

SUMMARY
Twenty-two tandemly arranged protocadherin-γ (Pcdh-γ) genes encode transmembrane proteins with distinct cadherin-related extracellular domains and a common intracellular domain. Genetic studies have implicated Pcdh-γs in regulation of neuronal survival and synapse formation. Because mice lacking the Pcdh-γ cluster die perinatally, we generated conditional mutants to analyze roles of Pcdh-γs in the development and function of neural circuits. Retina-specific deletion of Pcdh-γs led to accentuation of naturally occurring death of interneurons and retinal ganglion cells (RGCs) during the first two postnatal weeks. Nonetheless, many neuronal subtypes formed lamina-specific arbors. Blocking apoptosis by deletion of the pro-apoptotic gene Bax showed that even neurons destined to die formed appropriate connections. Moreover, electrophysiological analysis indicated that processing of visual information was largely normal in the absence of Pcdh-γs. These results suggest that Pcdh-γs are dispensable for elaboration of specific connections in retina, but play a primary role in sculpting neuronal populations to appropriate sizes or proportions during the period of naturally-occurring cell death.
Keywords: apoptosis, interneuron, laminar specificity, receptive field
INTRODUCTION
The assembly of neurons into complex, stereotyped circuits has been hypothesized to require large sets of cell-surface molecules that mediate cell-cell interactions. A group of genes called clustered protocadherins (Pcdhs) has intriguing features that suggest their involvement in these processes. First, they have a remarkable genomic organization in which 58 homologous genes are arranged in three subclusters (Pcdh-α, –β and -γ) arrayed in tandem on a single chromosome (Kohmura et al., 1998; Obata et al., 1998; Wu and Maniatis, 1999; Wu and Maniatis, 2000). Second, α– and γ–protocadherins arise by combination of distinct extracellular domains with a common cytoplasmic domain, suggesting a mechanism in which distinct recognition events promote a common cellular response. Third, Pcdhs are members of the cadherin superfamily, other members of which mediate selective intercellular interactions, including synapse formation (Takeichi, 2007). Fourth, Pcdhs are expressed predominantly in the nervous system, with individual family members expressed in combinatorial patterns (Esumi et al., 2005; Frank et al., 2005; Kohmura et al., 1998; Zou et al., 2007). Fifth, Pcdh proteins are associated at least in part with synaptic membranes (Kohmura et al., 1998; Phillips et al., 2003; Wang et al., 2002). Finally, clustered protocadherin orthologues are present in vertebrates but not invertebrates (Hill et al., 2001; Hirayama and Yagi, 2006; Noonan et al., 2004). Together, these features suggest that Pcdhs might underlie complex patterns of selective neural connectivity in vertebrates.
The first genetic test of this hypothesis led to an unexpected result: targeted mouse mutants lacking all 22 Pcdh–γ genes exhibited massive apoptosis of spinal interneurons during late fetal life and died within hours of birth (Wang et al., 2002). Synapse number was also reduced in mutant spinal cords. This was not a trivial consequence of decreased neuronal number, as synaptic defects and perinatal lethality persisted when apoptosis was blocked (Weiner et al., 2005). Thus, neural connectivity may be defective in the absence of Pcdh-γs, and apoptosis may be secondary to circuit defects. However, the associated lethality and the complexity of spinal circuitry have made it difficult to test these possibilities. In addition, it remains unknown whether Pcdh–γs are required for neuronal survival and synaptogenesis in other regions of the nervous system.
To address these issues, we generated conditional alleles of the Pcdh–γ cluster, restricting inactivation to defined neuronal populations and bypassing neonatal lethality. Here, we focus on retina, which has several advantages, including a stereotyped structure, markers for many neuronal and synaptic subtypes, and a clear understanding of the tissue's function (Masland, 2001; Wassle, 2004). We used Cre recombinase to delete Pcdh–γ genes from retinal neurons and glia, and assessed the consequences for neuronal structure and function. Surprisingly, lamina-specific arbors and complex functional circuits formed in the absence of Pcdh–γs, suggesting that these genes play limited roles in synaptic specificity. In contrast, loss of Pcdh–γs accentuated naturally occurring death of multiple retinal cell types. These results suggest a primary role for Pcdh–γs in neuronal population matching during development.
METHODS
Animals/Generation of targeted mice
Pcdh-γfusg and Pcdh-γdel mutants were described previously (Wang et al., 2002). Mice in which regulatory elements from the Chx10 gene drive expression of a Cre-GFP fusion protein linked by an internal ribosome entry site (IRES) to placental alkaline phosphatase (Chx10-Cre; Rowan and Cepko, 2004) were provided by Constance Cepko (Harvard). Mice in which a short enhancer fragment from the Pax6 gene drive expression of a Cre-IRES-GFP cassette (Paxα-Cre; Marquardt et al., 2001) were provided by Peter Gruss (Gottingen, Germany). Mice in which regulatory elements from the β-actin gene drive expression of Cre (Actin-Cre; Lewandoski et al., 1997) were provided by Gail Martin (UCSF). Bax−/− mutants (Knudson et al., 1995) and Z/EG reporter mice (Novak et al., 2000) were obtained from Jackson Laboratories.
The Pcdh-γfcon3 targeting vector was modified from the Pcdh-γfusg vector shown in Figure 2B of Wang et al (2002) by inserting a loxP sequence into an NheI site upstream of the final coding exon. The Pcdh-γfdel allele was generated by re-targeting the ES cells used to generate Pcdh-γfusg with the vector that had been used to generate Pcdh-γdel. This vector inserted a loxP sequence directly upstream of variable exon A1. Homologous recombinants and germ line chimeras were generated by standard methods. Mice were maintained on a C57/B6J background.
Figure 2. Pcdh-γs localize to retinal plexiform layers during postnatal development.

(A-D) Immunolabeling of Pcdh-γfusg/fusg retina at P0 (A), P3 (B), P7 (C), and P14 (D), with GFP antibody. At P0 and P3, Pcdh-γ-GFP proteins are present in the IPL and retinal axon layer (arrow), and are distributed around cell somata in the neuroblast layer (NBL; other abbreviations as in Figure 1). At P3, Pcdh-γs are also present in presumptive horizontal cells (asterisk; inset in B). By P7, Pcdh-γ-GFPs are detected in the emerging OPL, as it develops between the ONL and INL. By P14, the adult pattern of Pcdh-γ-GFP localization (see Figure 1) is attained. Scale bar, 20 μm; 10 μm in inset.
Histology
Mice were euthanized with intraperitoneal injection of Nembutal, and eye cups were fixed in 4% paraformaldehyde. Tissue was cryoprotected in sucrose, frozen, and sectioned at 20μm in a cryostat. Slides were incubated successively with blocking solution, primary antibodies (12h-16h at 4′C) and AlexaFluor-confugated secondary antibodies (Invitrogen; 3h at room temperature). Primary antibodies were: anti-GFP (Aves and Chemicon); anti-Calbindin (Swant); anti-choline acetyltransferase (Chemicon); anti-protein kinase Cα (AbCam); anti-neurokinin receptor 3 (Calbiochem); anti-synaptotagminII (Zebrafish International Resource Center); anti-Disabled-1 (gift from T.Curran); anti-Gγ13 (Santa Cruz); anti-Bassoon (Stressgen); anti-synaptophysin (Zymed); anti-Chx10 (Exalpha Biologicals); anti-Sox9 (Chemicon); anti-glutamine synthetase (BD Biosciences); anti-cleaved Caspase-3 (Cell Signaling Technology); anti-Brn-3a (Chemicon); anti-VGlut3 (Chemicon); anti-syntaxin (Sigma); anti-Thy1.2 (BD Pharmingen); anti-GlyT1 (Santa Cruz); and anti-tyrosine hydroxylase (Chemicon). Peanut agglutinin was from Invitrogen. Nuclei were labeled with DAPI, Po-pro1, or NeuroTrace Nissl 435/455 (Invitrogen).
For measurements of retinal layer thickness and cell number, areas were chosen at equivalent retinal eccentricities from the optic nerve head or ora serrata. Layer thickness was measured on single optical sections, adjacent to the optic nerve head. Two to four areas were measured from each retina and two sets of perpendicular measurements were made per area. Both Chx10-Cre; Pcdh-γfcon3/+ and Pcdh-γfcon3/+ littermates were used as controls for Chx10-Cre;Pcdh-γfcon3/fcon3 mutants, and similarly for Pcdh-γfdel. Immunolabeled cells were quantified from 0.13 mm2 (calbindin, ChAT, Brn3a, and Paxα-GFP), 0.05 mm2 (Chx10), 0.02 mm2 (Sox9) or 1280 μm2 (photoreceptors) optical sections. Apoptotic cells were counted on sections spanning the optic nerve head to the ora serrata. Cells were classified as apoptotic if cleaved caspase-3 immunoreactivity partially or completely surrounded a nucleus. Means were compared using ANOVA, Student's t test on condition of equivalent variances determined by F-test, or with Mann-Whitney non-parametric test.
In situ hybridization of retinal sections was performed as described previously (Wang et al., 2002).
Retinas were dissociated with papain by a modification of the protocol described by Meyer-Franke et al (Meyer-Franke et al., 1995). Dissociated cells were plated onto poly-D-lysine coated 8-well Permanox chamber slides (Nunc), then fixed with 4% paraformaldehyde/4% sucrose for 15 minutes, and immunostained. RGCs were enriched with CD90 magnetic Microbeads (Miltenyi-Biotec).
Electrophysiology
Dark-adapted retinas were isolated under an infrared microscope into Ringer's solution at room temperature. A piece of retina, ∼3-4 mm on a side, was placed with RGCs facing down on a 61-electrode array superfused with Ringer's (Kim et al., 2008). Extracellular action potentials were recorded, and single units identified by spike-sorting methods as described (Meister et al., 1994). White light stimuli were delivered from a computer-driven display projected on the retina.
