

Abstract
Matrix metalloproteinases (MMPs) are a class of extracellular matrix degrading enzymes over-expressed in many cancers and contribute to the metastatic ability of the cancer cells. We have recently demonstrated that liposomal contents can be released when triggered by the enzyme MMP-9. Herein, we report our results on the mechanistic studies of the MMP-9 triggered release of the liposomal contents. We synthesized peptides containing the cleavage site for MMP-9 and conjugated them with fatty acids to prepare the corresponding lipopeptides. By employing Circular Dichroism spectroscopy, we demonstrate that the lipopeptides, when incorporated in liposomes, are de-mixed in the lipid bilayers and generate triple helical structures. MMP-9 cleaves the triple helical peptides, leading to the release of the liposomal contents. Other MMPs, which cannot hydrolyze triple helical peptides, failed to release the contents from the liposomes. We also observed that the rate and the extent of release of the liposomal contents depend on the mismatch between acyl chains of the synthesized lipopeptide and phospholipid components of the liposomes. Circular Dichroism spectroscopic studies imply that the observed differences in the release reflect the ability of the liposomal membrane to anneal the defects following the enzymatic cleavage of the liposome-incorporated lipopeptides.
Keywords: Triggered release, liposomes, matrix metalloproteinases, phase separation, triple helical lipopeptides
Introduction
Matrix metalloproteinases (MMPs) are a class of Zn2+ and Ca2+ dependent metalloenzymes responsible for the degradation of extracellular matrix (ECM).1 These enzymes are normally involved in a variety of physiological processes, e.g., angiogenesis, wound healing, ovulation etc. The expression levels of these enzymes are tightly regulated at translational and at post-translational levels.2 Overexpression of the MMPs and changing patterns of ECM composition are correlated with many pathological conditions, notable among them are various types of cancers and cardiovascular diseases.1,3 MMP-2 and -9 are found at elevated levels in many types of metastatic tumors.4,5
Amongst the various drug carriers, liposomes offer several advantages as clinical drug delivery vehicles, and there are several liposome mediated drug delivery products approved for therapeutics and for clinical trials.6,7 Usually upon targeting, the encapsulated drugs are released passively to the selected tissue sites and the process is often rather slow. Triggered release of drugs and labeled molecules from liposomes has been recognized to be an attractive therapeutic approach. In this approach, the liposomes do not release contents until the membranes are destabilized by the external agents (trigger). Consequently, there are several reports in the literature on the synthesis of trigger cleavable lipids, formulation of liposomes and subsequent release of the encapsulated contents. Changes in pH,8 mechanical stress,9 metal ions,10 temperature,11 light,12 enzymes13 or conformational change in peptides14 have been successfully used as triggers to release liposomal contents.
For active targeting and subsequent release of liposomal contents, Terada et al. used liposomes functionalized with an MMP-2-cleavable polyethylene glycol (PEG)-peptide-DOPE lipid.15 Upon interaction with MMP-2, the PEG sheath was removed from the liposomes and subsequently, the liposomes were internalized by the carcinoma cells. Hatakeyama et al. showed the effectiveness of a similar system and have also used liposomes modified with anti-MMP antibodies to target tumors.16 Though these systems significantly enhance delivery to tumor sites, they may be susceptible to degradation by the general proteolytic enzymes and other MMPs, which are not localized to tumor tissues. Liposomes have been prepared with a triple helical peptide amphiphile that bind to CD44 on the cell surface and are internalized by metastatic melanoma cells.17 Recently, we have demonstrated the release of liposomal contents triggered by the enzyme MMP-9.18 Our approach is summarized as follows: a triple-helical lipopeptide containing the cleavage site for MMP-9 is synthesized and then incorporated in liposomes (as a minor lipid component). Upon interacting with recombinant human MMP-9, the peptide is cleaved, resulting in destabilization of the liposome bilayer and leakage of liposomal contents.18
Here in, we report our results on the mechanistic studies of the MMP-9 triggered release of the liposomal contents. The triple helical lipopeptides were synthesized employing an automated, microwave-assisted peptide synthesizer, resulting in drastic reduction in the synthesis time as well as major improvements in the quality of the crude product.19 Using fluorescence and circular dichroism (CD) spectroscopy, we demonstrate that the lipopeptides acquire triple helical conformations in the liposomes, and the release of the liposomal contents is due to enzymatic cleavage of the peptide by MMP-9. We observed that the rate and extent of leakage of the contents are dependent on MMP-9 concentration. In addition, we show that the degree of mismatch between the lipid acyl chains of the lipopeptides and the major phospholipid component of the liposomes determines the extent of leakage of contents. The lipid acyl chain dependence of release from the liposomes offers new insights into the mechanism of destabilization of the liposomes by MMP-9, and provides an additional method for fine-tuning the triggered release system. Taken together, these observations suggest that the enzyme triggered release of the liposomal contents can be controlled at multiple levels.
Experimental Section
Synthesis of the lipopeptides
The peptides were synthesized (0.1 mmole scale) using a microwave-assisted peptide synthesizer (Liberty Peptide Synthesizer, CEM Corporation, Matthews, NC) employing standard Fmoc-based solid phase chemistry. The reactions were conducted inside a 30 mL vessel enclosed within the microwave unit (CEM Discover) of the peptide synthesizer. The commercially available Fmoc-Gly-CLEAR acid resin (Peptides International, Louisville, KY) was used as the solid support and HBTU-HOBT as the coupling reagents. Each coupling step (except for Arg) was performed at 55 °C using 20 W of microwave power for 5 minutes with 5 fold excess of reagents. The amino acid Arg and the fatty acids at N-terminal end of the peptides were subjected to double coupling. The coupling of Arg was performed at 25 °C for 25 minutes without any microwave power. Activator base used was 5 equivalents of diisopropylethylamine in N-methylpyrrolidone solvent. The Fmoc groups were deprotected with 5% (by weight) piperazine in DMF with two treatments, 30 seconds and 3 minutes. A microwave-assisted cleavage from the solid support was performed using a mixture of trifluoroacetic acid/triisopropylsilane/water (95%−2.5%−2.5%) for 35 minutes. The crude peptides were precipitated by ice-cold ether, collected by centrifugation and washed with cold ether. The white solids obtained by precipitation were purified by reversed phase HPLC using a Vydac semipreparatory diphenyl column (RP 219TP510) with a linear gradient of 0−70% acetonitrile in water over 60 minutes at a rate of 8 mL/min (both solvents contained 0.1% TFA). The collected, purified fractions were analyzed using Vydac analytical diphenyl column (219TP5415) with a linear gradient of 0−70% acetonitrile in water as eluant, containing 0.1% TFA. Flow rate was maintained at 1.5 mL/min for 25 minutes. The purified peptides were characterized by CD and mass spectroscopy (MALDI-TOF).
Liposome preparation
Stock solutions of the lipopeptide in chloroform:methanol (9:1) were prepared by weighing appropriate amounts of the lipopeptide and dissolving in glass vials to concentrations of 1 mg/mL. The phospholipids were in chloroform stock solutions of concentration 2.5 mg/mL. Lipid thin films were prepared by transferring the appropriate volumes of the respective lipids into 10 mL round bottom flasks with Hamilton® gas tight syringes and adjusting the volume to 5 mL with chloroform. The organic solvents were removed under reduced pressure in a rotary evaporator (Yamato® RE 450) at 42 °C with rotation of the flask at 220 rpm. The thin lipid film was kept under vacuum for 24 hours to remove residual organic solvents.
The lipid thin film containing the phospholipid and lipopeptide was hydrated in the round bottom flask by addition of 2 mL of the appropriate buffer solution followed by rotation for 1 h at 220 rpm at 60 °C. The hydrated lipid suspension was then kept at 4 °C for 4 hours to ensure proper hydration and folding of the triple helical peptides after which it was extruded through a 100 nm pore size polycarbonate membrane at 60 °C to obtain large unilamellar vesicles. The size distribution and zeta potential of the liposomes were determined with a Mavern instruments Nano ZS 90® zetasizer.
The liposomes used for the carboxyfluorescein release experiments were hydrated as mentioned above with buffer containing 100 mM carboxyfluorescein. The unencapsulated carboxyfluorescein was separated from the liposomes by gel filtration through a Sephadex G100 column. Prior to passing the liposomes through the column, the column was equilibrated with 25 mM HEPES buffer pH = 8.0, whose osmolarity had been adjusted to match that of the carboxyfluorescein-containing buffer with which the liposomes were hydrated. Adjustment of osmolarity was performed by measuring the osmolarity of the solutions using an Advanced® Instruments Micro-Osmometer and adjusting the lower osmolarity solution with addition of solid NaCl. Thus, the buffers that were used to equilibrate the gel filtration columns and to elute the liposomes had matching osmolarity with that used to hydrate the liposomes. This ensures minimum background leakage of carboxyfluorescein from the liposomes.
