

Abstract
Glutathione (GSH) and GSH-associated metabolism provide the major line of defense for the protection of cells from oxidative and other forms of toxic stress. Of the three amino acids that comprise GSH, cysteine is limiting for GSH synthesis. Since extracellularly cysteine is readily oxidized to form cystine, cystine transport mechanisms are essential to provide cells with cysteine. Cystine uptake is mediated by system xc−, a Na+-independent cystine/glutamate antiporter. Inhibition of system xc− by millimolar concentrations of glutamate, a pathway termed oxidative glutamate toxicity, results in GSH depletion and nerve cell death. Recently, we described a series of compounds derived from the conjugation of epicatechin with cysteine and cysteine derivatives that protected nerve cells in culture from oxidative glutamate toxicity by maintaining GSH levels. In this paper, we characterize an additional epicatechin conjugate, cysteamine-epicatechin, that is 5-10 fold more potent than the earlier conjugates. In addition, we show that these epicatechin conjugates maintain GSH levels by enhancing the uptake of cystine into cells through induction of a disulfide exchange reaction, thereby uncoupling the uptake from system xc−. Thus, these novel epicatechin conjugates have the potential to enhance GSH synthesis under a wide variety of forms of toxic stress.
Keywords: oxidative stress, cysteine, cystine, cysteamine, epicatechin
Introductory Statement
Glutathione (GSH) and GSH-associated metabolism provide the major line of defense for the protection of cells from oxidative and other forms of toxic stress. GSH can scavenge free radicals, reduce peroxides and be conjugated with electrophilic compounds, thereby eliminating both reactive oxygen species (ROS) and their toxic by-products (Dickinson & Forman 2002, Hayes & McLellan 1999). In addition, the GSSG/GSH redox pair forms the major redox couple in cells and as such plays a critical role in regulating redox-dependent cellular functions (Schafer & Buettner 2001).
GSH is a tripeptide consisting of the amino acids cysteine, glutamate and glycine. Intracellular GSH levels are regulated by a complex series of mechanisms that include substrate availability and transport, rates of synthesis and regeneration, GSH utilization and GSH efflux to extracellular compartments (Meister & Anderson 1983). Because glutamate and glycine occur at relatively high intracellular concentrations, cysteine is limiting for GSH synthesis in many types of cells, including nerve cells (Wu et al. 2004). In the extracellular environment, cysteine is readily oxidized to form cystine, so for most cell types, cystine transport mechanisms are essential to provide them with the cysteine needed for GSH synthesis (Wu et al. 2004).
Cystine uptake in many types of cells is mediated by system xc−, a Na+-independent cystine/glutamate antiporter (Sato et al. 1999). System xc− is a member of the disulfide-linked heteromeric amino acid transporter family and consists of a light chain (xCT) that confers substrate specificity and a heavy chain (4F2hc) that is shared among a number of different amino acid transporters (Wagner et al. 2001). It transports cystine into cells in a 1:1 exchange with glutamate and is thus inhibited by high concentrations of extracellular glutamate (Murphy et al. 1989). The importance of system xc− for the maintenance of GSH levels in cells is demonstrated by the loss of GSH and subsequent cell death seen in nerve and other types of cells following exposure to millimolar concentrations of extracellular glutamate, a pathway termed oxidative glutamate toxicity or oxytosis (Tan et al. 2001). Furthermore, mice that lack system xc− function, either via truncation (Shih et al. 2006) or deletion show brain atrophy and redox imbalance (Sato et al. 2005), respectively.
GSH loss has been directly correlated with cell death following exposure of neural cells to a wide range of insults (Maher 2005). In addition, GSH loss is associated with the cell death seen in a variety of neurological disorders including Parkinson's disease, Alzheimer's disease, and stroke (Bains & Shaw 1997, Maher 2006). Thus, treatments that can maintain GSH levels in the presence of a GSH loss-inducing stress have a significant potential for the treatment of neurological diseases.
Recently, we described a series of compounds derived from the conjugation of the flavonoid epicatechin with cysteine and cysteine-related molecules that were significantly more effective than the underivatized flavonoid at protecting nerve cells in culture from oxidative glutamate toxicity (Torres et al. 2005). These compounds, which were obtained by acid depolymerization of proanthocyanidins in the presence of the thiol (Torres et al. 2002), protect nerve cells by maintaining GSH levels rather than scavenging free radicals through their phenolic moieties (Torres et al. 2005). However, they all had relatively low potencies with EC50 values ranging from 36-60 μM. In the work described below, we characterized an additional epicatechin conjugate, cysteamine-epicatechin (Cya-EC), that is 5-10 fold more potent than any of the earlier conjugates. In addition, we show that this novel epicatechin conjugate, as well as the previously described epicatechin conjugates, maintain GSH levels by enhancing the uptake of cystine/cysteine into cells by a mechanism that uncouples the uptake from system xc−. Thus, these flavonoid conjugates provide a novel mechanism for maintaining cellular redox homeostasis.
Experimental procedures
Materials
Quisqualate was from Tocris. Homocysteic acid, cystine, cysteine, cysteamine, glutamate, 2-aminobicyclo(2,2,1)heptane-2-carboxylic acid (BCH), methylaminoisobutyric acid (MAIBA), l-β-threo-benzyl-aspartate (TBOA), arginine, l-serine and d-serine as well as all other chemicals were from Sigma.