To map spatio-temporal receptive fields, we projected gratings of adjacent thin bars (8.3 or 16.6 μm width). Each bar flickered black or white according to a pseudo-random binary sequence (16.6 ms frame duration). For any given RGC, we computed the spike-triggered average of the flickering bar stimulus (Kim et al., 2008),
| (1) |
where s(x,t) is the stimulus intensity at location x and time t , with the time-averaged intensity subtracted, and the neuron fired a total of n spikes at times {tj}. Examples are shown in Supplemental Figure 2. We then approximated this function as the product of a spatial receptive field b(x) and a temporal integration function a(t),
| (2) |
These are the spatial and temporal components analyzed in Figure 9. For analysis of response threshold and gain, we fitted the time-dependent RGC firing rate r(t) by a Linear-Nonlinear model (Chichilnisky, 2001),
| (3) |
.
Figure 9. Visual processing in Pcdh-γmutant retina.
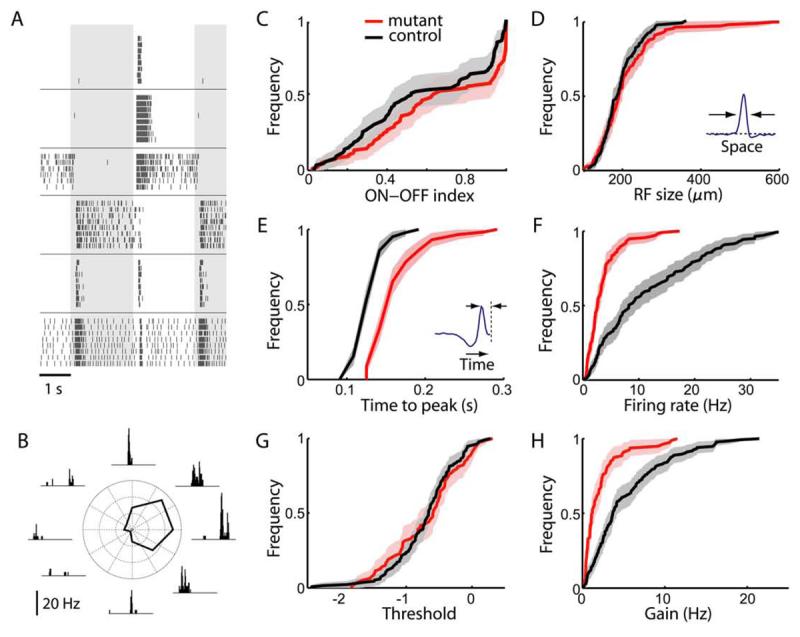
(A) Visual responses of RGCs in Pcdh-γ mutant retina to a flashing spot at photopic intensities. Background shading indicates periods of light On and Off. Each panel is a raster graph of firing from one neuron; each row is a repeat of the same stimulus; tick marks represent action potentials. Neurons vary greatly in whether they respond to light onset or offset, and whether firing is transient or sustained after the switch.
(B) Direction-selective response of an RGC in Pcdh-γ mutant retina to a grating stimulus moving in 8 different directions. Insets are histograms of spike times during one period of the grating (wavelength 664 μm, speed 664 μm/s). The polar plot reports the average firing rate as a function of direction.
(C-H) Distribution of response parameters in mutant and control retinas. Each panel inspects a different characteristic of the visual response, and plots a cumulative histogram of that quantity for RGCs in mutant retinas (red curve, 115 cells, Chx10-Cre; Pcdh-γf-del/f-del or the peripheral region of the Pax6-Cre; Pcdh-γf-del/f-del) and control retinas (black curve, 143 cells, genotype Pax6-Cre; Pcdh-γ+/f-del or Pcdh-γf-del/f-del). The shaded range indicates 95% confidence interval.
(C) Ratio of On and Off responses in mutant and control retinas. For each cell we computed an On-Off index from the experiment of panel A: (number of spikes fired during the on-phase of the spot)/(total number of spikes fired).
(D) Size of the receptive field center, measured as the full width at half maximum of the receptive field profile b(x) (inset, see Eqn 2).
(E) Speed of the response, measured as the time to peak of the temporal integration function a(t) (inset, see Eqn 2).
(F) Average firing rate observed during stimulation with flickering gratings.
(G,H) Threshold (G) and gain (H) of responses in a Linear-Nonlinear model (see Eqn 3).
RESULTS
Broad expression of gamma protocadherins in retina
We began our study by assessing the distribution of Pcdh-γs in retina. We used targeted mutant mice in which GFP is fused to the shared carboxyl-terminus, tagging all 22 Pcdh-γ isoforms (Pcdh-γfusg; Wang et al., 2002). Homozygous Pcdh-γfusg/fusg mutants are viable, fertile, and show none of the defects documented previously in Pcdh-γ mutants (Wang et al., 2002; Weiner et al., 2005). We therefore believe that GFP is a neutral reporter of endogenous Pcdh-γ localization.
The retina consists of three cellular layers separated by two synaptic or “plexiform” layers (Figure 1A). The cellular layers are the outer nuclear layer (ONL), containing photoreceptors; the inner nuclear layer (INL), containing interneurons (horizontal, bipolar and amacrine cells) and Müller glia; and the ganglion cell layer (GCL), containing RGCs and displaced amacrine cells. The outer plexiform layer (OPL) contains synapses of photoreceptors onto horizontal and bipolar cells, and the inner plexiform layer (IPL) contains synapses of bipolar and amacrine cells onto RGCs. As judged by localization of GFP in Pcdh-γfusg mice, Pcdh-γs are present in all five retinal layers (Figure 1A). In the ONL, Pcdh-γs are present in outer segments and around photoreceptor somata (Figure 1B); in the INL and GCL, Pcdh-γs outline neuronal somata (Figure 1C,D). Pcdh-γ levels are highest in the most membrane-rich layers—IPL, OPL, and the optic fiber layers that carry RGC axons to the brain (Figure 1A′,D). In situ hybridization confirmed Pcdh-γ expression by cells in the INL and GCL, though this method did not reliably detect Pcdh-γ RNA in photoreceptors (Figure 1E).
Figure 1. Pcdh-γs are broadly expressed in the retina.
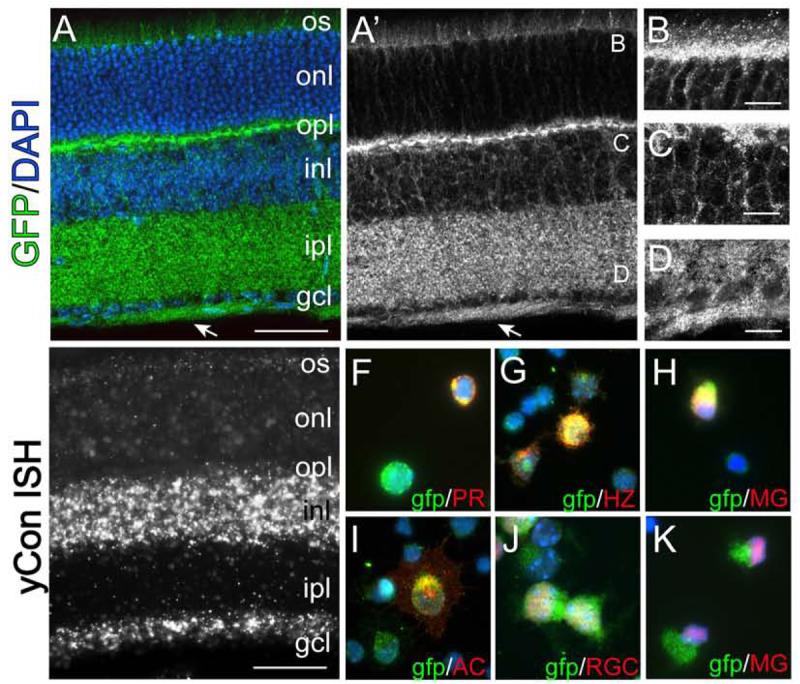
(A-D) Retinas from pcdh-γfusg/fusg mice, expressing a Pcdh-γ-GFP fusion, were stained with antibody to GFP (green in A) and DAPI (blue in A). Pcdh-γ–GFP fusion proteins are concentrated in the process-rich outer plexiform layer (OPL), inner plexiform layer (IPL) and retinal fiber layer (arrow) as well as in outer segments (OS) of photoreceptors, outer and inner nuclear layers (ONL, INL) and ganglion cell layer (GCL). B-D show high magnification images of areas labeled in A'. (E) In situ hybridization to P21 retina using a probe against the Pcdh-γ common intracellular domain. (F-K). Dissociated Pcdh-γfusg/fusg retinal cells immunolabeled with cell-type specific antibodies (red) and anti-GFP (green). Pcdh-γ–GFP-positive cells are labeled with photoreceptor (PR) marker recoverin, horizontal cell (HZ) marker calbindin, amacrine (AC) marker syntaxin, RGC marker Brn3a, and Müller Glia (MG) markers glutamine synthetase (H) and Sox9 (K). Scale bar: A, E, 50 μm; B-D, 10 μm.
To determine which retinal cell types express Pcdh-γs, we dissociated Pcdh-γfusg retinas and immunostained cells with antibodies to cell-type specific markers (Haverkamp and Wassle, 2000; Wahlin et al., 2004; Zhang et al., 2004). This method circumvented the difficulty of determining which of the cells abutting Pcdh-γ-rich membranes are themselves Pcdh-γ–positive. Markers included Brn3a and Thy-1 for RGCs, syntaxin-1 for amacrine cells, Chx10 for bipolar cells, calbindin for horizontal cells, recoverin for photoreceptors and glial fibrillary acidic protein, Sox9 and glutamine synthetase for Müller glia. All six cell types were Pcdh-γ-positive (Figures 1F-K and data not shown). Thus, Pcdh-γs are expressed in all cell types of the neural retina.