Buffer solutions
The lipid thin films were hydrated with different buffer solutions depending on the experiments for which they were used. For the Circular Dichroism experiments, the buffer used was 5 mM phosphate buffer maintained at pH = 8.0. For the carboxyfluorescein release experiments, the buffer solution used to hydrate the liposomes was 25 mM HEPES at pH = 8.0, containing 100 mM carboxyfluorescein, 10 mM CaCl2 and 10 μM ZnCl2. For equilibrating the column and elution of liposomes from the column, 25 mM HEPES buffer, pH = 8.0 was used, containing 10 mM CaCl2, 10 μM ZnCl2 and NaCl. The same buffer was used for the fluorescence measurements to determine carboxyfluorescein release.
Circular Dichroism (CD) Spectroscopy measurements
All CD spectra were collected on a Jasco® J815 CD spectrometer in a 1 mm path length quartz cuvette at 25 °C. Liposome solutions containing 1 mg/mL total lipid were transferred into the cuvette. The CD spectra were measured in the continuous scanning mode (270 − 180 nm) with a data pitch of 0.2 nm and a scan speed of 100 nm/min. The response time was set at 0.25 sec. For each sample, the average of 50 scans was recorded and corrected for the spectrum of buffer only. The Rpn values were calculated from the CD spectra as the ratio of maximum ellipticity (at 225 nm) and minimum ellipticity (at 200 nm).
Release studies
The rate and extent of release of carboxyfluorescein from the liposomes were monitored by following the fluorescence emission intensity of carboxyfluorescein at 518 nm with excitation wavelength set at 480 nm. In a typical experiment, recombinant human MMP-9 (20 μL from a 25 μM stock solution) was added to a solution of liposomes in buffer (25 mM HEPES, pH =8.0 containing 10 mM CaCl2 and 10 μM ZnCl2) in a total volume of 200 μL and the fluorescence intensity measured every 20 second over a period of 2 hours at 25 °C. The experiments were set up in 96-well fluorescent microplates in triplicates and the data were collected with a Molecular Devices SpectraMax® plate reader. The fractional release was determined by comparing the fluorescence intensity of the liposome solution to the intensity of a solution containing equal concentration of liposomes in the presence of 1% Triton X. Higher concentrations of triton did not cause further increase in the fluorescence intensity of the carboxyfluorescein. Hence, this concentration was deemed sufficient to cause complete release of the liposome contents. The fractional release was calculated as the ratio of the difference between the fluorescence intensity of carboxyfluorescein at time t (It) and its initial intensity at time zero (Io) and the difference between the intensity upon treatment with 1% Triton X (Itriton) and the intensity at time zero (Io) as shown in the equation below:
Results
Liposome characteristics
The lipopeptides used in these studies along with their calculated and observed molecular masses are shown in Table 1. Figure 1 shows the structure of LP1 and the cleavage site for MMP-9 in the peptide. By dynamic light scattering experiments, we found that the liposomes incorporating 30 mol% if the lipopeptides have a unimodal size distribution with mean diameter of 135 ± 30 nm. The zeta potential measurements revealed that the liposomes formulated with 30 mol% of lipopeptide LP1 or LP2 and 70 mol% of POPC, DSPC, DOPC or DMPC have essentially the same unimodal zeta potential of — 10 mV.
Table 1.
The amino acid sequences, acyl chains, calculated and observed molecular masses of the lipopeptides used in this study are shown.
| Lipopeptide | Peptide sequence | Acyl chain | Calcd. M+ | Obs. M+ |
|---|---|---|---|---|
| LP1 | GPQGIAGQR(GPO)4GG-OH | Stearic acid | 2332.24 | 2332.27 |
| LP2 | GPQGIAGQR(GPO)4GG-OH | Oleic acid | 2330.25 | 2331.20 (MH+) |
| LP3 | GPKGIAGQK(GPO)4GG-OH | Palmitic acid | 2276.21 | 2277.15 (MH+) |
Figure 1.

Structure of the lipopeptide LP1 with the cleavage site for MMP-9 is shown.
Mechanism of liposome destabilization
First, we sought to optimize the liposome formulation for minimum background leakage and high sensitivity to MMP-9. Carboxyfluorescein-encapsulated liposomes were prepared containing 10, 20 and 30 mol% of lipopeptide LP1 and 90, 80 and 70 mol% of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) respectively. In 25 mM HEPES buffer (pH = 8.0), the liposomes formulated with 10 and 20 mol% of LP1 released more than 20% of their contents in 2 hours in the absence of any added enzyme. In contrast, the liposomes formulated with 30 mol% of the LP1 released less than 3% of the dye in the absence of MMP-9. When we incorporated 40% of LP1 in the liposomes, the release of the encapsulated contents in the absence of MMP-9 was low (< 5%). However, in the presence of MMP-9, only 40% of the encapsulated dye was released. Hence, further experiments were conducted with the liposomes containing 30 mol% of the lipopeptide LP1. As will be shown later, the incorporated lipopeptides are de-mixed and form domains within the lipid bilayers of the liposomes. When we formulated liposomes containing 20% or less of the lipopeptide LP1, it is possible that large numbers of small domains are formed and the leakage takes place from the domain boundaries.
Next, we proceeded to determine whether the lipopeptides acquire the triple helical conformation when incorporated in the liposomes. In this endeavor, the CD spectrum of liposomes containing 30 mol% of lipopeptide LP1 and 70 mol% of POPC was recorded in 5 mM phosphate buffer (pH = 8.0). The spectrum (Figure 2, black trace) shows a pronounced negative peak at 200 nm and a less intense positive peak at 225 nm, characteristic of the triple helical collagen-related peptides and their fatty acid conjugates.17,20,21 The presence of these two peaks in the CD spectrum indicates that the lipopeptide LP1 displays a triple helix conformation even when incorporated in the liposomes.
Figure 2.
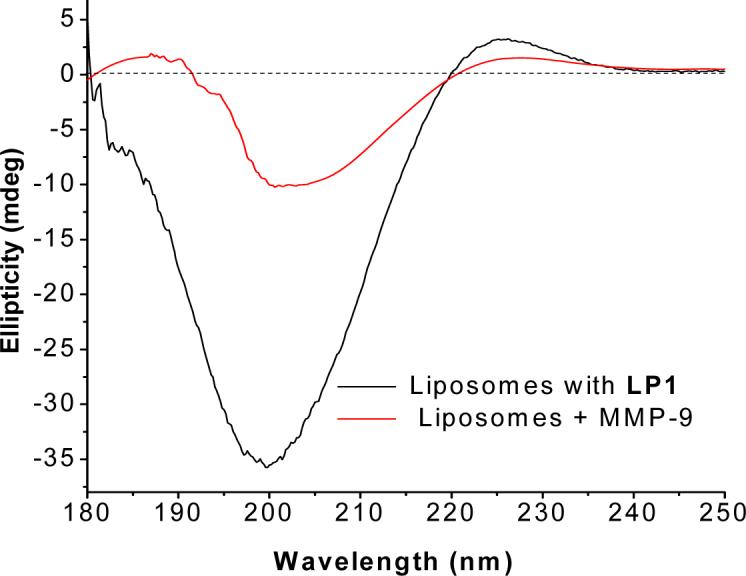
Far-UV circular dichroism spectra of the liposomes (5 mM phosphate buffer, pH = 8.0) formulated with 70 mol% of POPC and 30 mol% of lipopeptide LP1. The CD spectra of the liposomes were recorded before (black line) or 30 min after (red line) incubation with MMP-9. Each spectrum represents the average of 50 scans.
Quantitative analysis reveals that the Rpn value of the lipopeptide (i.e., the ratio of the intensity of CD minimum at 200 nm and maximum at 225 nm)21 is 0.07. This indicates that a substantial fraction of the lipopeptides within the liposomes acquire the triple helical conformation.20,21 When the liposomes were incubated with 3 μM recombinant human MMP-9 for 30 minutes (in 5 mM phosphate buffer, pH = 8.0), pronounced reductions in the intensities for both 200 nm and 225 nm peaks were observed (Figure 2, red trace). In the latter experiment, the CD spectrum of MMP-9 was subtracted to remove the contribution of the enzyme spectrum. These observations suggest that MMP-9 is able to cleave the liposome-incorporated lipopeptide LP1 within 30 minutes. Unfortunately, we could not obtain a kinetic trace of the rate of reduction of the peak at 200 nm (or at 225 nm) with good signal-to-noise ratio.