Preparation of the thioconjugates
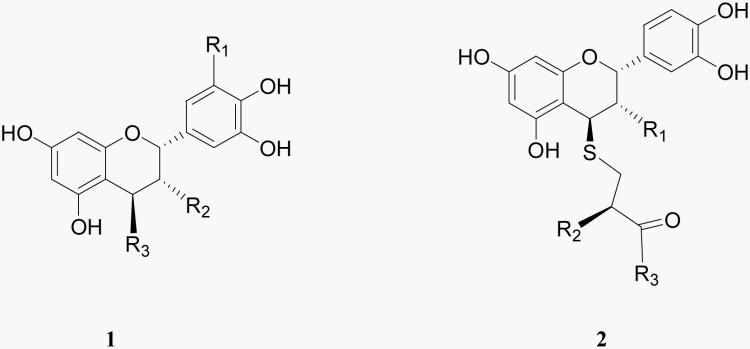
Conjugates of epicatechin, epigallocatechin, epicatechin gallate and epigallocatechin gallate with cysteamine, cysteine or cysteine derivatives were obtained by cleavage of polymeric flavanols (proanthocyanidins) essentially as described (Torres & Bobet 2001, Lozano et al. 2006). The sources of epicatechins and epigallocatechins were grape pomace and witch hazel stems, respectively. Briefly, the plant material was treated with the corresponding thiol in 0.25% aqueous HCl at 90°C for 2h. The resulting conjugates were purified by cation-exchange and reversed-phase liquid chromatography and characterized by electrospray mass spectrometry (ESI-MS), high resolution mass spectrometry (HR-MS) and nuclear magnetic resonance (NMR). ES-MS spectra were recorded on an Agilent 1100/API 3000 (Waldbronn, Germany) system and HR-MS spectra were recorded on a LC/MSD-TOF (Agilent Technologies, Santa Clara, CA) system. 1H-NMR spectra were observed with an Inova-500 MHz (Varian, Palo Alto, CA) apparatus for (CD3)OD and D2O solutions. The analytical data for Cya-EC, Cya-ECG, Cys-EC, ECys-EC and AMCys-EC were measured and compared with those already published (Torres & Bobet 2001, Lozano et al. 2006). The analytical data for Cya-EGC and Cya-EGCG are reported here for the first time.
4ß-(2-aminoethylthio) epigallocatechin (Cya-EGC)
ES-MS positive ions, m/z 382.1 [M + 1]+; LC/MSD-TOF positive ions, m/z 382,09595 calculated for C17H20NO7S [M + H]+ 382.09605; 1H-NMR (CD3)OD, 500 MHz): δ 3.06 (1H, dt J=15, 7 Hz, S-CH2-CH2-NH3+); 3.29 (1H, dt J=15, 5.5 Hz, S-CH2-CH2-NH3+); 3.47-3.58 (2H, m, S-CH2-CH2-NH3+); 4.04 (1H, d J=2.5 Hz, 4-H 3, 4-trans configuration); 4.20 (1H, d J=1.5 Hz, 3-H); 5.28 (1H, s, 2-H); 6.09 (1H, d J=2.0 Hz, 8-H); 6.16 (1H, d J=2.0 Hz, 6-H); 6.72 (2H, m, 2′-H, 6′-H).
4ß-(2-aminoethylthio) epigallocatechin 3-O-gallate(Cya-EGCG)
ES-MS positive ions, m/z 534.1 [M + 1]+; LC/MSD-TOF positive ions, m/z 534.10651 calculated for C24H24NO11S [M + H]+ 534.10701; 1H-NMR (D2O, 500 MHz): δ 2.98 (1H, dt J=15, 7.5 Hz, S-CH2-CH2-NH3+); 3.28 (1H, dt J=15, 5.5 Hz, S-CH2-CH2-NH3+); 3.38-3.41 (2H, m, S-CH2-CH2-NH3+); 4.05 (1H, d J=2.0 Hz, 4-H 3, 4-trans configuration); 5.10 (1H, bs, 3-H); 5.32 (1H, s, 2-H); 5.94 (1H, d J=2.0 Hz, 8-H); 5.96 (1H, d J=2.5 Hz, 6-H); 6.57 (2H, s, 2′-H, 6′-H); 6.84 (2H, s, galloyl-H).
Cell Culture and Viability Assays
HT22 cells were grown on tissue culture dishes in DMEM-high glucose supplemented with 10% FCS as described (Davis & Maher 1994). Primary rat cortical neurons were prepared and grown as described (Maher 2001). Cell viability was determined by a modified version of the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay based on the standard procedure (Hansen et al. 1989). The cells were plated into 96 well dishes at 5 × 103 cells/dish in complete medium and 18 hr later the medium was replaced with DMEM-high glucose supplemented with 7.5% dialyzed FCS (DFCS) and the experimental agents were added. In some experiments, cystine-free DMEM-high glucose supplemented with 7.5% DFCS was used in order to deplete cystine from cells. 24 hr after the addition of the experimental agents, the cell culture medium in each dish was aspirated and replaced with 100 μl DMEM-high glucose with 7.5% DFCS containing 2.5 μg/ml MTT. After 4 hours of incubation at 37°C, the cells were solubilized by the addition of 100 μl of a solution containing 50% dimethylformamide and 20% SDS (pH 4.7). The absorbance at 570 nm was measured on the following day with a microplate reader. Results obtained from the MTT assay correlated directly with the extent of cell death as confirmed visually. Controls employing wells without cells and cells without the experimental agents were used to determine the effects of agents upon the assay chemistry or cell viability, respectively.
Total Intracellular GSH/GSSG
Cells were washed twice with ice-cold PBS, collected by scraping, and lysed with 3% sulfosalicylic acid. Lysates were incubated on ice for 10 minutes and supernatants were collected after centrifugation in an Eppendorf microfuge. Upon neutralization of the supernatant with triethanolamine, the concentration of total glutathione (reduced and oxidized) was determined by the method of Tietze (Tietze 1969) with modifications (Ishige et al. 2001). The protein content of each sample was determined using the BCA Protein Assay kit from Pierce (Rockford, IL) with BSA as a standard.
35S-Cystine Uptake
Cells were plated into 24 well dishes at 3 × 105 cells/well. 24 hr later the cells were rinsed 3 times with sodium-free (sodium replaced with choline chloride) or sodium-containing Hank's Balanced Salt Solution (HBSS) and incubated with 25 μM 35S-cystine (Amersham) for 10-60 min in the same buffer as used for washing. The uptake was terminated by placing the dish on ice and washing 3 times with ice-cold HBSS. The cells were solubilized by overnight incubation in 500 μl 0.2 M NaOH. The radioactivity in a 150 μl aliquot was determined by scintillation counting and normalized to protein determined using the Bradford Assay (Pierce).
Thiol exchange
Stock solutions of Cya-EC and l-Cystine (1 mg/ml) were used. l-Cystine (60 μl) was added to Cya-EC (120 μl) and taken up to a final volume of 5 ml with Hanks balanced solution. The final concentration of each compound was 50 μM. The solution was incubated at 37°C for 50 min. Samples were taken and kept frozen until the HPLC analysis.
