displays the raw calorimetric data whereas the bottom panel contains a plot of the total heat generated per injection as a function of the molar ratio of actinonin to enzyme. Note that in the top panel of Fig. 2, the initial injections produced exothermic peaks (downward spikes), as was noted during our preliminary experiments. However, as the titration progressed, the exothermic peaks decreased in magnitude (as expected for the increased occupancy of PDFEc sites by actinonin), but to our surprise, subsequent injections of actinonin started producing endothermic peaks (upward spikes). As apparent from the data of Fig. 2 (panel A), both the exothermic and endothermic peaks exhibit saturating profiles at appropriate concentrations of actinonin. Since the heat peaks at the end of the titration corresponded to those obtained for the injection of actinonin to the buffer (i.e., blank titration; data not shown), the ITC titration profile was deemed to be complete.
Figure 2.
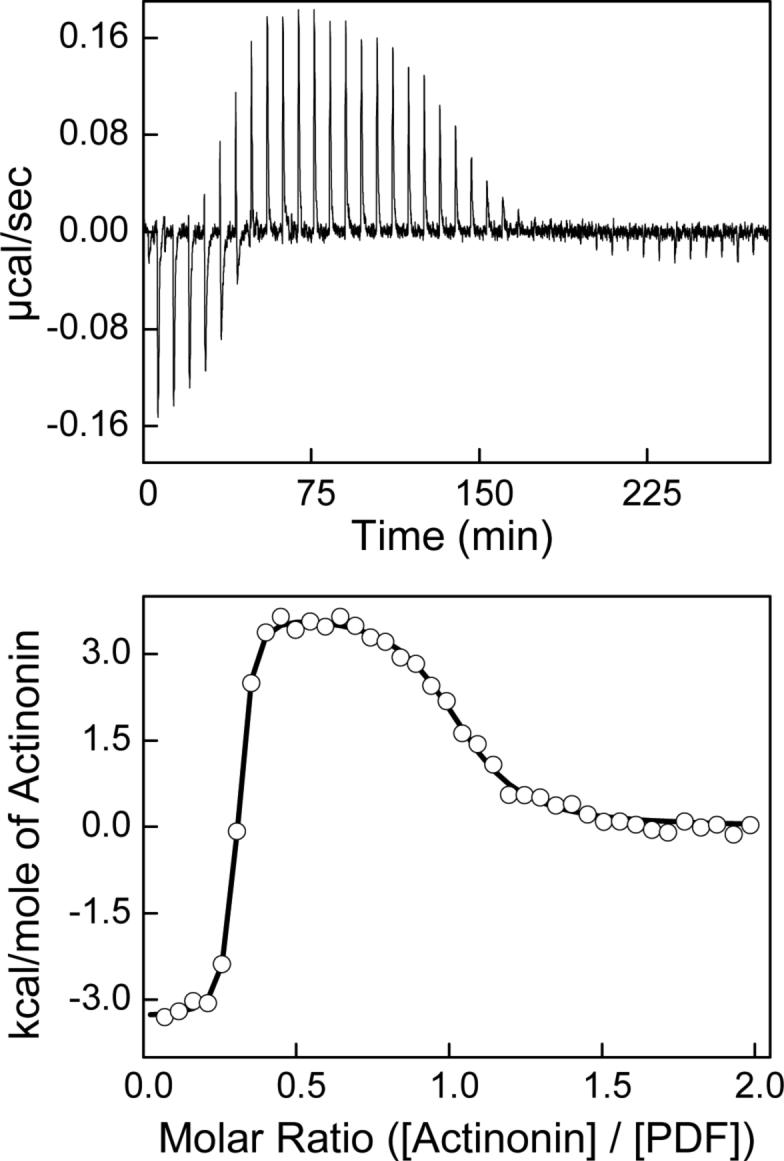
Titration of 31.5 μM Ni2+-PDFEc with 325 μM actinonin in 50 mM potassium phosphate, pH 7.0 containing 1 mM NiCl2 at 25 °C. Top panel shows the raw calorimetric data as a function of time whereas the bottom panel shows the integrated heat per injection versus the molar ratio of actinonin to PDF. The solid smooth line corresponds to the best fit of the experimental data to an independent two-site model providing stoichiometries (n) of 0.29 and 0.73 (moles bound actinonin per mole PDFEc), association constants (Ka) of 8.4 × 108 and 1.9 × 106 M−1 a and standard enthalpy changes (ΔH°) of −3.3 and 3.8 kcal mol−1 for the higher and lower-affinity binding sites, respectively.
The bottom panel of Figure 2 shows the amount of heat released/absorbed per mol of actinonin as a function of the molar ratio of actinonin to PDFEc. Note that the binding isotherm conforms to both exothermic and endothermic phases, which is consistent with a minimal model for the two independent binding sites of actinonin with PDFEc (42). The solid smooth line in the lower panel of Fig. 2 is the best fit of the experimental data for the two independent binding sites model with stoichiometry (n), association constant (Ka) and standard enthalpy changes (ΔH°) of 0.29 ± 0.002, 8.4 (± 1.7) × 108 M−1 and −3.3 ± 0.06 kcal mol−1 for the exothermic phase, and 0.73 ± 0.007, 1.9 (± 0.2) × 106 M−1 and 3.8 ± 0.06 kcal mol−1 for the endothermic phase, respectively. With the standard state being equal to 1.0 M, the free energy changes for the binding of actinonin to PDFEc was calculated (ΔG° = −RT ln Ka) to be −12.2 and −8.6 kcal mol−1 for the exothermic and endothermic phases, respectively. From the determined values of ΔG° and ΔH°, the standard entropy changes (ΔS°) of the exothermic and endothermic binding sites can be calculated to be 29.7 and 41.5 cal mol−1 K−1, respectively.
The thermodynamic parameters derived from the data of Figure 2 reveal that the binding of actinonin to both sites is primarily dominated by favorable entropic changes. At 25 °C, the entropic contribution (TΔS°) to the overall free energy of interaction is equal to −8.9 kcal mol−1 for the higher-affinity exothermic site and −12.4 kcal mol−1 for the lower-affinity endothermic site. In the case of the higher-affinity site, a comparison of the magnitude of −TΔS° with ΔG° (−12.2 kcal mol−1) shows that only a small fraction of favorable binding energy arises from the enthalpic changes (−3.3 kcal mol−1). Moreover, consistent with its endothermic binding profile, the magnitude of −TΔS° is larger than that of ΔG° (−12.4 kcal mol−1 versus −8.6 kcal mol−1, respectively) at the lower-affinity binding site. Thus, the binding of actinonin at this site is driven entirely by entropy with unfavorable enthalpic contribution.
