

Abstract
Eukaryotic cytochrome c oxidase (COX) is the terminal enzyme of the mitochondrial respiratory chain. COX is a multimeric enzyme formed by subunits of dual genetic origin whose assembly is intricate and highly regulated. In addition to the structural subunits, a large number of accessory factors are required to build the holoenzyme. The function of these factors is required in all stages of the assembly process. They are relevant to human health because devastating human disorders have been associated with mutations in nuclear genes encoding conserved COX assembly factors. The study of yeast strains and human cell lines from patients carrying mutations in structural subunits and COX assembly factors has been invaluable to attain the current state of knowledge, even if still fragmentary, of the COX assembly process. After the identification of the genes involved, the isolation and characterization of genetic and metabolic suppressors of COX assembly defects, reviewed here, have become a profitable strategy to gain insight into their functions and the pathways in which they operate. Additionally, they have the potential to provide useful information for devising therapeutic approaches to combat human disorders associated with COX deficiency.
Keywords: Mitochondria, cytochrome c oxidase, suppression, mitochondrial disorder, COX assembly
1. Eukaryotic Cytochrome c oxidase assembly in health and disease
The fundamental mechanism by which respiratory energy transduction occurs in mitochondria and aerobic bacteria is the coupling of electron transfer reactions to the formation of transmembrane electrochemical gradients [1]. In eukaryotes, cytochrome c oxidase (COX) or complex IV, the terminal enzyme of the mitochondrial respiratory chain, is one of the primary coupling sites. COX is a multimeric copper-heme A metalloenzyme that functions as an electron-driven proton pump. It catalyzes the transfer of electrons from ferrocytochrome c to molecular oxygen via its four redox active metal cofactors. Fig. 1 summarizes the catalytic metal centers and the electron transfer pathway in COX. Electrons enter COX through a mixed valence dinuclear copper center, the CuA site, located in subunit 2. Electrons are transferred from CuA to a low spin heme a located in subunit 1 and are subsequently transferred intra-molecularly to the active site where a high spin heme a3 and CuB form a binuclear center for oxygen binding. The mechanism of electron transfer through COX has been extensively studied (reviewed in [2–4]). From an evolutionary point of view, the presence of the CuA center defines COX and distinguishes this enzyme from other terminal oxidases that use quinol rather than cytochrome c as the electron donor [2, 5, 6]. The electron transfer reaction is coupled to the vectorial transfer of protons from the matrix to the intermembrane space thus contributing to the generation of the proton gradient which is subsequently used by the F1F0 mitochondrial ATPase to drive the synthesis of ATP. The mechanism of proton pumping has been the focus of intense study and has been recently reviewed elsewhere ([7] and [8]).
Fig. 1.

Cytochrome c oxidase. Representation of the complex, showing the two catalytic subunits (subunits 1 and 2) involved in electron transfer from cytochrome c to molecular oxygen. The mechanism of electron transport, coupled to proton pumping, is depicted. Subunit 2 contains the binuclear CuA center that receives electrons from cytochrome c. In subunit I, a low-spin heme (heme a) accepts electrons from CuA and transfers them to a binuclear center consisting of a high-spin heme (heme a3) and a copper atom (CuB). Within the binuclear center, molecular oxygen is bound to heme a3 and sequentially reduced to water. IMS, intermembrane space; MIM, mitochondrial inner membrane.
Eukaryotic COX is formed by 11–13 subunits (11 in the yeast Saccharomyces cerevisiae and 13 in mammals) of dual genetic origin. Subunits 1, 2 and 3 form the catalytic core of the enzyme and in the majority of eukaryotes are encoded in the mitochondrial DNA. The core is surrounded by a set of nuclear-encoded small subunits that are important for both the assembly and function of the enzyme as well as its dimerization (reviewed in [9, 10]). These subunits are also involved in the modulation of the catalytic activity and in the protection of the core from reactive oxygen species. A list of COX homologue subunits in yeast and mammals is shown in Table 1.
Table 1.
Homologue COX subunits and COX assembly factors in yeast and mammals. Genes involved in expression of the mitochondrial DNA-encoded subunits and in mitochondrial import of nuclear encoded subunits are not included.
| YEAST | MAMMALS | FUNCTION | ||
|---|---|---|---|---|
| GENE | PROTEIN | GENE | PROTEIN | |
|
Catalytic core (mtDNA encoded structural subunits)
| ||||
| COX1 | Cox1p | MTCOXI | COX1 | Catalytic subunits |
| COX2 | Cox2p | MTCOXII | COX2 | |
| COX3 | Cox3p | MTCOXIII | COX3 | Catalytic core stability? |
|
| ||||
|
Core protective shield (nDNA encoded structural subunits)
| ||||
| COX4 | Cox4p | COXVb | COX5b | Subunits required for COX assembly and function |
| COX5a | Cox5ap | COXIV-1 | COX4-1 | |
| COX5b | Cox5bp | COXIV-2 | COX4-2 | |
| COX6 | Cox6p | COXVa | COX5a | |
| COX7 | Cox7p | COXVIIa | COX7a | |
| COX8 | Cox8p | COXVIIc | COX7c | |
| COX9 | Cox7ap | COXVIc | COX6c | |
| -- | -- | COXVIIb | COX7b | |
| -- | -- | COXVIII | COX8 | |
|
| ||||
| COX12 | Cox9p | COXVIb | COX6b | Non-essential subunits |
| COX13 | Cox10p | COXVIa | COX6a | |
|
| ||||
|
Membrane insertion and processing of catalytic core subunits
| ||||
| OXA1 | Oxa1p | OXA1 | OXA1 | Membrane insertion of COX subunits, cytochrome b and ATPase proteolipid |
| COX20 | Cox20p | COX20 | COX20 | Cox2p chaperone. Presentation of Cox2p-precursor to the IMP complex |
| COX18 | Cox18p | COX18 | COX18 | Export of the Cox2p C-terminus tail |
| MSS2 | Mss2p | -- | -- | Export of the Cox2p C-terminus tail |
| PNT1 | Pnt1p | -- | -- | Export of the Cox2p C-terminus tail |
| IMP1 | Imp1p | -- | -- | Responsible for the maturation of Cox2p. |
| IMP2 | Imp2p | IMMP2L | IMMP2L | Necessary for the stability and activity of Imp1. |
| SOM1 | Som1p | -- | -- | Third component of the yeast IMP complex. It could play a role in substrate recognition |
|
| ||||
|
Copper Metabolism and Insertion
| ||||
| COX17 | Cox17p | COX17 | COX17 | Delivery of copper to Sco1p and Cox11p |
| SCO1 | Sco1p | SCO1 | SCO1 | Transfer of copper to COX and/or reduction of |
| SCO2 | SCO2 | cysteine residues in subunit 2 | ||
| COX11 | Cox11p | COX11 | COX11 | Stable formation of the Cu(B) and magnesium centers |
|
| ||||
| COX19 | Cox19p | COX19 | COX19 | CX9C proteins. They could play roles in redox control and copper trafficking in the intermembrane space |
| COX23 | Cox23p | COX23 | COX23 | |
| PET191 | Pet191p | PET191 | PET191 | |
| CMC1 | Cmc1p | CMC1 | CMC1 | |
|
| ||||
|
Heme A Biosynthesis
| ||||
| COX10 | Cox10p | COX10 | COX10 | Farnesylation of protoheme |
| COX15 | Cox15p | COX15 | COX15 | Hydroxylation of heme O |
|
| ||||
|
Assembly/Unknown
| ||||
| COX16 | Cox16p | COX16 | COX16 | Unknown function |
| PET117 | Pet117p | -- | -- | Unknown function |
| PET100 | Pet100p | -- | -- | Formation of assembly intermediates containing Cox7p, Cox8p, and Cox9p |
| SHY1 | Surf1p | SURF1 | SURF1 | Catalyzes an assembly step in which Cox1p is one of the partners |
| MSS51 | Mss51p | -- | -- | Required for translation of COX1 mRNA. Additionally, binds Cox1p and is required for its stability/maturation/assembly |
| COX14 | Cox14p | -- | -- | Binds Cox1p and is required for its stability/maturation/assembly |
| COA1 | Coa1p | -- | -- | Binds Cox1p and is required for its stability/maturation/assembly |
As a consequence of its central role in oxidative metabolism, COX has been intensively studied by biochemical, genetic, spectroscopic, and crystallographic means [11–13]. From these studies it is known that other metals such as zinc and magnesium are also bound to the enzyme, although the basis for their specific requirements are largely unknown. The zinc atom could play a role in structural stability of the complex [14]. The magnesium/manganese site is in close proximity to the H2O exit channel and is thought to aid in the stability and release of H2O produced during the reduction of O2 [15].
COX biogenesis probably occurs with the different subunits and cofactors being added in an ordered manner. Data obtained from analyses of the human enzyme performed by Blue-Native electrophoresis suggests an assembly pathway characterized by the sequential incorporation of COX subunits. Assembly is initiated around a seed formed by subunit 1 and proceeds with the formation of several discrete assembly intermediates probably representing rate-limiting steps in the process [16].
Analysis of assembly intermediates has given insights into the overview of the assembly process, but it has provided very limited information about additional players involved and their specific roles. To disclose the non-structural ancillary factors involved in COX assembly, a profitable strategy has been to systematically analyze yeast mutants defective in COX assembly. This approach was followed with the goal of identifying the functions of the gene products responsible for the COX defective phenotype to subsequently reconstruct the different steps of the assembly pathway. Screens of nuclear respiratory deficient mutants have revealed the existence of a large number of nuclear genes coding for accessory factors that selectively affect expression of this respiratory complex in yeast [17, 18]. Their functions, required for all steps of the process and significantly conserved from yeast to humans, are summarized in Table 1 and have been previously reviewed [9, 19, 20].
Over the last 15 years, COX biogenesis has received significant attention because of its medical relevance. Defective COX biogenesis results in mitochondrial diseases frequently involving brain, skeletal muscle and heart tissues (reviewed in [21–23]). To date, all Mendelian disorders presenting COX deficiency have been assigned to mutations in ancillary factors. Specifically, mutations have been found in SURF1, required for the formation of early assembly intermediates [24, 25], SCO1 and SCO2, required for COX copper metallation [26–30], COX10 and COX15, essential for heme A biosynthesis [31–33], and LRPPRC, required for the expression of COX subunits [34].
Mutant fibroblast cell lines from patients suffering from some of these disorders have been used to refine the proposed sequential model for COX assembly by studying the accumulation of subassembly intermediates in the absence of specific COX assembly factors [35–37]. These studies, described below, have provided information concerning the assembly step either catalyzed or affected by the mutated factor. In contrast, the study of assembly intermediates in yeast has been restricted since they do not seem to accumulate in detectable amounts [37]. The accumulation of assembly intermediates in most yeast COX mutants is probably limited by the small amount of Cox1p that is newly synthesized in the absence of fully assembled COX [38]. However, subassemblies have been recently detected in yeast cox2 [39] and pet100 mutants [40] and seem to be similar to those observed in mammalian cells.