Statistical Analysis
All statistical analyses were performed using InStat 3. The results were analyzed for statistically significant differences at the p < 0.05 level using the analysis of variance (ANOVA) test and Tukey's post test for individual group means comparisons. A minimum of three independent experiments were used for statistical analyses.
Results
Similar to the previously described epicatechin conjugates (Torres et al. 2005) (Table 1), cysteamine-epicatechin (Cya-EC) dose dependently protected the HT22 nerve cell line (Fig. 1A) from oxidative glutamate toxicity with an EC50 of ∼ 6 μM. Cya-EC also protected primary neurons from glutamate (Fig. 1B). In addition, Cya-EC protected nerve cells from homocysteic acid and quisqualate (Fig. 1C), two additional compounds that kill nerve cells by blocking cystine uptake through system xc−. The cysteine and cysteine derivative epicatechin conjugates were shown previously to protect cells via the maintenance of GSH levels (Torres et al. 2005). Similarly, as shown in Figure 2A, Cya-EC dose-dependently maintains GSH levels in the presence of glutamate. Even after 8 hr of glutamate treatment when the levels of GSH have fallen below the critical 20% level, HT22 cells treated with 10 μM Cya-EC maintained at least 50% of the control level of GSH (Fig. 2B). The effects of Cya-EC on both GSH levels and cell survival in glutamate-treated cells is blocked by treatment with BSO, an inhibitor of GSH synthesis (Griffith & Meister 1979) (Fig. 2C), demonstrating that Cya-EC acts at or before GSH synthesis to increase GSH levels.
Table 1.
 | ||||
|---|---|---|---|---|
| Compounds | R1 | R2 | R3 | EC50 (glutamate toxicity) |
|
1 4β-(S-aminoethylthio)epicatechin (Cya- EC) |
H | OH | SCH2CH2NH3 + | 6 |
|
1 4β-(S-aminoethylthio)epicatechin-3-O- gallate (Cya-ECG) |
H | gallate | SCH2CH2NH3 + | 9 |
|
1 4β-(S-aminoethylthio)epigallocatechin (Cya-EGC) |
OH | OH | SCH2CH2NH3 + | 12 |
|
1 4β-(S-aminoethylthio)epigallocatechin- 3-O-gallate (Cya-EGCG) |
OH | gallate | SCH2CH2NH3 + | 20 |
| 2 4β-(S-cysteinyl)epicatechin (Cys-EC) | OH | NH3 + | O− | 40 |
|
2 4β-[(S-(O-ethyl-cysteinyl)]epicatechin (ECys-EC) |
OH | NH3 + | OCH2CH3 | 36 |
|
2 4β-[(S-(N-acetyl-O-methyl- cysteinyl)]epicatechin (AMCys-EC) |
OH | NHCOCH3 | OCH3 | 50 |
gallate: 
Figure 1.

(A) Dose dependent effect of Cya-EC on the survival of glutamate-treated HT22 cells. Cells were untreated or treated with increasing concentrations of Cya-EC in the presence of 5 mM glutamate. 24 hr later cell survival was measured by the MTT assay. The results are the average ± SEM of five independent experiments. (B) Cya-EC protects primary cortical cultures from oxidative glutamate toxicity. 2 day rat primary cortical cultures were treated with 5 or 10 mM glutamate alone or in the presence of 25 μM Cya-EC. 24 hr later cell survival was measured by the MTT assay. The results are the average ± SEM of three independent experiments. (C) Cya-EC protects HT22 cells from homocysteic acid (HCA) and quisqualate. Cells were treated with 1 mM HCA or 250 μM quisqualate in the absence or presence of 10 μM Cya-EC. 24 hr later cell survival was measured by the MTT assay. The results are the average ± SEM of three independent experiments.
Figure 2.

(A) Dose dependent effect of Cya-EC on the maintenance of GSH levels in glutamate-treated HT22 cells. Cells were treated with 5 mM glutamate alone or in the presence of increasing concentrations of Cya-EC. GSH levels were measured after 8 hr and normalized to total protein. The results are the average ± SEM of three independent experiments. (B) Time dependent effect of Cya-EC on the maintenance of GSH levels in glutamate-treated HT22 cells. Cells were treated with 5 mM glutamate alone or in the presence of 10 μM Cya-EC. GSH levels were measured after 2-8 hr and normalized to total protein. The results are the average ± SEM of three independent experiments. (C) The glutamate cysteine ligase inhibitor, BSO, blocks the effects of Cya-EC on the maintenance of GSH levels and cell survival. Cells were treated with 5 mM glutamate alone, 5 mM glutamate + 10 μM Cya-EC or 5 mM glutamate + 10 μM Cya-EC in the presence of increasing concentrations of BSO. Cell survival was measured after 24 hr by the MTT assay. GSH levels were measured after 8 hr and normalized to total protein. The results are the average ± SEM of three independent experiments.
To understand how Cya-EC maintains GSH levels, we decided to examine its effects on each of the steps in GSH metabolism, beginning with cystine uptake. Surprisingly, we found that Cya-EC enhanced cystine uptake in a time- (Fig. 3A) and dose-dependent (Fig. 3B) manner. There was an excellent correlation between increases in cystine uptake and increases in GSH levels (Fig. 3C). Furthermore, in contrast to the results with glutamate toxicity, Cya-EC failed to protect cells from cystine depletion (Fig. 3E). In the uptake experiments, Cya-EC was mixed with 35S-cystine and then added directly to the cells for the 60 min uptake time point or after incubation at 37°C for 10-50 min so that while the time for the treatment of the cells with the mixture ranged from 10-60 min, the total incubation time for the mixture was always 60 min. However, unexpectedly, when we began studies to tease out the mechanisms underlying the effect of Cya-EC on cystine uptake and used only a 10 min total incubation time, we saw no effect (data not shown). In contrast, if we pre-incubated Cya-EC with 35S-cystine at 37°C before adding the mixture to the cells for 10 min, then the enhancement of cystine uptake was restored. Indeed, further studies showed a time dependent increase in 35S-cystine uptake following pre-incubation with Cya-EC that reached a plateau after ∼50 min at 37°C (Fig. 3D). These results suggested that there was a direct interaction between Cya-EC and cystine that was not instantaneous and perhaps generated a product that was transported into cells by a pathway distinct from system xc.
Figure 3.