Metal dependence of the two binding modes of actinonin to PDFEc
Based on the structural and binding data, it is clearly evident that actinonin binds to PDFEc in a competitive manner with a 1:1 stoichiometry of actinonin to the enzyme (refer to Figure 1). As two independent binding sites were observed in our calorimetric titration experiments, it appeared plausible that the origin of these sites was based on the enzyme population containing two types of metal ions. Should this be the case, the removal of the active site Ni2+ would result in the loss of one of the binding phases (corresponding to PDFEc-Ni2+) while leaving the phase associated with PDFEc-Zn2+ (since it is considered nearly impossible to remove bound Zn2+). As a test of our hypothesis, freshly prepared recombinant PDFEc was exhaustively dialyzed against metal-free buffer (50 mM potassium phosphate, pH 7.0 treated with Chelex® 100) and the Ni2+ content was quantified by the UV/Vis spectroscopy. The spectral data revealed that the Cys90-Ni2+ charge-transfer complex was non-existent, suggesting that Ni2+ was no longer present in the active site of PDFEc. The lack of Ni2+ in the PDFEc preparation was further confirmed via ICPE spectroscopy. Results indicated that whereas the Ni2+ content of PDFEc was negligible after dialysis against the metal-free buffer (0.01 mol fraction), the Zn2+ content decreased only slightly from 0.51 to 0.43 mol/mol of enzyme.
The upper panel of Figure 3 depicts the ITC titration of the Ni2+-free form of PDFEc with actinonin at 25 °C. As is evident from the figure, the higher-affinity binding site has disappeared, leaving only the lower-affinity binding site. The solid line conforms to the best fit of the experimental data to a single binding site model and provides values of n, Ka and ΔH°, respectively of 0.41 ± 0.004, 2.7 (± 0.32) × 106 M−1 and 3.6 ± 0.05 kcal mol−1. Using the standard state being equal to 1.0 M, ΔG° and ΔS° for the binding of actinonin to PDFEc were calculated to be −8.8 kcal mol−1 and 41.4 cal mol−1 K−1, respectively.
Figure 3.

Comparison of the binding isotherms for Ni2+-free (panel A) and ZnCl2-treated PDFEc (panel B) titrated with 325 μM actinonin in 50 mM potassium phosphate, pH 7.0 at 25 °C. Total protein concentrations were 31.5 μM in both experiments and the best fit of single binding site model (solid line in panel A) provided n, Ka and ΔH° of 0.41, 2.7 × 106 and 3.6 kcal mol−1, respectively for Ni2+-free PDFEc. The solid line in panel B corresponds to the best fit of the ZnCl2-treated PDFEc experimental data to an independent two-site model providing n of 0.31 and 0.42, Ka of 3.9 × 108 and 5.7 × 106 M−1 and ΔH° of −2.8 and 2.0 kcal mol−1 for the high and low affinity binding sites, respectively.
Comparison of the derived thermodynamic parameters between Ni2+-free PDFEc and those of the mixed preparation show relatively little change at the endothermic site in any of the thermodynamic parameters, except for the stoichiometry. It should be emphasized that among different parameters, derived from the best fit of the calorimetric data, stoichiometry is most sensitive to variations in the enzyme concentrations. Since the concentration determined for the PDFEc preparation takes into account all species including those that do not bind actinonin, the stoichiometry for the endothermic phase of the mixed preparation of the enzyme is 0.7 and that of the Ni2+-free form is 0.4. We believe the lower stoichiometry observed during the above titration is due to the absence of heat signals upon binding of actinonin to the metal-free form of the enzyme.
As a further test of whether the endothermic phase for the binding of actinonin to the lower affinity site is solely a consequence of the metal identity, we incubated the Ni2+- free PDFEc with excess ZnCl2 and repeated the titration. In this case, Zn2+ would occupy the active site of PDFEc from which Ni2+ had been removed, and we predicted that the thermodynamic parameters of Ka and ΔH° for the endothermic binding phase would remain unchanged but the stoichiometry would increase to a value approaching 1. However, we were surprised to note that the titration of ZnCl2-treated PDFEc with actinonin resulted in the return of the higher-affinity binding site, as shown in the bottom panel of Fig. 3. The data was readily fitted by the two independent binding sites model (solid line) and values of n, Ka and ΔH° were derived to be 0.32 ± 0.004, 3.4 (± 1.5) × 108 M−1 and −2.8 ± 0.09 kcal mol−1, respectively, for the exothermic binding site. The latter values are in good agreement with the thermodynamic parameters obtained for the titration of the native enzyme (Fig. 2). Hence, contrary to our initial hypothesis, this finding unraveled that the two binding modes of actinonin to PDFEc were due to the distinct conformational states of the enzyme, whose distribution was modulated by the presence or absence of the enzyme bound metal cofactor irrespective of its nature (i.e., Ni2+ versus Zn2+).
Influence of the ionization enthalpy of the buffer (ΔH°ion) on the observed thermodynamic parameters
It has been established that the standard enthalpic changes observed calorimetrically (such as in Fig. 2) are considered to be the net sum of the enthalpic changes due to several processes (43). The contributions of bond formation and/or breaking as well as the changes in the conformational state of the protein may be grouped together macroscopically as integral to the binding process, and thus are a part of the intrinsic standard enthalpic changes (ΔH°int). In contrast, heat associated with the enthalpies of dilution and ionization occur simultaneously and must be deconvoluted from the observed standard enthalpic changes (ΔH°obs) (44). Although the heats of dilution are readily removed via subtraction of either a buffer blank or the heats remaining upon saturation of the protein, the enthalpic contributions that occur from protonation/deprotonation of the buffer species must be quantified by comparing the difference in ΔH°obs (ΔΔH°obs) between buffers of known ionization enthalpies (ΔH°ion) (41; 44; 45). The change in proton inventory (p) can be quantified by taking the ratio of (ΔΔH°obs)/(ΔΔH°ion) in the two buffers. For example, if a single proton is taken up upon binding, the ΔH°obs will be more positive in the buffer with the larger ionization enthalpy by a value similar to ΔΔH°ion and the resulting ratio would be approximately 1 and vice versa for release of a single proton. If there is no change in the proton inventory, the ΔH°obs should remain unchanged and the ratio would be equal to zero. However, a possible complication of the above deduction may arise at different pH values. Depending on the pKa of the interacting species (which is protonated or deprotonated upon binding), only a fraction of such species would be in the proper ionization state to take up or release protons at a particular pH. To account for this feature, we chose phosphate and Tris as buffers with known ionization enthalpies of 1.22 and 11.5 kcal mol−1, respectively (41), at pH 6.3 and 7.0.