We asked whether Pcdh-γs are present in the retina during early postnatal life, when neural circuits form. At postnatal day (P) 0, the retina contains ganglion cell and neuroblast layers, separated by a nascent IPL. All RGCs have been born by this time, while neurogenesis and migration of newborn interneurons and photoreceptors continue in the neuroblast layer. At this time, Pcdh-γ is present on cells in the neuroblast layer, in the IPL, and on RGC axons (Figure 2A). At P3, Pcdh-γ is apparent in the layer of horizontal cells that prefigures the OPL (Figure 2B). By P7, Pcdh-γ appears in the OPL, as it divides the neuroblast layer into INL and ONL (Figure 2C). By P14, the adult pattern described above is established (Figure 2D).
Synaptic localization of Pcdh-γ proteins in the retina
To evaluate the subcellular localization of Pcdh-γs, we focused on the OPL, because its synapses are larger than those in the IPL. We labeled photoreceptor terminals with antibodies to Bassoon, present in both rod terminals (spherules) and cone terminals (pedicles), and labeled spherules and pedicles selectively with anti-PSD-95 and Peanut Agglutinin, respectively (Blanks et al., 1987; Koulen et al., 1998; tom Dieck and Brandstatter, 2006). Pcdh-γ was associated with both spherules and pedicles (Figures 3A-C). We labeled bipolar cell dendrites with antibodies to Protein Kinase Cα and Neurokinin Receptor 3, which mark rod and cone bipolars, respectively, and to G protein γ13 (Gγ13), which is present in subsets of both rod and cone bipolars (Haverkamp et al., 2003; Huang et al., 2003). Pcdh-γ was associated with both rod and cone bipolar dendrites (Figure 3D and data not shown). Thus, Pcdh-γs were present in pre- and post-synaptic compartments of rod and cone synapses. In contrast, although horizontal cell processes labeled with anti-calbindin (Sharma et al., 2003) were Pcdh-γ-positive, little Pcdh-γ was present in their synaptic varicosities (Figure 3E).
Figure 3. Subcellular localization of Pcdh-γs in the synaptic plexiform layers.
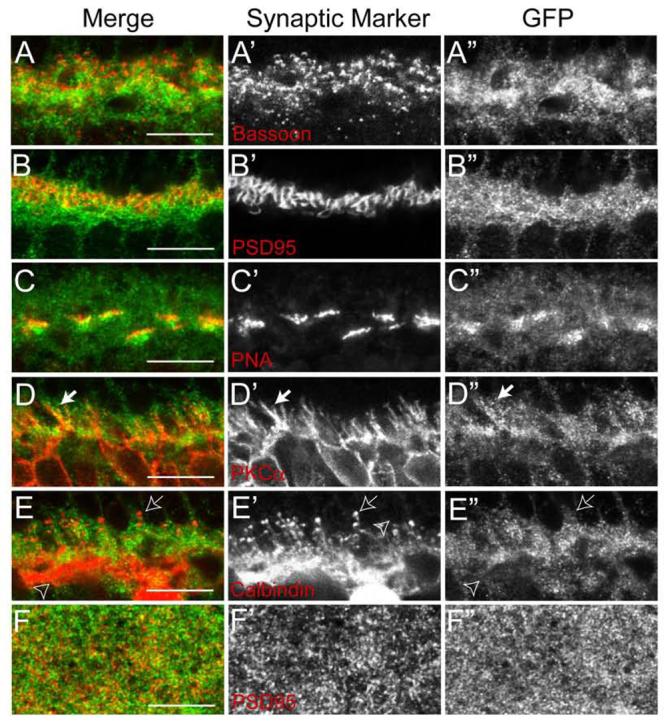
(A-E) Confocal images of the OPL of Pcdh-γfusg/fusg retinas double labeled with anti-GFP (green) and antibodies against proteins present in the synapses that photoreceptors form on horizontal and bipolar cells (red). (A) Bassoon, a component of synaptic ribbons in all photoreceptor nerve terminals. (B) PSD-95, a component of rod terminals (spherules). (C) PKCα, a component of rod bipolar dendrites (arrow). (D) Calbindin, a component of horizontal cell processes (open arrow) that terminate onto rod spherules as well as horizontal cell processes that stratify in the inner OPL (open arrowhead). (E) Peanut agglutinin (PNA) labels cone terminals (pedicles). Pcdh-γs are present in both spherules and pedicles and in bipolar and amacrine processes, but are not seen in horizontal cell varicosities (arrow). (F) A single confocal plane of the IPL of Pcdh-γfusg/fusg retinas labeled with anti-GFP (green) and antibodies to PSD-95 (red), which marks excitatory postsynaptic sites in this lamina. Fine Pcdh-γ–GFP puncta are distributed throughout the IPL, and are present at but not restricted to synapses. Scale bars, 10 μm.
Pcdh-γ was also present throughout the IPL. GFP-positive puncta often overlapped with Bassoon-positive presynaptic ribbons in bipolar cells, glutamate decarboxylase- and GlyT1-positive terminals of inhibitory amacrines, and PSD-95-positive postsynaptic membranes of excitatory synapses (Figure 3F, data not shown). Taken together, these results suggest that Pcdh-γs are present at many synapses in the retina, although they are not confined to synapses.
Inactivation of Pcdh-γs in the retina leads to neuronal and synaptic loss
Pcdh-γ null and hypomorphic mice die shortly after birth (Wang et al., 2002; Weiner et al., 2005). We examined retinas of Pcdh-γ null mutants at late embryonic stages (embryonic day [E] 17-18; birth is at E19) but found no obvious defects in retinal structure (see below). However, since the development of retinal circuitry occurs largely during postnatal life, roles of Pcdh-γ in circuit formation and function could not be studied in these mutants. We therefore generated two conditional inactivation alleles to bypass neonatal lethality (Figure 4A). In Pcdh-γfdel, loxP sites flank the entire Pcdh-γ locus, such that Cre-mediated recombination generates a null allele. In Pcdh-γfcon3, the carboxy-terminal exon shared by all isoforms is flanked by loxP sites, such that Cre truncates all Pcdh-γs. In both alleles, GFP is fused to this carboxy-terminal exon, allowing us to use loss of GFP as an indicator of Cre-mediated Pcdh-γ excision. The Pcdh-γ-GFP fusion protein was identical to that in the Pcdh-γfusg allele described above.
Figure 4. Thinned INL and IPL in retinae lacking Pcdh-γs.

(A) Diagram of conditional Pcdh-γ inactivation alleles. Each Pcdh-γ protein is encoded by an mRNA, comprising one of 22 variable exons and the 3 constant “C” exons. In Pcdh-γfdel, loxP sites flanking the entire pcdh-γ locus result in deletion of all Pcdh-γs upon Cre-mediated recombination. In Pcdh-γfcon3, loxP sites flank the final C3-GFP exon, resulting in truncated forms of Pcdh-γs. In both alleles, GFP is fused to the carboxyl-terminus of the C3 exon. (B-G) Sections from P18 control and Pcdh-γ-deficient retinas, labeled with DAPI or anti-synaptophysin to highlight nuclear and synaptic layers, respectively. Retinal lamination is normal, and OPL and ONL are normal in thickness but IPL and INL are markedly thinned in mutants. (H) Quantification of nuclear and plexiform layer thickness in control (black), Chx10-Cre; Pcdh-γfdel/fdel (white) and Chx10-Cre; pcdh-γfcon3/fcon3 (grey) retinas at P18. Bars show mean +/− SEM from 3-4 animals of each genotype. **, p < 0.01, ANOVA and post-hoc Tukey Test. (I) Quantification of retinal cell types in Paxα6-Cre; Pcdh-γ+/fcon3 (black) and Paxα6-Cre; Pcdh-γfcon3/fcon3 (grey) P18 retinas. Bars show mean +/− SEM from 6-8 animals of each genotype. PR, photoreceptors by Po-pro1; HZ, calbindin+ horizontal cells; BP, Chx10+ bipolar cells; AC, Paxα6+ amacrines; RGC, Brn3a+ retinal ganglion cells; MG, Sox9+ Müller Glia. Other abbreviations as in Figure 1. Error bars indicate SEM. ***, p <0.0001, Mann-Whitney non-parametric test. Scale bars, 50 μm.
In initial tests, we excised the floxed segments of the Pcdh-γfdel and Pcdh-γfcon3 alleles in the germline by mating them to transgenic mice in which Cre is expressed ubiquitously (Actin-cre; (Lewandoski et al., 1997). Homozygotes generated from these animals died at birth and exhibited spinal cord phenotypes similar to those described previously in null mutants (Wang et al., 2002), indicating that recombination inactivates the Pcdh-γ gene (data not shown). Western blotting reported by Prasad et al. (submitted) failed to detect Pcdh-γ protein in Actin-Cre;Pcdh-γfcon3/fcon3 mice, indicating that this allele is effectively a protein null. The truncation in Pcdh-γfcon3 is more extensive than the hypomorphic allele described previously (Pcdh-γtr), in which Pcdh-γ levels were decreased several-fold (Weiner et al., 2005). We speculate that increased truncation of Pcdh-γfcon3 and lack of a polyadenylation signal led to greater destabilization of Pcdh-γ protein and mRNA, respectively.