(A) Time dependent effect of Cya-EC on 35S-cystine uptake in HT22 cells. Cells were treated with 25 μM 35S-cystine alone or in the presence of 10 μM Cya-EC. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. Results are presented as cpm taken up per μg cellular protein. Similar results were obtained in three independent experiments. (B) Dose dependent effect of Cya-EC on 35S-cystine uptake in HT22 cells. Cells were treated for 50 min with 25 μM 35S-cystine alone or in the presence of increasing concentrations of Cya-EC. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. Results are presented as cpm taken up per μg cellular protein. Similar results were obtained in three independent experiments. (C) Correlation between the dose dependent effects of Cya-EC on 35S-cystine uptake and GSH maintenance. (D) Effect of pretreatment time on the Cya-EC-mediated increase in 35S-cystine uptake. Cya-EC was added to 35S-cystine immediately before addition to the cells or incubated with 35S-cystine at 37oC for 10-50 min before addition to the cells for 10 min. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. Results are the average ± SEM of three independent experiments. (E) Cya-EC does not protect from cystine deprivation. HT22 cells were treated with 5 mM glutamate or cystine-free DME alone or in the presence of 10 μM Cya-EC. 24 hr later cell survival was measured by the MTT assay. The results are the average ± SEM of three independent experiments.
To examine the fate of Cya-EC upon incubation with cystine we mixed the compounds together in HBSS and analyzed the mixture by reversed-phase high performance liquid chromatography (RP-HPLC) at different times over a total period of 60 min. We observed a decrease in the peak corresponding to Cya-EC and the appearance of a new, small peak corresponding to the cysteine conjugate, Cys-EC (Fig 4). The same behavior was observed with the dimer of O-ethyl-cysteine (data not shown). These results indicate that the cysteamine moiety of Cya-EC can be replaced by other sulfur containing compounds which are present in the form of disulfides.
Figure 4.

RP-HPLC profiles of Cys-EC (A), Cya-EC (B) and the incubation mixture of cystine and Cya-EC in Hank's balanced solution at 37°C at t = 0 (C) and t = 50 min (D). Column: Kromasil 100 C18 (250 × 4 mm i.d.) 5 μm particle size. Eluents: [A] 0.1% (v/v) aqueous TFA; [B] 0.09% TFA in water-acetonitrile (1:4). Gradient elution: 8-30% [B] over 30 min, at a flow rate of 1 ml/min. Load: 90 μl. Detection at 214 nm.
To provide evidence for the idea that sulfur-containing compounds distinct from cystine can be transported into cells upon treatment with Cya-EC, we first looked at the ability of glutamate and quisqualate to inhibit cystine uptake. In control cells, 5 mM glutamate or 250 μM quisqualate, a more specific inhibitor of system xc−, inhibited 35S-cystine uptake by ∼80% (Fig. 5). In contrast, in cells treated with Cya-EC, both glutamate and quisqualate only reduced 35S-cystine uptake by ∼30% (Fig. 5).
Figure 5.

Cysteine but not glutamate or quisqualate inhibit Cya-EC-mediated 35S-cystine uptake. Cells were treated with 25 μM 35S-cystine alone or in the presence of 10 μM Cya-EC along with either 5 mM glutamate, 250 μM quisqualate, 5 mM cysteine or 5 mM cystine, added to the labeling mixture just before addition to the cells for 10 min. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. Results are presented as the percent of uptake in the absence of glutamate, quisqualate, cysteine or cystine. Similar results were obtained in three independent experiments. * indicates significantly different from control.
If the interaction of Cya-EC with cystine results in the generation of cysteine and eventually, a mixed disulfide, cysteine could be taken up by cells through a variety of ubiquitously expressed Na+–independent and –dependent transport systems distinct from system xc− (Wagner et al. 2001, Hyde et al. 2003) while mixed disulfides could be taken up by the Na+-independent system L (Ishii et al. 1981). Indeed, high concentrations of cysteamine have been shown to increase 35S-cystine uptake into cells by this mechanism (Issels et al. 1988). To test this idea, we first looked at the ability of cysteine to block the Cya-EC-dependent increase in 35S-cystine uptake into cells. In these experiments, cysteine was added to the mixture of Cya-EC and 35S-cystine after the 50 min incubation period and immediately before the addition of the mixture to cells to preclude any effects on the interaction between Cya-EC and 35S-cystine. As shown in Figure 5, 5 mM cysteine inhibited by 75% the Cya-EC dependent increase in 35S-cystine uptake but had no effect on 35S-cystine uptake in control cells. In contrast, 5 mM cystine inhibited by ∼85% 35S-cystine uptake in control cells but had a much lesser effect on 35S-cystine uptake in Cya-EC-treated cells (Fig. 5).
To determine which uptake systems for cysteine were responsible for the enhanced Cya-EC-mediated uptake of 35S-cystine, we tested inhibitors of each of the different uptake systems that have been shown to transport cysteine and/or the mixed disulfide for their effects on Cya-EC-stimulated 35S-cystine uptake. Both system asc and system L can transport cysteine by Na+-independent mechanisms (Wagner et al. 2001) and system L can also transport mixed disulfides (Ishii et al. 1981). However, only an inhibitor of system L (BCH) but not an inhibitor of system asc (d-serine) blocked the enhancement of Na+-independent 35S-cystine uptake by Cya-EC (Fig. 6). System A, system ASC, EAATs and system Bo,+ can transport cysteine by Na+-dependent mechanisms (Hyde et al. 2003). However, while an inhibitor of system ASC (l-serine) blocked the enhancement of Na+-dependent 35S-cystine uptake by Cya-EC, inhibitors of system A (methylaminoisobutyric acid (MAIBA)), EAATs (TBOA) and system Bo,+ (arginine) had little or no effect. Although d-serine has been reported to inhibit system ASC, this inhibition is specific for ASCT2 (Shafqat et al. 1993, Utsunomiya-Tate et al. 1996) while ASCT1 is the ASC transporter expressed in neurons (Yamamoto et al. 2004).
Figure 6.