A summary of the results of PDFEc titration with actinonin at 25 °C in selected buffers is presented in Table 1. The data of Table 1 reveals that the association constants remained comparable between different buffers and represent only minor changes in the difference in free energies (ΔΔG°) by 0.02 and 0.7 kcal/mol for the higher-affinity site and 0.2 and 0.7 kcal mol−1 for the lower-affinity site at pH 6.3 and 7.0, respectively. It was observed that whereas the binding affinity of actinonin at the lower-affinity site remains relatively constant upon changing the pH from 7.0 to 6.3, that of the higher-affinity site is favored at pH 6.3 compared to pH 7.0 by a ΔΔG° of −0.9 to −1.6 kcal mol−1. In terms of enthalpy, at pH 7.0 the ΔH°obs exhibited no change at the higher-affinity site and varied slightly (ΔΔH°obs ≈ 0.7 kcal mol−1) at the lower-affinity site between the phosphate and Tris buffers. The above variation is not significant as it translates into a release of 0.07 protons from the enzyme phase. At pH 6.3, the ΔH°obs again showed minor variation by 1.2 kcal mol−1 at the lower-affinity site, whereas the higher-affinity site exhibited a much lower ΔH°obs in Tris versus phosphate buffer with a difference of 6.7 kcal mol−1. The above ΔΔH°obs values at pH 6.3 correspond to the release of 0.12 and 0.65 protons from the enzyme phase at the lower and higher-affinity sites, respectively.
Table 1.
Effect of pH and Buffers on the Binding of Actinonin to PDFa
| Buffer | Exothermic Site | Endothermic Site | ||||||
|---|---|---|---|---|---|---|---|---|
| pH 7.0 | n | Ka × 109 (M−1) | ΔH° obs (kcal mol−1) | p | n | Ka × 106 (M−1) | ΔH° obs (kcal mol−1) | p |
| Phosphate | 0.19 ± 0.002 | 0.54 ± 0.1 | −2.2 ± 0.1 | 0.0 | 0.86 ± 0.006 | 2.8 ± 0.3 | 4.0 ± 0.05 | −0.07 |
| Tris | 0.16 ± 0.001 | 0.17 ± 0.03 | −2.2 ± 0.07 | 0.77 ± 0.007 | 0.83 ± 0.06 | 3.3 ± 0.05 | ||
| pH 6.3 | ||||||||
| Phosphate | 0.10 ± 0.01 | 2.6 ± 5.6 | 3.8 ± 0.1 | −0.65 | 0.94 ± 0.01 | 2.3 ± 0.3 | 4.8 ± 0.05 | −0.12 |
| Tris | 0.09 ± 0.001 | 2.5 ± 1.0 | −2.9 ± 0.1 | 0.94 ± 0.006 | 1.7 ± 0.2 | 3.6 ± 0.03 | ||
n refers to the stoichiometry of actinonin-PDFEc complex (mol/mol) and p refers to the change in proton inventory.
Effect of Temperature on the Thermodynamic Parameters
To investigate the effect of temperature on the binding of actinonin to both binding sites of PDFEc, we performed titrations of the mixed metal form of the enzyme with actinonin over the temperature range of 10 to 35 °C in 50 mM potassium phosphate, pH 7.0 containing 1 mM NiCl2 (Table 2). The data of Table 2 indicates that the stoichiometry of the two binding sites remains essentially constant as a function of temperature. The temperature-independence of the stoichiometry for both sites indicates little to no interconversion from one form of the enzyme to the other, which is consistent with thermal unfolding experiments (see below). As it is highly improbable that both forms would have similar stabilizing free energies at all temperatures, it is likely that the conformations are essentially independent due to high transition state energies between the putative enzyme populations.
Table 2.
| Temperature (°C) | n | ΔH° (kcal mol−1) | ΔG° (kcal mol−1) | TΔS° (kcal mol−1) |
|---|---|---|---|---|
| 10 | 0.26 ± 0.003 | −0.3 ± 0.1 | −10.5 | 10.2 |
| 0.76 ± 0.009 | 6.3 ± 0.1 | −7.7 | 14.1 | |
| 15 | 0.29 ± 0.002 | −1.1 ± 0.08 | −11.5 | 10.3 |
| 0.76 ± 0.007 | 5.5 ± 0.07 | −8.1 | 13.6 | |
| 20 | 0.29 ± 0.002 | −2.1 ± 0.07 | −12.0 | 9.9 |
| 0.78 ± 0.008 | 4.4 ± 0.07 | −8.4 | 12.8 | |
| 25 | 0.29 ± 0.002 | −3.3 ± 0.06 | −12.2 | 8.9 |
| 0.73 ± 0.007 | 3.8 ± 0.06 | −8.6 | 12.4 | |
| 30 | 0.32 ± 0.001 | −3.9 ± 0.03 | −12.5 | 8.6 |
| 0.76 ± 0.005 | 2.8 ± 0.04 | −9.0 | 11.6 | |
| 35 | 0.29 ± 0.002 | −5.1 ± 0.08 | −12.4 | 7.2 |
| 0.76 ± 0.02 | 1.7 ± 0.06 | −9.2 | 10.8 |
in 50 mM potassium phosphate buffer, 1mM NiCl2, pH 7.0. n refers to the stoichiometry of actinonin-PDFEc complex (mol/mol).
For each temperature the upper row lists thermodynamic parameters for the higher-affinity site whereas the bottom row lists those for the lower-affinity site.
The temperature dependent thermodynamic parameters revealed that the magnitudes of ΔG°, ΔH°, and TΔS° all decrease as a function of temperature. Evidently, the increase in temperature favors ΔG° and ΔH° values for the binding of actinonin to PDFEc, but disfavors the entropic contribution (TΔS° value). This feature (as depicted by the data of Figure 4) obviates the emergence of the enthalpy-entropy compensation effect, a widely observed phenomenon during the course of enzyme-ligand interactions (46; 47). The linear increase in ΔG° and ΔH° as a function of TΔS° led to suggestion that the binding of actinonin to PDFEc is presumably dictated by somewhat stronger interactions; i.e., interactions which are energetically more favorable than those given by weaker interactions such as hydrogen bonding.
Figure 4.