After the identification of the genes involved, the further exploitation of the yeast paradigm by isolating and characterizing genetic and metabolic suppressors of COX assembly defects, reviewed here, has become a rewarding effort to gain insight into the functions of these genes and the pathways in which they operate. The genetic suppressors can be classified in two general groups, extragenic and intragenic. Intragenic suppressors restore function to a protein at a site different from the primary mutation. They are very useful for example to understand the roles of several domains in a protein or the role of a particular residue and their tertiary structure neighbors within a protein. Extragenic suppressors, instead, reconstitute function in a system either by mutating a protein to work with another mutated protein or by bypassing the requirement of the first protein. Here, we will review exclusively some cases of extragenic suppression of COX assembly defects. Extragenic suppressors of strains carrying a null allele of a particular gene can arise naturally by a spontaneous mutation in a gene that compensates for the absence of the first gene. These natural suppressors are rare. In the cases of the respiratory deficient COX assembly mutants, only a few yeast strains spontaneously revert to a respiratory competent phenotype (Table 2). The hunt for genetic suppressors is designed to identify additional mutations that restore a particular aspect of COX assembly and/or its respiratory capacity to the initial mutant. The characterization of these suppressor mutations has lead to the identification of new genes with functions related to the initial mutated gene. These studies have also suggested interactions between proteins that participate in a common pathway. Another often-used means for uncovering interacting proteins in a common pathway is suppression by overexpression of wild-type alleles. Suppression caused by overexpression can occur by many different mechanisms but in general the necessity of the mutated protein is bypassed. Additionally, the characterization of these genetic suppressors in combination with the identification of pharmacological suppressors have the potential to provide useful information for devising therapeutic approaches to combat human disorders associated with COX deficiency.
Table 2.
Genetic and metabolic suppressors of COX deficiencies in cellular and animal models as well as in human patients. Intragenic suppressors are not listed.
| GENETIC SUPPRESSORS
|
|||
|---|---|---|---|
| MUTATED GENE | SPONTANEOUS EXTRAGENIC SUPPRESSORS | HIGH COPY SUPPRESSORS | METABOLIC/PHARMACOLOGIC SUPPRESSORS |
|
Yeast cultures
| |||
| COX5a | ROX1 [51] | ||
| ORD1 [52] | |||
| OXA1 | CYT1[144] | HAP4 [132] | |
| OMS1[145] | |||
| RMD9[146] | |||
| IMP1 | SOM1[147] | ||
| COX17 | SCO1 [71] | Copper [61] | |
| SCO2 + Copper [71] | |||
| SCO1 | SCO2 [71] | ||
| COX23 | COX17 + Copper [68] | ||
| SHY1 | MSS51 [112] | MSS51 [112] | Copper [128] |
| HAP4 [128] | |||
| COX5a + COX6 [128] | |||
| COA1 | MSS51 [123] | ||
| COX10 [123] | |||
| MDJ1 [123] | |||
|
| |||
|
Human cell cultures
| |||
| SURF1 | NF-YA [128] | ||
| SCO2 | Copper [91, 148] | ||
| COX10 | PPAR agonists [104] | ||
| COX1 | PGC1-α[143] | PGC1-α[143] | |
|
| |||
|
Human patients
| |||
| SCO2 | Copper [93] | ||
The aim of this review is to summarize some of the understanding concerning COX assembly that has been obtained by studying different classes of suppressors. The information obtained by the study of suppressors of mitochondrial gene expression [41] and of defects in the membrane insertion and processing of mtDNA-encoded COX subunits ([42] and listed on Table 2) has been reviewed elsewhere. We have focused on describing some of the knowledge obtained by studying suppressors of structural, heme A biosynthesis, copper metabolism and insertion defects and alterations in the formation of assembly intermediates.
2. Suppression of cytochrome c oxidase biogenesis defects in yeast and human
2.1. Suppressors of structural defects
Some nuclear encoded subunits have evolved isoforms that allow the enzyme to regulate its activity depending on environmental factors. For example, subunit Cox5p in the yeast S. cerevisiae exists in two isoforms, Cox5ap and Cox5bp, which are expressed according to oxygen availability. In humans, tissue specific isoforms have been reported for four nuclear encoded subunits, including COX4, the homologue of yeast Cox5p [43, 44]. Studies on suppressors of yeast cox5a mutants have given insight into oxygen-regulation of COX subunits and their switch to modulate COX enzymatic activity.
Yeast Cox5ap and Cox5bp share 66% sequence homology [45]. The presence of either one or the other isoform confers different kinetic properties to the holoenzyme. Moreover, the transmembrane α-helix of Cox5p interacts with Cox1p and it is able to alter, depending on the isoform, the protein environment around the binuclear center. The Cox5bp-containing enzyme has a higher maximal turnover, since electron transfer from heme a to heme a3 occurs three to four times faster than in the Cox5ap-containing isoenzyme [46]. The expression of COX5a and COX5b genes is regulated by oxygen availability [47]. In normoxia, the expression of COX5a is induced by the heme-dependent transcriptional activator Hap2p/3p/4p/5p complex, while COX5b expression is repressed, also in a heme-dependent fashion [48]. When the environmental oxygen concentration decreases below 0.5 μM, a switch in Cox5p isoform expression takes place [49]. The change results from the consequence of low heme biosynthesis, a process that requires oxygen [50]. Due to the lack of induction of COX5a and the lack of repression of COX5b, Cox5bp is expressed and used to assemble a more efficient COX holoenzyme. By this means, ATP production is maximized under hypoxia while minimizing ROS generation. In this way, by regulating COX isoforms composition, yeast cells can rapidly respond to changes in environmental oxygen concentration.
The oxygen regulation of these genes has been exhaustively studied in the yeast S. cerevisiae taking advantage of the ability of a respiratory deficient Δcox5a strain to spontaneously revert to a respiratory competent phenotype. Two different revertant complementation groups were isolated and analyzed [51, 52]. In both cases, the reversion was accounted for by recessive mutations in a single nuclear gene. These mutations do not bypass the subunit 5 requirement. The mechanism of reversion rather consists of an increase in COX5b expression in normoxia. The cloning of the suppressor genes allowed the identification of two new players in the oxygen regulatory pathway: Reo1p/Rox1p and Ordlp/Ixr1p, two transcriptional repressors of COX5b gene [51, 52]. Rox1p is a general hypoxic regulator that represses a large group of hypoxic genes, including COX5b, during aerobic growth. In normoxia, ROX1 is induced by the heme-binding transcriptional activator Hap1p [53]. In the absence of oxygen, ROX1 expression is down-regulated and the repression of COX5b is released [51]. Rox1p levels are tightly regulated in the cell and additional regulatory factors of ROX1 have been described [54]. Furthermore, the synergistic activity of Rox1p and Mot3p, another general repressor of hypoxic genes, has been recently reported, enlarging our understanding of the complex pathway of oxygen-dependent gene regulation [55]. In contrast to Rox1p, Ord1p is an oxygen dependent but Hap1p independent specific COX5b repressor [52]. Both Rox1p and Ord1p bind to the same 44bp Upstream Repression Sequence in the promoter of COX5b gene [52]. Currently, it remains unknown how Ord1p expression or function is regulated by oxygen and which kind of functional interactions (competition/cooperation) exist between Rox1p and Ord1p. The physiological significance for the existence of multiple COX5b repressors probably lies in the necessity to achieve a large range of gene expression variations in response to oxygen availability.
Noteworthy, COX isoform oxygen-dependent expression is highly conserved from lower to higher eukaryotes. In mammals, COX subunit 4, the homologue of yeast subunit 5, also exists in two isoforms, COX4-1 and COX4-2. As their yeast counterparts, they are regulated by oxygen and confer different kinetic properties to the holoenzyme [56]. The regulation of the mammalian isoform switch seems to be regulated differently than in yeast. Homologues of the main yeast regulatory elements have not been found in mammals. The regulation of COX4 isoform expression in response to oxygen availability is instead mediated by the hypoxia-inducible factor 1α (HIF-1 α) (reviewed in [57]). In aerobic conditions, HIF-1α is hydroxylated in an oxygen-dependent reaction and degraded by the proteosome [58]. During hypoxia, hydroxylation is inhibited; HIF-1α is not degraded and accumulates in the cell. Consequently, HIF-1α induces transcription of both, the COXIV-2 gene and the gene encoding for the mitochondrial protease LON, responsible for the degradation of COX4-1 [59]. Like in yeast, the COX4 isoform switch in mammals is a fundamental cellular adaptive response to maintain the efficiency of respiration under conditions of reduced oxygen availability. Additionally, while COXIV-1 is ubiquitously expressed in all tissues, COXIV-2 is expressed at particularly high levels in the lungs and trachea [56]. Huttemann and colleagues have proposed that the expression of COX4-2 isoform in these highly oxygenated tissues can have a physiological protective function by increasing electron transfer efficiency and consequently decreasing ROS formation [60].
2.2. Suppressors of copper insertion defects
The analyses of suppressors of copper insertion defects have allowed for the identification of factors involved in mitochondrial copper metabolism and the COX copper insertion pathway as well as for the development of therapeutic approaches.
COX copper metallation involves the function of several conserved proteins located in the mitochondrial intermembrane space (IMS) that were initially identified in yeast. The first protein involved in this function was the IMS chaperone Cox17p [61], a small soluble hydrophilic protein which binds copper(I) ions [62] through a CCxC metal binding motif. Cox17p also contains a twin Cx9C structural motif. Mutational analyses in yeast have shown that while the first cysteine in the Cx9C motif is part of the copper binding motif, the remaining three conserved cysteines are not important for Cox17p copper-binding function [63]. However, structural studies on human COX17 have recently shown that the two adjacent cysteines in the CCxC domain are actually the copper binding cysteines [64]. Cox17p has a dual location in the IMS and in the cytoplasm. The respiratory deficient phenotype of cox17 null mutant strains can be rescued by supplementation of high copper concentrations to the media or by significantly lower concentrations in combination with over-expression of CTR1 (the structural gene for the plasma membrane copper pump) [61]. This suppression was the basis to propose that Cox17p could be shuttling copper from the cytoplasm into the mitochondrial intermembrane space [61, 62]. This hypothesis has been challenged by the fact that the tethering of Cox17p to the inner mitochondrial membrane does not affect COX assembly [65]. Nevertheless, the exogenous copper suppression studies were very useful to place Cox17p in the COX copper delivery pathway. The copper source for COX metallation seems to be a matrix pool of copper bound by a low molecular weight non-proteinaceous ligand [65, 66]. The ligand from the copper complex has been found in the cytoplasm and it has been suggested that it may recruit, in place of a copper chaperone, the copper that is translocated and stored within the mitochondrial matrix [66].
At least two homologues of Cox17p are present in the IMS, the small soluble proteins Cox19p [67] and Cox23p [68], which also contain twin Cx9C metal binding motifs and are required for COX assembly. Cox23p does not physically interact with Cox17p in a stable complex but the COX defect in cox23 mutants is suppressed by exogenous copper in combination with COX17 overexpression. These data have suggested that both proteins function in a common pathway with Cox17p acting downstream of Cox23p [68]. Cox19p is a copper binding protein [69] and could be part of the same copper distribution pathway.
Cox17p transfers copper ions to two additional chaperones [70] that facilitate copper insertion into the COX CuA and CuB active sites, Sco1p [71] and Cox11p [72, 73], respectively. These proteins are anchored to the mitochondrial inner membrane (MIM) through a transmembrane α-helix and expose their copper binding sides in the IMS where copper transfer occurs [62, 74]. In vitro experiments have shown that soluble truncated forms of both Cox11p and Sco1p are able to bind copper donated from Cox17p [75, 76]. It is still not clear, however, how the transfer of copper occurs between Cox17p and these proteins because physical interactions among them have not been detected [70].
High copy suppressor screens were essential to place Sco1p into the COX copper delivery pathway. SCO1 was originally identified as essential for COX assembly in yeast [77] and subsequently as a multicopy suppressor of a cox17 null mutant [61]. Sco1p transfers copper from Cox17p to Cox2p and has been shown to directly interact with Cox2p [78]. Sco1p has a metal binding thioredoxin-like CX3C motif analogous to the copper binding motif of Cox2p and this motif is essential for its function as demonstrated by site-direct mutagenesis [79]. The mechanism of copper transfer from Cox17p and to Cox2p has been recently reviewed [80]. Although the experimental data could support a role for Sco1p in copper insertion, considering its structural similarity to the protein family of disulfide reductases, it has been proposed that it could rather be involved in the reduction of cysteines in the Cox2p copper binding site [81, 82]. This reduction is necessary for the co-factor incorporation [83, 84]. Sco1p has the ability to form homodimeric complexes [78] which could facilitate the performance of both functions by the collaborative action of each monomer.