We proceeded to determine the effect of lipopeptide cleavage on the stability of the liposomes. Liposomes containing 30 mol% of LP1 and 70 mol% of POPC were prepared encapsulating the self-quenching dye, carboxyfluorescein.22 The kinetics of carboxyfluorescein release from these liposomes was measured in the presence of 2.3 μM MMP-9 (25 mM HEPES buffer, pH = 8.0). We observed complete release of the encapsulated dye within 80 minutes in the presence of MMP-9 (Figure 3, black squares). In order to demonstrate that the release of the liposomal contents is due to the cleavage of the lipopeptide LP1 by MMP-9 and subsequent destabilization of the membrane, we conducted the following control experiments. Liposomes encapsulating carboxyfluorescein were formulated with POPC only and the dye release from these liposomes was measured in the presence of 2.3 μM MMP-9. We observed that only 2% of the encapsulated dye was released after 2 hours (Figure 3, blue trace). Next, we formulated the liposomes encapsulating carboxyfluorescein with 30 mol% of the LP1 and 70 mol% POPC, but CaCl2 and ZnCl2 were omitted from the buffer. The release of carboxyfluorescein from these liposomes in the presence of 2.3 μM MMP-9, measured in Ca2+ and Zn2+-free buffer is also shown in Figure 2 (red trace). Less than 20% of the liposomal contents were released when the buffer did not contain any ZnCl2 and CaCl2. Since MMP-9 is known to require Ca2+ ions to maintain its full catalytic activity,23 the reduced leakage is likely due to low catalytic activity of the enzyme (which results in its inability to cleave the lipopeptide and subsequently destabilize the liposomes).
Figure 3.
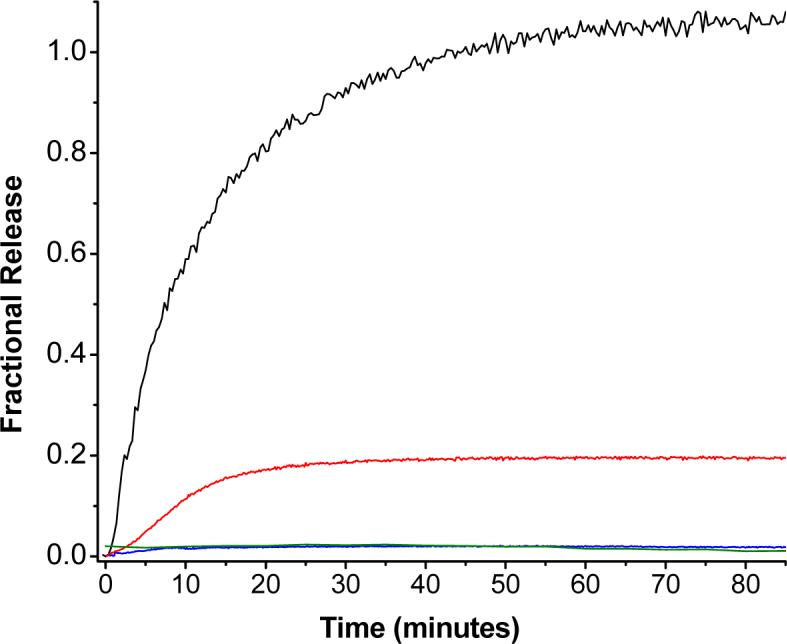
MMP-9 triggered leakage of encapsulated carboxyfluorescein from liposomes formulated with POPC and MMP-9-cleavable lipopeptides. The kinetic trace of carboxyfluorescein fluorescence (λex = 480 nm, λem = 518 nm) was monitored for 85 minutes for liposomes formulated with 70 mol% POPC and 30 mol% of lipopeptide LP1 in presence of 2.3
As a further test of our hypothesis, we formulated liposomes with 70 mol% POPC and the 30 mol% of lipopeptide LP3. This peptide has two lysine residues in its sequence replacing a glutamine and an arginine residue of lipopeptide LP1 (Table 1). MMP-9 cleaves triple helical polypeptide substrates between the glycine and isoleucine residues.24 It has been demonstrated that in the P2 position (where a lysine was substituted for a glutamine), nonpolar residues are preferred, and that introduction of basic or acidic residues in this position leads to reduction in cleavage activity of the MMP by up to fourteen-fold.25 Hence, we expected that MMP-9 will demonstrate reduced catalytic activity for the lipopeptide LP3 and consequently cause less dye release from liposomes formulated with this lipopeptide compared to liposomes prepared with LP1. The release studies with liposomes incorporating LP3 in the presence of 2.3 μM MMP-9 reveal that less than 5% of the contents were released after 2 hours (Figure 3, green trace). These results demonstrate that the carboxyfluorescein release in the presence of MMP-9 is indeed due to destabilization of the lipopeptide-containing liposomes, triggered by the cleavage of the lipopeptides by the enzyme. μM MMP-9. The reactions were conducted at 25 °C in 25 mM HEPES buffer, pH = 8.0 in the presence (black trace) or absence (red trace) of 10 mM CaCl2 and 10 μM ZnCl2. The blue trace shows the control experiment using liposomes formulated with 100 mol% POPC. The green trace (partially overlapping with the blue trace) represents the leakage from liposomes formulated with 70 mol% POPC and 30 mol% lipopeptide LP3. Each trace shown is the average of three experiments.
Dependence of liposomal contents release on MMP-9 concentration
A precondition for successful targeted delivery of drugs or diagnostic agents to sites overexpressing MMP-9 is that the release of the liposomal contents be dependent on the concentration of MMP-9. To test if our system demonstrates this desirable property, we measured the release of encapsulated carboxyfluorescein from the liposomes (formulated with 70 mol% of POPC and 30 mol% of lipopeptide LP1) as a function of the concentration of MMP-9. Figure 4 shows the kinetic traces for the release of carboxyfluorescein from the liposomes in the presence of 0.35, 0.70 and 2.30 μM of enzyme.
Figure 4.
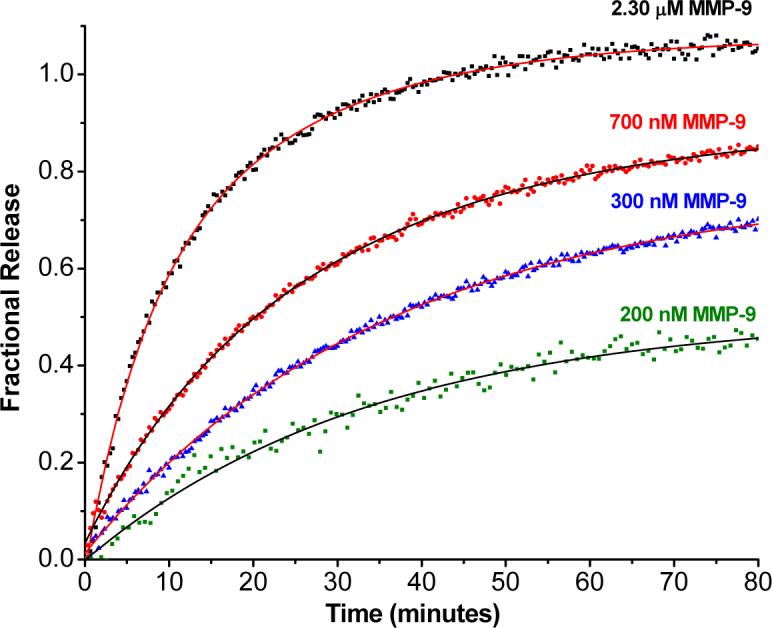
Dependence of the liposomal contents release on the concentration of MMP-9. The kinetic traces of carboxyfluorescein fluorescence (λex = 480 nm, λem = 518 nm) were recorded for 85 minutes for liposomes formulated with 70 mol% POPC and 30 mol% of lipopeptide LP1 in the presence of 2.3 μM (black squares), 700 nM (red diamonds), 300 nM (blue triangles) and 200 nM (green squares) MMP-9. The experiments were conducted at 25 °C in 25 mM HEPES buffer, pH = 8.0 containing 10 mM CaCl2 and 10 μM ZnCl2. The solid lines are single exponential fits of the data by non-linear regression.
We observed that both the extent and rates of contents release from the liposomes are increased by increasing the concentration of MMP-9. The traces of Figure 4 could be fit by the single exponential rate equation: F = F0 — A* e-kt, to obtain the rate constants ‘k’ for the dye release and the amplitudes ‘A’ (which represent the extent of release from the liposomes). ‘F’ represents fractional release. The values obtained for the release rates and the amounts released from these traces are summarized in Table 2. Clearly, the rate and the extent of contents release from the liposomes are determined by the concentration of the active MMP-9. In the presence of 200 nM MMP-9, we observed that 50% of the encapsulated dye was released. Given that the MMP-9 expression is several-fold higher in tumors and in arthritic joints than in normal tissues (for some lung cancer patients, the concentration of MMP-9 in bronchial lavage fluid can be as high as 100 nM; in arthritic joints, the MMP-9 concentration is reported to be in the range 100 − 200 nM).26 we expect that the contents release from our liposomes will be more prevalent in the former.