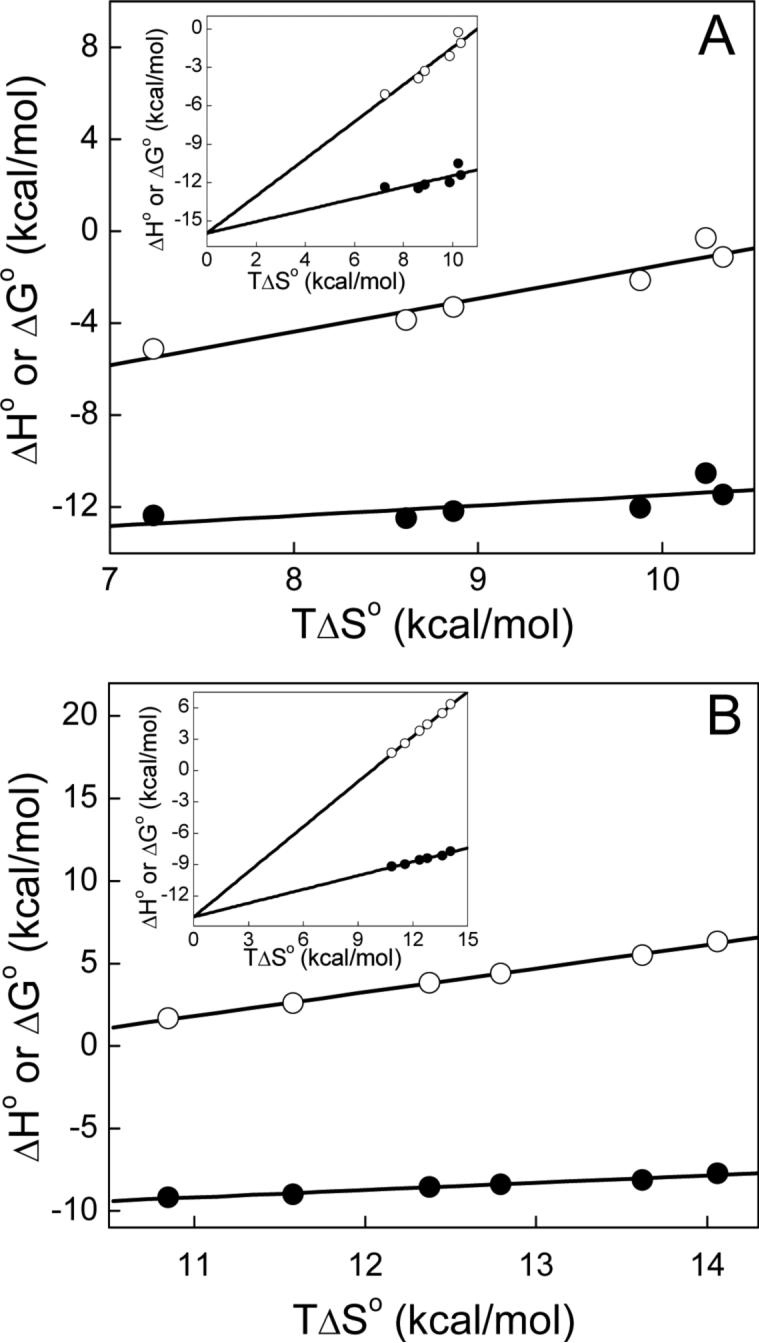
Enthalpy-entropy compensation plots for the binding of actinonin to Ni2+-substituted PDFEc in 50 mM potassium phosphate, pH 7.0 containing 1 mM NiCl2. The thermodynamic parameters were obtained as described in Figure 2 as well as in the text with panel A depicting the higher-affinity site parameters and panel B depicting those of the lower-affinity site. The dependence of ΔG° and ΔH° on TΔS° is shown by closed and open circles, respectively, and the solid lines represent linear regression analysis of the plotted data. For the higher-affinity site slopes and Y-axis intercepts are 0.45 ± 0.2 and −16.0 ± 1.9 for ΔG° and 1.45 ± 0.2 and −16.0 ± 2.0 kcal mol−1 for ΔH° and those of the lower-affinity site are 0.44 ± 0.03 and −14.0 ± 0.4 for ΔG° and 1.44 ± 0.032 and −14.0 ± 0.4 kcal mol−1 for ΔH°, respectively. Insets depict the mutual intersection of the fitted lines at the Y-axis.
The temperature dependence of the thermodynamic data of Table 2 led us to determine the change in heat capacity (ΔCp°) that occurs upon binding of actinonin to PDFEc. Using the assumption that ΔCp° is invariant as a function of temperature, a plot of ΔH° versus temperature was expected to be linear over a limited temperature range with the slope being equal to ΔCp°. Figure 5 shows the plot of ΔH° as a function of temperature. Note the linear dependence of such a plot, yielding the magnitudes of ΔCp° of −0.192 ± 0.007 and −0.187 ± 0.007 kcal mol−1 K−1 and Y-axis intercepts at 0 K of 54.1 and 59.2 kcal mol−1 for the high and low affinity sites, respectively. It should be emphasized that the calculated ΔCp° for both sites is within the error of each other and thus can be considered to be essentially equal. The strongly negative ΔCp° value suggests that the binding of actinonin to PDFEc is dominated by hydrophobic forces as will be elaborated in the Discussion section.
Figure 5.

Dependence of ΔH° on the experimental temperature for the binding of actinonin to Ni2+-substituted PDFEc. The experimental conditions were the same as described in Figure 4. The ΔH° for the higher and lower-affinity sites are depicted as closed and open circles, respectively, and linear regression analysis (solid lines) provide ΔCp° (slope) values of −0.192 and −0.187 kcal mol−1 K−1 and intercepts (at 0 K) of 54.1 and 59.2 kcal mol−1 for the high and low-affinity sites, respectively.
Conformational States of PDFEc Revealed by Circular Dichroic Spectroscopy
Based on the nearly constant stoichiometries (n) for the binding of actinonin to PDFEc as a function of temperature, we surmised that the two binding sites are a consequence of distinct and independent conformational states of PDFEc. To probe this, we determined the temperature-induced unfolding of PDFEc and monitored the changes in the secondary structure of the enzyme via circular dichroic spectroscopy (Figure 6). Panel A of Figure 6 shows the dependence of the ellipticity (θ) at 223 nm of the mixed metal PDFEc on temperature at a heating rate of 2 °C per minute. It can be seen that the unfolding of PDFEc proceeds via two distinct phases characterized by transition temperatures (Tm) of 56 ± 3 and 79 ± 1 °C, suggesting the homogeneous preparation of PDFEc shows two distinct conformational states. When heated at 60 °C for 10 minutes, cooled on ice and centrifuged to remove the precipitate, it was found that the lower transition peak did not re-appear (Figure 6, panel B). Clearly the first transition was due to a heat sensitive form of PDFEc. This same heat treatment of the mixed metal PDFEc prior to titration with actinonin resulted in the loss of the exothermic binding phase (our unpublished results), similar to that observed when Ni2+ was removed from the enzyme preparation. Moreover, no charge-transfer complex peak was observed in the heat treated samples and metal analysis led to the suggestion that although there was no Ni2+ present, approximately 0.45 mol fraction Zn2+ remained bound to the enzyme. The dependence of the two conformations on the identity of the catalytic metal center will be discussed in the next section.