To selectively inactivate Pcdh-γs in retina, we crossed Pcdh-γfdel and Pcdh-γfcon3 mutants with mice in which a GFP-Cre recombinase fusion protein is expressed under the control of regulatory elements from the Chx10 gene (Chx10-Cre; Rowan and Cepko, 2004). These elements drive expression of GFP-Cre transiently in embryonic retinal progenitors and postnatally in bipolar cells. To assay recombination in retinas of Chx10-Cre mice, we crossed them to a reporter line in which β-galactosidase (LacZ) and GFP label non-recombined and recombined cells, respectively (Z/EG; Novak et al., 2000). Recombination was extensive (>90%) in the INL and ONL but occasional columns of cells were spared (Supplementary Figure 1A). In contrast, approximately half of the cells in the GCL were GFP-negative and LacZ-positive (Supplementary Figure 1B). This pattern may reflect the fact that many RGCs are born by E12 (Farah and Easter, 2005), before Cre accumulates in progenitors. We then used loss of GFP to assay Chx10-Cre-mediated loss of Pcdh-γ-GFP from the Pcdh-γ alleles. This method did not allow us to assess excision in bipolars, in which GFP was expressed from the Chx10 transgene (see Methods). Nonetheless, Chx10-Cre excised Pcdh-γfdel, Pcdh-γfcon3 and Z/EG genes in similar patterns and to a similar extent (Supplementary Figure 1C-E). The efficacious excision of the Pcdh-γfdel allele is surprising given the length of the floxed segment.
Chx10-Cre;Pcdh-γfdel/fdel mice are healthy and outwardly normal. We first examined these mutants at P18, by which time the retinal architecture is well-developed. Labeling of nuclear layers with DAPI and plexiform layers with antibodies to the synaptic vesicle protein synaptophysin revealed that mutant retinas were properly laminated (Figures 4B-E). However, mutant retinas were ∼25% thinner than those of wild-type mice or heterozygote littermates. The difference resulted from a selective reduction of ∼40% in the thickness of the INL and the IPL (Figure 4H). Thus, Pcdh-γs are required for development or maintenance of retinal interneurons and the layer in which they form synapses. The INL and IPL were thinned to the same extent in Chx10-Cre;Pcdh-γfcon3/fcon3 and Chx10-Cre;Pcdh-γfdel/fdel mice (Fig. 4D-G), consistent with the idea that Pcdh-γfcon3 is functionally a null. In subsequent studies, we used the two alleles interchangeably, but most of the results reported here are from Pcdh-γfcon3 mice.
The mosaicism described above for Chx10-Cre retinas made quantification of cell loss imprecise in the GCL. We therefore used a second transgenic line, Pax6α-Cre, in which Cre is expressed under the control of retina-specific sequences from the Pax6 gene (Marquardt et al., 2001). The Pax6α-Cre transgene drives expression of Cre transiently in embryonic retinal progenitors, leading to essentially complete (>99%) inactivation in peripheral retina; a sector in central retina is spared, as described below. Postnatally, Pax6α-Cre is expressed in a subset of amacrines, which can be identified by a GFP reporter within the transgene.
We used cell type-specific markers (see above) to quantify cell loss from peripheral regions of Pax6α-Cre;Pcdh-γfcon3/fcon3 retinas at P18. Numbers of bipolar, amacrine, and retinal ganglion cells were reduced by 45-65% (Figure 4I). Müller glia were also decreased, but only by ∼20%. In contrast, numbers of horizontal cells and photoreceptors differed little between mutants and controls (Fig. 4I). Together, these results demonstrate that Pcdh-γs are essential for the survival of many but not all retinal cell types.
Increased postnatal apoptosis in the absence of Pcdh-γs
We next asked when retinal defects arise in Pcdh-γ mutants, and whether they are progressive. We detected no differences in laminar arrangement or thickness between mutant and control retinas perinatally (E17.5-P3; Figure 5A,B,G,H and data not shown). By P7, however, shortly after the ONL and INL form, mutant retinas were thinner than those of controls (Figure 5C,D). We used Chx10-Cre; Pcdh-γfcon3 mice for quantification of these defects. Both layers were ∼40% thinner in mutants than in controls by P14, then changed little over the following several months (Figure 5E-I). Thus, the difference between mutant and control retinas appears during the first postnatal week, is maximal by the end of the second postnatal week, and neither abates nor worsens substantially thereafter.
Figure 5. Thinning of Pcdh-γ mutant retina reflects increased apoptosis during a restricted postnatal period.
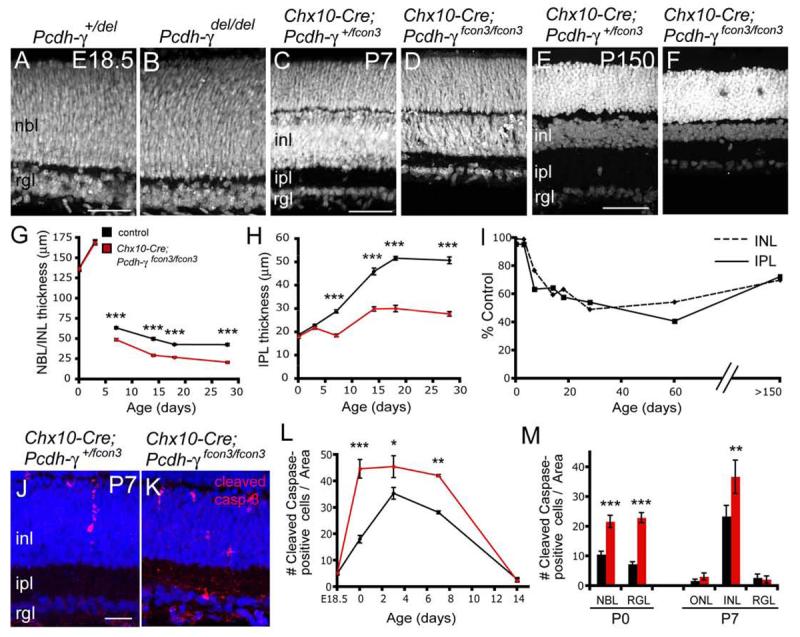
(A-F) Nuclear labeling of Pcdh-γdel/del, Pcdh-γfcon3/fcon3 and control retinas at E18.5 (A,B), P7 (C.D) and P150 (E,F). Mutant and control retinas are indistinguishable at E18.5 but INL and IPL are thinner in mutant retinas than controls by P7. Although the INL becomes thinner in both mutants and controls over the subsequent 5 months, the difference between them is not progressive. (G,H) Thickness of NBL/INL (NBL at P0 and P3, INL at later stages) and IPL in in control (black) and Chx10-Cre; pcdh-γfcon3/fcon3 mutant retina sections (red). The mutant NBL/INL and IPL develop normally through P3, then decline in thickness relative to controls over the next few weeks. Graphs show mean +/− SEM from 3-4 animals. ***, p < 0.001, Student's t-test. (I) NBL/INL and IPL thickness in Pcdh-γfcon3/fcon mutant retinas, expressed as percent of control. (J,K) Increased numbers of apoptotic cells, marked by cleaved caspase-3 immunoreactivity (red), in Pcdh-γfcon3/fcon3 mutant retinas at P7 compared to controls. (M,N) Quantification of cleaved caspase-3 immunopositive cells in control (black) and Pcdh-γfcon3/fcon3 mutant (red) retinas. Differences between genotypes are significant in the NBL/IPL at P0 and P7 but in the ganglion cell layer only at P7. Results from 3-6 animals per stage. ***, p < 0.001; **, p < 0.01; *, p < 0.05; Student's t-test or Mann-Whitney test. Abbreviations as in Figure 1. Scale bars, 20 μm.
A process of naturally-occurring programmed cell death eliminates many retinal neurons during the first two postnatal weeks (Pequignot et al., 2003; Young, 1984). Apoptosis followed a similar time course in Pcdh-γ deficient retinas, but levels were significantly higher in mutants than in controls (Figure 5K,L). Although increased apoptosis was seen in both neuroblast and ganglion cell layers at P0, it was confined to the INL at P7 (Figure 5M). This pattern is consistent with the finding that naturally occurring cell death in the GCL is complete several days before that in the INL (Farah and Easter, 2005; Pequignot et al., 2003; Young, 1984). These results suggest that Pcdh-γs regulate neuronal survival during the period of naturally occurring programmed cell death.
Cell autonomy of Pcdh-γ-dependent cell survival in retina
Are Pcdh-γs required for cell survival in cells that express them, in neighboring cells, or both? As a first step to distinguish among these possibilities, we capitalized on the recombination pattern in Pax6α-Cre transgenic mice. As noted above, Cre is expressed in all progenitors in peripheral retina as well as in a subset of amacrine cells marked by the GFP in the transgene. In a large dorso-ventrally oriented swath of central retina, however, Cre is expressed in amacrines but not progenitors (Marquardt et al., 2001; Stacy et al., 2005). Thus in central retina of Paxα6-Cre; Pcdh-γfcon3/fcon3 mice, GFP-positive amacrine cells lack Pcdh-γs whereas all other cells, including Müller glia, are Pcdh-γ-positive (Figure 6A-E).
Figure 6. Cell autonomous and non-autonomous components of Pcdh-γ–dependent cell survival.