Table 2.
Dependence of the carboxyfluorescein release from the liposomes on MMP-9 concentration.
| MMP-9 concentration (μM) | Rate constant (min−1) | Fractional release |
|---|---|---|
| 0.20 | 0.029 ± 0.008 | 0.51 ± 0.01 |
| 0.35 | 0.028 ± 0.001 | 0.76 ± 0.03 |
| 0.70 | 0.039 ± 0.002 | 0.84 ± 0.02 |
| 2.30 | 0.075 ± 0.001 | 1.03 ± 0.09 |
Effect of non-collagenase MMPs
The premise for selecting a collagen related, triple helical peptide for our strategy is the susceptibility of such peptides to MMP-9 cleavage, while resistance to cleavage by non-collagenase MMPs or other proteolytic enzymes. Hence, the release of the liposomal contents should not be triggered by MMPs −7 and −10 (which lack the ability to hydrolyze triple-helical peptide substrates27) or by trypsin. In order to test this hypothesis, the release of carboxyfluorescein from the liposomes (formulated with 70 mol% of POPC and 30 mol% of lipopeptide LP1) was measured in the presence of these enzymes (Figure 5). We observed that 2.3 μM MMP-7 released only 15% of the encapsulated dye (Figure 5, red triangles) and 2.3 μM MMP-10 was able to release about 20% of the dye in 85 minutes (Figure 5, blue diamonds). Trypsin also released less than 10% of the encapsulated dye from the liposomes (green squares).
Figure 5.

Comparison of release of liposomal contents triggered by MMPs −7, −9, −10 and trypsin. The kinetic traces for the carboxyfluorescein fluorescence (λex = 480 nm, λem = 518 nm) were recorded for 85 minutes for liposomes formulated with 70 mol% POPC and 30 mol% of lipopeptide LP1. The reactions were conducted at 25 °C in 25 mM HEPES buffer, pH = 8.0 containing 10 mM CaCl2 and 10 μM ZnCl2 with 2.3 μM concentration of MMP-9 (black squares), MMP-10 (blue diamonds), MMP-7 (red triangles) or trypsin (green squares).
Effect of lipid acyl chain mismatch on carboxyfluorescein release
In biomembranes, the distribution of lipid and protein components is not random and components may form domains whose lifetimes may be very short (on the order of 10 μs) or much longer.28 In model membranes containing two lipids, the formation and stability of the domains depend on differential interactions between the two lipid components.29 The difference in the interaction energy between two similar lipids and two different lipids provide the thermodynamic driving force for the formation of the domains.29,30 For our contents release studies, the liposomal membrane is composed of 70 mol% POPC and 30 mol% of lipopeptide LP1. Considerable hydrophobic mismatch exists between the stearoyl chain (18:0) of LP1 and the acyl chains of the major lipid component of these liposomes (POPC). POPC has a palmitoyl chain (16:0) and an oleoyl chain (18:1) in the structure. Hence, we expected some degree of demixing of the POPC and lipopeptides within the lipid bilayers of the liposomes.31
When incorporated in the liposomes, the formation of collagen-like triple helices by lipopeptide molecules is subject to availability of lipopeptide neighbors in the immediate vicinity. The increase in number of lipopeptide nearest neighbors shifts the monomer:trimer equilibrium towards the latter.20 In addition, upon enzymatic cleavage of the lipopeptides, the membrane destabilization should be affected by the amount of demixing of the phospholipids and the lipopeptides. The cleavage of isolated lipopetide molecules in the liposomal membrane should produce “transient” destabilization of the membrane which can be “healed” quickly by the surrounding phospholipid molecules. However, the cleavage of “patches” of lipopeptides in the areas with high local concentration of lipopeptide molecules will produce larger defect areas on the membrane; the surrounding phospholipid molecules will take longer time to “anneal” these larger defects. Thus, we hypothesized that the major phospholipid used in the liposomal formulation will significantly affect the MMP-9 triggered leakage of the contents.
To study the effect of the acyl chains of the major lipids on MMP-9 triggered release, liposomes encapsulating carboxyfluorescein were formulated with 30 mol% of lipopeptide LP1 and the major lipid component (70 mol%) of the liposomes was varied [POPC, 1, 2-distearoyl-sn-glycero-3-phosphocholine (DSPC) or 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)]. This will ensure that the concentrations of the major lipids (i.e., POPC, DOPC and DSPC) in the liposomes are equivalent in these formulations. Figure 6 shows the kinetic traces for the release of the encapsulated dye from these liposomes in the presence of 0.7 μM MMP-9.
Figure 6.

Dependence of MMP-9-triggeredrelease from liposomes on the major phospholipid component. The kinetic traces for carboxyfluorescein fluorescence (λex = 480 nm, λem = 518 nm) were recorded for 85 minutes for liposomes formulated with 30 mol% of lipopeptide LP1 and 70 mol% POPC (black squares), DOPC (red diamonds) or DSPC (blue triangles). The reactions were conducted at 25 °C in 25 mM HEPES buffer, pH = 8.0 containing 10 mM CaCl2 and 10 μM ZnCl2 in the presence of 700 nM MMP-9. The solid black lines are single exponential fits of the data by non-linear regression.
For the liposomes formulated with the lipopeptide LP1, the highest extent of contents release was observed when POPC was used as the major lipid (Figure 6, black squares). Using DOPC as the major lipid reduced the rate as well as the amount of dye release (Figure 6, red diamonds). When we used DSPC as the major lipid component of the liposomes (which contains stearoyl chains, the same acyl chain as the lipopeptide LP1), the extent of release was very small (Figure 6, blue circles). The kinetic traces for DOPC and DSPC liposomes could not be fit by the single exponential rate equation. Biexponential fit of the release data for the DSPC liposomes showed total amplitude of 0.1, indicating that only 10% of the encapsulated dye was released from the liposomes over the 2 hours interval, compared to 87% and 72% for the POPC and DOPC liposomes respectively (Table 3). Release from the DOPC liposomes in the presence of MMP-9 proceeded with a lag phase; we do not yet understand the molecular mechanism for the origin of this lag phase. The discrepancy between the observed data points and the fitted curve for the DOPC liposomes indicates that the release from these liposomes is kinetically more complex than a biexponential rate equation.
Table 3.
Summary of the kinetic parameters for liposomes formulated with stearic and oleic acid lipopeptide conjugates LP1 and LP2 with different major phospholipid components.
| Lipopeptides | POPC liposomes | DOPC liposomes | DSPC liposomes | |||
|---|---|---|---|---|---|---|
| Rate (min−1) | Amplitude | Rate (min−1) | Amplitude | Rate (min−1) | Amplitude | |
| LP1 (stearic acid conjugate) | 0.036 ± 0.02 | 0.87 ± 0.02 | 0.063 ± 0.006 | 0.72 ± 0.05 | 0.13 ± 0.01 0.004 ± 0.001 |
0.016 ± 0.003 0.086 ± 0.001 |
| LP2 (oleic acid conjugate) | 0.16 ± 0.01 | 0.22 ± 0.02 | 0.09 ± 0.01 | 0.27 ± 0.02 | 0.09 ± 0.001 | 0.70 ± 0.01 |
The data suggests that acyl chain matching between the lipopeptide and major phospholipid components plays a significant role in determining the rate and the extent of MMP-9-triggered release of the liposomal contents. Another factor that may also contribute to the noted differences in release is the difference in the phase composition of the liposome membranes. DSPC bilayers (Tm = 55 °C) are expected to be in the gel phase at room temperature, while DOPC (Tm = − 20 °C) and POPC (Tm = − 3 °C) bilayers are in the liquid crystalline states at room temperature.32,33
In order to determine if the lipid phase affects the liposomal contents release by MMP-9, we synthesized the lipopeptide LP2. LP2 has the same peptide sequence as LP1, but the acyl chain contains the oleoyl moiety (Table 1). Liposomes encapsulating carboxyfluorescein were formulated with 30 mol% of LP2 and 70 mol% of POPC, DOPC or DSPC and the contents release studies were conducted in the presence of 700 nM of MMP-9 (Figure 7). When POPC was used as the major lipid, 22% of the encapsulated dye was released from the liposomes in 1 hour (Figure 7, blue squares). Similarly, for the liposomes with DOPC as the major lipid component, we observed 27% release of the contents in 1 hour (Figure 7, red circles). However, when the major lipid had the acyl chains mismatched compared to the lipopeptide LP2 (i.e., DSPC as the major lipid), 70% of the dye was released in 1 hour (Figure 7, black squares). The amplitudes and single exponential rate constants obtained from quantitative analyses of the data are given in Table 3. The data presented in Figures 6 and 7 provide confirmation that the triggered release of the liposomal contents is favored by greater degree of acyl chain mismatch between the lipopeptide and major phospholipid component of the liposomes.