Figure 6.
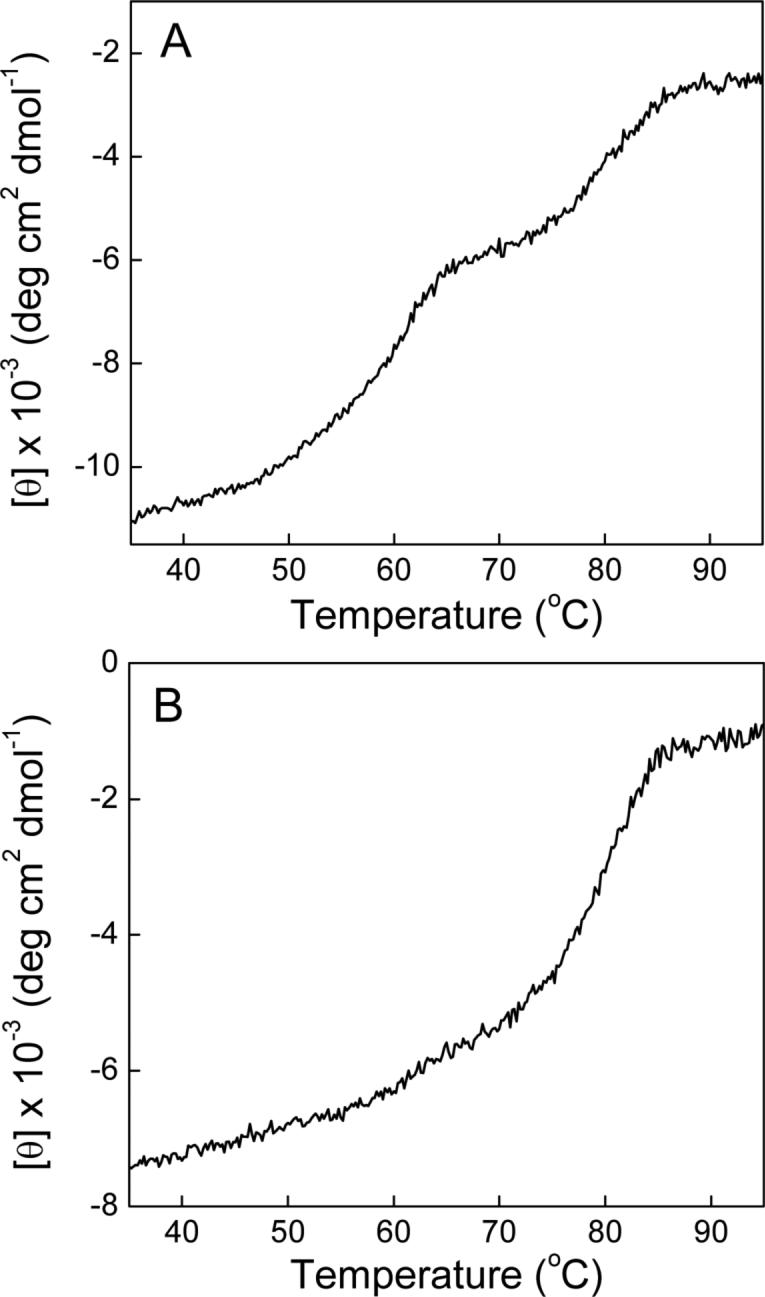
Thermal unfolding of normal Ni2+-substituted PDFEc (panel A) and Ni2+-substituted PDFEc after incubation at 60 °C for 10 min followed by cooling on ice (panel B) as monitored by circular dichroism. Enzyme (29 μM initial concentration) in 5 mM HEPES, pH 7.5 containing 5 mM NiCl2 was heated at a rate of 2 °C min−1 and the ellipticity (θ) at 223 nm monitored as a function of temperature. Experimental data is corrected for effective concentration and is plotted as mean residue ellipticity [θ].
DISCUSSION
The experimental data presented herein lead to the conclusion that the recombinant form of E. coli peptide deformylase (PDFEc) exists in two alternative conformational states, and such conformational states are not solely dictated by the nature (viz., Zn2+ versus Ni2+) of the bound metal ions to the active site of the enzyme. The above conclusion has been derived from our detailed isothermal titration calorimetric data for the binding of actinonin to the differently metal ligated forms of the enzyme and is supported by the following features: (1) The overall binding isotherm is comprised of a high-affinity exothermic and a low-affinity endothermic phase, and both these phases are entropically driven, (2) Although the E. coli expressed and purified form of the recombinant enzyme contains both Zn2+ and Ni2+ with nearly equal molar ratios, the exothermic phase is not dependent on the nature of these metal ions. Whereas the removal of the enzyme bound Ni2+ abolishes the exothermic phase, the above phase reappears upon substitution of Ni2+ by Zn2+, (3) The binding of actinonin to the lower and higher-affinity sites results in the release of about 0.12 and 0.65 mole of proton per mole of PDFEc-actinonin complex, respectively, at pH 6.3 but not at pH 7.0, (4) The stoichiometric ratio for the binding of actinonin to PDFEc remains unaltered upon variation of temperature, suggesting that the putative conformational states (corroborated by the changes in the secondary structural features of the protein during the course of the thermal denaturation) are either “static” or that they possess a high transition-state energy barrier for their interconversion, (5) Although both exothermic and endothermic phases do not conform to the enthalpy-entropy compensation effect (46; 47), they yield highly negative ΔCp° values.
Based on the structural data, it is apparent that actinonin binds to PDFEc both via hydrophobic and hydrophilic moieties, and like many other metalloproteinases (48), the hydroxamate group of actinonin interacts with the active site resident metal ion. Hence, it is not surprising that the binding of actinonin to PDFEc yields pronounced heat signals during the ITC experiments. However, the emergence of both exothermic and endothermic peaks upon binding of actinonin to PDFEc was not expected. A simplistic explanation of the origin of the above observation can be that actinonin binds to PDFEc in two orientations; in one orientation, the hydroxamate group of the inhibitor makes direct contact with the active site resident metal ion (yielding exothermic peaks), but in the other orientation, the binding involves significant adjustment in the enzyme's active site pocket (yielding endothermic peaks). Nevertheless, since both these binding modes are dominated by entropic contributions to their overall binding free energies, they are likely to originate, at least in part, by desolvation of the enzyme's active site pocket. The latter is supported by considerably negative ΔCp° values for the binding of actinonin to PDFEc in both of the above binding modes (Figure 5).