Effects of cysteine transporter inhibitors on Cya-EC-mediated increases in 35S-cystine uptake and the maintenance of GSH levels and cell survival in glutamate-treated HT22 cells. For the measurement of 35S-cystine uptake, cells were treated with 25 μM 35S-cystine in the presence of 10 μM Cya-EC alone or along with 5 mM BCH, 5 mM d-serine, 5 mM arginine, 5 mM serine, 100 μM TBOA or 5 mM MAIBA which were added to the labeling mixture just before addition to the cells for 10 min. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. For the measurement of GSH, cells were treated with 5 mM glutamate alone or in the presence of 10 μM Cya-EC alone or along with 5 mM BCH, 5 mM d-serine, 5 mM arginine, 5 mM serine, 100 μM TBOA or 5 mM MAIBA. GSH levels were measured after 8 hr by a chemical assay and normalized to total protein. For the measurement of cell survival, cells were treated with 5 mM glutamate alone or in the presence of 10 μM Cya-EC alone or along with 5 mM BCH, 5 mM d-serine, 5 mM arginine, 5 mM serine, 100 μM TBOA or 5 mM MAIBA. Cell survival was measured after 24 hr by the MTT assay. Results are the average ± SEM of three independent experiments. * indicates significantly different from Cya-EC alone.
To further validate these findings, we looked at the effects of the different inhibitors of cysteine transport on both Cya-EC-mediated increases in GSH and cell survival in the presence of glutamate. As shown in Figure 6, only the system ASC inhibitor l-serine significantly decreased Cya-EC-mediated increases in GSH levels. l-serine also significantly decreased the effect of Cya-EC on cell survival in the presence of toxic concentrations of glutamate while inhibitors of the other transport systems, d-serine, MAIBA, TBOA and arginine, had little or no effect. BCH by itself increased GSH levels and cell survival in the presence of glutamate and so its effect on Cya-EC-mediated increases in GSH levels and protection from glutamate toxicity could not be evaluated.
If Cya-EC increases 35S-cystine uptake by releasing cysteine from cystine through a mechanism involving the cleavage of cystine with the eventual formation of a mixed disulfide by the departing cysteamine moiety, then we would expect to see similar effects on 35S-cystine uptake and cell survival with equivalent concentrations of cysteamine (Cya). As shown in Figure 7A, this is indeed the case. 10 μM Cya enhances 35S-cystine uptake and this enhancement is inhibited by both l-serine and BCH but not by MAIBA, TBOA, arginine or d-serine (Fig. 7B). Cya also protects cells from oxidative glutamate toxicity and this protection is inhibited by l-serine but not by MAIBA, TBOA, arginine or d-serine (Fig. 7B).
Figure 7.

Effect of cysteamine (Cya) on cell survival (A) and 35S-cystine uptake and cell survival in glutamate-treated HT22 cells (B). For the measurement of cell survival, cells were treated with 5 mM glutamate alone or in the presence of increasing doses of (A) Cya or (B) 10 μM Cya alone or along with 5 mM BCH, 5 mM d-serine, 5 mM arginine, 5 mM serine, 100 μM TBOA or 5 mM MAIBA. Cell survival was measured after 24 hr by the MTT assay. For the measurement of 35S-cystine uptake, cells were treated with 25 μM 35S-cystine in the presence of 10 μM Cya alone or along with 5 mM BCH, 5 mM d-serine, 5 mM arginine, 5 mM serine, 100 μM TBOA or 5 mM MAIBA which were added to the labeling mixture just before addition to the cells for 10 min. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. Results are the average ± SEM of three independent experiments. * indicates significantly different from Cya alone.
Several other EC conjugates have been shown to increase GSH levels and cell survival in the HT22 cells (Torres et al. 2005). In addition, we made additional Cya conjugates including Cya-epicatechin gallate (Cya-ECG), Cya-epigallocatechin (Cya-EGC) and Cya-epigallocatechin gallate (Cya-EGCG) (Table 1), to determine if modification of epicatechin affected the ability of the Cya conjugate to enhance GSH levels and protect cells from oxidative stress. While Cya-ECG was reasonably effective at enhancing GSH levels (Fig. 8B) and protecting HT22 cells from oxidative glutamate toxicity (Fig. 8B), the other modifications led to significant reductions in potency and efficacy. Because the less effective cysteamine derivatives included moieties (ECG, EGC, EGCG) more active than EC at scavenging free radicals, these results confirmed our previous observation that scavenging of mitochondrial ROS does not play a significant role in the protection against oxidative glutamate toxicity by these particular kinds of compounds (Torres et al. 2005). We also looked at the ability of the cysteamine conjugates, as well as the previously described EC conjugates (Cys-EC, AMCys-EC and ECys-EC) (Table 1), to enhance cystine uptake. Using a fixed concentration of 10 μM, we found that Cya-EC was the most effective of all of the EC conjugates at increasing 35S-cystine uptake although several of the other conjugates were also quite good including Cya-ECG (Fig. 8A). In contrast, both Cya-EGC and Cya-EGCG were almost completely ineffective. The effects of the conjugates on 35S-cystine uptake showed a good correlation with their effects on GSH levels strongly suggesting that they increase GSH levels by a mechanism very similar to that of Cya-EC.
Figure 8.

Effect of different EC conjugates on 35S-cystine uptake (A) and the maintenance of GSH levels and cell survival in glutamate-treated HT22 cells (B). For the measurement of 35S-cystine uptake, cells were treated with 25 μM 35S-cystine in the presence of 10 μM of each of the conjugates or EC for 10 min following a 50 min pre-incubation at 37°C. After solubilization of the cells in 0.2 M NaOH, aliquots were taken for scintillation counting and protein determination. For the measurement of GSH, cells were treated with 5 mM glutamate alone or in the presence of 10 μM of each of the conjugates. GSH levels were measured after 8 hr by a chemical assay and normalized to total protein. For the measurement of cell survival, cells were treated with 5 mM glutamate alone or in the presence of 10 μM of each of the conjugates. Cell survival was measured after 24 hr by the MTT assay. Results are the average ± SEM of three independent experiments. * indicates significantly different from glutamate alone.
Discussion
In these studies we describe a novel epicatechin derivative, Cya-EC, that enhances GSH levels and cell survival by increasing cystine uptake into cells. It does so not by increasing the activity of system xc−, but rather by reacting with cystine and releasing cysteine which can react with the leaving cysteamine moiety to form the mixed disulfide Cya-Cys (Fig. 9), thereby allowing these sulfur-containing compounds to enter cells via multiple transport systems. We have preliminary data suggesting that the same reactions occur under physiological conditions. Cya-EC is partially converted into Cys-EC when absorbed and metabolized in the small intestine, probably through reaction with endogenous cystine (unpublished results).