Figure 7.

Dependence of MMP-9-triggered release from liposomes formulated with lipopeptide LP2 on the acyl chains of the major phospholipid. The kinetic traces of carboxyfluorescein fluorescence (λex = 480 nm, λem = 518 nm) were recorded for 60 minutess for liposomes formulated with 30 mol% of lipopeptide LP2 and 70 mol% DSPC (black squares), DOPC (red circles) or POPC (blue squares). The reactions were conducted at 25 °C in 25 mM HEPES buffer, pH = 8.0 containing 10 mM CaCl2 and 10 μM ZnCl2 in the presence of 700 nM MMP-9. The solid black lines are single exponential fits of the data by non-linear regression.
Circular Dichroism studies
As discussed above, MMP-9-triggered carboxyfluorescein release from the liposomes is favored when the acyl chain of lipopeptide does not match with that of the major phospholipid in the liposomes. We considered that this could be due to any or a combination of two related mechanisms. The incorporation of phospholipids having mismatched chains with the lipopeptides in the lipid bilayer could favor the formation of domains and tight collagen-like triple helices in the lipopeptide domains (due to high local concentration of the lipopeptides). Concentration-dependence of the triple helix formation by collagen-related peptides has been previously demonstrated.20,21 Such higher triple helicity will, in turn, create an additional kinetic barrier for unwinding and subsequent cleavage of the lipopeptides by MMP-9. It is also possible that the higher carboxyfluorescein release from the acyl chain mismatched liposomes is due to lower annealing ability (after the cleavage of the lipopeptides by MMP-9) of these membranes relative to those with matched acyl chains. To determine which of these two possible mechanisms is responsible for the observed acyl chain dependence of liposomal release, we determined the Rpn values20 of the lipopeptides in different liposome formulations by CD spectroscopy (Table 4). The Rpn value indicates the presence of triple helical conformations of peptides with higher values implying higher degree of triple helicity.20,21
Table 4.
Rpn values for lipopeptides when incorporated in the liposomes.
| POPC liposomes | DSPC liposomes | DOPC liposomes | |
|---|---|---|---|
| LP1 | 0.067 | 0.085 | 0.120 |
| LP2 | 0.065 | 0.097 | 0.083 |
| LP3 | 0.039 | 0.085 | 0.041 |
For the liposome-incorporated lipopeptide LP1, the Rpn values are 0.067 and 0.085 respectively for liposomes with POPC and DSPC as the major lipids. For liposomal LP2, the Rpn value is 0.097 when the major phopholipid is DSPC and 0.065 when the major phospholipid is POPC. Clearly, there is no correlation between the acyl chain mismatch and the calculated Rpn values for the lipopeptides in the liposomes. Thus, it appears that the higher triple helicity of the incorporated lipopeptides is not contributing to reduced extent of contents release for the acyl chain mismatched liposomes. It is likely that when the acyl chains of the lipopeptides and the major lipid component of the liposomes are matched, the “defects” created by the enzymatic cleavage of the lipopeptides are “annealed” quickly, leading to the reduction of the extent of the encapsulated dye release from these liposomes.
Discussion
For most of the literature reports on MMP-directed tumor targeting using liposomes, the goal was to enhance the uptake of the intact liposomes into the cells to deliver the encapsulated cytotoxic agents.15,16 Uptake of liposomes by endocytosis usually exposes the liposomes to the acidic lysosomal environment which often degrades the encapsulated drug.34 Our goal is to deliver the drugs (especially inhibitors for MMPs and other extracellular cancer associated enzymes) in the extracellular matrix employing the extracellular MMPs as the triggers. In addition, the compounds released in tumor extracellular matrix by employing our methodology should be able to diffuse through the membranes of nearby cells if the molecules have favorable partition coefficients.35
The triple helical conformation for collagen-mimetic lipopeptides has been observed in solution previously.20,21 However, incorporation of the lipopeptides in liposomes may not render them triple helical. For instance, due to restrictions imposed by the neighboring phospholipid or lipopeptide molecules in the membrane, the conformational entropy of the lipopeptides may be reduced upon incorporation in the lipid bilayers of the liposomes.36 The CD spectra of the lipopeptide-containing liposomes (Figure 1) show that this is not the case. For both lipopeptides LP1 and LP2, the CD spectra show pronounced minima at 200 nm and positive peaks at 225 nm, characteristic of the triple helical conformation.20,21 Upon incubation of the liposomes with MMP-9, we observed reductions of the intensity for both negative and positive peaks (Figure 1). Clearly, upon cleavage of the liposome incorporated lipopeptides, the triple helicity is reduced. Due to high background noise and light scattering from the liposomes, the CD spectra shown in Figure 1 were recorded using a 1 mm pathlength cuvette and averaged over 50 scans. Because of the unfavorable signal to noise ratio, our attempts to follow the kinetics of enzymatic cleavage of the liposome-incorporated lipopeptides by CD spectroscopy were unsuccessful.
To determine the effects of peptide cleavage and the subsequent release of the liposomal contents, we employed the fluorescence emission signal from the self-quenching dye, carboxyfluorescein.22 Carboxyfluorescein has been used extensively to monitor the kinetics of release of encapsulated aqueous contents from liposomes.37,38 At high concentration (such as those within the liposomes), the fluorescence emission of the dye is self-quenched. Release of the dye from the liposomes to the surrounding medium results in dequenching of its fluorescence emission.22 Consequently, a large enhancement in the fluorescence emission intensity of carboxyfluorescein is observed. We determined that the increase in emission intensity of carboxyfluorescein is linear in the concentration range of 3 μM to 30 μM (data not shown) and the dye is not photobleached during the course of our release studies (90 minutes). The complete release of the liposome encapsulated carboxyfluorescein was achieved by the addition of 1% of a detergent (Triton X) which destabilizes the membrane.39
Our studies with liposomes formulated with POPC and the lipopeptide LP1 show that complete release of the contents occurs in 40 minutes with 2.3 μM MMP-9. During these experiments, we included 10 mM CaCl2 and 10 μM ZnCl2 in the assay buffer to maintain the optimal enzyme activity.23 However, some peptides and proteins are known to partition into or cross the membranes of liposomes. Such transient membrane perturbations can cause the release of contents from the liposomes. For example, δ-lysin (a hemolytic Staphylococcal peptide), causes leakage of carboxyfluorescein from liposomes by partitioning into the liquid-disordered ‘ld’ regions in the membrane.40 To determine if such a mechanism is operating for our MMP-9-triggered release of liposomal contents (i.e., the enzyme is getting embedded in the lipid bilayers of the liposomes and causing contents release), we conducted the appropriate control experiments. Liposomes were prepared with POPC only and incubated with 2.3 μM MMP-9 in a buffer containing CaCl2 and ZnCl2. These liposomes showed no significant leakage of the contents in 85 minutes (Figure 3, blue trace). In interpreting these results, we noted that the liposome phase composition is expected to be different when the lipopeptide is excluded from the lipid bilayers. This difference may be sufficient to alter the membrane properties such that the added MMP-9 cannot get imbedded in the bilayers of the liposomes formulated with POPC only.40 However, our results from the other control experiment using the liposomes formulated with POPC and lipopeptide LP1 rule out this mechanistic possibility. When we incubated these liposomes with 2.3 μM MMP-9 in a buffer that does not contain any Ca2+ or Zn2+ ions, only 15% of the encapsulated dye was released (Figure 3, red trace). The minimal leakage observed in this case is likely due to residual enzyme activity caused by any contaminant Ca2+ and Zn2+ or other metal ions in the buffer solution or due to the presence of these two ions in the stock solutions of MMP-9.