The question arises as to why the exothermic profile of PDFEc-actinonin interaction disappears upon removal of loosely-bound Ni2+ from the enzyme site, and it reappears upon substitution of Ni2+ by Zn2+. In pursuit of answering this question, we realized that both the Ni2+ and Zn2+-containing enzyme yields an exothermic profile upon binding with actinonin. This observation led to the suggestion that the nature of the enzyme bound metal ions (particularly Ni2+ versus Zn2+) is not the sole determinant of the exothermic versus endothermic ITC profiles. This is not surprising since the structural data reveals that PDFEc containing either Ni2+ or Zn2+ interacts with actinonin in similar orientations with the hydroxamate group of the inhibitor coordinating the enzyme bound metal ion. In addition, since the titration of Ni2+-free PDFEc by actinonin neither produces the exothermic profile nor enhances the magnitude of the endothermic profile, it implies that the inhibitor does not interact with the metal-free form of the enzyme. However, paradoxically, the binding of actinonin to PDFEc which contains the intrinsically-bound Zn2+ (acquired during the de novo folding process) yields the endothermic profile whereas the binding of actinonin to the enzyme in which the bound Ni2+ was replaced by Zn2+ yields the exothermic profile. Clearly, these two enzyme forms are not the same, and these forms produce markedly different thermodynamic signatures (viz., exothermic versus endothermic profiles) upon binding with actinonin.
We believe the overall proton inventory of PDFEc-actinonin interaction (see Table 1) is given by the protonation/deprotonation of the metal bound water which is partially compensated by the protonation/deprotonation of the hydroxamic acid –OH of actinonin (depicted in Scheme II). It has been known that the pKa of the –OH group of hydroxamic acids such as that of actinonin decreases by approximately 3 pH units upon binding to the enzyme (49; 50). Under this scenario, the PDFEc-actinonin interaction would result in the release of one mole of proton/mol of the complex at both pH 7.0 and pH 6.3 (given by H+ in Scheme 2). However, at pH 7.0 the deprotonation of actinonin is counteracted by the protonation of the metal-bound hydroxide ion (of water) prior to release in the bulk solvent. As consequence, at pH 7.0, there is no net change in the proton inventory. On the other hand, at pH 6.3, since a fraction of the metal-bound hydroxide ion of water remains in the protonated form, the proton released from actinonin is incompletely compensated by the protonation of the hydroxide ion. Hence, the release of 0.65 protons at pH 6.3 represents the fraction of the metal-bound hydroxide that is in the acid (water) form, since only 0.35 protons are abstracted from the buffer.
Scheme 2.

Changes in proton inventory upon formation of PDFEc-actinonin complex.
Temperature dependence of the enzyme-actinonin interaction
Despite marked differences in the two alternative conformational states of the PDFEc-actinonin complex, the temperature dependence of ΔH° yields similar ΔCp° values. From the classical thermodynamic point of view, such similarity implies that the binding of actinonin to either of the PDFEc conformational states produces similar changes in the water accessible polar and non-polar surface areas (51). However, the above similarity can also arise if the heat capacity contribution arising from the changes in the non-polar surface areas in one conformational state is compensated by those from the polar surface areas in the other conformational states. Irrespectively, as the X-ray crystallographic structure of the PDFEc-actinonin complex could not be arbitrarily assigned as representative of either of the experimentally derived conformational states, we chose not to predict the magnitudes of the ΔCp° values from the structural coordinates of the complex (20).
Another interesting aspect of the temperature-dependent thermodynamic data for the binding of actinonin to PDFEc is the lack of enthalpy-entropy compensation effect in both exothermic and endothermic phases. This was unexpected since the enthalpy-entropy compensation is frequently observed in case of enzyme-ligand interactions, where the origin of such compensation is believed to lie in the weaker (e.g., van der Waals interaction, hydrogen bonding etc.) interactions between the species. The enthalpy-entropy compensation effect is not expected in processes which involve the formation of stronger interactions (47; 52). In this regard, it is noteworthy that the interaction between PDFEc and actinonin is partially dominated by the formation of coordination bonds between the metal and the hydroxamate; an interaction which may not adhere to the enthalpy-entropy compensation effect observed for other enzyme-ligand interactions.
The detailed thermodynamic experimental data presented herein provide convincing evidence that the recombinant form of PDFEc exists in two alternative conformational states, and those states have remained elusive during previous physicochemical characterization of the enzyme (23-29). The above deduction has been possible via the ITC based thermodynamic analyses of the PDFEc-actinonin interactions, which has been further supported by the changes in the secondary structure of the protein during the course of thermal unfolding. The prevalence of alternative conformational states and their differential avidity for inhibitors will prompt developing strategy for designing newer compounds against PDFEc as potential antibiotics.
ACKNOWLEDGMENTS
We would like to thank Dr. Stefan Balaz (Department of Pharmaceutical Sciences, North Dakota State University) for his generous support in the use of the VP-ITC microcalorimeter.
ABBREVIATIONS
peptide deformylase
- Ec
E. coli
- ITC
isothermal titration calorimetry
- ICPE
Inductively coupled plasma emission
- CD
circular dichroism
- n
stoichiometry
- p
change in proton inventory
- Tm
transition temperature
- EDTA
ethylenediaminetetraacetic acid
- Ka
association constant
- ΔH°ion
standard enthalpy of ionization
Footnotes
This research was supported by the National Institutes of Health grant CA113746
While this paper was being finalized, a preliminary account of the ITC studies for the binding of ligand to PDF was uncovered in an undergraduate thesis from Ohio State University (35). However, these studies do not alter our experimental outcomes and conclusions presented herein.