Yeast SCO1 has a highly conserved homologue, SCO2 [85]. However, deletion of sco2 does not affect COX assembly [71]. SCO2 over-expression does not suppress the COX assembly defect of a sco1 null mutant strain but it is able to partially rescue a sco1 point mutant [71]. SCO2 over-expression also suppresses cox17 mutations, although less efficiently than SCO1, and only when the growth media is supplemented with copper [71]. These data were interpreted to indicate that yeast Sco1p and Sco2p have overlapping but not identical functions [71].
Remarkably, humans also have two homologues of yeast Sco1p, SCO1 and SCO2 [86], both of which are essential for COX assembly. Mutations in SCO1 [26] and SCO2 [27–29] result in severe mitochondrial disorders described below. Functional complementation studies have shown that expression of either human SCO1 or SCO2 does not complement a yeast strain carrying a null allele of sco1 [87]. Interestingly, the expression of a chimeric protein with the N terminus of yeast and the C terminus (that contains the CX3C copper binding domain) of human SCO1 (but not of SCO2) is able to complement the yeast mutant [87]. The functional differences among the two yeast and human isoforms is explained by the fact that the two genes probably originated from a duplication that occurred separately in the two organisms [27].
In contrast with their yeast homologues, human SCO1 and SCO2 have been shown to perform independent, cooperative functions in the process of COX assembly [88]. This is suggested by the fact that overexpression of each SCO protein in fibroblasts from patients with mutation in the other SCO protein results in a dominant negative phenotype [88]. In a proposed model, human COX17 delivers copper to SCO2 which in turn transfers it directly to the CuA site in COX subunit II in a reaction facilitated by SCO1 [88]. SCO2 could form a heterodimer with SCO1, which could be required to create the Cu(II)-Cu(I) dinuclear CuA site [89]. Recently, SCO1 and SCO2 were shown to have additional regulatory roles in the maintenance of cellular copper homeostasis cooperating to regulate copper efflux under conditions of excessive cellular copper [90].
A better understanding of the pathophysiology of human disorders associated with mutations in genes required for copper insertion in COX has suggested new avenues for treatment of these disorders. Specifically, mutations in SCO2 produce childhood onset cardioencephalomyopathy [27, 28, 30]. The addition of CuCl2 [91] or copper-histidine (Cu-His) [91] to cultured cells from patients with SCO2 mutations improved the biochemical defect. The mechanism of suppression remains unclear. As pointed out by Jaksch and colleagues [91], suppression could result from the very low residual levels of SCO2 in the patient cells, direct addition of copper to the CuA site, or by another copper-binding protein. Whatever the mechanism, copper supplementation has suggested a possible therapy for this mitochondrial disease. Cu-His has been used in the treatment of Menke’s disease (a neurological disorder produced by the loss of the copper-transporting ATPase ATP7A) with some success [92] and the same treatment may be effective for children with SCO2 mutations. Actually, subcutaneous injection of Cu-His, a physiological compound found in blood, in a patient affected by severe hypertrophic cardiomyopathy associated with SCO2 mutations did induce hypertrophic regression and ventricular function improvement with a significantly longer survival in comparison with all previously reported patients [93].
The CuB site located in Cox1p is formed by one copper ion coordinated by three histidine ligands and present in close proximity to the heme a moiety. The metallochaperone for the formation of the CuB site of Cox1p is the product of COX11 [72, 73]. Cox11p was initially described in yeast as necessary for COX assembly [94] but its role in CuB formation was first shown in the prokaryote Rhodobacter sphaeroides [72]. The COX assembly defect in cox11 yeast mutants is not suppressed by exogenous copper and genetic suppressors of the null mutants have not been obtained. However, Cox11p was shown to bind Cu(I) [95]. The soluble C-terminal domain of Cox11p forms a dimer that coordinates one Cu(I) per monomer. The two Cu(I) ions in the dimer exist in a binuclear cluster and appear to be ligated by three conserved cysteine residues [95]. The mechanism of copper transfer to CuB remains to be elucidated.
2.3. Suppression of heme A biogenesis and insertion defects
The study of suppressors of heme A biosynthesis defects in yeast and human cell lineshas provided information of biological and medical relevance.
Heme A is a unique heme compound present exclusively in COX. It differs from protoheme (heme B) because it has a farnesyl instead of a vinyl group at carbon C2 and a formyl instead of a methyl group at carbon C8 [96]. The first step of the heme A biosynthetic pathway is the conversion of heme B to heme O, a reaction that is catalyzed by the farnesyl-transferase Cox10p [97]. The subsequent oxidation of heme O to heme A occurs in two discrete monooxygenase steps. The first consists of a monooxygenase-catalyzed hydroxylation of the methyl group at carbon position 8 resulting in an alcohol that would be further oxidized to the aldehyde by a dehydrogenase. The first step is catalyzed by Cox15p. In S. cerevisiae, Cox15p acts in concert with ferredoxin (Yah1p) and the putative ferredoxin reductase Arh1p. The two latter enzymes are probably necessary for the supply of electrons to the oxygenase Cox15p [98–100]. Interestingly, in the yeast Schizosaccharomyces pombe COX15 and YAH1 are fused in a single gene [98]. At present, the identity of the putative gene product involved in the oxidation of the alcohol resulting from the Cox15p action to the corresponding aldehyde to yield heme A remains unknown.
The biosynthesis of heme A is regulated by downstream events in the COX assembly process. By measuring the amount of heme A and heme O in different yeast COX mutants it was observed that all the mutants analyzed showed a drastic reduction of steady-state levels of heme A, with the exception of shy1, cox20 and cox5a null mutants, in which heme A was still detectable at 10–25%. This amount is probably associated with residual assembled COX [101]. The overexpression of COX15 acted as a suppressor of the heme A accumulation defect in COX mutants including mutants in which Cox1p was not synthesized. This observation suggested that the absence of heme A in the mutants is not due to a rapid turnover of the cofactor in the absence of COX subunit 1, but rather to a feedback regulation of the heme A synthesis when the COX assembly process is blocked. The COX mutants analyzed, with the obvious exception of cox10, also showed an accumulation of heme O, indicating that this compound is stable. In addition, the cox15 null mutant presented a very low amount of heme O, a phenotype that was not rescued by the overexpression of COX10. This observation suggested that the first step of the heme A biosynthesis is also positively regulated in a Cox15p dependent manner [101]. This regulatory system could be different in mammalian cells. Analyses of mitochondrial heme content in COX15 deficient fibroblasts from a human patient suffering from hypertrophic cardiomyopathy showed levels of heme O significantly higher than in control fibroblasts [33].
Pharmacological suppressor studies of the respiratory defect of human fibroblasts from patients with defects in components of the mitochondrial respiratory chain, including mutants of COX10 [31], have allowed the identification of agents for possible use in therapeutic approaches to combat these mitochondrial diseases. Specifically, it was recently reported that treatment with bezafibrate significantly rescues the respiratory defects in the mutant cells. Bezafibrate is a widely used hypolipidemic drug that acts as a highly specific Peroxisome Proliferator-Activated Receptor (PPAR) agonist [102]. PPARs are ligand-activated nuclear receptors involved in the regulation of several energy metabolism genes, such as the ones encoding mitochondrial fatty acid β–oxidation enzymes (reviewed in [103]). Supplementation with 400μM bezafibrate for 72 hours significantly increases COX activity in COX10 mutant fibroblasts. This partial restoration of COX activity is accompanied by complete restoration of cell respiration to normal levels [104]. In the bezafibrate-treated COX10 deficient cells, the levels of both mRNA and protein of the COX mitochondrial encoded subunit COX2 and the COX nuclear encoded subunit COX4 are increased to normal levels. More importantly, the expression of the COX10 mutated gene, encoding for a partially functional protein, is also increased suggesting that the increase in COX activity is due to improved COX assembly. Bastin and colleagues dissected the bezafibrate-mediated suppression mechanism. It was previously reported that fibrates are able to induce PPARδ-mediated expression of PGC-1α (PPAR coactivator-1α) in mice muscle [105]. This is in agreement with the existence of a PPAR response element in the PGC-1α promoter [106]. PGC-1α up-regulates a large group of nuclear genes encoding respiratory chain components or proteins involved in mitochondria biogenesis, such as the mitochondrial transcription factor A (Tfam). PGC-1α does not bind to the DNA, but rather activates the nuclear respiratory factors 1 and 2 (NRF1 and NRF2) already bound to the promoter of the target genes, namely genes involved in oxidative phosphorylation (review in [107]). It is also known that PGC-1α induces the expression of NRF1 and NRF2 [108]. In the bezafibrate-treated COX10 deficient cells, PGC-1α expression increases about 2.5 fold compared to untreated cells while an additional slight increase in the mRNA levels of NRF1 and Tfam is also observed [104]. Bezafibrate treatment activates PPARs which in turn directly induces PCG-1α expression and thereby starts an activation cascade of nuclear and mitochondrial transcriptional activators leading to increased COX structural and assembly gene expression. It seems clear that bezafibrate suppression of COX10 defects does not bypass COX10 function, but it is mediated by the increased amounts of the partially functional COX10 concomitant with a general increase in the expression of COX structural subunits. These results support the possibility that therapeutic approaches aiming to increase mitochondrial biogenesis can rescue to some extent partial oxidative phosphorylation defects, including COX assembly deficiencies.
2.4. Suppression of defects affecting the formation of assembly intermediates
Suppression studies of defects affecting the formation of assembly intermediates have provided novel information concerning COX assembly regulation and interactions among several COX assembly factors.
COX assembly is characterized by a concerted accumulation of its constitutive subunits. Data mainly obtained from studies performed with yeast mutants indicate that most unassembled subunits, particularly the ones forming the catalytic core, are post-translationally degraded [110, 111]. Recently, another contribution to the stoichiometric accumulation of subunits during COX biogenesis in the yeast Saccharomyces cerevisiae has been reported. It consists of a regulatory mechanism by which the synthesis of subunit 1 is down-regulated in the absence of its assembly partners [112–115]. Decreased Cox1p synthesis relative to other mitochondrial translation products was previously reported as a secondary observation in several yeast COX assembly mutants [116, 117]. This phenotype could not be accounted for by a defect in the translation system because the mutations were not in proteins related to this mitochondrial activity [116, 117]. A major step in the characterization of this regulatory mechanism came from studies aiming to understand the function of the COX assembly factor Shy1p [118]. Shy1p function is of significant interest because the human homologue of SURF1 is responsible for cases of Leigh’s syndrome (LS) associated with COX deficiency, a severe neurological disorder of childhood [24, 25].
The study of respiratory deficient strains carrying a null allele of shy1 that spontaneously reverted to a respiratory-competent phenotype allowed the identification of MSS51 as a extragenic suppressor of shy1 mutations. Both mutant forms of mss51 and overexpression of the wild type gene act as suppressors of shy1 null mutants by increasing the levels of newly synthesized Cox1p [112]. Mss51p is required for COX1 mRNA translation [114, 119] and also plays a post-translational role [113, 114] as discussed below. Screens of a collection of strains carrying null mutations of COX biogenesis factors showed that the amount of newly synthesized Cox1p is not only reduced in shy1 mutants, but also in most COX assembly mutants. In all these mutants, the Cox1p synthesis defect is restored by the mss51 suppressors of shy1 or by mutations in cox14, which codes for another COX assembly factor [120]. Mss51p and Cox1p were shown to form a transient complex [113, 114] that is stabilized by Cox14p [38]. These interactions have been postulated to down-regulate Cox1p synthesis when COX assembly is impaired [38]. According to this model, the release of Mss51p from the ternary complex and its availability for additional Cox1p synthesis occur at a downstream step in the assembly pathway, and is likely catalyzed by Shy1p [113, 121]. The unique properties of this regulatory mechanism offer a means to catalyze multiple-subunit assembly [113, 114, 121]. In addition, reduced Cox1p synthesis in the absence of COX assembly and resulting degradation of the other core subunits will also limit the accumulation of partially matured core subunits that, as recently proposed for yeast subunit 1, could contribute to the production of unstable pro-oxidant intermediates [122].