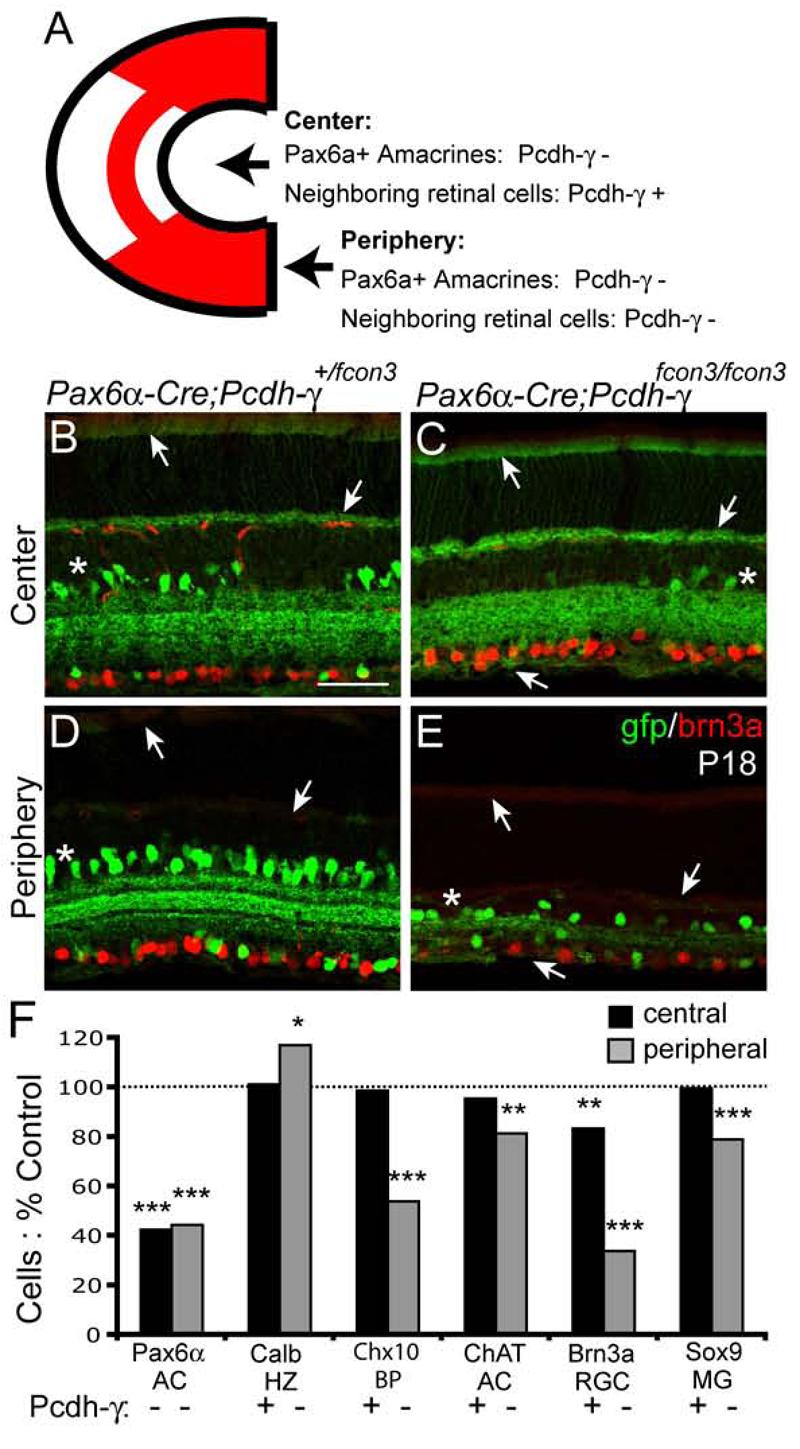
(A) Schematic of the recombination pattern in Pax6α-Cre retina. In Pax6α-Cre; pcdh-γfcon3/fcon3 mutants, all cells are Pcdh-γ-negative in peripheral retina, but only Pax6α+ amacrine cells are Pcdh-γ-negative in the central sector. (B-E) Immunolabeling of central and peripheral regions of Pax6α-Cre; pcdh-γ+/fcon3 and Pax6α-Cre; pcdh-γfcon3/fcon3 mutant retinas with GFP and brn3a antibodies. Anti-GFP labels both Pax6α-Cre-ires-GFP positive amacrine cells (asterisks) and Pcdh-α–GFP proteins (arrows). In unrecombined portions of the central Pax6α-Cre; Pcdh-γfcon3/+ and Pax6α-Cre; pcdh-γfcon3/fcon3 retinas, Pcdh-γ–GFP proteins are visible in the outer segments, OPL, and retinal axons (arrows). In peripheral regions, Pcdh-γ–GFPs are absent. In both regions, mutant retinas have reduced numbers of Pax6α-positive amacrine cells; Brn3a-positive RGCs are dramatically decreased in the mutant peripheral sector while slightly decreased in the central sector. (F) Quantification of retinal cell types in central regions of Pax6α-Cre; Pcdh-γfcon3/fcon3 retinas, expressed as % cells in Paxα6-Cre; Pcdh-αfcon3/+ littermates. 6 to 8 animals per genotype were analyzed; * p < 0.05; ** p < 0.01; *** p< 0.0001, by Student's t-test or Mann-Whitney test. Scale bar, 50μm.
We asked whether Pax6α-positive (that is, GFP-positive) amacrines were lost from central retina of Pax6α-Cre; Pcdh-γfcon3/fcon3 mice, despite being surrounded by Pcdh-γ-positive cells. The number of Pax6α-amacrines in central retina was 58% lower in mutants than in controls (Figure 6B,C,F). Because multiple, neighboring amacrines are Pcdh-γ-deficient in the central region, this result does not demonstrate cell autonomy sensu strictu, but does indicate that loss of Pcdh-γs from a single cell type impairs its survival, even when the majority of its synaptic inputs (bipolar cells) and targets (RGCs) are wild-type. This result also rules out the possibility that neuronal apoptosis in the absence of Pcdh-γ is secondary to a defect in surrounding glial cells. Furthermore, loss of Pax6α-positive amacrines is equivalent in peripheral and central retina (Figure 6F), indicating that Pcdh-γ–negative cells are not protected from apoptosis when surrounded by Pcdh-γ–positive cells.
We also asked whether loss of Pcdh-γs from Pax6α-amacrines was detrimental to survival of neighboring cells (Figure 6F). Loss of Pcdh-γs from the Pax6α-positive amacrines had no detectable effect on the survival of bipolar cells, horizontal cells or Müller glia. Likewise, survival of a distinct, intermingled subpopulation of amacrines, the cholinergic starburst cells was unaffected in central retina. In contrast, we detected a small (∼15%) but significant loss of Brn3a-positive RGCs from central retina of Pax6α-Cre; Pcdhγfcon3/fcon3 mice. We do not know whether this loss reflects absence of Pcdh-γs per se, or death of amacrines, which regulate at least some aspects of RGC development (Goldberg et al., 2002).
Pcdh-γs are dispensable for laminar targeting of retinal neurons
Although the width of the IPL is dramatically reduced in Pcdh-γ-deficient retina, it nonetheless contains synapses, as judged by the presence of pre- and postsynaptic markers such as synaptophysin and Bassoon (Figures 4E-G and 8A,B). Are they appropriate synapses? The retina is well-suited to test specificity, because discrete subsets of bipolar and amacrine cells arborize and synapse in just one or a few of ≥10 closely spaced, parallel sublaminae within the IPL (Pang et al., 2002; Roska and Werblin, 2001; Wassle, 2004).
Figure 8. Laminar specificity and synapse formation by Pcdh-γ-deficient neurons rescued from apoptosis.
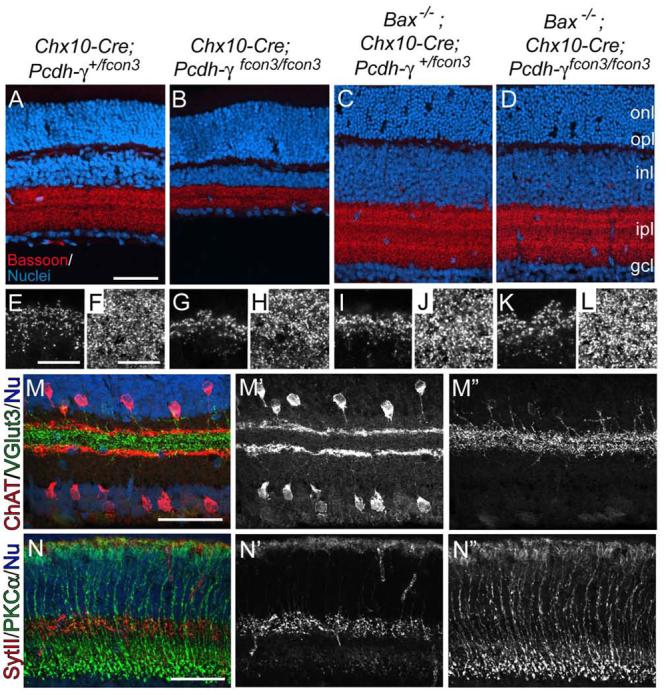
(A-D) Sections from retinas mutant for Bax (Chx10-Cre; Pcdh-γfcon3/+ Bax−/−), Pcdh-γ (Chx10-Cre; Pcdh-γfcon3/fcon3 Bax+/−), both (Chx10-Cre; Pcdh-γfcon3-fcon3 Bax−/−). or neither (Chx10-Cre; Pcdh-γfcon3/+ Bax−/+). Sections were stained with anti-Bassoon (red) and Po-pro1 (blue). Thickness of IPL and INL are similar in Bax mutants and Bax, Pcdh-γ double mutants; both are thicker than those in Pcdh-γ mutants. (E-L) High power images of OPL (E.G.I.K) and IPL (F,H,J,L) from retinas in A-D. Density of synaptic puncta is similar in all four genotypes. (M,N) Chx10-Cre; Pcdh-γfcon3/fcon3 Bax−/− mutants immunostained for ChAT (red) and vGluT3 (green) (M) or SynaptotagminII (red), and Gγ13 (green) (N). All processes make lamina-specific arbors (compare with Figure 7) and disruptions seen in Pcdh-γ single mutants are absent in double mutants. Scale bars, 100 μm (A-D), 10 μm (E-L), 50 μM (M,N).