Figure 9.

Proposed mechanism for the conversion of cystine to cysteine and a mixed disulfide by Cya-EC.
Although a similar pathway was described for the enhancement of cystine uptake by cysteamine in CHO cells (Issels et al. 1988) and N-acetyl cysteine (Phelps et al. 1992) in pulmonary artery endothelial cells, relatively high concentrations of cysteamine (0.4 mM) or NAC (1 mM) were required for an effect to be seen in these studies. In contrast, we find that Cya-EC is effective below 10 μM. However, in our system we also see a strong effect of cysteamine at this concentration. Nevertheless, we believe that Cya-EC provides a better option for enhancing cystine uptake into cells than cysteamine for several reasons. First, the presence of the polyphenol group in Cya-EC is likely to modify the ability of the compound to cross the blood-brain barrier or the blood-retina barrier, thereby allowing it access to two tissues, the brain and the eye, where the ability to maintain GSH levels is of utmost importance. Indeed, a glucuronidated and methylated metabolite of epicatechin has been detected in the brains of rats and mice after oral ingestion of the intact polyphenol (El-Mohsen et al. 2002, van Praag et al. 2007). This metabolite may be involved in the retention of spatial memory in mice fed with EC (van Praag et al. 2007). While there could be some problems with Cya-EC crossing membranes due to its charge, we also saw significant effects on 35S-cystine uptake with uncharged cysteine derivatives such as AMCys-EC. Second, solutions of cysteamine tend to oxidize. Cystamine, the oxidized form of cysteamine, is much less effective at promoting cystine uptake (Wood et al. 2007). In contrast, cysteamine in the form of the epicatechin conjugate cannot oxidize because it does not have a free thiol group. Furthermore, Cya-EC is a potent inhibitor of bacterial lipopolysaccharide (LPS)-induced cytokine production (Mitjans et al. 2004), a model for anti-inflammatory activity, with > 80% inhibition seen at 70 μM whereas a similar level of anti-inflammatory activity was only seen in the presence of millimolar concentrations of cysteamine (Ozaki et al. 2007).
The ability to enhance cystine uptake by converting it to cysteine and thereby shifting its uptake to alternative transport systems could have a number of potential benefits. First, as indicated in the Introductory Statement, system xc− is an antiporter, expelling one molecule of glutamate for every molecule of cystine taken up into cells. There are a number of conditions, especially in the brain, where increases in extracellular glutamate are not desirable because of its interaction with ionotropic glutamate receptors. Therefore, a mechanism for increasing cysteine, the rate limiting precursor for GSH synthesis, without increasing extracellular glutamate, could be highly beneficial (e.g. Piani & Fontana 1994, Fogal et al. 2007). Second, when brain glutamate is high, cystine uptake through system xc− is inhibited. The EC derivatives allow cells to bypass this system. Furthermore, since cysteine uptake can occur through a number of different transporters, it is less likely to be impacted by a specific insult. Third, system xc− works in conjunction with glutamate transporters (Lewerenz et al. 2006). Glutamate transporters have been shown to be quite sensitive to oxidative stress (Trotti et al. 1998). Thus, under conditions where it may be critical to increase GSH levels, system xc− may not function efficiently. Cya-EC could provide an alternative for maintaining the cysteine needed for GSH synthesis. Fourth, Cya-EC might be particularly useful under conditions of lactacidosis which occurs during ischemia, head trauma, hyperglycemia and seizures (Lipton 1999). System xc− is impaired under these conditions (Bannai & Kitamura 1981, Koyama et al. 2000), potentially leading to a deficiency in cystine uptake across the blood-brain barrier where xCT expression is highest (Burdo et al. 2006). Cya-EC could ameliorate this impairment and would not have to cross the blood-brain barrier to do so.
Cysteamine is neuroprotective in a variety of in vitro and in vivo models of neurodegenerative diseases (for review see Wood et al. 2007). Although a number of mechanisms underlying this neuroprotection have been proposed, it is likely that an important aspect is the ability of cysteamine to increase intracellular thiol pools. This, in turn, will enhance the synthesis of GSH and may also have additional protective effects (Wood et al. 2007).
The ability of cysteamine to react with cystine to produce cysteine and a mixed disulfide is well known (Meier & Issels 1995). Other thiols share this property (Meier & Issels 1995), but all of these compounds contain a free thiol group. Indeed, oxidation of the thiol or addition of a phosphate group abolished the ability of the compounds to promote cystine uptake (Meier & Issels 1995). It is therefore somewhat surprising that the Cya-EC conjugate is able to promote cystine uptake. However, the studies with the different cysteamine-epicatechin conjugates suggest that epicatechin allows the cysteamine to readily react with disulfides but other epicatechin-related compounds do not appear to be as effective at transferring their attached sulfur-containing moiety.
Using inhibitors of a variety of ubiquitously expressed Na+–independent and –dependent transport systems that are known to transport cysteine into cells, we found that only inhibitors of the Na+–independent system L and the Na+-dependent system ASC altered the increase in cyst(e)ine uptake promoted by treatment of the HT22 cells with Cya-EC. This is somewhat surprising in that other studies that looked at the mechanisms underlying cysteine uptake in nerve cells showed that glutamate transporters (EAATs) provided the major route for transport (Shanker et al. 2001, Chen & Swanson 2003). While EAATs are expressed by the HT22 cells (Lewerenz et al. 2006) both glutamate and the EAAT inhibitor, TBOA, had little or no effect on cyst(e)ine transport. However, since Cya-EC is also able to promote the survival of primary neurons in response to glutamate treatment it is unlikely that the promotion of cysteine transport by Cya-EC is specific to HT22 cells.
In summary, we have characterized an epicatechin derivative that increases GSH synthesis and protects cells from oxidative stress by virtue of its ability to enhance cystine uptake through its conversion to cysteine and eventually a mixed disulfide. This allows uptake through a variety of pathways which are not subject to the same problems and constraints as system xc−. Thus, this derivative has the potential to enhance GSH synthesis under a wide variety of forms of toxic stress.
Acknowledgements
Mass spectrometry analyses were performed at the Servei d'Espectrometria de Mases de la Universitat de Barcelona. The assistance of Dr. Irene Fernández is gratefully acknowledged.
