

Abstract
Colon cancer arises through a multistep process involving inactivation of tumor suppressor proteins and activation of oncogene-encoded proteins. Development of colon cancer frequently involves mutation of the APC (adenomatous polyposis coli) tumor suppressor. The activity of the proto-oncogene-encoded Src tyrosine kinase is commonly elevated in colon cancer, with higher activity observed as tumors progress and metastasize. Both APC and Src are multifunctional proteins that have been implicated in the control of cell proliferation, but also as regulators of cytoskeletal changes associated with cell motility and invasion. To investigate the potential for biological cooperativity between APC partial loss-of-function and Src gain-of-function, oncogenic Src was stably expressed in mouse colon epithelial cell lines IMCE (APC+/min) and YAMC (APC+/+). Under permissive growth conditions, these lines are conditionally immortalized through inactivation of p53. Irrespective of the APC genotype or p53 status, oncogenic Src expression led to morphologic transformation associated with loss of cell-cell junctions, cytoskeletal disorganization, and acquisition of invasive properties. However IMCE cells that carry one copy of the mutant APCmin allele exhibited increased capacity for Src-mediated anchorage-independent proliferation as compared to the YAMC cells, and this property was enhanced under permissive growth conditions. β-catenin levels and transcriptional activity were also elevated in the Src-transformed IMCE cells. The selective Src inhibitor, AZD0530, was found to be effective in blocking both cell invasion and anchorage-independent proliferation. These findings suggest that the combined effects of elevated Src activity and APC partial loss-of-function may contribute to the growth of colon tumors.
Keywords: beta-Catenin, Colon Cancer, Haploinsufficiency, Invasiveness
Introduction
Colon cancer arises through a multi-step process of genetic and epigenetic alterations resulting in aberrant functions of tumor suppressors, oncogene-encoded proteins, and proteins involved in maintaining genomic stability [1]. Inactivation of the tumor suppressor gene APC (adenomatous polyposis coli) is an early and prevalent event in the development of human colorectal carcinoma, and the APC protein is regarded as the “gatekeeper” of colorectal tumorigenesis [reviewed in 2, 3]. The APC gene was first identified [4, 5] in patients with Familial Adenomatous Polyposis (FAP), a dominant autosomal disease that results in the formation of multiple colorectal polyps. FAP patients inherit one inactive mutant APC allele, and polyp development is commonly associated with a second somatic mutation or loss of heterozygosity leading to functional loss of the other wild-type (WT) APC allele [6]. APC mutations are also commonly associated with sporadic colorectal cancers, again with the majority showing mutational “hits” to both APC alleles [7]. As a tumor suppressor, the APC protein functions as a scaffold in the canonical WNT pathway by targeting soluble β-catenin for degradation, thus inhibiting β-catenin/TCF-mediated transcription [reviewed in 8]. In colorectal tumors lacking APC mutations, β-catenin mutations resulting in a non-degradable form of the protein have been frequently observed [9]. APC also acts as a regulator of microtubule stability and cytoskeletal organization, and the loss of these functions could negatively impact cell division and migration during tumorigenesis [reviewed in 10].
A small fraction of colorectal tumors may retain one WT APC allele [discussed in 11], and in these cases the single mutant APC allele may contribute to disease pathogenesis in combination with alterations to other genes/proteins that act in pathways related to APC function. Such haploinisufficiency for APC was suggested by a study that found ∼50% reduction of APC transcript levels in a subset of adenomatous polyposis patients without apparent APC mutations [12]. Further evidence for APC haploinsufficiency in tumorigenesis has come from studies using a model of conditionally immortalized colon epithelial cell lines: YAMC [13] (with two WT APC alleles), and IMCE [14] (with one WT APC allele and one mutant APCmin allele). The YAMC/IMCE model has been employed to show synergy between the single APCmin allele with either oncogenic Ras [15] or mutant non-degradable β-catenin [16] in assays for anchorage-independent proliferation and tumor growth.
Another alteration, observed in ∼80% of colon adenocarcinomas, is the enhanced activity of the protooncogene-encoded nonreceptor tyrosine kinase Src [reviewed in 17]. Elevated Src activity is detected in early colonic polyps where it correlates with high malignant potential [18]. Further enhancement of Src activity is observed with progression toward the metastatic phenotype [19], and high Src activity is an indicator of poor clinical prognosis in colon carcinoma [20]. Elevated Src activity can contribute to malignant progression by impacting on multiple receptor signaling centers including cell-cell adhesions, cell-extracellular matrix (ECM) adhesions, and receptor tyrosine kinases [reviewed in 21, 22, 23].