We used markers of 10 lamina-specified amacrine and bipolar subtypes to assess lamina-specific arborization and connectivity in the absence of Pcdh-γs. We follow a scheme in which 5 sublaminae of equal width are numbered, from the border of the INL (S1) to the border of the ganglion cell layer (Ghosh et al., 2004; Yamagata and Sanes, 2008). Populations examined were starburst amacrines, labeled by choline acetyltransferase; glutamatergic amacrines (anti-vGlut3); GABAergic amacrines (anti-GAD65/67); dopaminergic amacrines (anti-tyrosine hydroxylase); type AII amacrines (anti-disabled); calbindin-positive amacrines and RGCs ; OFF bipolar cells (anti-synaptotagmin 2); ON bipolar cells (anti-Gγ13); OFF bipolars (anti-NK3R); and rod bipolars (anti-protein kinase Cα; Figure 7E). In all ten cases, processes were arrayed in appropriate sublaminae in mutant retinas at P18, although disruptions or gaps were sometimes present (Figure 7A-D and data not shown). We also observed proper laminar targeting of amacrine subsets at P7, and of bipolar subsets at P14, in each case soon after these synapses formed in controls (data not shown).
Figure 7. Sublamina-specific targeting of amacrine and bipolar processes in the IPL in the absence of Pcdh-γs.
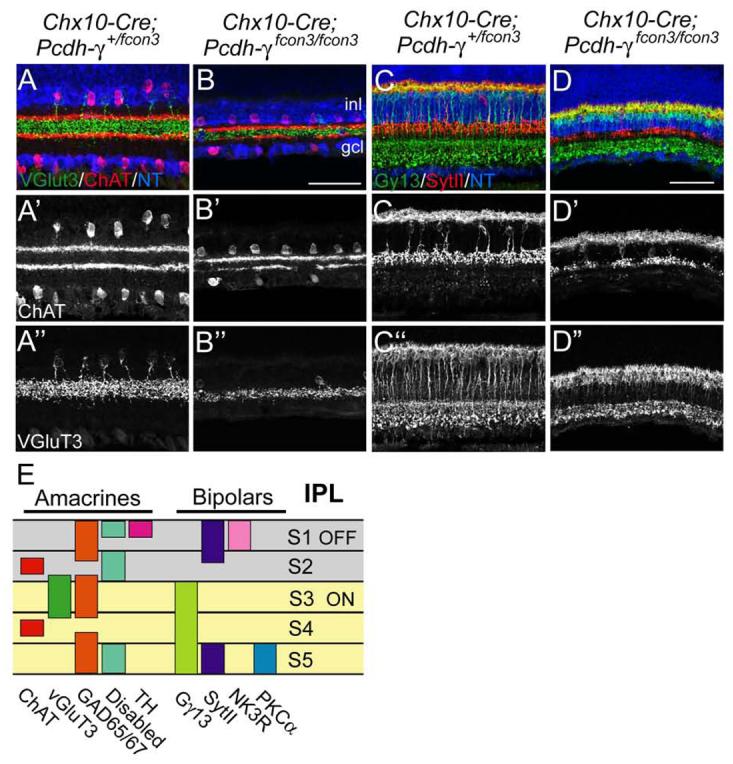
(A,B) ChAT- (red) and vGluT3-positive amacrine subsets (green) in Chx10-Cre; Pcdh-γfcon3/+ (control) and Chx10-Cre; Pcdh-γfcon3/fcon3 retinas. ChAT-positive processes ramify in sublaminae (S) 2 and 4, and vGlut3-positive processes ramify in S3. (C,D) Synaptotagmin II- (SytII-) positive OFF bipolar processes (red), and Gγ13-positive ON bipolar processes (green) ramify in the outer and inner portions of the IPL, respectively. In all cases, laminar specificity is retained in mutants, but marker-laminae are reduced and disrupted. (E) Sketch of IPL sublaminae stained by the markers used in this study. Scale bar, 50 μm.
Synapses and arbors of Pcdh-γ-deficient neurons destined to die
Results illustrated in Figure 7 are consistent with the idea that Pcdh-γs are dispensable for formation of neural circuits in retina. Alternatively, however, some INL interneurons might fail to target appropriate sublaminae and then die. In this case, neuronal apoptosis in Pcdh-γ mutants might be secondary to circuit defects. To test this possibility, we blocked apoptosis in Pcdh-γfcon3/fcon3 mice by deleting the pro-apoptotic gene, Bax. Naturally occurring death in many regions of the nervous system, including retina, is dramatically reduced in Bax−/− mice (Mosinger Ogilvie et al., 1998; Pequignot et al., 2003; White et al., 1998), and deletion of Bax preserves spinal interneurons that would otherwise die in Pcdh-γ null spinal cord (Weiner et al., 2005). Likewise, Bax deletion rescued neurons destined to die in Pcdh-γ mutant retina: the thickness of the INL and GCL and the number of Chx10-positive bipolar cells were indistinguishable in Chx10-Cre; Pcdh-γ+/fcon3; Bax−/− and Chx10-Cre; Pcdh-γfcon3/fcon3; Bax−/− retinas (Figure 8A-D and data not shown).
Deletion of Bax also resulted in expansion of the IPL. The IPL in Chx10-Cre; Pcdh-γfcon3/fcon3; Bax−/− mice was thicker than that in Pcdh-γ mutants and indistinguishable from that in Pcdh-γ-positive Bax−/− mutants (Figure 8A-D). The density of synapses in the IPL and OPL, as judged by staining for PNA or Bassoon, did not differ significantly between Pcdh-γ-positive and Pcdh-γ-negative Bax−/− retinas (Figure 8E-L; Supplementary Figure 2). Thus, loss of Pcdh-γ had little effect on synapse number in the IPL when apoptosis was prevented. This result is consistent with the idea that much of the synapse loss in the IPL of Pcdh-γ–deficient retina is a consequence of decreased neuron number.
To assess the laminar targeting of interneurons that would have died in the presence of Bax, we stained Chx10-Cre; Pcdh-γfcon3/fcon3; Bax−/− retinae with the panel of markers listed above. In all cases, targeting of processes to appropriate laminae was as specific in double mutants as in Pcdh-γ single mutants (Figure 8M,N and data not shown). Moreover, thinning and disruptions of layers observed in Pcdh-γ single mutants were rescued in Pcdh-γ−/−; Bax−/− double mutants (compare Figures 7A,B and 8M,N). We therefore conclude that the gaps observed in Pcdh-γ deficient retina are secondary to the loss of cells rather than a manifestation of improper laminar targeting.
Functional visual circuits form in the absence of Pcdh-γs
To test whether circuits that form in Pcdh-γ-deficient retina are functional, we recorded light responses from RGCs. These cells integrate signals from amacrine and bipolar interneurons and send the resulting spike trains to the brain. RGCs differ in their responses to visual stimuli, depending on the synaptic inputs they receive. Thus, ON RGCs, which respond primarily to light onset, receive synapses from ON bipolar cells in the inner half of the IPL (nearest the GCL); OFF RGCs receive synapses from OFF bipolars in the outer IPL; and ON-OFF RGCs receive both types of synapses. Further specializations, such as responses that are transient, sustained or selective for moving objects, result from innervation by specific subsets of bipolar and amacrine cells (Masland, 2001; Wassle, 2004). Accordingly, the presence of diverse, specific responses from RGCs is a sensitive indicator of precisely patterned synaptic connectivity. We therefore monitored action potentials simultaneously from large populations of RGCs in control and Pcdh-γ mutant retinas, using a multielectrode array (Meister et al., 1994). Results from Chx10-Cre; Pcdh-γfcon3/fcon3 retinas and peripheral regions of Pax6α-Cre; Pcdh-γfcon3/fcon3 retinas were similar, so they are combined here. We did not use Pcdh-γ;Bax double mutants for this study, because visual responses are compromised in Bax single mutants (Pequignot et al., 2003).
RGCs in Pcdh-γ mutant and control retinas showed a similar variety of responses to small flashing spots, including sustained and transient ON, OFF and ON-OFF responses (Figure 9A). Proportions of ON- and OFF-dominated responses were identical in mutants and controls (Figure 9C). Mutant RGCs responded to very dim flashes, which excite only rods, and also to bright flashes, which predominantly excite cones (data not shown), indicating that both rod- and cone-activated pathways were functional. We also probed the retina with moving bars and gratings to elicit direction-selective responses, known to depend on specific patterns of connectivity in the IPL (Masland, 2001). Both ON and ON-OFF direction-selective cells were encountered in mutant retinas (Figure 9B and data not shown). Some control and mutant RGCs had receptive field surrounds, where light has the opposite effect of the center (Supplementary Figure 3), indicating that lateral inhibitory connections are functional in these circuits.
To survey response properties quantitatively, we stimulated the retina with randomly flickering bars and applied a reverse correlation method (Chichilnisky, 2001; Meister et al., 1994). This measures spatio-temporal receptive fields, revealing how RGCs respond to light intensity at different points on the retina and at different times in the past (Supplemental Figure 2). The size of the receptive field center varied greatly among RGCs, but the distribution was similar in control and mutant retinas (Figure 9D). On the other hand, the time course of the light response was significantly slower in mutant retina (Figure 9E). Moreover, mutant RGCs fired at much lower rates in reponse to flicker stimuli (Figure 9F). In principle, this could result from an elevated response threshold; alternatively, the gain of the response might be lower once the threshold is crossed. Based on fitting with a Linear-Nonlinear model (Chichilnisky, 2001), we found that the threshold is unaltered, but the gain is reduced in mutants (Figures 9G-H).