Financial support of the NIH (PM) (grant AG025337) and the Spanish Ministry of Education and Science (JLT) (research grants PPQ2003-06602-C04-01 and AGL2006-12210-C03-02/ALI) is acknowledged.
References
- Bains JS, Shaw CA. Neurodegenerative disorders in humans: the role of glutathione in oxidative stress-mediated neuronal death. Brain Res. Rev. 1997;25:335–358. doi: 10.1016/s0165-0173(97)00045-3. [DOI] [PubMed] [Google Scholar]
- Bannai S, Kitamura E. Role of proton dissociation in the transport of cystine and glutamate in human diploid fibroblasts in culture. J. Biol. Chem. 1981;256:5770–5772. [PubMed] [Google Scholar]
- Burdo J, Dargusch R, Schubert D. Distribution of the cystine/glutamate antiporter system Xc− in the brain, kidney and duodenum. J. Histochem. Cytochem. 2006;54:549–557. doi: 10.1369/jhc.5A6840.2006. [DOI] [PubMed] [Google Scholar]
- Chen Y, Swanson RA. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003;84:1332–1339. doi: 10.1046/j.1471-4159.2003.01630.x. [DOI] [PubMed] [Google Scholar]
- Davis JB, Maher P. Protein kinase C activation inhibits glutamate-induced cytotoxicity in a neuronal cell lines. Brain Res. 1994;652:169–173. doi: 10.1016/0006-8993(94)90334-4. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Forman HJ. Glutathione in defense and signaling. Ann. N.Y. Acad. Sci. 2002;973:488–504. doi: 10.1111/j.1749-6632.2002.tb04690.x. [DOI] [PubMed] [Google Scholar]
- El-Mohsen A, Kuhnle MM, Rechner AR, Schroeter H, Rose S, Jenner P, Rice-Evans CA. Uptake and metabolism of epicatechin and its access to the brain after oral ingestion. Free Rad. Biol. Med. 2002;33:1693–1702. doi: 10.1016/s0891-5849(02)01137-1. [DOI] [PubMed] [Google Scholar]
- Fogal B, Li J, Lobner D, McCullough LD, Hewett SJ. System Xc- activity and astrocytes are necessary for interleukin-1b-mediated hypoxic neuronal injury. J. Neurosci. 2007;27:10094–10105. doi: 10.1523/JNEUROSCI.2459-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith OW, Meister A. Potent and specific inhibition of glutathione synthesis by buthionine sulfoximine (S-n-butyl homocysteine sulfoximine) J. Biol. Chem. 1979;254:7558–7560. [PubMed] [Google Scholar]
- Hansen MB, Nielsen SE, Berg K. Re-examination and further development of a precise and rapid dye method for measuring cell growth/cell kill. J. Immunol. Methods. 1989;119:203–210. doi: 10.1016/0022-1759(89)90397-9. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Rad. Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- Hyde R, Taylor PM, Hundal HS. Amino acid transporters: roles in amino acid sensing and signaling in animal cells. Biochem. J. 2003;373:1–18. doi: 10.1042/bj20030405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishige K, Schubert D, Sagara Y. Flavonoids protect neuronal cells from oxidative stress by three distinct mechanisms. Free Radic. Biol. Med. 2001;30:433–446. doi: 10.1016/s0891-5849(00)00498-6. [DOI] [PubMed] [Google Scholar]
- Ishii T, Bannai S, Sugita Y. Mechanism of growth stimulation of L1210 cells by 2-mercaptoethanol in vitro. J. Biol. Chem. 1981;256:12387–12392. [PubMed] [Google Scholar]
- Issels RD, Nagele A, Eckert K-G, Wilmann W. Promotion of cystine uptake and its utilization for glutathione biosynthesis induced by cysteamine and N-acetylcysteine. Biochem. Pharmacol. 1988;37:881–888. doi: 10.1016/0006-2952(88)90176-1. [DOI] [PubMed] [Google Scholar]
- Koyama Y, Kimura Y, Hashimoto H, Matsuda T, Baba A. L-Lactate inhibits L-cystine/L-glutamate exchange transport and decreases glutathione content in cultured astrocytes. J. Neurosci. Res. 2000;59:685–691. doi: 10.1002/(SICI)1097-4547(20000301)59:5<685::AID-JNR12>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- Lewerenz J, Klein M, Methner A. Cooperative action of glutamate transporters and cystine/glutamate antiporter system Xc- protects from oxidative glutamate toxicity. J. Neurochem. 2006;98:916–925. doi: 10.1111/j.1471-4159.2006.03921.x. [DOI] [PubMed] [Google Scholar]
- Lipton P. Ischemic death in brain neurons. Physiol. Rev. 1999;79:1431–1568. doi: 10.1152/physrev.1999.79.4.1431. [DOI] [PubMed] [Google Scholar]
- Lozano C, Juliá L, Jiménez A, Touriño S, Centelles JJ, Cascante M, Torres JL. Electron transfer capacity of catechin derivatives and influence on the cell cycle and apoptosis in HT29 cells. FEBS J. 2006;273:2475–2486. doi: 10.1111/j.1742-4658.2006.05255.x. [DOI] [PubMed] [Google Scholar]
- Maher P. How protein kinase C activation protects nerve cells from oxidative stress-induced cell death. J. Neurosci. 2001;21:2929–2938. doi: 10.1523/JNEUROSCI.21-09-02929.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 2005;4:288–314. doi: 10.1016/j.arr.2005.02.005. [DOI] [PubMed] [Google Scholar]
- Maher P. Redox control of neural function: Background, mechanisms and significance. Antioxid. Redox Signal. 2006;8:1941–1970. doi: 10.1089/ars.2006.8.1941. [DOI] [PubMed] [Google Scholar]
- Meier T, Issels RD. Promotion of cyst(e)ine uptake. Meth. Enzymol. 1995;252:103–112. doi: 10.1016/0076-6879(95)52013-9. [DOI] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Ann. Rev. Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Mitjans M, Del Campo J, Abajo C, Martinez V, Selga A, Lozano C, Torres JL, Vinardell MP. Immunomodulatory activity of a new family of antioxidants obtained from grape polyphenols. J. Agric. Food Chem. 2004;52:7297–7299. doi: 10.1021/jf049403z. [DOI] [PubMed] [Google Scholar]