Coa1p was recently described as a new player in the subunit 1 synthesis regulatory loop [121, 123]. Coa1p is a protein associated with the inner mitochondrial membrane where it is part of the high molecular weight complex containing Cox14p, Mss51p and newly synthesized Cox1p [121, 123]. The interaction Cox14p-Mss51p is unaffected in a Δcoa1 mutant. In the absence of Cox14p, however, the binding of Coa1p-Mss51p is disrupted suggesting that the interaction of Coa1p with the Cox1p-Cox14p-Mss51p complex is mediated through Cox14p (27). Shy1p is not part of the Cox1p-Cox14p-Mss51p-Coa1p complex (27), but it has been shown to co-precipitate with Coa1p [121, 123], suggesting that these two factors probably interact once Mss51p has disengaged from the complex (27). As for cox14 mutants, mutations in coa1 do not affect the synthesis but the accumulation of Cox1p in the holoenzyme suggesting that Coa1p also plays a role in the feedback regulation of Cox1p expression [121, 123]. The COX assembly defect in a coa1 null mutant strain can be suppressed by overexpression of Mss51p and Cox10p [97]. Both suppressors can act synergistically when the two proteins are co-expressed and thus link Coa1p to both translational regulation and maturation of Cox1p [123]. Pierrel and colleagues have speculated that Coa1p could stabilize the Cox1p-Cox14p-Mss51p complex until Shy1p interacts with Coa1p in a step involving heme A insertion into Cox1p and further progression in the assembly process [123]. While Cox1p maturation certainly occurs within these initial Cox1p complexes, the precise role of each factor remains to be elucidated.
At present, it is not clear if Cox1p translation in other organisms is also subject to regulation by downstream events. Mammalian homologues of Mss51p, Cox14p and Coa1p have not been identified to date.
The precise function of SURF1/Shy1p in COX assembly is currently unknown. However, data gathered by studying spontaneous suppressor mutations and high copy suppressors of yeast shy1 mutants have indicated that SURF1/Shy1p plays a role in the formation of an early COX assembly intermediate containing subunit 1 as mentioned above. Analysis of COX by Blue Native gel electrophoresis has indicated that assembly of COX in SURF1 deficient fibroblasts is blocked at an early step, most likely before the incorporation of subunit II into the nascent intermediates composed of subunit 1 alone or subunit 1 plus subunit 4 and 5a [124–126]. In the COX assembly model originally proposed by Nitjmans and colleagues [16], the first subassembly (S1) is formed exclusively by subunit 1, which acts as a seed for sequential incorporation of COX subunits. In this model, the addition of subunits COX4 and COX5a (yeast Cox5ap and Cox6p, respectively) to S1 results in the progression to the second assembly intermediate (S2). Insertion of heme A into COX1 probably occurs before the addition of subunits 4 and 5a, as suggested by the accumulation of the COX1-COX4-COX5a intermediate in SCO1 and SCO2 mutant fibroblasts [36, 126] but not in COX10 and COX15 mutant cells [32, 33, 126]. These findings suggest that the presence of heme A in COX1 might stabilize its binding to COX4 and COX5a. After the formation of the COX1-COX4-COX5a subassembly, the COX assembly process continues with the formation of the third proposed intermediate (S3) by the addition of the remaining structural subunits with the exception of COX6a and COX7a/b (yeast subunits 10 and 7). These subunits are added later to complete the holoenzyme [16, 126]. This model has been refined by recent observations. New analysis of subassemblies in fibroblasts from patients with SCO2 and SURF1 suggested that mammalian COX4 and COX5a subunits form a dimer before their incorporation into S1 [36]. Noticeably, the equivalent yeast Cox5p-Cox6p dimer was also detected in a yeast COX mutant in which assembly is compromised in the latter stages of the process [40]. This result is consistent with the observation that in yeast the presence of subunit 6 is required for subunit 5 stability [127]. An additional intermediate seems to exist between S2 and S3 and consists of at least mammalian subunits COX1-COX2-COX4-COX5a [36]. COX2 may associate with the COX1-COX4-COX5a intermediate upon its copper metallation by the specific SCO1-SCO2 copper chaperones as suggested by the observation that SCO1 and SCO2 mutant fibroblasts from patients with COX deficiency accumulate the COX1-COX4-COX5a intermediate [36, 126].
Recent studies of novel high copy suppressors of shy1 mutants have provided insight into SURF1/Shy1p role within the COX assembly process. However, it remains unclear whether the primary Shy1p function is required for either Cox1p maturation, assembly or both. Mss51p mediated suppression of yeast shy1 mutants [112] has been shown to be enhanced by both co-overexpression of COX10 [123] and co-overexpression of the COX subunits Cox5p and Cox6p [128]. The presence of SURF1 orthologues in terminal oxidase operons of several prokaryotes [129] in which COX contains the evolutionary conserved core subunits (Cox1p, Cox2p, Cox3p) but not the other structural subunits such as Cox5p and Cox6p suggests a role for SURF1/Shy1p in forming the catalytic core of the enzyme. Studies in Rhodobacter sphaeroides have suggested that bacterial SURF1 is required for either insertion of heme A at the a3 center or stabilization of the a3-CuB binuclear center in COX1 [130]. The respiratory defect of yeast shy1 mutants is not rescued by supplementation of the growth media with hemin [128]. Analyses of human COX assembly intermediates have shown that the SURF1 deficient fibroblasts accumulate the S2 assembly intermediate. They are similar in this respect to SCO1 and SCO2 deficient fibroblast but differ from COX10 and COX15 deficient fibroblasts in which S2 is not formed and only S1 accumulates as mentioned above [36, 125, 126]. These observations apparently argue against a primary role of Surf1p in heme A insertion. Exogenous copper, however, partially suppresses the respiratory defect of a yeast shy1 null mutant strain by a mechanism that remains to be characterized [128]. Although Shy1p could play a direct or indirect role in Cox1p maturation, the possibility of a SURF1/Shy1p role in facilitating the addition of subunit 2 to the Cox1p-5p-6p complex can not be excluded. Further support for the role of Shy1p as an assembly factor was recently reported by showing that Shy1p promotes COX biogenesis through association with different protein modules, which are potential COX assembly intermediates containing Cox1p and Cox5p, among other proteins [121]. In addition, Shy1p and Cox14p were also found to interact with partially and fully assembled forms of COX associated with complex III of the mitochondrial respiratory chain [121], which suggests that these two COX assembly factors play chaperone roles beyond the early stages of COX assembly.
Finally, our group has recently reported that overexpression of HAP4 suppresses the respiratory deficient phenotype of yeast shy1 null and point mutant strains [128]. Hap4p is the catalytic subunit of the CCAAT binding site transcriptional activator Hap2p,3p,4p,5p (HAP) complex which globally activates transcription of nuclear genes involved in mitochondrial respiration during the transition from fermentation to respiration [131]. HAP4 over-expression in strains carrying null alleles of cox10, cox11, cox14, cox15, cox16, cox17, oxa1, mss51 and pet191 failed to induce any suppression of their respiratory growth defect. This observation indicated that the suppression by HAP4 was specific to the shy1 null mutant and suggested a specific mechanism of HAP4 suppression in this strain.
It was previously reported that over-expression of the HAP4 gene was able to suppress the respiratory defect of a strain carrying a deletion of the matrix domain of Oxa1p although it did not compensate for the total absence of Oxa1p [132], in agreement with our results. Oxa1p is a key component of the machinery for the insertion of membrane proteins in mitochondria [133]. The protein interacts with nascent mitochondrial polypeptides [133] and it has been proposed to mediate the co-translational insertion of mitochondrially encoded subunits through an interaction of its C-terminal tail located in the mitochondrial matrix with the mitochondrial ribosome [134, 135]. Additionally, Oxa1p seems essential for the translocation of the hydrophilic domain of Cox2p [136–138]. The mechanism of the oxa1 suppression by HAP4 remains to be elucidated. Hlavacek and co-workers reported an increase in the expression of Cox2p and proposed that this fact alone or in combination with an increase in the expression of other respiratory subunits by HAP4 over-expression could compensate for the defect in co-translational membrane insertion of these subunits due to the oxa1 mutation [132].
In the case of the shy1 null mutants, the specificity of HAP4 suggested that it could act by either increasing the expression of a protein with an overlapping function with Shy1p or the amount of specific COX subunits and/or assembly factors favoring a more efficient assembly of COX. In this case, the shy1 suppression by HAP4 was demonstrated to be mediated by a specific increase in the expression of subunits Cox5p and Cox6p, the partners of Cox1p in early COX subassemblies [128]. These results give support to the notion that Shy1p could act by promoting/stabilizing the formation of assembly intermediates. Some of the described suppressor mechanisms of shy1 mutants are summarized in Fig. 2.
Fig. 2.
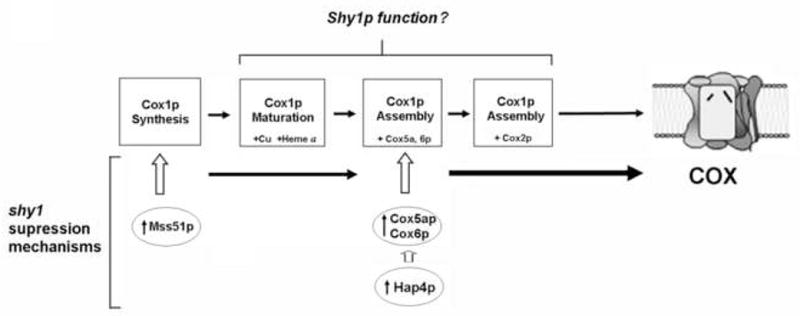
Mechanisms of suppression of the COX assembly defect of yeast shy1 mutants. After Cox1p synthesis, this subunit is matured by insertion of heme A and copper prosthetic groups, before or during the formation of an assembly intermediate containing Cox5ap and Cox6p. This ternary complex will subsequently assemble with metallated Cox2p. Shy1p could play a role in either Cox1p maturation or assembly. Mutant and additional copies of mss51 act as shy1 suppressors by increasing the amount of newly synthesized Cox1p, while overexpression of Hap4p suppresses largely by increasing the amount of Cox5ap-Cox6p. This figure has been reproduced with permission from [128].
The HAP complex is conserved from yeast to mammals where the human complex, termed NF-Y, contains three subunits, NF-YA, B and C. The proteins share homology to Hap2p, 3p and 5p, respectively. Some of the subunits are interchangeable across species. For example, human NF-YA, which contains both a DNA binding domain and a Q-rich activation domain, complements the respiratory defect of a hap2 yeast mutant [139]. Interestingly, overexpression of NF-YA, the catalytic subunit of the human NF-Y complex, is able to increase mitochondrial COX activity in SURF1 deficient fibroblasts through a mechanism that remains to be characterized [128].