DISCUSSION
Protocadherins and neural specificity
Interest in the clustered protocadherins has centered on the tantalizing idea that their molecular diversity may underlie the extraordinary synaptic specificity of the brain (Benson et al., 2001; Hamada and Yagi, 2001; Hirayama and Yagi, 2006; Kohmura et al., 1998; Morishita and Yagi, 2007; Serafini, 1999; Shapiro and Colman, 1999; Washbourne et al., 2004; Wu and Maniatis, 1999; Yagi and Takeichi, 2000). Several observations that led to this notion are summarized in the Introduction. Moreover, Hasegawa et al. (Hasegawa et al., 2008) recently showed that olfactory axons bearing a single type of receptor fail to coalesce properly onto glomeruli in olfactory bulbs of mice lacking Pcdh-αs.
These considerations, coupled with the finding that most retinal cells express Pcdh-γs, led us to expect that retinal circuitry might be grossly defective in their absence. Surprisingly, it was not. Synaptic specializations were present in the OPL and IPL of Pcdh-γ mutants, and the light-responsiveness of RGCs indicates that synapses in both laminae were functional. Moreover, synapses in the IPL were sublamina-specific as judged by distribution of arbors. This distribution provides a stringent test of targeting, in that 10 or more IPL sublaminae are separated by only a few tens of microns (Roska and Werblin, 2001; Wassle, 2004).
The loss of neurons in Pcdh-γ-deficient retinas potentially complicates this interpretation: neurons making improper arbors or connections could be selectively eliminated, so only neurons that wired up properly would be retained. The ability to block apoptosis in Pcdh-γ mutant retinas by deleting the Bax gene allowed us to test this possibility. Lamina-specific targeting was, if anything, more precise in the absence of Bax than in its presence, in that disruptions and irregularities seen in Pcdh-γ−/− laminae were absent in double mutants. Therefore, IPL disruptions observed in Pcdh-γ−/− retinas presumably reflected loss of cells rather than mistargeting of neurites.
The ability of mutant retinas to process visual information was also remarkably preserved. RGCs exhibited a wide range of complex responses, and their receptive field sizes were normal. Because the spatial extent of RGC receptive field centers are largely determined by their dendritic fields, which collect input from bipolar cells (Wassle, 2004), this result suggests that mutant RGC arbors are normal in size. In that bipolar cells provide the main excitation to RGCs, their decreased number could account for the lower firing rate and response gain in mutant RGCs. Most likely to result from lack of Pcdh-γ rather than from decreased cell number are the defects in response dynamics, which are controlled by synaptic properties in the OPL (DeVries, 2000) and IPL (Nirenberg and Meister, 1997). One way to distinguish which defects are due to loss of interneurons and altered ratios of cell types and which to loss of Pcdh-γs per se will be to record from retinas lacking both Pcdh-γṣ and Bax. This work is underway, but is complicated by the facts that naturally occurring cell death is also blocked by Bax deletion, and that visual responses are compromised in these mutants (Pequignot et al., 2003).
Synaptic circuitry and neuronal survival
Patterns of apoptosis in Pcdh-γ-deficient retina are similar to those in spinal cord (Wang et al., 2002; Weiner et al., 2005) Prasad et al., submitted) in several respects. First, approximately half of the interneurons in each region are lost in the absence of Pcdh-γs. Second, some interneuronal subtypes and primary sensory neurons (dorsal root ganglion cells and photoreceptors) are spared in both regions, even though they express Pcdh-γ genes. Third, the loss of neurons in Pcdh-γ mutants occurs during the period of naturally-occurring cell death. One apparent difference is that the output neurons of the spinal cord, motor neurons, are spared in Pcdh-γ-deficient mice, whereas those of retina, RGCs, are affected. However, at least some apoptosis of RGCs is cell-nonautonomous, reflecting either loss of Pcdh-γ from presynaptic cells or loss of input cells themselves. It is possible that in the mutants analyzed to date, motor neurons retain a larger fraction of their inputs than do RGCs, and that this contributes to their survival.
Retina and spinal cord phenotypes are also similar in that loss of Pcdh-γs leads to decreased numbers of synapses in both tissues. In spinal cord, synapse loss does not result simply from neuron loss, as shown by analysis of Pcdh–γ-deficient mice in which apoptosis was blocked: neuronal number was normal in these animals, but synapse number was still reduced (Weiner et al., 2005). This result is consistent with the idea that failure of synapse formation or function impairs neuronal survival (see also Prasad et al., submitted). In fact, complete blockade of synaptic function in embryonic brain leads to increased apoptosis (Verhage et al., 2000). In contrast, deletion of Pcdh-γ in a Bax−/− background does not substantially decrease synapse density in retina. Synapses in the IPL are small and densely packed, so we could not determine their number accurately. Given the electrophysiological evidence for maintained synaptic function, however, it seems unlikely that any synaptic defect is sufficient in magnitude to explain the massive apoptosis we observe. Likewise, synaptic patterns in the IPL of Pcdh–γ mutants are at least as well preserved in the absence of Bax as in its presence, ruling out the possibility that apoptosis reflects selective elimination of inappropriate synapses.
Thus, synapses can be lost in the absence of neuronal loss in the spinal cord, and neurons can be lost in the absence of major synaptic defects in retina. These results suggest that Pcdh–γ regulates neuronal survival and synaptic maturation by distinct mechanisms, and that effects on these two processes differ in severity among brain regions. The combinatorial diversity provided by the Pcdh–γ may therefore be useful for selectively controlling the size of diverse neuronal populations.
Supplementary Material
ACKNOWLEDGMENTS
We thank Joshua Weiner for sharing data. This work was supported by grants from the National Institutes of Health to M.M. and J.R.S, a NARSAD Young Investigator award to J.L., and Damon Runyons Cancer Research Foundation Fellowship to Y-F.Z.
REFERENCES
- Benson DL, Colman DR, Huntley GW. Molecules, maps and synapse specificity. Nat Rev Neurosci. 2001;2:899–909. doi: 10.1038/35104078. [DOI] [PubMed] [Google Scholar]
- Blanks JC, Hageman GS, Johnson LV. Appearance of PNA-binding cells within the outer nuclear layer coinciding with photoreceptor degeneration in rd mice. Prog Clin Biol Res. 1987;247:229–42. [PubMed] [Google Scholar]
- Chichilnisky EJ. A simple white noise analysis of neuronal light responses. Network. 2001;12:199–213. [PubMed] [Google Scholar]
- DeVries SH. Bipolar cells use kainate and AMPA receptors to filter visual information into separate channels. Neuron. 2000;28:847–56. doi: 10.1016/s0896-6273(00)00158-6. [DOI] [PubMed] [Google Scholar]
- Esumi S, Kakazu N, Taguchi Y, Hirayama T, Sasaki A, Hirabayashi T, Koide T, Kitsukawa T, Hamada S, Yagi T. Monoallelic yet combinatorial expression of variable exons of the protocadherin-alpha gene cluster in single neurons. Nat Genet. 2005;37:171–6. doi: 10.1038/ng1500. [DOI] [PubMed] [Google Scholar]
- Farah MH, Easter SS., Jr. Cell birth and death in the mouse retinal ganglion cell layer. J Comp Neurol. 2005;489:120–34. doi: 10.1002/cne.20615. [DOI] [PubMed] [Google Scholar]
- Frank M, Ebert M, Shan W, Phillips GR, Arndt K, Colman DR, Kemler R. Differential expression of individual gamma-protocadherins during mouse brain development. Mol Cell Neurosci. 2005;29:603–16. doi: 10.1016/j.mcn.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Ghosh KK, Bujan S, Haverkamp S, Feigenspan A, Wassle H. Types of bipolar cells in the mouse retina. J Comp Neurol. 2004;469:70–82. doi: 10.1002/cne.10985. [DOI] [PubMed] [Google Scholar]
- Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–4. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- Hamada S, Yagi T. The cadherin-related neuronal receptor family: a novel diversified cadherin family at the synapse. Neurosci Res. 2001;41:207–15. doi: 10.1016/s0168-0102(01)00281-4. [DOI] [PubMed] [Google Scholar]
- Hasegawa S, Hamada S, Kumode Y, Esumi S, Katori S, Fukuda E, Uchiyama Y, Hirabayashi T, Mombaerts P, Yagi T. The protocadherin-alpha family is involved in axonal coalescence of olfactory sensory neurons into glomeruli of the olfactory bulb in mouse. Mol Cell Neurosci. 2008;38:66–79. doi: 10.1016/j.mcn.2008.01.016. [DOI] [PubMed] [Google Scholar]
- Haverkamp S, Ghosh KK, Hirano AA, Wassle H. Immunocytochemical description of five bipolar cell types of the mouse retina. J Comp Neurol. 2003;455:463–76. doi: 10.1002/cne.10491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverkamp S, Wassle H. Immunocytochemical analysis of the mouse retina. J Comp Neurol. 2000;424:1–23. [PubMed] [Google Scholar]
- Hill E, Broadbent ID, Chothia C, Pettitt J. Cadherin superfamily proteins in Caenorhabditis elegans and Drosophila melanogaster. J Mol Biol. 2001;305:1011–24. doi: 10.1006/jmbi.2000.4361. [DOI] [PubMed] [Google Scholar]