REFERENCES
- 1.Kunin CM. Resistance to antimicrobial drugs--A worldwide calamity. Ann. Intern. Med. 1993;118:557–561. doi: 10.7326/0003-4819-118-7-199304010-00011. [DOI] [PubMed] [Google Scholar]
- 2.Black MT, Hodgson J. Novel target sites in bacteria for overcoming antibiotic resistance. Adv. Drug Deliv. Rev. 2005;57:1528–1538. doi: 10.1016/j.addr.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Walsh C. Where will new antibiotics come from?. Nat. Rev. Microbiol. 2003;1:65–70. doi: 10.1038/nrmicro727. [DOI] [PubMed] [Google Scholar]
- 4.Waller AS, Clements JM. Novel approaches to antimicrobial therapy: Peptide deformylase. Curr Opin Drug Discov Devel. 2002;5:785–792. [PubMed] [Google Scholar]
- 5.Marcker K, Sanger F. N-Formyl-methionyl-S-RNA. J. Mol. Biol. 1964;8:835–840. doi: 10.1016/s0022-2836(64)80164-9. [DOI] [PubMed] [Google Scholar]
- 6.Meinnel T, Mechulam Y, Blanquet S. Methionine as translation start signal: A review of the enzymes of the pathway in Escherichia coli. Biochimie. 1993;75:1061–1075. doi: 10.1016/0300-9084(93)90005-d. [DOI] [PubMed] [Google Scholar]
- 7.Giglione C, Meinnel T. Organellar peptide deformylases: Universality of the N-terminal methionine cleavage mechanism. Trends Plant Sci. 2001;6:566–572. doi: 10.1016/s1360-1385(01)02151-3. [DOI] [PubMed] [Google Scholar]
- 8.Giglione C, Boularot A, Meinnel T. Protein N-terminal methionine excision. Cell. Mol. Life Sci. 2004;61:1455–1474. doi: 10.1007/s00018-004-3466-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Giglione C, Serero A, Pierre M, Boisson B, Meinnel T. Identification of eukaryotic peptide deformylases reveals universality of N-terminal protein processing mechanisms. EMBO J. 2000;19:5916–5929. doi: 10.1093/emboj/19.21.5916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Serero A, Giglione C, Sardini A, Martinez-Sanz J, Meinnel T. An unusual peptide deformylase features in the human mitochondrial N-terminal methionine excision pathway. J. Biol. Chem. 2003;278:52953–52963. doi: 10.1074/jbc.M309770200. [DOI] [PubMed] [Google Scholar]
- 11.Nguyen KT, Hu X, Colton C, Chakrabarti R, Zhu MX, Pei D. Characterization of a human peptide deformylase: Implications for antibacterial drug design. Biochemistry. 2003;42:9952–9958. doi: 10.1021/bi0346446. [DOI] [PubMed] [Google Scholar]
- 12.Lee MD, Antczak C, Li Y, Sirotnak FM, Bornmann WG, Scheinberg DA. A new human peptide deformylase inhibitable by actinonin. Biochem. Biophys. Res. Commun. 2003;312:309–315. doi: 10.1016/j.bbrc.2003.10.123. [DOI] [PubMed] [Google Scholar]
- 13.Lee MD, She Y, Soskis MJ, Borella CP, Gardner JR, Hayes PA, Dy BM, Heaney ML, Philips MR, Bornmann WG, Sirotnak FM, Scheinberg DA. Human mitochondrial peptide deformylase, a new anticancer target of actinonin-based antibiotics. J. Clin. Invest. 2004;114:1107–1116. doi: 10.1172/JCI22269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Walker JE, Lutter R, Dupuis A, Runswick MJ. Identification of the subunits of F1F0-ATPase from bovine heart mitochondria. Biochemistry. 1991;30:5369–5378. doi: 10.1021/bi00236a007. [DOI] [PubMed] [Google Scholar]
- 15.Yagi T, Hatefi Y. Identification of the dicyclohexylcarbodiimide-binding subunit of NADH-ubiquinone oxidoreductase (Complex I). J. Biol. Chem. 1988;263:16150–16155. [PubMed] [Google Scholar]
- 16.Meinnel T, Blanquet S. Characterization of the Thermus thermophilus locus encoding peptide deformylase and methionyl-tRNA(fMet) formyltransferase. J. Bacteriol. 1994;176:7387–7390. doi: 10.1128/jb.176.23.7387-7390.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meinnel T, Blanquet S, Dardel F. A new subclass of the zinc metalloproteases superfamily revealed by the solution structure of peptide deformylase. J. Mol. Biol. 1996;262:375–386. doi: 10.1006/jmbi.1996.0521. [DOI] [PubMed] [Google Scholar]
- 18.Chan MK, Gong W, Rajagopalan PT, Hao B, Tsai CM, Pei D. Crystal structure of the Escherichia coli peptide deformylase. Biochemistry. 1997;36:13904–13909. doi: 10.1021/bi9711543. [DOI] [PubMed] [Google Scholar]
- 19.Becker A, Schlichting I, Kabsch W, Groche D, Schultz S, Wagner AF. Iron center, substrate recognition and mechanism of peptide deformylase. Nat. Struct. Biol. 1998;5:1053–1058. doi: 10.1038/4162. [DOI] [PubMed] [Google Scholar]
- 20.Clements JM, Beckett RP, Brown A, Catlin G, Lobell M, Palan S, Thomas W, Whittaker M, Wood S, Salama S, Baker PJ, Rodgers HF, Barynin V, Rice DW, Hunter MG. Antibiotic activity and characterization of BB-3497, a novel peptide deformylase inhibitor. Antimicrob. Agents Chemother. 2001;45:563–570. doi: 10.1128/AAC.45.2.563-570.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 22.Adams JM. On the release of the formyl group from nascent protein. J. Mol. Biol. 1968;33:571–589. doi: 10.1016/0022-2836(68)90307-0. [DOI] [PubMed] [Google Scholar]
- 23.Rajagopalan PT, Datta A, Pei D. Purification, characterization, and inhibition of peptide deformylase from Escherichia coli. Biochemistry. 1997;36:13910–13918. doi: 10.1021/bi971155v. [DOI] [PubMed] [Google Scholar]
- 24.Meinnel T, Blanquet S. Evidence that peptide deformylase and methionyl-tRNA(fMet) formyltransferase are encoded within the same operon in Escherichia coli. J. Bacteriol. 1993;175:7737–7740. doi: 10.1128/jb.175.23.7737-7740.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Meinnel T, Blanquet S. Enzymatic properties of Escherichia coli peptide deformylase. J. Bacteriol. 1995;177:1883–1887. doi: 10.1128/jb.177.7.1883-1887.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopalan PTR, Yu XC, Pei D. Peptide deformylase: A new type of mononuclear iron protein. J. Am. Chem. Soc. 1997;119:12418–12419. [Google Scholar]