In contrast to the role of the HAP complex in yeast, the NF-Y complex is not the main activator of transcription of nuclear genes involved in mitochondrial biogenesis and oxidative phosphorylation in mammals. The mammalian respiratory genes are known to be regulated by a variety of transcription factors, most notably NRF-1 and NRF-2 [140]. However, expression of at least two key proteins of the mammalian mitochondrial translational machinery, mitoribosomal protein S12 (Mrps12) and mitochondrial seryl-tRNA ligase, were recently shown to be regulated by NF-Y [141]. Since NF-Y regulates expression of genes in the fatty acid synthesis and oxidation pathways, its stimulatory effect on mitochondrial biogenesis could be indirect as reported for other transcriptional factors such as the PPARδ in skeletal muscle [142]. Other examples include the transcriptional co-activators PGC-1α and PGC-1βwhich overexpression in osteosarcoma cybrid cells carrying MTCOX1 mutations (amount of PGC-1α transcripts were 44-fold higher than in the controls) stimulated mitochondrial biogenesis and respiration [143]. As mentioned in the previous section for the treatments with PPAR agonists, these results suggest therapeutic approaches to combat COX and other oxidative phosphorylation partial defects. These approaches aim to increase mitochondrial biogenesis thus compensating for the partial respiratory defect. However, it is important to note that in some cases, pharmacological agents can fail to mimic the effects of specific transcriptional activators overexpression probably because their milder effect. For example, bezafibrate treatment, which increases PGC-1α expression by 2.5 fold as mentioned earlier, is unable to suppress the COX lesion of mutant human SURF1 deficient fibroblasts [104]. These cells carry a homozygous mutation in SURF1 that produces a truncated unstable protein [109]. Bezafibrate treatment to SURF1 deficient cells leads to increased SURF1 mRNA, but the translated SURF1 protein does not accumulate [104]. It is possible to speculate that bezafibrate failed to suppress the COX defect in these cells because the enhanced expression of COX structural subunits induced by the 2.5-fold increased amounts of PGC-1α did not reach the threshold of accumulated proteins required to effectively bypass the absence of SURF1.
Concluding remarks
The isolation and characterization of genetic and metabolic suppressors of COX assembly defects in yeast strains and cell lines derived from human patients has provided significant amount of information concerning the assembly of this enzyme. Suppressor studies have made possible the identification of many COX assembly ancillary factors, the characterization of their functions and the understanding of the pathways in which they operate. The information gathered from these studies is also proving to be useful for devising therapeutic approaches for the management of human disorders associated with COX deficiency.
Acknowledgments
We thank Dr. Francisca Diaz for critically reading the manuscript. Our research is supported by National Institutes of Health Research Grant GM071775A (to A.B.) and a Research Grant from the Muscular Dystrophy Association (to A.B.). F.F. is supported by Telethon-Italy, Fellowship n° GFP05008.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nicholls DG. Bioenergetics, An introduction to the chemiosmotic theory. Academic Press; New York: 1982. [Google Scholar]
- 2.Hill BC. Modeling the sequence of electron transfer reactions in the single turnover of reduced, mammalian cytochrome c oxidase with oxygen. J Biol Chem. 1994;269:2419–2425. [PubMed] [Google Scholar]
- 3.Brunori M, Giuffre A, Malatesta F, Sarti P. Investigating the mechanism of electron transfer to the binuclear center in Cu-heme oxidases. J Bioenerg Biomembr. 1998;30:41–45. doi: 10.1023/a:1020503410377. [DOI] [PubMed] [Google Scholar]
- 4.Brunori M, Giuffre A, Sarti P. Cytochrome c oxidase, ligands and electrons. J Inorg Biochem. 2005;99:324–336. doi: 10.1016/j.jinorgbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- 5.Babcock GT, Wikstrom M. Oxygen activation and the conservation of energy in cell respiration. Nature. 1992;356:301–309. doi: 10.1038/356301a0. [DOI] [PubMed] [Google Scholar]
- 6.Saraste M. Structure and evolution of cytochrome oxidase. Antonie Van Leeuwenhoek. 1994;65:285–287. doi: 10.1007/BF00872214. [DOI] [PubMed] [Google Scholar]
- 7.Belevich I, Verkhovsky MI, Wikstrom M. Proton-coupled electron transfer drives the proton pump of cytochrome c oxidase. Nature. 2006;440:829–832. doi: 10.1038/nature04619. [DOI] [PubMed] [Google Scholar]
- 8.Yoshikawa S, Muramoto K, Shinzawa-Itoh K, Aoyama H, Tsukihara T, Shimokata K, Katayama Y, Shimada H. Proton pumping mechanism of bovine heart cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1110–1116. doi: 10.1016/j.bbabio.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 9.Barrientos A, Barros MH, Valnot I, Rotig A, Rustin P, Tzagoloff A. Cytochrome oxidase in health and disease. Gene. 2002;286:53–63. doi: 10.1016/s0378-1119(01)00803-4. [DOI] [PubMed] [Google Scholar]
- 10.Fontanesi F, Soto IC, Horn D, Barrientos A. Assembly of mitochondrial cytochrome c oxidase, a complicated and highly regulated cellular process. Am J Physiol Cell Physiol. 2006;291:C1129–1147. doi: 10.1152/ajpcell.00233.2006. [DOI] [PubMed] [Google Scholar]
- 11.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 A. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 12.Ostermeier C, Harrenga A, Ermler U, Michel H. Structure at 2.7 A resolution of the Paracoccus denitrificans two-subunit cytochrome c oxidase complexed with an antibody FV fragment. Proc Natl Acad Sci U S A. 1997;94:10547–10553. doi: 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yoshikawa S, Shinzawa-Itoh K, Nakashima R, Yaono R, Yamashita E, Inoue N, Yao M, Fei MJ, Libeu CP, Mizushima T, Yamaguchi H, Tomizaki T, Tsukihara T. Redox-coupled crystal structural changes in bovine heart cytochrome c oxidase. Science. 1998;280:1723–1729. doi: 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- 14.Coyne HJ, 3rd, Ciofi-Baffoni S, Banci L, Bertini I, Zhang L, George GN, Winge DR. The characterization and role of zinc binding in yeast Cox4. J Biol Chem. 2007;282:8926–8934. doi: 10.1074/jbc.M610303200. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt B, McCracken J, Ferguson-Miller S. A discrete water exit pathway in the membrane protein cytochrome c oxidase. Proc Natl Acad Sci U S A. 2003;100:15539–15542. doi: 10.1073/pnas.2633243100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nijtmans LG, Taanman JW, Muijsers AO, Speijer D, Van den Bogert C. Assembly of cytochrome-c oxidase in cultured human cells. Eur J Biochem. 1998;254:389–394. doi: 10.1046/j.1432-1327.1998.2540389.x. [DOI] [PubMed] [Google Scholar]
- 17.McEwen JE, Ko C, Kloeckner-Gruissem B, Poyton RO. Nuclear functions required for cytochrome c oxidase biogenesis in Saccharomyces cerevisiae. Characterization of mutants in 34 complementation groups. J Biol Chem. 1986;261:11872–11879. [PubMed] [Google Scholar]
- 18.Tzagoloff A, Dieckmann CL. PET genes of Saccharomyces cerevisiae. Microbiol Rev. 1990;54:211–225. doi: 10.1128/mr.54.3.211-225.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Khalimonchuk O, Rodel G. Biogenesis of cytochrome c oxidase. Mitochondrion. 2005;5:363–388. doi: 10.1016/j.mito.2005.08.002. Epub 2005 Sep 2029. [DOI] [PubMed] [Google Scholar]
- 20.Herrmann JM, Funes S. Biogenesis of cytochrome oxidase-sophisticated assembly lines in the mitochondrial inner membrane. Gene. 2005;354:43–52. doi: 10.1016/j.gene.2005.03.017. [DOI] [PubMed] [Google Scholar]
- 21.Solans A, Zambrano A, Barrientos A. Cytochrome c oxidase deficiency: from yeast to human. Preclinica. 2004;2:336–348. [Google Scholar]
- 22.Pecina P, Houstkova H, Hansikova H, Zeman J, Houstek J. Genetic defects of cytochrome c oxidase assembly. Physiol Res. 2004;53:S213–223. [PubMed] [Google Scholar]
- 23.Shoubridge EA. Cytochrome c oxidase deficiency. Am J Med Genet. 2001;106:46–52. doi: 10.1002/ajmg.1378. [DOI] [PubMed] [Google Scholar]
- 24.Zhu Z, Yao J, Johns T, Fu K, De Bie I, Macmillan C, Cuthbert AP, Newbold RF, Wang J, Chevrette M, Brown GK, Brown RM, Shoubridge EA. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 25.Tiranti V, Hoertnagel K, Carrozzo R, Galimberti C, Munaro M, Granatiero M, Zelante L, Gasparini P, Marzella R, Rocchi M, Bayona-Bafaluy MP, Enriquez JA, Uziel G, Bertini E, Dionisi-Vici C, Franco B, Meitinger T, Zeviani M. Mutations of SURF-1 in Leigh disease associated with cytochrome c oxidase deficiency. Am J Hum Genet. 1998;63:1609–1621. doi: 10.1086/302150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valnot I, Osmond S, Gigarel N, Mehaye B, Amiel J, Cormier-Daire V, Munnich A, Bonnefont JP, Rustin P, Rotig A. Mutations of the SCO1 gene in mitochondrial cytochrome c oxidase deficiency with neonatal-onset hepatic failure and encephalopathy. Am J Hum Genet. 2000;67:1104–1109. doi: 10.1016/s0002-9297(07)62940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papadopoulou LC, Sue CM, Davidson MM, Tanji K, Nishino I, Sadlock JE, Krishna S, Walker W, Selby J, Glerum DM, Coster RV, Lyon G, Scalais E, Lebel R, Kaplan P, Shanske S, De Vivo DC, Bonilla E, Hirano M, DiMauro S, Schon EA. Fatal infantile cardioencephalomyopathy with COX deficiency and mutations in SCO2, a COX assembly gene. Nat Genet. 1999;23:333–337. doi: 10.1038/15513. [DOI] [PubMed] [Google Scholar]
- 28.Jaksch M, Ogilvie I, Yao J, Kortenhaus G, Bresser HG, Gerbitz KD, Shoubridge EA. Mutations in SCO2 are associated with a distinct form of hypertrophic cardiomyopathy and cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:795–801. doi: 10.1093/hmg/9.5.795. [DOI] [PubMed] [Google Scholar]
- 29.Sue CM, Karadimas C, Checcarelli N, Tanji K, Papadopoulou LC, Pallotti F, Guo FL, Shanske S, Hirano M, De Vivo DC, Van Coster R, Kaplan P, Bonilla E, DiMauro S. Differential features of patients with mutations in two COX assembly genes, SURF-1 and SCO2. Ann Neurol. 2000;47:589–595. [PubMed] [Google Scholar]