Lipopeptide LP3 (Table 1) was synthesized and incorporated in the liposomes to determine the influence of the peptide sequence on the enzymatic cleavage and subsequent destabilization of the liposomal membranes. LP3 [sequence: GPKGIAGQK(GPO)4GG] contains the MMP-9 cleavage site (glycine-isoleucine) in the structure.24 The glutamine and the arginine residues of LP1 are replaced with lysine in LP3. The rationale for this replacement is that MMP-9 prefers nonpolar residues flanking the cleavage site25 and substitution with a polar residue is expected to reduce the activity of the enzyme for this substrate. The ε—amino groups of the lysine residues of LP3 (pKR = 10.5) will have positive charges under our experimental conditions (pH = 8.0). When incubated with MMP-9, the liposomes formulated with the 30 mol% of lipopeptide LP3 and 70 mol% of POPC released less than 5% of their contents (Figure 3, green trace), confirming that the enzymatic cleavage of the peptide by MMP-9 is the trigger for membrane perturbation and release of liposomal contents. By CD spectroscopy, we observed that in solution, lipopeptide LP3 is not as triple helical as the LP1 (data not shown), possibly due to the repulsion caused by the positive charges of the lysine residues. In addition, after cleavage of the liposome-incorporated LP3 by MMP-9, the cleaved product will have a positively charged head group and may not cause enough perturbation of the liposomal membranes for efficient release of the encapsulated dye.
A triggered release system should exhibit release characteristics that are dependent on the concentration of the triggering enzyme. Our results indicate that both the rate and extent of release of the encapsulated dye consistently increase with increasing concentration of MMP-9 (Figure 4 and Table 2). Between 100 nM and 2.3 μM concentrations of MMP-9, the kinetic traces could be fit by the single exponential rate equation. The rates obtained from these fits (Table 1) are representative of the average of a series of microscopic rate constants encompassing several processes, e.g., unwinding of the triple helix, cleavage of the peptide, perturbation of the membrane and annealing of the defect in the membrane.40,41 Clearly, the extent of contents release from our liposomes can be controlled by the amount (activity) of the MMP-9 present. In addition, if the liposome contents include inhibitors of MMP-9, our controlled release methodology will function akin to a “negative-feedback” system. The released inhibitors will inhibit the cleavage of the liposome incorporated lipopeptides by MMP-9; this, in turn, will slow the rate as well as the extent of the release of the encapsulated contents. Thus, the liposomes can act as a self-adjusting, triggered release system for delivering drugs and other molecules.
Our recombinant MMP-9 has the catalytic and the fibronectin domains of the full-length enzyme. It is hypothesized in the literature that the fibronectin domain of MMP-9 is important for the unwinding of triple helical peptide substrates prior to the cleavage by the catalytic domain of the enzyme.41 MMP-7 and −10 lack the fibronectin domains in their structures.41 We observed that less than 20% of the liposome-encapsulated dye was released in the presence of recombinant MMP-7 and −10 (Figure 5). This small release observed may be attributed (in part) to cleavage of the monomeric lipopeptide which is in equilibrium with the trimer.17,20 Similarly, even though our triple helical peptides contain the cleavage site for trypsin, the enzyme failed to release significant amounts of the encapsulated carboxyfluorescein from the liposomes (Figure 5, green squares) due to the inability of the enzyme to unwind the triple helix.
The question arises: why does the enzyme-triggered release from the liposomes depend on the major phospholipid in the formulations? Our results (Figures 6, 7 and Table 3) show that when the acyl chains of the lipopeptides and phospholipids are similar, small amounts of the liposomal contents are released. When the corresponding acyl chains are different, amounts of contents release also increase. However, the Rpn values (i.e., triple helicity) measured for the liposome-incorporated lipopeptides (Table 4) do not show any obvious correlation between the extent of triple helicity and the corresponding extents of contents release.
In order to explain these contradicting observations, we noted that after enzymatic cleavage of the liposome-incorporated triple helical lipopeptides, one of the products will contain a short peptide linked to the acyl chain. For example, enzymatic cleavage of LP1 results in the liberation of the polypeptide sequence IAGQR(GPO)4GG-OH while the stearoyl moiety remains conjugated to the peptide GPQG. The estimated isoelectric points (pI) for this remnant four-residue polypeptide-strearic acid conjugate is ∼ 5.5 and for the uncleaved lipopeptide is ∼ 9.5.42 Hence, under our experimental conditions (pH = 8.0), the cleavage of lipopeptide results in a reversal of the net charge on the lipopeptide from positive to negative. It is also possible that the reduced headgroup size of the cleaved lipopeptide product will induce curvature stress in the bilayer since the enzyme does not have access to the inner lipid layer of the liposomes.43 In order to minimize the total free energy due to these changes in the lipid membranes, domain reorganization is likely to occur. For example, domains in raft-containing membranes exhibit a circular shape in order to minimize line tension. If these domains are perturbed by mechanical or chemical means, they quickly reassume the circular shape to minimize the boundary length.44
The microscopic details of the domain reorganization following the MMP-9 mediated cleavage of our liposome-incorporated lipopeptides are complex, and we do not yet understand the molecular mechanism. However, it is likely that this step will lead to defects in the membrane that are sufficient for contents of the liposome to leak. The membrane changes that result from the hydrolysis of the lipopeptides are expected to be similar to the membrane perturbation that occurs due to phospholipase A2 mediated hydrolysis of the headgroups of phosphocholines which also leads to the leakage of encapsulated contents from liposomes.45
In the liposomes formulated with lipopeptide and phosphocholine of mismatched acyl chains, it is expected that the extent of a priori demixing of the lipopeptides into lipopeptide-rich domains will be higher.46 Consequently, in these liposomes, upon enzymatic cleavage of the lipopeptides, the perturbation in the membrane will be larger (the effect is localized in domains rather than distributed over the entire membrane surface). In this scenario, the probability of reorganization of surrounding lipids to “anneal” the defects is less. When the liposomes are formulated with the lipopeptides bearing similar acyl chains as the major phospholipid, the lipopeptides are more mixed with the phosphocholine.46 In this situation, the membrane defects generated by the cleavage of the lipopeptides can be “annealed” rapidly.47 While the amplitudes obtained from exponential fits of the acyl chain dependence release data (Table 3) are consistent with this hypothesis, the rate constants are not. For example, for the oleic acid conjugated peptide (LP2), the rate constant for dye release from the liposomes are 0.09 min−1 (with DSPC as the major lipid), 0.09 min−1 (with DOPC as the major lipid) and 0.16 min−1 (with POPC as the major lipid). It should be noted that the amplitudes for these dye release traces are very different: 0.70 for the DSPC liposomes and 0.27 and 0.22 for DOPC and POPC liposomes respectively. With such different amplitudes, it is unrealistic to compare the rate constants obtained from the fits of the release data.48 The first exponential phase rate constant (i.e., k1) calculated for the release from the liposomes incorporating DSPC and lipopeptide LP1 may also be attributed to a similar artifact, as the total amplitude in this case is only 0.1 (Table 3). For the liposomes formulated with DOPC as the major lipid, a lag phase was observed in the kinetic trace, creating a noticeable deviation from a single exponential process (Figure 6). As discussed previously, the enzyme triggered release of the liposomal contents is a complex, multi-step process and analyzing the rates as single exponential processes is an approximation, except when one of the steps is clearly rate-limiting.
Conclusions
In conclusion, we have designed liposomes whose contents are released upon triggering with human MMP-9. The liposomes are formulated with phospholipids and a lipopeptide that contains a collagen-related peptide sequence with the MMP-9 cleavage site. Because of the amino acid sequence, the lipopeptide is able to acquire triple helical conformation (similar to collagen) when incorporated in the liposomes. The encapsulated dye is released from these liposomes in the presence of MMP-9 in a concentration dependent manner. We observed that the dye can be completely released in 40 minutes in the presence of 2.3 μM of MMP-9. MMP-7, -10 and trypsin failed to release the liposome encapsulated dye due to their inability to unwind the triple helical lipopeptides. Interestingly, the extent of MMP-triggered release is strongly dependent on the acyl chain mismatch between the lipopeptides and the major phospholipid components of the liposomes. Our results led to the conclusion that formation of lipopeptide-rich domains in the liposomal membrane facilitates MMP-9-triggered release of the liposome contents. It is likely that as the local, ‘effective’ concentration of lipopeptides exceeds a certain critical mole fraction in the membranes, repair of defects caused by the enzymatic cleavage of the peptide becomes inefficient, resulting in complete release of the encapsulated contents. The results demonstrate that the sensitivity of the liposomal release to MMP-9 can be altered simply by changing the major phospholipid used in the formulation. Due to the ease of purification of the lipopeptide LP1 and the observed release rates, it appears that the LP1-POPC liposomes are better compared to the LP2-DSPC liposomes in releasing the encapsulated contents. Overall, our methodology will find applications in drug delivery and in diagnostics systems that are based on the release of liposomal contents using enzymatic as well as other triggers.
Acknowledgments
This research was supported by the NIH grant 1R01 CA113746 and NSF DMR-0705767 to SM and DKS. AJH was supported by the NSF-EPSCoR award EPS-0447679. AIE was supported by the NSF EPSCoR award EPS-047679. XL thanks the support of the proteomics core facility by NIH Grant P20 RR016741 from the INBRE Program of the NCRR.