- Murphy TH, Miyamoto M, Sastre A, Schnaar RL, Coyle JT. Glutamate toxicity in a neuronal cell line involves inhibition of cystine transport leading to oxidative stress. Neuron. 1989;2:1547–1558. doi: 10.1016/0896-6273(89)90043-3. [DOI] [PubMed] [Google Scholar]
- Ozaki T, Kaibori M, Matsui K, Tokuhara K, Tanaka H, Kamiyama Y, Nishizawa M, Ito S, Okumura T. Effect of thiol-containing molecule cysteamine on the induction of inducible nitric oxide synthase in hepatocytes. J. Parenter. Enteral. Nutr. 2007;31:366–371. doi: 10.1177/0148607107031005366. [DOI] [PubMed] [Google Scholar]
- Phelps DT, Deneke SM, Daley DL, Fanburg BL. Elevation of glutathione levels in bovine pulmonary artery endothelial cells by N-acetylcysteine. Amer. J. Respir. Cell Mol. Biol. 1992;7:293–299. doi: 10.1165/ajrcmb/7.3.293. [DOI] [PubMed] [Google Scholar]
- Piani D, Fontana A. Involvement of the cystine transport system xc- in the macrophage-induced glutamate-dependent cytotoxicity to neurons. J. Immunol. 1994;152:3578–3585. [PubMed] [Google Scholar]
- Sato H, Shiiya A, Kimata M, et al. Redox imbalance in cystine/glutamate transporter-deficient mice. J. Biol. Chem. 2005;280:37423–37429. doi: 10.1074/jbc.M506439200. [DOI] [PubMed] [Google Scholar]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J. Biol. Chem. 1999;274:11455–11458. doi: 10.1074/jbc.274.17.11455. [DOI] [PubMed] [Google Scholar]
- Schafer FQ, Buettner GR. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Rad. Biol. Med. 2001;30:1191–1212. doi: 10.1016/s0891-5849(01)00480-4. [DOI] [PubMed] [Google Scholar]
- Shafqat S, Tamarappoo BK, Kilberg MS, Puranam RS, McNamara JO, Guadano-Ferraz A, Fremeau RT. Cloning and expression of a novel Na+-dependent neutral amino acid transporter structurally related to mammalian Na+/glutamate cotransporters. J. Biol. Chem. 1993;268:15351–15355. [PubMed] [Google Scholar]
- Shanker G, Allen JW, Mutkus LA, Aschner M. The uptake of cysteine in cultured primary astrocytes and neurons. Brain Res. 2001;902:156–163. doi: 10.1016/s0006-8993(01)02342-3. [DOI] [PubMed] [Google Scholar]
- Shih AY, Erb H, Sun X, Toda S, Kalivas P, Murphy TH. Cystine/glutamate exchange modulates glutathione supply for neuroprotection from oxidative stress and cell proliferation. J. Neurosci. 2006;26:10514–10523. doi: 10.1523/JNEUROSCI.3178-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan S, Schubert D, Maher P. Oxytosis: a novel form of programmed cell death. Curr. Top. Med. Chem. 2001;1:497–506. doi: 10.2174/1568026013394741. [DOI] [PubMed] [Google Scholar]
- Tietze F. Enzymic method for quantitative determination of nanogram amounts of total and oxidized glutathione:applications to mammalian blood and other tissues. Anal. Biochem. 1969;106:207–212. doi: 10.1016/0003-2697(69)90064-5. [DOI] [PubMed] [Google Scholar]
- Torres JL, Bobet R. New flavonol derivatives from grape (Vitis vinifera) byproducts. Antioxidant amnioethylthio-flavan-3-ol conjugates from a polymeric waste fraction used as a source of flavanols. J. Agric. Food Chem. 2001;49:4627–4634. doi: 10.1021/jf010368v. [DOI] [PubMed] [Google Scholar]
- Torres JL, Lozano C, Julia L, Sanchez-Baeza FJ, Anglada JM, Centelles JJ, Cascante M. Cysteinyl-flavan-3-ol conjugates from grape procyanidins. Antioxidant and antiproliferative properties. Bioorg. Med. Chem. 2002;10:2497–2509. doi: 10.1016/s0968-0896(02)00127-x. [DOI] [PubMed] [Google Scholar]
- Torres JL, Lozano C, Maher P. Conjugation of catechins with cysteine generates antioxidant compounds with enhanced neuroprotective activity. Phytochem. 2005;66:2032–2037. doi: 10.1016/j.phytochem.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Trotti D, Danbolt C, Volterra A. Glutamate transporters are oxidant-vulnerable: a molecular link between oxidative and excitotoxic neurodegeneration? Trends Pharmacol. Sci. 1998;19:328–334. doi: 10.1016/s0165-6147(98)01230-9. [DOI] [PubMed] [Google Scholar]
- Utsunomiya-Tate N, Endou H, Kanai Y. Cloning and functional characterization of a system ASC-like Na+-dependent neutral amino acid transporter. J. Biol. Chem. 1996;271:14883–14890. doi: 10.1074/jbc.271.25.14883. [DOI] [PubMed] [Google Scholar]
- van Praag H, Lucero MJ, Yeo GW, et al. Plant-derived flavanol (−)epicatechin enhances angiogenesis and retention of spatial memory in mice. J. Neurosci. 2007;27:5869–5878. doi: 10.1523/JNEUROSCI.0914-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner CA, Lang F, Broer S. Function and structure of heterodimeric amino acid transporters. Amer. J. Physiol. Cell Physiol. 2001;281:C1077–C1093. doi: 10.1152/ajpcell.2001.281.4.C1077. [DOI] [PubMed] [Google Scholar]
- Wood PL, Khan MA, Moskal JR. Cellular thiol pools are responsible for sequestration of cytotoxic reactive aldehydes: Central role of free cysteine and cysteamine. Brain Res. 2007;1158:158–163. doi: 10.1016/j.brainres.2007.05.007. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang Y-Z, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J. Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nishizaki I, Nukada T, et al. Functional identification of ASCT1 neutral amino acid transporter as the predominant system for the uptake of L-serine in rat neurons in primary culture. Neurosci. Res. 2004;49:101–111. doi: 10.1016/j.neures.2004.02.004. [DOI] [PubMed] [Google Scholar]