- 30.Salviati L, Sacconi S, Rasalan MM, Kronn DF, Braun A, Canoll P, Davidson M, Shanske S, Bonilla E, Hays AP, Schon EA, DiMauro S. Cytochrome c oxidase deficiency due to a novel SCO2 mutation mimics Werdnig-Hoffmann disease. Arch Neurol. 2002;59:862–865. doi: 10.1001/archneur.59.5.862. [DOI] [PubMed] [Google Scholar]
- 31.Valnot I, von Kleist-Retzow JC, Barrientos A, Gorbatyuk M, Taanman JW, Mehaye B, Rustin P, Tzagoloff A, Munnich A, Rotig A. A mutation in the human heme A:farnesyltransferase gene (COX10) causes cytochrome c oxidase deficiency. Hum Mol Genet. 2000;9:1245–1249. doi: 10.1093/hmg/9.8.1245. [DOI] [PubMed] [Google Scholar]
- 32.Antonicka H, Leary SC, Guercin GH, Agar JN, Horvath R, Kennaway NG, Harding CO, Jaksch M, Shoubridge EA. Mutations in COX10 result in a defect in mitochondrial heme A biosynthesis and account for multiple, early-onset clinical phenotypes associated with isolated COX deficiency. Hum Mol Genet. 2003;12:2693–2702. doi: 10.1093/hmg/ddg284. [DOI] [PubMed] [Google Scholar]
- 33.Antonicka H, Mattman A, Carlson CG, Glerum DM, Hoffbuhr KC, Leary SC, Kennaway NG, Shoubridge EA. Mutations in COX15 produce a defect in the mitochondrial heme biosynthetic pathway, causing early-onset fatal hypertrophic cardiomyopathy. Am J Hum Genet. 2003;72:101–114. doi: 10.1086/345489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mootha VK, Lepage P, Miller K, Bunkenborg J, Reich M, Hjerrild M, Delmonte T, Villeneuve A, Sladek R, Xu F, Mitchell GA, Morin C, Mann M, Hudson TJ, Robinson B, Rioux JD, Lander ES. Identification of a gene causing human cytochrome c oxidase deficiency by integrative genomics. Proc Natl Acad Sci U S A. 2003;100:605–610. doi: 10.1073/pnas.242716699. Epub 2003 Jan 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Williams SL, Valnot I, Rustin P, Taanman JW. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J Biol Chem. 2004;279:7462–7469. doi: 10.1074/jbc.M309232200. Epub 2003 Nov 7467. [DOI] [PubMed] [Google Scholar]
- 36.Stiburek L, Vesela K, Hansikova H, Pecina P, Tesarova M, Cerna L, Houstek J, Zeman J. Tissue-specific cytochrome c oxidase assembly defects due to mutations in SCO2 and SURF1. Biochem J. 2005;392:625–632. doi: 10.1042/BJ20050807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nijtmans LG, Artal Sanz M, Bucko M, Farhoud MH, Feenstra M, Hakkaart GA, Zeviani M, Grivell LA. Shy1p occurs in a high molecular weight complex and is required for efficient assembly of cytochrome c oxidase in yeast. FEBS Lett. 2001;498:46–51. doi: 10.1016/s0014-5793(01)02447-4. [DOI] [PubMed] [Google Scholar]
- 38.Barrientos A, Zambrano A, Tzagoloff A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004;23:3472–3482. doi: 10.1038/sj.emboj.7600358. Epub 2004 Aug 3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Horan S, Bourges I, Taanman JW, Meunier B. Analysis of COX2 mutants reveals cytochrome oxidase subassemblies in yeast. Biochem J. 2005;390:703–708. doi: 10.1042/BJ20050598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Church C, Goehring B, Forsha D, Wazny P, Poyton RO. A role for Pet100p in the assembly of yeast cytochrome c oxidase: interaction with a subassembly that accumulates in a pet100 mutant. J Biol Chem. 2005;280:1854–1863. doi: 10.1074/jbc.M410726200. Epub 2004 Oct 1826. [DOI] [PubMed] [Google Scholar]
- 41.Towpik J. Regulation of mitochondrial translation in yeast. Cell Mol Biol Lett. 2005;10:571–594. [PubMed] [Google Scholar]
- 42.Stuart R. Insertion of proteins into the inner membrane of mitochondria: the role of the Oxa1 complex. Biochim Biophys Acta. 2002;1592:79–87. doi: 10.1016/s0167-4889(02)00266-5. [DOI] [PubMed] [Google Scholar]
- 43.Kadenbach B, Stroh A, Becker A, Eckerskorn C, Lottspeich F. Tissue- and species-specific expression of cytochrome c oxidase isozymes in vertebrates. Biochim Biophys Acta. 1990;1015:368–372. doi: 10.1016/0005-2728(90)90042-3. [DOI] [PubMed] [Google Scholar]
- 44.Linder D, Freund R, Kadenbach B. Species-specific expression of cytochrome c oxidase isozymes. Comp Biochem Physiol B Biochem Mol Biol. 1995;112:461–469. doi: 10.1016/0305-0491(95)00093-3. [DOI] [PubMed] [Google Scholar]
- 45.Burke PV, Poyton RO. Structure/function of oxygen-regulated isoforms in cytochrome c oxidase. J Exp Biol. 1998;201:1163–1175. doi: 10.1242/jeb.201.8.1163. [DOI] [PubMed] [Google Scholar]
- 46.Allen LA, Zhao XJ, Caughey W, Poyton RO. Isoforms of yeast cytochrome c oxidase subunit V affect the binuclear reaction center and alter the kinetics of interaction with the isoforms of yeast cytochrome c. J Biol Chem. 1995;270:110–118. doi: 10.1074/jbc.270.1.110. [DOI] [PubMed] [Google Scholar]
- 47.Hodge MR, Kim G, Singh K, Cumsky MG. Inverse regulation of the yeast COX5 genes by oxygen and heme. Mol Cell Biol. 1989;9:1958–1964. doi: 10.1128/mcb.9.5.1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trueblood CE, Wright RM, Poyton RO. Differential regulation of the two genes encoding Saccharomyces cerevisiae cytochrome c oxidase subunit V by heme and the HAP2 and REO1 genes. Mol Cell Biol. 1988;8:4537–4540. doi: 10.1128/mcb.8.10.4537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burke PV, Raitt DC, Allen LA, Kellogg EA, Poyton RO. Effects of oxygen concentration on the expression of cytochrome c and cytochrome c oxidase genes in yeast. J Biol Chem. 1997;272:14705–14712. doi: 10.1074/jbc.272.23.14705. [DOI] [PubMed] [Google Scholar]
- 50.Saltzgaber-Muller J, Schatz G. Heme is necessary for the accumulation and assembly of cytochrome c oxidase subunits in Saccharomyces cerevisiae. J Biol Chem. 1978;253:305–310. [PubMed] [Google Scholar]
- 51.Trueblood CE, Poyton RO. Identification of REO1, a gene involved in negative regulation of COX5b and ANB1 in aerobically grown Saccharomyces cerevisiae. Genetics. 1988;120:671–680. doi: 10.1093/genetics/120.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lambert JR, Bilanchone VW, Cumsky MG. The ORD1 gene encodes a transcription factor involved in oxygen regulation and is identical to IXR1, a gene that confers cisplatin sensitivity to Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1994;91:7345–7349. doi: 10.1073/pnas.91.15.7345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Keng T. HAP1 and ROX1 form a regulatory pathway in the repression of HEM13 transcription in Saccharomyces cerevisiae. Mol Cell Biol. 1992;12:2616–2623. doi: 10.1128/mcb.12.6.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Deckert J, Perini R, Balasubramanian B, Zitomer RS. Multiple elements and auto-repression regulate Rox1, a repressor of hypoxic genes in Saccharomyces cerevisiae. Genetics. 1995;139:1149–1158. doi: 10.1093/genetics/139.3.1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sertil O, Kapoor R, Cohen BD, Abramova N, Lowry CV. Synergistic repression of anaerobic genes by Mot3 and Rox1 in Saccharomyces cerevisiae. Nucleic Acids Res. 2003;31:5831–5837. doi: 10.1093/nar/gkg792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huttemann M, Kadenbach B, Grossman LI. Mammalian subunit IV isoforms of cytochrome c oxidase. Gene. 2001;267:111–123. doi: 10.1016/s0378-1119(01)00385-7. [DOI] [PubMed] [Google Scholar]
- 57.Semenza GL. Oxygen-dependent regulation of mitochondrial respiration by hypoxia-inducible factor 1. Biochem J. 2007;405:1–9. doi: 10.1042/BJ20070389. [DOI] [PubMed] [Google Scholar]
- 58.Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1alpha is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci U S A. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukuda R, Zhang H, Kim JW, Shimoda L, Dang CV, Semenza GL. HIF-1 regulates cytochrome oxidase subunits to optimize efficiency of respiration in hypoxic cells. Cell. 2007;129:111–122. doi: 10.1016/j.cell.2007.01.047. [DOI] [PubMed] [Google Scholar]
- 60.Huttemann M, Lee I, Liu J, Grossman LI. Transcription of mammalian cytochrome c oxidase subunit IV-2 is controlled by a novel conserved oxygen responsive element. FEBS J. 2007;274:5737–5748. doi: 10.1111/j.1742-4658.2007.06093.x. [DOI] [PubMed] [Google Scholar]
- 61.Glerum DM, Shtanko A, Tzagoloff A. Characterization of COX17, a yeast gene involved in copper metabolism and assembly of cytochrome oxidase. J Biol Chem. 1996;271:14504–14509. doi: 10.1074/jbc.271.24.14504. [DOI] [PubMed] [Google Scholar]
- 62.Beers J, Glerum DM, Tzagoloff A. Purification, characterization, and localization of yeast Cox17p, a mitochondrial copper shuttle. J Biol Chem. 1997;272:33191–33196. doi: 10.1074/jbc.272.52.33191. [DOI] [PubMed] [Google Scholar]
- 63.Heaton D, Nittis T, Srinivasan C, Winge DR. Mutational analysis of the mitochondrial copper metallochaperone Cox17. J Biol Chem. 2000;275:37582–37587. doi: 10.1074/jbc.M006639200. [DOI] [PubMed] [Google Scholar]
- 64.Banci L, Bertini I, Ciofi-Baffoni S, Janicka A, Martinelli M, Kozlowski H, Palumaa P. A structural-dynamical characterization of human Cox17. J Biol Chem. 2007 doi: 10.1074/jbc.M708016200. E-published ahead of print Dec 19. [DOI] [PubMed] [Google Scholar]
- 65.Cobine PA, Ojeda LD, Rigby KM, Winge DR. Yeast contain a non-proteinaceous pool of copper in the mitochondrial matrix. J Biol Chem. 2004;279:14447–14455. doi: 10.1074/jbc.M312693200. [DOI] [PubMed] [Google Scholar]
- 66.Cobine PA, Pierrel F, Bestwick ML, Winge DR. Mitochondrial matrix copper complex used in metallation of cytochrome oxidase and superoxide dismutase. J Biol Chem. 2006;281:36552–36559. doi: 10.1074/jbc.M606839200. [DOI] [PubMed] [Google Scholar]
- 67.Nobrega MP, Bandeira SC, Beers J, Tzagoloff A. Characterization of COX19, a widely distributed gene required for expression of mitochondrial cytochrome oxidase. J Biol Chem. 2002;277:40206–40211. doi: 10.1074/jbc.M207348200. [DOI] [PubMed] [Google Scholar]
- 68.Barros MH, Johnson A, Tzagoloff A. COX23, a homologue of COX17, is required for cytochrome oxidase assembly. J Biol Chem. 2004;279:31943–31947. doi: 10.1074/jbc.M405014200. [DOI] [PubMed] [Google Scholar]
- 69.Rigby K, Zhang L, Cobine PA, George GN, Winge DR. Characterization of the cytochrome c oxidase assembly factor Cox19 of Saccharomyces cerevisiae. J Biol Chem. 2007;282:10233–10242. doi: 10.1074/jbc.M610082200. [DOI] [PubMed] [Google Scholar]
- 70.Horng YC, Cobine PA, Maxfield AB, Carr HS, Winge DR. Specific copper transfer from the Cox17 metallochaperone to both Sco1 and Cox11 in the assembly of yeast cytochrome C oxidase. J Biol Chem. 2004;279:35334–35340. doi: 10.1074/jbc.M404747200. [DOI] [PubMed] [Google Scholar]