- 27.Rajagopalan PT, Pei D. Oxygen-mediated inactivation of peptide deformylase. J. Biol. Chem. 1998;273:22305–22310. doi: 10.1074/jbc.273.35.22305. [DOI] [PubMed] [Google Scholar]
- 28.Groche D, Becker A, Schlichting I, Kabsch W, Schultz S, Wagner AF. Isolation and crystallization of functionally competent Escherichia coli peptide deformylase forms containing either iron or nickel in the active site. Biochem. Biophys. Res. Commun. 1998;246:342–346. doi: 10.1006/bbrc.1998.8616. [DOI] [PubMed] [Google Scholar]
- 29.Rajagopalan PT, Grimme S, Pei D. Characterization of cobalt(II)-substituted peptide deformylase: Function of the metal ion and the catalytic residue Glu-133. Biochemistry. 2000;39:779–790. doi: 10.1021/bi9919899. [DOI] [PubMed] [Google Scholar]
- 30.Boularot A, Giglione C, Artaud I, Meinnel T. Structure-activity relationship analysis and therapeutic potential of peptide deformylase inhibitors. Curr Opin Investig Drugs. 2004;5:809–822. [PubMed] [Google Scholar]
- 31.Chen DZ, Patel DV, Hackbarth CJ, Wang W, Dreyer G, Young DC, Margolis PS, Wu C, Ni ZJ, Trias J, White RJ, Yuan Z. Actinonin, a naturally occurring antibacterial agent, is a potent deformylase inhibitor. Biochemistry. 2000;39:1256–1262. doi: 10.1021/bi992245y. [DOI] [PubMed] [Google Scholar]
- 32.Van Aller GS, Nandigama R, Petit CM, DeWolf WEJ, Quinn CJ, Aubart KM, Zalacain M, Christensen SB, Copeland RA, Lai Z. Mechanism of time-dependent inhibition of polypeptide deformylase by actinonin. Biochemistry. 2005;44:253–260. doi: 10.1021/bi048632b. [DOI] [PubMed] [Google Scholar]
- 33.Chen D, Yuan Z. Therapeutic potential of peptide deformylase inhibitors. Expert Opin Investig Drugs. 2005;14:1107–1116. doi: 10.1517/13543784.14.9.1107. [DOI] [PubMed] [Google Scholar]
- 34.Berg AK, Manokaran S, Eiler D, Kooren J, Mallik S, Srivastava DK. Energetic rationale for an unexpected and abrupt reversal of guanidinium chloride-induced unfolding of peptide deformylase. Protein Sci. 2008;17:11–15. doi: 10.1110/ps.073270608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Simmons A. Inhibition and thermodynamics of Escherichia coli peptide deformylase. Ohio State University; 2007. [Google Scholar]
- 36.Ragusa S, Blanquet S, Meinnel T. Control of peptide deformylase activity by metal cations. J. Mol. Biol. 1998;280:515–523. doi: 10.1006/jmbi.1998.1883. [DOI] [PubMed] [Google Scholar]
- 37.Wei Y, Pei D. Continuous spectrophotometric assay of peptide deformylase. Anal. Biochem. 1997;250:29–34. doi: 10.1006/abio.1997.2194. [DOI] [PubMed] [Google Scholar]
- 38.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 39.Wiseman T, Williston S, Brandts JF, Lin LN. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989;179:131–137. doi: 10.1016/0003-2697(89)90213-3. [DOI] [PubMed] [Google Scholar]
- 40.Leavitt S, Freire E. Direct measurement of protein binding energetics by isothermal titration calorimetry. Curr. Opin. Struct. Biol. 2001;11:560–566. doi: 10.1016/s0959-440x(00)00248-7. [DOI] [PubMed] [Google Scholar]
- 41.Morin PE, Freire E. Direct calorimetric analysis of the enzymatic activity of yeast cytochrome c oxidase. Biochemistry. 1991;30:8494–8500. doi: 10.1021/bi00098a030. [DOI] [PubMed] [Google Scholar]
- 42.Lin LN, Mason AB, Woodworth RC, Brandts JF. Calorimetric studies of the binding of ferric ions to ovotransferrin and interactions between binding sites. Biochemistry. 1991;30:11660–11669. doi: 10.1021/bi00114a008. [DOI] [PubMed] [Google Scholar]
- 43.Fisher HF, Singh N. Calorimetric methods for interpreting proteinligand interactions. Meth. Enzymol. 1995;259:194–221. doi: 10.1016/0076-6879(95)59045-5. [DOI] [PubMed] [Google Scholar]
- 44.Srivastava DK, Wang S, Peterson KL. Isothermal titration microcalorimetric studies for the binding of octenoyl-CoA to medium chain acyl-CoA dehydrogenase. Biochemistry. 1997;36:6359–6366. doi: 10.1021/bi9700734. [DOI] [PubMed] [Google Scholar]
- 45.Beschiaschvili G, Seelig J. Peptide binding to lipid bilayers. Nonclassical hydrophobic effect and membrane-induced pK shifts. Biochemistry. 1992;31:10044–10053. doi: 10.1021/bi00156a026. [DOI] [PubMed] [Google Scholar]
- 46.Lumry R, Rajender S. Enthalpy-entropy compensation phenomena in water solutions of proteins and small molecules: A ubiquitous property of water. Biopolymers. 1970;9:1125–1227. doi: 10.1002/bip.1970.360091002. [DOI] [PubMed] [Google Scholar]
- 47.Dunitz JD. Win some, lose some: Enthalpy-entropy compensation in weak intermolecular interactions. Chem. Biol. 1995;2:709–712. doi: 10.1016/1074-5521(95)90097-7. [DOI] [PubMed] [Google Scholar]
- 48.Muri EMF, Nieto MJ, Sindelar RD, Williamson JS. Hydroxamic acids as pharmacological agents. Curr Med Chem. 2002;9:1631–1653. doi: 10.2174/0929867023369402. [DOI] [PubMed] [Google Scholar]
- 49.Cross JB, Duca JS, Kaminski JJ, Madison VS. The active site of a zinc-dependent metalloproteinase influences the computed pK(a) of ligands coordinated to the catalytic zinc ion. J. Am. Chem. Soc. 2002;124:11004–11007. doi: 10.1021/ja0201810. [DOI] [PubMed] [Google Scholar]
- 50.Wang D, Helquist P, Wiest O. Zinc binding in HDAC inhibitors: A DFT study. J. Org. Chem. 2007;72:5446–5449. doi: 10.1021/jo070739s. [DOI] [PubMed] [Google Scholar]
- 51.Spolar RS, Livingstone JR, Record MTJ. Use of liquid hydrocarbon and amide transfer data to estimate contributions to thermodynamic functions of protein folding from the removal of nonpolar and polar surface from water. Biochemistry. 1992;31:3947–3955. doi: 10.1021/bi00131a009. [DOI] [PubMed] [Google Scholar]
- 52.Gallicchio E, Kubo MM, Levy RM. Entropy-enthalpy compensation in solvation and ligand binding revisited. J. Am. Chem. Soc. 1998;120:4526–4527. [Google Scholar]