References
- 1.a Verma RP, Hansch C. Bioorg. Med. Chem. 2007;15:2223–2268. doi: 10.1016/j.bmc.2007.01.011. [DOI] [PubMed] [Google Scholar]; b Vartak DG, Gemeinhart RA. J. Drug targeting. 2007;15:1–20. doi: 10.1080/10611860600968967. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Malemud CJ. Frontiers in Bioscience. 2006;11:1696–1701. doi: 10.2741/1915. [DOI] [PubMed] [Google Scholar]
- 2.Yan C, Boyd DDJ. Cellular Physiology. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 3.a Fingleton B. Curr. Pharm. Design. 2007;13:333–346. doi: 10.2174/138161207779313551. [DOI] [PubMed] [Google Scholar]; b Brauer PR. Frontiers in Bioscience. 2006;11:447–478. doi: 10.2741/1810. [DOI] [PubMed] [Google Scholar]; c Gallagher GL, Jackson CJ, Hunyor SN. Frontiers in Bioscience. 2007;12:1410–1419. doi: 10.2741/2157. [DOI] [PubMed] [Google Scholar]
- 4.a Verma RP, Hansch C. Bioorg. Med. Chem. 2007;15:2223–2268. doi: 10.1016/j.bmc.2007.01.011. [DOI] [PubMed] [Google Scholar]; b Vihinen P, Ala-aho R, Kaehaeri VM. Current Cancer Drug Targets. 2005;5:203–220. doi: 10.2174/1568009053765799. [DOI] [PubMed] [Google Scholar]
- 5.Demers M, Couillard J, Belanger S, St-Pierre Y. Critical Rev. Immunology. 2005;25:493–523. doi: 10.1615/critrevimmunol.v25.i6.30. [DOI] [PubMed] [Google Scholar]
- 6.For a list of FDA approved liposomal drugs to treat cancer, see: http://www.icare.org/fda.htm. Liposomal drugs formulations have been approved by FDA for the treatment of AIDS, acute pain after surgery, bacterial infections etc.
- 7.There are currently 251 clinical trials in progress on the use of liposomes to treat various diseases, including cancer [February 26, 2008]; ( http://www.clinicaltrials.gov. For literature reports, see:Torchilin VP. Adv. Drug Delivery Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009.Gabizon AA, Shmeeda H, Zalipsky S. J. Liposome Res. 2006;16:175–183. doi: 10.1080/08982100600848769.Torchilin VP. Nature Rev. Drug Discovery. 2005;4:145–160. doi: 10.1038/nrd1632.
- 8.a Karanth H, Murthy RSR. J. Pharmacy Pharmacology. 2007;59:469–483. doi: 10.1211/jpp.59.4.0001. [DOI] [PubMed] [Google Scholar]; b Kale AA, Torchilin VP. J. Liposome Res. 2007;17:97–203. doi: 10.1080/08982100701525035. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sawant RM, Hurley JP, Salmaso S, Kale A, Tolcheva E, Levchenko TS, Torchilin VP. Bioconjugate Chem. 2006;17:943–949. doi: 10.1021/bc060080h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a Jain S, Tiwary AK, Jain NK. Current Drug Delivery. 2006;3:157–166. doi: 10.2174/156720106776359221. [DOI] [PubMed] [Google Scholar]; b Karoonuthaisiri N, Titiyevskiy K, Thomas JL. Coll. Surfaces B: Biointerfaces. 2003;27:365–375. [Google Scholar]
- 10.Davis SC, Szoka FC. Bioconjugate Chem. 1998;9:783–792. doi: 10.1021/bc980047y. [DOI] [PubMed] [Google Scholar]
- 11.Dromi S, Frenkel V, Luk A, Traughber B, Angstadt M, Bur M, Poff J, Xie J, Libutti SK, Li KCP, Wood BJ. Clinical Cancer Research. 2007;13:2722–2727. doi: 10.1158/1078-0432.CCR-06-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ponce AM, Wright A, Dewhirst MW, Needham D. Future Lipidology. 2006;1:25–34. [Google Scholar]; c Hauck ML, LaRue SM, Petros WP, Poulson JM, Yu D, Spasojevic I, Pruitt AF, Klein A, Case B, Thrall DE, Needham D, Dewhirst MW. Clin. Cancer Res. 2006;12:4004–4010. doi: 10.1158/1078-0432.CCR-06-0226. [DOI] [PubMed] [Google Scholar]
- 12.a Chandra B, Mallik S, Srivastava DK. Org. Biomol. Chem. 2006;4:1730–1740. doi: 10.1039/b518359f. [DOI] [PubMed] [Google Scholar]; b Chandra B, Mallik S, Srivastava DK. J. Chem. Soc. Chem. Commun. 2005:3021–3023. doi: 10.1039/b503423j. [DOI] [PubMed] [Google Scholar]; c Li Z, Wan Y, Kutateladze AG. Langmuir. 2003;19:6381–6391. [Google Scholar]
- 13.a Foged C, Nielsen HM, Frokjaer S. Intl. J. Pharmaceutics. 2007;331:160–166. doi: 10.1016/j.ijpharm.2006.11.010. [DOI] [PubMed] [Google Scholar]; b Foged C, Nielsen HM, Frokjaer S. J. Liposome Res. 2007;17:191–196. doi: 10.1080/08982100701530373. [DOI] [PubMed] [Google Scholar]; c Boyer C, Zasadzinski JA. ACS Nano. 2007;1:176–182. doi: 10.1021/nn7002025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerasimov OV, Boomer JA, Qualls MM, Thompson DH. Adv. Drug Delivery Rev. 1999;38:317–338. doi: 10.1016/s0169-409x(99)00035-6. [DOI] [PubMed] [Google Scholar]
- 15.Terada T, Iwai M, Kawakami S, Yamashita F, Hashida M. J. Controlled Release. 2006;111:333–342. doi: 10.1016/j.jconrel.2005.12.023. [DOI] [PubMed] [Google Scholar]
- 16.a Hatakeyama H, Akita H, Kogure K, Oishi M, Nagasaki Y, Kihira Y, Ueno M, Kobayashi H, Kikuchi H, Harashima H. Gene Ther. 2007;14:68–77. doi: 10.1038/sj.gt.3302843. [DOI] [PubMed] [Google Scholar]; b Hatakeyama H, Akita H, Ishida E, Hashimoto K, Kobayashi H, Aoki T, Yasuda J, Obata K, Kikuchi H, Ishida T, Kiwada H, Harashima H. Int. J. Pharmaceutics. 2007;342:194–200. doi: 10.1016/j.ijpharm.2007.04.037. [DOI] [PubMed] [Google Scholar]
- 17.Rezler EM, Khan DR, Lauer-Fields J, Cudic M, Baronas-Lowell D, Fields GB. J Am. Chem. Soc. 2007;129:4961–4972. doi: 10.1021/ja066929m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.a Sarkar N, Banerjee J, Hanson AJ, Elegbede AI, Rosendahl T, Krueger AB, Banerjee AL, Tobwala S, Wang R, Lu X, Mallik S, Srivastava DK. Bioconjugate Chem. 2008;19:57–64. doi: 10.1021/bc070081p. [DOI] [PubMed] [Google Scholar]; b Sarkar NR, Rosendahl T, Krueger AB, Banerjee AL, Benton K, Mallik S, Srivastava DK. J. Chem. Soc. Chem. Commun. 2005:999–1001. doi: 10.1039/b416827e. [DOI] [PubMed] [Google Scholar]
- 19.a Diaz-Mochon JJ, Fara MA, Sanchez-Martin RM, Bradley M. Tetrahedron Lett. 2008;49:923–926. [Google Scholar]; b Murray JK, Gellman SH. Nature Protocols. 2007;2:624–631. doi: 10.1038/nprot.2007.23. [DOI] [PubMed] [Google Scholar]; c Collins JM, Leadbeater NE. Org. Biomol. Chem. 2007;5:1141–1150. doi: 10.1039/b617084f. [DOI] [PubMed] [Google Scholar]; d Palasek SA, Cox ZJ, Collins JM. J. Peptide Sci. 2007;13:143–148. doi: 10.1002/psc.804. [DOI] [PubMed] [Google Scholar]