- 71.Glerum DM, Shtanko A, Tzagoloff A. SCO1 and SCO2 act as high copy suppressors of a mitochondrial copper recruitment defect in Saccharomyces cerevisiae. J Biol Chem. 1996;271:20531–20535. doi: 10.1074/jbc.271.34.20531. [DOI] [PubMed] [Google Scholar]
- 72.Hiser L, Di Valentin M, Hamer AG, Hosler JP. Cox11p is required for stable formation of the Cu(B) and magnesium centers of cytochrome c oxidase. J Biol Chem. 2000;275:619–623. doi: 10.1074/jbc.275.1.619. [DOI] [PubMed] [Google Scholar]
- 73.Carr HS, George GN, Winge DR. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J Biol Chem. 2002;277:31237–31242. doi: 10.1074/jbc.M204854200. Epub 32002 Jun 31212. [DOI] [PubMed] [Google Scholar]
- 74.Carr HS, Maxfield AB, Horng YC, Winge DR. Functional analysis of the domains in Cox11. J Biol Chem. 2005;280:22664–22669. doi: 10.1074/jbc.M414077200. Epub 22005 Apr 22619. [DOI] [PubMed] [Google Scholar]
- 75.Nittis T, George GN, Winge DR. Yeast Sco1, a protein essential for cytochrome c oxidase function is a Cu(I)-binding protein. J Biol Chem. 2001;276:42520–42526. doi: 10.1074/jbc.M107077200. Epub 42001 Sep 42526. [DOI] [PubMed] [Google Scholar]
- 76.Carr HS, Maxfield AB, Horng YC, Winge DR. Functional analysis of the domains in Cox11. J Biol Chem. 2005;280:22664–22669. doi: 10.1074/jbc.M414077200. [DOI] [PubMed] [Google Scholar]
- 77.Krummeck G, Rodel G. Yeast SCO1 protein is required for a post-translational step in the accumulation of mitochondrial cytochrome c oxidase subunits I and II. Curr Genet. 1990;18:13–15. doi: 10.1007/BF00321109. [DOI] [PubMed] [Google Scholar]
- 78.Lode A, Kuschel M, Paret C, Rodel G. Mitochondrial copper metabolism in yeast: interaction between Sco1p and Cox2p. FEBS Lett. 2000;485:19–24. doi: 10.1016/s0014-5793(00)02176-1. [DOI] [PubMed] [Google Scholar]
- 79.Rentzsch A, Krummeck-Weiss G, Hofer A, Bartuschka A, Ostermann K, Rodel G. Mitochondrial copper metabolism in yeast: mutational analysis of Sco1p involved in the biogenesis of cytochrome c oxidase. Curr Genet. 1999;35:103–108. doi: 10.1007/s002940050438. [DOI] [PubMed] [Google Scholar]
- 80.Bertini I, Cavallaro G. Metals in the “omics” world: copper homeostasis and cytochrome c oxidase assembly in a new light. J Biol Inorg Chem. 2008;13:3–14. doi: 10.1007/s00775-007-0316-9. [DOI] [PubMed] [Google Scholar]
- 81.Chinenov YV. Cytochrome c oxidase assembly factors with a thioredoxin fold are conserved among prokaryotes and eukaryotes. J Mol Med. 2000;78:239–242. doi: 10.1007/s001090000110. [DOI] [PubMed] [Google Scholar]
- 82.Banci L, Bertini I, Calderone V, Ciofi-Baffoni S, Mangani S, Martinelli M, Palumaa P, Wang S. A hint for the function of human Sco1 from different structures. Proc Natl Acad Sci U S A. 2006;103:8595–8600. doi: 10.1073/pnas.0601375103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Abajian C, Rosenzweig AC. Crystal structure of yeast Sco1. J Biol Inorg Chem. 2006;11:459–446. doi: 10.1007/s00775-006-0096-7. [DOI] [PubMed] [Google Scholar]
- 84.Ye Q, Imriskova-Sosova I, Hill BC, Jia Z. Identification of a disulfide switch in BsSco, a member of the Sco family of cytochrome c oxidase assembly proteins. Biochemistry. 2005;44:2934–2942. doi: 10.1021/bi0480537. [DOI] [PubMed] [Google Scholar]
- 85.Smits PH, De Haan M, Maat C, Grivell LA. The complete sequence of a 33 kb fragment on the right arm of chromosome II from Saccharomyces cerevisiae reveals 16 open reading frames, including ten new open reading frames, five previously identified genes and a homologue of the SCO1 gene. Yeast. 1994;10:S75–80. doi: 10.1002/yea.320100010. [DOI] [PubMed] [Google Scholar]
- 86.Petruzzella V, Tiranti V, Fernandez P, Ianna P, Carrozzo R, Zeviani M. Identification and characterization of human cDNAs specific to BCS1, PET112, SCO1, COX15, and COX11, five genes involved in the formation and function of the mitochondrial respiratory chain. Genomics. 1998;54:494–504. doi: 10.1006/geno.1998.5580. [DOI] [PubMed] [Google Scholar]
- 87.Paret C, Ostermann K, Krause-Buchholz U, Rentzsch A, Rodel G. Human members of the SCO1 gene family: complementation analysis in yeast and intracellular localization. FEBS Lett. 1999;447:65–70. doi: 10.1016/s0014-5793(99)00266-5. [DOI] [PubMed] [Google Scholar]
- 88.Leary SC, Kaufman BA, Pellecchia G, Guercin GH, Mattman A, Jaksch M, Shoubridge EA. Human SCO1 and SCO2 have independent, cooperative functions in copper delivery to cytochrome c oxidase. Hum Mol Genet. 2004;13:1839–1848. doi: 10.1093/hmg/ddh197. [DOI] [PubMed] [Google Scholar]
- 89.Horng YC, Leary SC, Cobine PA, Young FB, George GN, Shoubridge EA, Winge DR. Human Sco1 and Sco2 function as copper-binding proteins. J Biol Chem. 2005;280:34113–34122. doi: 10.1074/jbc.M506801200. [DOI] [PubMed] [Google Scholar]
- 90.Leary SC, Cobine PA, Kaufman BA, Guercin GH, Mattman A, Palaty J, Lockitch G, Winge DR, Rustin P, Horvath R, Shoubridge EA. The human cytochrome c oxidase assembly factors SCO1 and SCO2 have regulatory roles in the maintenance of cellular copper homeostasis. Cell Metab. 2007;5:9–20. doi: 10.1016/j.cmet.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 91.Jaksch M, Paret C, Stucka R, Horn N, Muller-Hocker J, Horvath R, Trepesch N, Stecker G, Freisinger P, Thirion C, Muller J, Lunkwitz R, Rodel G, Shoubridge EA, Lochmuller H. Cytochrome c oxidase deficiency due to mutations in SCO2, encoding a mitochondrial copper-binding protein, is rescued by copper in human myoblasts. Hum Mol Genet. 2001;10:3025–3035. doi: 10.1093/hmg/10.26.3025. [DOI] [PubMed] [Google Scholar]
- 92.Christodoulou J, Danks DM, Sarkar B, Baerlocher KE, Casey R, Horn N, Tumer Z, Clarke JT. Early treatment of Menkes disease with parenteral copper-histidine: long-term follow-up of four treated patients. Am J Med Genet. 1998;76:154–164. [PubMed] [Google Scholar]
- 93.Freisinger P, Horvath R, Macmillan C, Peters J, Jaksch M. Reversion of hypertrophic cardiomyopathy in a patient with deficiency of the mitochondrial copper binding protein Sco2: is there a potential effect of copper? J. Inherit Metab Dis. 2004;27:67–79. doi: 10.1023/B:BOLI.0000016614.47380.2f. [DOI] [PubMed] [Google Scholar]
- 94.Tzagoloff A, Capitanio N, Nobrega MP, Gatti D. Cytochrome oxidase assembly in yeast requires the product of COX11, a homolog of the P denitrificans protein encoded by ORF3. EMBO J. 1990;9:2759–2764. doi: 10.1002/j.1460-2075.1990.tb07463.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Carr HS, George GN, Winge DR. Yeast Cox11, a protein essential for cytochrome c oxidase assembly, is a Cu(I)-binding protein. J Biol Chem. 2002;277:31237–31242. doi: 10.1074/jbc.M204854200. [DOI] [PubMed] [Google Scholar]
- 96.Caughey WS, Smythe GA, O’Keeffe DH, Maskasky JE, Smith MI. Heme A of cytochrome c oxicase. Structure and properties: comparisons with hemes B, C, and S and derivatives. J Biol Chem. 1975;250:7602–7622. [PubMed] [Google Scholar]
- 97.Tzagoloff A, Nobrega M, Gorman N, Sinclair P. On the functions of the yeast COX10 and COX11 gene products. Biochem Mol Biol Int. 1993;31:593–598. [PubMed] [Google Scholar]
- 98.Barros MH, Carlson CG, Glerum DM, Tzagoloff A. Involvement of mitochondrial ferredoxin and Cox15p in hydroxylation of heme O. FEBS Lett. 2001;492:133–138. doi: 10.1016/s0014-5793(01)02249-9. [DOI] [PubMed] [Google Scholar]
- 99.Barros MH, Nobrega FG, Tzagoloff A. Mitochondrial ferredoxin is required for heme A synthesis in Saccharomyces cerevisiae. J Biol Chem. 2002;277:9997–10002. doi: 10.1074/jbc.M112025200. Epub 12002 Jan 10011. [DOI] [PubMed] [Google Scholar]
- 100.Brown KR, Allan BM, Do P, Hegg EL. Identification of novel hemes generated by heme A synthase: evidence for two successive monooxygenase reactions. Biochemistry. 2002;41:10906–10913. doi: 10.1021/bi0203536. [DOI] [PubMed] [Google Scholar]
- 101.Barros MH, Tzagoloff A. Regulation of the heme A biosynthetic pathway in Saccharomyces cerevisiae. FEBS Lett. 2002;516:119–123. doi: 10.1016/s0014-5793(02)02514-0. [DOI] [PubMed] [Google Scholar]
- 102.Tenenbaum A, Motro M, Fisman EZ. Dual and pan-peroxisome proliferator-activated receptors (PPAR) co-agonism: the bezafibrate lessons. Cardiovasc Diabetol. 2005;4:14. doi: 10.1186/1475-2840-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee CH, Olson P, Evans RM. Minireview: lipid metabolism, metabolic diseases, and peroxisome proliferator-activated receptors. Endocrinology. 2003;144:2201–2207. doi: 10.1210/en.2003-0288. [DOI] [PubMed] [Google Scholar]
- 104.Bastin J, Aubey F, Rotig A, Munnich A, Djouadi F. Activation of peroxisome proliferator activated receptor pathway stimulates the mitochondrial respiratory chain and can correct deficiencies in patients’ cells lacking its components. J Clin Endocrinol Metab. 2008 doi: 10.1210/jc.2007-1701. E-pub Jan 22. [DOI] [PubMed] [Google Scholar]
- 105.Hondares E, Pineda-Torra I, Iglesias R, Staels B, Villarroya F, Giralt M. PPARdelta, but not PPARalpha, activates PGC-1alpha gene transcription in muscle. Biochem Biophys Res Commun. 2007;354:1021–1027. doi: 10.1016/j.bbrc.2007.01.092. [DOI] [PubMed] [Google Scholar]
- 106.Schuler M, Ali F, Chambon C, Duteil D, Bornert JM, Tardivel A, Desvergne B, Wahli W, Chambon P, Metzger D. PGC1alpha expression is controlled in skeletal muscles by PPARbeta, whose ablation results in fiber-type switching, obesity, and type 2 diabetes. Cell Metab. 2006;4:407–414. doi: 10.1016/j.cmet.2006.10.003. [DOI] [PubMed] [Google Scholar]
- 107.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 108.Xu F, Morin C, Mitchell G, Ackerley C, Robinson BH. The role of the LRPPRC (leucine-rich pentatricopeptide repeat cassette) gene in cytochrome oxidase assembly: mutation causes lowered levels of COX (cytochrome c oxidase) I and COX III mRNA. Biochem J. 2004;382:331–336. doi: 10.1042/BJ20040469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Von Kleist-Retzow JC, Yao J, Taanman JW, Chantrel K, Chretien D, Cormier-Daire V, Rotig A, Munnich A, Rustin P, Shoubridge EA. Mutations in SURF1 are not specifically associated with Leigh syndrome. J Med Genet. 2001;38:109–113. doi: 10.1136/jmg.38.2.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nakai T, Mera Y, Yasuhara T, Ohashi A. Divalent metal ion-dependent mitochondrial degradation of unassembled subunits 2 and 3 of cytochrome c oxidase. J Biochem. 1994;116:752–758. doi: 10.1093/oxfordjournals.jbchem.a124592. [DOI] [PubMed] [Google Scholar]