- Hirayama T, Yagi T. The role and expression of the protocadherin-alpha clusters in the CNS. Curr Opin Neurobiol. 2006;16:336–42. doi: 10.1016/j.conb.2006.05.003. [DOI] [PubMed] [Google Scholar]
- Huang L, Max M, Margolskee RF, Su H, Masland RH, Euler T. G protein subunit G gamma 13 is coexpressed with G alpha o, G beta 3, and G beta 4 in retinal ON bipolar cells. J Comp Neurol. 2003;455:1–10. doi: 10.1002/cne.10396. [DOI] [PubMed] [Google Scholar]
- Kim IJ, Zhang Y, Yamagata M, Meister M, Sanes JR. Molecular identification of a retinal cell type that responds to upward motion. Nature. 2008;452:478–82. doi: 10.1038/nature06739. [DOI] [PubMed] [Google Scholar]
- Knudson CM, Tung KS, Tourtellotte WG, Brown GA, Korsmeyer SJ. Bax-deficient mice with lymphoid hyperplasia and male germ cell death. Science. 1995;270:96–9. doi: 10.1126/science.270.5233.96. [DOI] [PubMed] [Google Scholar]
- Kohmura N, Senzaki K, Hamada S, Kai N, Yasuda R, Watanabe M, Ishii H, Yasuda M, Mishina M, Yagi T. Diversity revealed by a novel family of cadherins expressed in neurons at a synaptic complex. Neuron. 1998;20:1137–51. doi: 10.1016/s0896-6273(00)80495-x. [DOI] [PubMed] [Google Scholar]
- Koulen P, Fletcher EL, Craven SE, Bredt DS, Wassle H. Immunocytochemical localization of the postsynaptic density protein PSD-95 in the mammalian retina. J Neurosci. 1998;18:10136–49. doi: 10.1523/JNEUROSCI.18-23-10136.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandoski M, Meyers EN, Martin GR. Analysis of Fgf8 gene function in vertebrate development. Cold Spring Harb Symp Quant Biol. 1997;62:159–68. [PubMed] [Google Scholar]
- Marquardt T, Ashery-Padan R, Andrejewski N, Scardigli R, Guillemot F, Gruss P. Pax6 is required for the multipotent state of retinal progenitor cells. Cell. 2001;105:43–55. doi: 10.1016/s0092-8674(01)00295-1. [DOI] [PubMed] [Google Scholar]
- Masland RH. The fundamental plan of the retina. Nat Neurosci. 2001;4:877–86. doi: 10.1038/nn0901-877. [DOI] [PubMed] [Google Scholar]
- Meister M, Pine J, Baylor DA. Multi-neuronal signals from the retina: acquisition and analysis. J Neurosci Methods. 1994;51:95–106. doi: 10.1016/0165-0270(94)90030-2. [DOI] [PubMed] [Google Scholar]
- Meyer-Franke A, Kaplan MR, Pfrieger FW, Barres BA. Characterization of the signaling interactions that promote the survival and growth of developing retinal ganglion cells in culture. Neuron. 1995;15:805–19. doi: 10.1016/0896-6273(95)90172-8. [DOI] [PubMed] [Google Scholar]
- Morishita H, Yagi T. Protocadherin family: diversity, structure, and function. Curr Opin Cell Biol. 2007;19:584–92. doi: 10.1016/j.ceb.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Mosinger Ogilvie J, Deckwerth TL, Knudson CM, Korsmeyer SJ. Suppression of developmental retinal cell death but not of photoreceptor degeneration in Bax-deficient mice. Invest Ophthalmol Vis Sci. 1998;39:1713–20. [PubMed] [Google Scholar]
- Nirenberg S, Meister M. The light response of retinal ganglion cells is truncated by a displaced amacrine circuit. Neuron. 1997;18:637–50. doi: 10.1016/s0896-6273(00)80304-9. [DOI] [PubMed] [Google Scholar]
- Noonan JP, Grimwood J, Schmutz J, Dickson M, Myers RM. Gene conversion and the evolution of protocadherin gene cluster diversity. Genome Res. 2004;14:354–66. doi: 10.1101/gr.2133704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novak A, Guo C, Yang W, Nagy A, Lobe CG. Z/EG, a double reporter mouse line that expresses enhanced green fluorescent protein upon Cre-mediated excision. Genesis. 2000;28:147–55. [PubMed] [Google Scholar]
- Obata S, Sago H, Mori N, Davidson M, John T, Suzuki ST. A common protocadherin tail: multiple protocadherins share the same sequence in their cytoplasmic domains and are expressed in different regions of brain. Cell Adhes Commun. 1998;6:323–33. doi: 10.3109/15419069809010791. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Gao F, Wu SM. Segregation and integration of visual channels: layer-by-layer computation of ON-OFF signals by amacrine cell dendrites. J Neurosci. 2002;22:4693–701. doi: 10.1523/JNEUROSCI.22-11-04693.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pequignot MO, Provost AC, Salle S, Taupin P, Sainton KM, Marchant D, Martinou JC, Ameisen JC, Jais JP, Abitbol M. Major role of BAX in apoptosis during retinal development and in establishment of a functional postnatal retina. Dev Dyn. 2003;228:231–8. doi: 10.1002/dvdy.10376. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Tanaka H, Frank M, Elste A, Fidler L, Benson DL, Colman DR. Gamma-protocadherins are targeted to subsets of synapses and intracellular organelles in neurons. J Neurosci. 2003;23:5096–104. doi: 10.1523/JNEUROSCI.23-12-05096.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roska B, Werblin F. Vertical interactions across ten parallel, stacked representations in the mammalian retina. Nature. 2001;410:583–7. doi: 10.1038/35069068. [DOI] [PubMed] [Google Scholar]
- Rowan S, Cepko CL. Genetic analysis of the homeodomain transcription factor Chx10 in the retina using a novel multifunctional BAC transgenic mouse reporter. Dev Biol. 2004;271:388–402. doi: 10.1016/j.ydbio.2004.03.039. [DOI] [PubMed] [Google Scholar]
- Serafini T. Finding a partner in a crowd: neuronal diversity and synaptogenesis. Cell. 1999;98:133–6. doi: 10.1016/s0092-8674(00)81008-9. [DOI] [PubMed] [Google Scholar]
- Shapiro L, Colman DR. The diversity of cadherins and implications for a synaptic adhesive code in the CNS. Neuron. 1999;23:427–30. doi: 10.1016/s0896-6273(00)80796-5. [DOI] [PubMed] [Google Scholar]
- Sharma RK, O'Leary TE, Fields CM, Johnson DA. Development of the outer retina in the mouse. Brain Res Dev Brain Res. 2003;145:93–105. doi: 10.1016/s0165-3806(03)00217-7. [DOI] [PubMed] [Google Scholar]
- Stacy RC, Demas J, Burgess RW, Sanes JR, Wong RO. Disruption and recovery of patterned retinal activity in the absence of acetylcholine. J Neurosci. 2005;25:9347–57. doi: 10.1523/JNEUROSCI.1800-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeichi M. The cadherin superfamily in neuronal connections and interactions. Nat Rev Neurosci. 2007;8:11–20. doi: 10.1038/nrn2043. [DOI] [PubMed] [Google Scholar]
- tom Dieck S, Brandstatter JH. Ribbon synapses of the retina. Cell Tissue Res. 2006;326:339–46. doi: 10.1007/s00441-006-0234-0. [DOI] [PubMed] [Google Scholar]
- Verhage M, Maia AS, Plomp JJ, Brussaard AB, Heeroma JH, Vermeer H, Toonen RF, Hammer RE, van den Berg TK, Missler M, et al. Synaptic assembly of the brain in the absence of neurotransmitter secretion. Science. 2000;287:864–9. doi: 10.1126/science.287.5454.864. [DOI] [PubMed] [Google Scholar]
- Wahlin KJ, Lim L, Grice EA, Campochiaro PA, Zack DJ, Adler R. A method for analysis of gene expression in isolated mouse photoreceptor and Muller cells. Mol Vis. 2004;10:366–75. [PubMed] [Google Scholar]
- Wang X, Weiner JA, Levi S, Craig AM, Bradley A, Sanes JR. Gamma protocadherins are required for survival of spinal interneurons. Neuron. 2002;36:843–54. doi: 10.1016/s0896-6273(02)01090-5. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Dityatev A, Scheiffele P, Biederer T, Weiner JA, Christopherson KS, El-Husseini A. Cell adhesion molecules in synapse formation. J Neurosci. 2004;24:9244–9. doi: 10.1523/JNEUROSCI.3339-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wassle H. Parallel processing in the mammalian retina. Nat Rev Neurosci. 2004;5:747–57. doi: 10.1038/nrn1497. [DOI] [PubMed] [Google Scholar]
- Weiner JA, Wang X, Tapia JC, Sanes JR. Gamma protocadherins are required for synaptic development in the spinal cord. Proc Natl Acad Sci U S A. 2005;102:8–14. doi: 10.1073/pnas.0407931101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–39. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Q, Maniatis T. A striking organization of a large family of human neural cadherin-like cell adhesion genes. Cell. 1999;97:779–90. doi: 10.1016/s0092-8674(00)80789-8. [DOI] [PubMed] [Google Scholar]
- Wu Q, Maniatis T. Large exons encoding multiple ectodomains are a characteristic feature of protocadherin genes. Proc Natl Acad Sci U S A. 2000;97:3124–9. doi: 10.1073/pnas.060027397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi T, Takeichi M. Cadherin superfamily genes: functions, genomic organization, and neurologic diversity. Genes Dev. 2000;14:1169–80. [PubMed] [Google Scholar]
- Yamagata M, Sanes JR. Dscam and Sidekick proteins direct lamina-specific synaptic connections in vertebrate retina. Nature. 2008;451:465–9. doi: 10.1038/nature06469. [DOI] [PubMed] [Google Scholar]
- Young RW. Cell death during differentiation of the retina in the mouse. J Comp Neurol. 1984;229:362–73. doi: 10.1002/cne.902290307. [DOI] [PubMed] [Google Scholar]
- Zhang J, Gray J, Wu L, Leone G, Rowan S, Cepko CL, Zhu X, Craft CM, Dyer MA. Rb regulates proliferation and rod photoreceptor development in the mouse retina. Nat Genet. 2004;36:351–60. doi: 10.1038/ng1318. [DOI] [PubMed] [Google Scholar]
- Zou C, Huang W, Ying G, Wu Q. Sequence analysis and expression mapping of the rat clustered protocadherin gene repertoires. Neuroscience. 2007;144:579–603. doi: 10.1016/j.neuroscience.2006.10.011. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.