- 20.a Gauba V, Hartgerink JD. J. Am. Chem. Soc. 2007;129:15034–15041. doi: 10.1021/ja075854z. [DOI] [PubMed] [Google Scholar]; b Gauba V, Hartgerink JD. J. Am. Chem. Soc. 2007;129:2683–2690. doi: 10.1021/ja0683640. [DOI] [PubMed] [Google Scholar]; c Cejas MA, Kinney WA, Chen C, Leo GC, Tongue BA, Vinter JG, Joshi PP, Maryanoff BE. J. Am. Chem. Soc. 2007;129:2202–2203. doi: 10.1021/ja066986f. [DOI] [PubMed] [Google Scholar]; d Kusebauch U, Cadamuro SA, Musiol HJ, Lenz MO, Wachtveitl J, Moroder L, Renner C. Angew. Chem. Int. Ed. 2006;45:7015–7018. doi: 10.1002/anie.200601432. [DOI] [PubMed] [Google Scholar]; e Shoulders MD, Hodges JA, Raines RT. J. Am. Chem. Soc. 2006;128:8112–8113. doi: 10.1021/ja061793d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rezler EM, Khan DR, Tu R, Tirrell M, Fields GB. Methods Mol. Biol. 2007;386:269–298. doi: 10.1007/978-1-59745-430-8_10. [DOI] [PubMed] [Google Scholar]
- 22.a Brochu H, Vermette P. Langmuir. 2007;23:7679–7686. doi: 10.1021/la7003547. [DOI] [PubMed] [Google Scholar]; b Pajewski R, Ferdani R, Pajewska J, Djedovic N, Schlesinger PH, Gokel GW. Org. Biomol Chem. 2005;3:619–625. doi: 10.1039/b417009a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Underwood CK, Min D, Lyons JG, Hambley TW. J. Inorg. Biochem. 2003;95:165–170. doi: 10.1016/s0162-0134(03)00100-4. [DOI] [PubMed] [Google Scholar]
- 24.Minond D, Lauer-Fields JL, Cudic M, Overall CM, Pei D, Brew K, Visse R, Nagase H, Fields GB. J. Biol. Chem. 2006;281:38302–38313. doi: 10.1074/jbc.M606004200. [DOI] [PubMed] [Google Scholar]
- 25.Netzel-Arnett S, Sang QX, Moore WG, Navre M, Birkedal-Hansen H, Van Wart HE. Biochemistry. 1993;32:6427–6432. doi: 10.1021/bi00076a016. [DOI] [PubMed] [Google Scholar]
- 26.a Turpeenniemi-Hujanen T. Biochimie. 2005;87:287–297. doi: 10.1016/j.biochi.2005.01.014. [DOI] [PubMed] [Google Scholar]; b Koc M, Ediger D, Budak F, Karadag M, Oral HB, Uzaslan E, Ege E, Gozu RO. Tumori. 2006;92:149–154. doi: 10.1177/030089160609200211. [DOI] [PubMed] [Google Scholar]; c Seki M, Uzuki M, Ohmoto H, Yoshino K, Maeda S, Kokubun S, Sakurai M, Sawal T. Ryumachi. 1995;35:792–801. [PubMed] [Google Scholar]
- 27.Lauer-Fields JL, Sritharan T, Stack MS, Nagase H, Fields GB. J. Biol. Chem. 2003;278:18140–18145. doi: 10.1074/jbc.M211330200. [DOI] [PubMed] [Google Scholar]
- 28.Marguet D, Lenne PF, Rigneault H, He HT. EMBO J. 2006;25:3446–3457. doi: 10.1038/sj.emboj.7601204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veatch SL, Soubias O, Keller SL, Gawrisch K. Proc. Natl. Acad. Sci. USA. 2007;104:17650–17655. doi: 10.1073/pnas.0703513104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Almeida PF, Pokorny A, Hinderliter A. Biochim. Biophys. Acta. 2005;1720:1–13. doi: 10.1016/j.bbamem.2005.12.004. [DOI] [PubMed] [Google Scholar]
- 31.Khanna K, Chang TT, Kindt JT. J. Chem. Phys. 2006;124:036102. doi: 10.1063/1.2162535. [DOI] [PubMed] [Google Scholar]
- 32.a Leekumjorn S, Sum AK. J. Phys. Chem. B. 2007;111:6026–6033. doi: 10.1021/jp0686339. [DOI] [PubMed] [Google Scholar]; b Kaneshina S, Ichimori H, Hata T, Matsuki H. Biochim. Biophys. Acta. 1998;1374:1–8. doi: 10.1016/s0005-2736(98)00122-9. [DOI] [PubMed] [Google Scholar]
- 33.For the phase transition temperatures of these and other glycerophospholipids, see: http://www.avantilipids.com/PhaseTransitionTemperaturesGlycerophospholipids.asp?T=Phase%20Transition%20Temperatures%20for%20Glycerophospholipids.
- 34.Tarragó-Trani MT, Storrie B. Adv. Drug Deliv. Rev. 2007;59:782–797. doi: 10.1016/j.addr.2007.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Avdeef A. Curr.Top. Med. Chem. 2001;1:277–351. doi: 10.2174/1568026013395100. [DOI] [PubMed] [Google Scholar]
- 36.Tsamaloukas A, Szadkowska H, Heerklotz H. Biophys. J. 2006;90:4479–4487. doi: 10.1529/biophysj.105.080127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.a Volodkin D, Arntz Y, Schaaf P, Moehwald H, Voegel JC, Ball V. Soft Matter. 2008;4:122–130. doi: 10.1039/b713563g. [DOI] [PubMed] [Google Scholar]; b Gabel D, Awad D, Schaffran T, Radovan D, Daraban D, Damian L, Winterhalter M, Karlsson G, Edwards K. ChemMedChem. 2007;2:51–53. doi: 10.1002/cmdc.200600227. [DOI] [PubMed] [Google Scholar]
- 38.a Nagahama M, Otsuka A, Sakurai J. Biochim. Biophys. Acta. 2006;1762:110–114. doi: 10.1016/j.bbadis.2005.10.002. [DOI] [PubMed] [Google Scholar]; b De Maria P, Filippone P, Fontana A, Gasbarri C, Siani G, Velluto D. Colloids Surf. B: Biointerfaces. 2005;40:11–18. doi: 10.1016/j.colsurfb.2004.09.007. [DOI] [PubMed] [Google Scholar]
- 39.a Marcelino J, Lima JLFC, Reis S, Matos C. Chem. Phys. Lipids. 2007;146:94–103. doi: 10.1016/j.chemphyslip.2006.12.008. [DOI] [PubMed] [Google Scholar]; b Chern CS, Chiu HC, Yang YS. J. Colloid and Interface Sci. 2006;302:335–340. doi: 10.1016/j.jcis.2006.06.028. [DOI] [PubMed] [Google Scholar]
- 40.Pokorny A, Almeida PF. Biochemistry. 2005;44:9538–9544. doi: 10.1021/bi0506371. [DOI] [PubMed] [Google Scholar]
- 41.a Minond D, Lauer-Fields JL, Cudic M, Overall CM, Pei D, Brew K, Moss ML, Fields GB. Biochemistry. 2007;46:3724–3733. doi: 10.1021/bi062199j. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Xu X, Wang Y, Lauer-Fields JL, Fields GB, Steffensen B. Matrix Biol. 2004;23:171–181. doi: 10.1016/j.matbio.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 42.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. Nucleic Acids Res. 2003;31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lee YC, Zheng YO, Taraschi TF, Janes N. Biochemistry. 1996;35:3677–3684. doi: 10.1021/bi9517502. [DOI] [PubMed] [Google Scholar]
- 44.García-Sáez AJ, Chiantia S, Schwille P. J. Biol. Chem. 2007;282:33537–33544. doi: 10.1074/jbc.M706162200. [DOI] [PubMed] [Google Scholar]
- 45.a Burack WR, Dibble AR, Allietta MM, Biltonen RL. Biochemistry. 1997;36:10551–10557. doi: 10.1021/bi970509f. [DOI] [PubMed] [Google Scholar]; b Burack WR, Yuan Q, Biltonen RL. Biochemistry. 1993;32:583–589. doi: 10.1021/bi00053a025. [DOI] [PubMed] [Google Scholar]
- 46.a Coste V, Puff N, Lockau D, Quinn PJ, Angelova MI. Biochim. Biophys. Acta. 2006;1758:460–467. doi: 10.1016/j.bbamem.2006.03.003. [DOI] [PubMed] [Google Scholar]; b Hungerford G, Castanheira EMS, Baptista ALF, Coutinho PJG, Real Oliveira MECD. J. Fluorescence. 2005;15:835–840. doi: 10.1007/s10895-005-0014-3. [DOI] [PubMed] [Google Scholar]
- 47.Black SG, Dixon GS. Biochemistry. 1981;20:6740–6744. doi: 10.1021/bi00526a033. [DOI] [PubMed] [Google Scholar]
- 48.Pokorny A, Yandek LE, Elegbede AI, Hinderliter A, Almeida PF. Biophys. J. 2006;91:2184–2197. doi: 10.1529/biophysj.106.085027. [DOI] [PMC free article] [PubMed] [Google Scholar]