- 111.Nijtmans LG, Spelbrink JN, Van Galen MJ, Zwaan M, Klement P, Van den Bogert C. Expression and fate of the nuclearly encoded subunits of cytochrome-c oxidase in cultured human cells depleted of mitochondrial gene products. Biochim Biophys Acta. 1995;1265:117–126. doi: 10.1016/0167-4889(94)00203-q. [DOI] [PubMed] [Google Scholar]
- 112.Barrientos A, Korr D, Tzagoloff A. Shy1p is necessary for full expression of mitochondrial COX1 in the yeast model of Leigh’s syndrome. EMBO J. 2002;21:43–52. doi: 10.1093/emboj/21.1.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Barrientos A, Zambrano A, Tzagoloff A. Mss51p and Cox14p jointly regulate mitochondrial Cox1p expression in Saccharomyces cerevisiae. EMBO J. 2004;23:3472–3482. doi: 10.1038/sj.emboj.7600358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Perez-Martinez X, Broadley SA, Fox TD. Mss51p promotes mitochondrial Cox1p synthesis and interacts with newly synthesized Cox1p. EMBO J. 2003;22:5951–5961. doi: 10.1093/emboj/cdg566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zambrano A, Fontanesi F, Solans A, de Oliveira RL, Fox TD, Tzagoloff A, Barrientos A. Aberrant translation of cytochrome c oxidase subunit 1 mRNA species in the absence of Mss51p in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2007;18:523–535. doi: 10.1091/mbc.E06-09-0803. Epub 2006 Nov 2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Poutre CG, Fox TD. PET111, a Saccharomyces cerevisiae nuclear gene required for translation of the mitochondrial mRNA encoding cytochrome c oxidase subunit II. Genetics. 1987;115:637–647. doi: 10.1093/genetics/115.4.637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Calder KM, McEwen JE. Deletion of the COX7 gene in Saccharomyces cerevisiae reveals a role for cytochrome c oxidase subunit VII in assembly of remaining subunits. Mol Microbiol. 1991;5:1769–1777. doi: 10.1111/j.1365-2958.1991.tb01926.x. [DOI] [PubMed] [Google Scholar]
- 118.Mashkevich G, Repetto B, Glerum DM, Jin C, Tzagoloff A. SHY1, the yeast homolog of the mammalian SURF-1 gene, encodes a mitochondrial protein required for respiration. J Biol Chem. 1997;272:14356–14364. doi: 10.1074/jbc.272.22.14356. [DOI] [PubMed] [Google Scholar]
- 119.Decoster E, Simon M, Hatat D, Faye G. The MSS51 gene product is required for the translation of the COX1 mRNA in yeast mitochondria. Mol Gen Genet. 1990;224:111–118. doi: 10.1007/BF00259457. [DOI] [PubMed] [Google Scholar]
- 120.Glerum DM, Koerner TJ, Tzagoloff A. Cloning and characterization of COX14, whose product is required for assembly of yeast cytochrome oxidase. J Biol Chem. 1995;270:15585–15590. doi: 10.1074/jbc.270.26.15585. [DOI] [PubMed] [Google Scholar]
- 121.Mick DU, Wagner K, van der Laan M, Frazier AE, Perschil I, Pawlas M, Meyer HE, Warscheid B, Rehling P. Shy1 couples Cox1 translational regulation to cytochrome c oxidase assembly. EMBO J. 2007;26:4347–4358. doi: 10.1038/sj.emboj.7601862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Khalimonchuk O, Bird A, Winge DR. Evidence for a pro-oxidant intermediate in the assembly of cytochrome oxidase. J Biol Chem. 2007;282:17442–17449. doi: 10.1074/jbc.M702379200. [DOI] [PubMed] [Google Scholar]
- 123.Pierrel F, Bestwick ML, Cobine PA, Khalimonchuk O, Cricco JA, Winge DR. Coa1 links the Mss51 post-translational function to Cox1 cofactor insertion in cytochrome c oxidase assembly. EMBO J. 2007;26:4335–4346. doi: 10.1038/sj.emboj.7601861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Coenen MJ, van den Heuvel LP, Nijtmans LG, Morava E, Marquardt I, Girschick HJ, Trijbels FJ, Grivell LA, Smeitink JA. SURFEIT-1 gene analysis and two-dimensional blue native gel electrophoresis in cytochrome c oxidase deficiency. Biochem Biophys Res Commun. 1999;265:339–344. doi: 10.1006/bbrc.1999.1662. [DOI] [PubMed] [Google Scholar]
- 125.Tiranti V, Galimberti C, Nijtmans L, Bovolenta S, Perini MP, Zeviani M. Characterization of SURF-1 expression and Surf-1p function in normal and disease conditions. Hum Mol Genet. 1999;8:2533–2540. doi: 10.1093/hmg/8.13.2533. [DOI] [PubMed] [Google Scholar]
- 126.Williams SL, Valnot I, Rustin P, Taanman JW. Cytochrome c oxidase subassemblies in fibroblast cultures from patients carrying mutations in COX10, SCO1, or SURF1. J Biol Chem. 2004;279:7462–7469. doi: 10.1074/jbc.M309232200. [DOI] [PubMed] [Google Scholar]
- 127.Glerum DM, Tzagoloff A. Submitochondrial distributions and stabilities of subunits 4, 5, and 6 of yeast cytochrome oxidase in assembly defective mutants. FEBS Lett. 1997;412:410–414. doi: 10.1016/s0014-5793(97)00799-0. [DOI] [PubMed] [Google Scholar]
- 128.Fontanesi F, Jin C, Tzagoloff A, Barrientos A. Transcriptional Activators HAP/NF-Y Rescue a Cytochrome c Oxidase Defect in Yeast and Human Cells. Hum Mol Genet Nov. 2007;27 doi: 10.1093/hmg/ddm349. [DOI] [PubMed] [Google Scholar]
- 129.Poyau A, Buchet K, Godinot C. Sequence conservation from human to prokaryotes of Surf1, a protein involved in cytochrome c oxidase assembly, deficient in Leigh syndrome. FEBS Lett. 1999;462:416–420. doi: 10.1016/s0014-5793(99)01571-9. [DOI] [PubMed] [Google Scholar]
- 130.Smith D, Gray J, Mitchell L, Antholine WE, Hosler JP. Assembly of cytochrome-c oxidase in the absence of assembly protein Surf1p leads to loss of the active site heme. J Biol Chem. 2005;280:17652–17656. doi: 10.1074/jbc.C500061200. [DOI] [PubMed] [Google Scholar]
- 131.Forsburg SL, Guarente L. Identification and characterization of HAP4: a third component of the CCAAT-bound HAP2/HAP3 heteromer. Genes Dev. 1989;3:1166–1178. doi: 10.1101/gad.3.8.1166. [DOI] [PubMed] [Google Scholar]
- 132.Hlavacek O, Bourens M, Salone V, Lachacinski N, Lemaire C, Dujardin G. The transcriptional activator HAP4 is a high copy suppressor of an oxa1 yeast mutation. Gene. 2005;354:53–57. doi: 10.1016/j.gene.2005.03.016. [DOI] [PubMed] [Google Scholar]
- 133.Hell K, Neupert W, Stuart RA. Oxa1p acts as a general membrane insertion machinery for proteins encoded by mitochondrial DNA. EMBO J. 2001;20:1281–1288. doi: 10.1093/emboj/20.6.1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Szyrach G, Ott M, Bonnefoy N, Neupert W, Herrmann JM. Ribosome binding to the Oxa1 complex facilitates co-translational protein insertion in mitochondria. EMBO J. 2003;22:6448–6457. doi: 10.1093/emboj/cdg623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Jia L, Dienhart M, Schramp M, McCauley M, Hell K, Stuart RA. Yeast Oxa1 interacts with mitochondrial ribosomes: the importance of the C-terminal region of Oxa1. EMBO J. 2003;22:6438–6447. doi: 10.1093/emboj/cdg624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.He S, Fox TD. Membrane translocation of mitochondrially coded Cox2p: distinct requirements for export of N and C termini and dependence on the conserved protein Oxa1p. Mol Biol Cell. 1997;8:1449–1460. doi: 10.1091/mbc.8.8.1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Hell K, Herrmann J, Pratje E, Neupert W, Stuart RA. Oxa1p mediates the export of the N- and C-termini of pCoxII from the mitochondrial matrix to the intermembrane space. FEBS Lett. 1997;418:367–370. doi: 10.1016/s0014-5793(97)01412-9. [DOI] [PubMed] [Google Scholar]
- 138.Herrmann JM, Bonnefoy N. Protein export across the inner membrane of mitochondria: the nature of translocated domains determines the dependence on the Oxa1 translocase. J Biol Chem. 2004;279:2507–2512. doi: 10.1074/jbc.M310468200. [DOI] [PubMed] [Google Scholar]
- 139.Becker DM, Fikes JD, Guarente L. A cDNA encoding a human CCAAT-binding protein cloned by functional complementation in yeast. Proc Natl Acad Sci U S A. 1991;88:1968–1972. doi: 10.1073/pnas.88.5.1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Scarpulla RC. Nuclear control of respiratory gene expression in mammalian cells. J Cell Biochem. 2006;97:673–683. doi: 10.1002/jcb.20743. [DOI] [PubMed] [Google Scholar]
- 141.Zanotto E, Shah ZH, Jacobs HT. The bidirectional promoter of two genes for the mitochondrial translational apparatus in mouse is regulated by an array of CCAAT boxes interacting with the transcription factor NF-Y. Nucleic Acids Res. 2007;35:664–677. doi: 10.1093/nar/gkl1037. Epub 2006 Dec 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM. Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity. Cell. 2003;113:159–170. doi: 10.1016/s0092-8674(03)00269-1. [DOI] [PubMed] [Google Scholar]
- 143.Srivastava S, Barrett JN, Moraes CT. PGC-1alpha/beta upregulation is associated with improved oxidative phosphorylation in cells harboring nonsense mtDNA mutations. Hum Mol Genet. 2007;16:993–1005. doi: 10.1093/hmg/ddm045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Hamel P, Lemaire C, Bonnefoy N, Brivet-Chevillotte P, Dujardin G. Mutations in the membrane anchor of yeast cytochrome c1 compensate for the absence of Oxa1p and generate carbonate-extractable forms of cytochrome c1. Genetics. 1998;150:601–611. doi: 10.1093/genetics/150.2.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Lemaire C, Guibet-Grandmougin F, Angles D, Dujardin G, Bonnefoy N. A yeast mitochondrial membrane methyltransferase-like protein can compensate for oxa1 mutations. J Biol Chem. 2004;279:47464–47472. doi: 10.1074/jbc.M404861200. [DOI] [PubMed] [Google Scholar]
- 146.Nouet C, Bourens M, Hlavacek O, Marsy S, Lemaire C, Dujardin G. Rmd9p controls the processing/stability of mitochondrial mRNAs and its overexpression compensates for a partial deficiency of oxa1p in Saccharomyces cerevisiae. Genetics. 2007;175:1105–1115. doi: 10.1534/genetics.106.063883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Esser K, Pratje E, Michaelis G. SOM 1, a small new gene required for mitochondrial inner membrane peptidase function in Saccharomyces cerevisiae. Mol Gen Genet. 1996;252:437–445. doi: 10.1007/BF02173009. [DOI] [PubMed] [Google Scholar]
- 148.Salviati L, Hernandez-Rosa E, Walker WF, Sacconi S, DiMauro S, Schon EA, Davidson MM. Copper supplementation restores cytochrome c oxidase activity in cultured cells from patients with SCO2 mutations. Biochem J. 2002;363:321–327. doi: 10.1042/0264-6021:3630321. [DOI] [PMC free article] [PubMed] [Google Scholar]