Src has been implicated in the regulation of β-catenin function in epithelial cell-cell junctions. Expression of oncogenic Src with constitutive kinase activity in MDCK kidney epithelial cells is associated with elevated tyrosine phosphorylation of E-cadherin and β-catenin, disruption of cell-cell junctions, and enhanced invasiveness [24]. Phosphorylation of β-catenin Tyr-654 by Src was shown to negatively affect the interaction with E-cadherin and enhance transcriptional activity of β-catenin targets [25, 26].
Given the potential for cross-talk between APC and Src, we used the YAMC/IMCE model to investigate the potential for APC haploinsufficiency in combination with elevated Src signaling in the neoplastic transformation of colonic epithelial cells. Under permissive culture conditions associated with p53 inactivation, both YAMC (APC+/+) and IMCE (APC+/min) cells could be transformed by oncogenic Src expression, with similar morphologic and invasive properties observed. However the Src-transformed IMCE cells, in comparison to the counterpart YAMC cells, exhibited a higher capacity for anchorage-independent growth with increased β-catenin transcriptional activity.
Materials and methods
Materials
Mouse monoclonal anti-phosphotyrosine antibody 4G10 was obtained from Millipore (Billerica, MA). Rabbit polyclonal phosphospecific antibody against Src pTyr416 (mouse Src pTyr-418) was from Cell Signaling Technology (Beverly, MA). Mouse monoclonal antibody clone 327 ascites against Src was a generous gift from Christine Cartwright (Stanford University). Mouse monoclonal anti-pan-actin antibody was from LabVison Corp. (Fremont, CA). Mouse monoclonal antibody against β-catenin was from BD Biosciences (San Jose, CA). Rabbit phosphospecific polyclonal antibody against ERK pThr183/pTyr185 was from Promega Corp. (Madison, WI). Rabbit polyclonal anti-ERK 2 (C14) and mouse monoclonal anti-histone H1 (AE-4) antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit polyclonal antibody against full length APC was a generous gift from Kristi Neufeld (University of Kansas). Secondary antibodies for immunoblotting were goat anti-rabbit AlexaFluor 680 from Molecular Probes (Eugene, OR) and goat anti-mouse IPDye 800 from Rockland Immunochemicals (Gilbertsville, PA). Secondary antibody for immunostaining was goat anti-mouse AlexaFluor 488 (Molecular Probes).
AZD0530, a highly selective inhibitor for Src-family kinases [27], was provided by AstraZeneca (Alderly Park, UK). From a 1 mM stock solution prepared in DMSO, the compound was added to the cell culture media to a final concentration of 1 μM.
Cells and cell culture
Parental YAMC (APC+/+) cells [13] derived from Immortomouse, parental IMCE (APC+/min) cells [14] derived from an F1 Immortomouse/Min mouse hybrid and IMCE cells stably expressing oncogenic Ras [15] were all kindly provided by Bob Whitehead (Vanderbilt University). Stable expression of the oncogenic mouse Src variant Y529F in YAMC and IMCE cells was achieved using the bicistronic retroviral construct pLZRS-MS-SrcF-GFP and sorting for equivalent GFP levels [28]. Control cell lines expressing GFP only were similarly established using the empty pLZRS-MS-IRES-GFP vector.
YAMC and IMCE cells (and derivatives) were routinely cultured in 5% CO2 in RPMI medium 1640 containing L-glutamine and HEPES (Invitrogen-GIBCO, Grand Island, NY) supplemented with 5% FBS (Atlanta Biologicals, Lawrenceville, GA), 1% nonessential aminoacids (GIBCO), 1% insulin-transferin selenium (GIBCO), 1% antimycotic/antibiotic (Mediatech, Herndon, VA), and 5μg/mL plasmocin (InvivoGen, San Diego, CA). Under the standard “permissive” condition, YAMC and IMCE cells were maintained at 33°C and the culture medium was further supplemented with 5 U/ml γ-interferon (Roche Diagnostics, Mannheim, Germany) to achieve expression and activation of temperature-sensitive Simian Virus 40 (SV40) large T-antigen that acts to immortalize the cells through inactivation of p53. To shift to the “restrictive” growth condition, the cells were grown for at least 1 day at 37°C in the absence of γ-interferon.
Immunoblotting
For immunoblotting of total cell lysates, cells cultures under permissive or restrictive conditions, as indicated, were lysed in modified RIPA buffer (50 mM Tris-HCl (pH 7.4), 150 mM NaCl, 5 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 50 mM NaF, 1% aprotinin and 0.1 mM Na3VO4). For immunoblotting of subcellular fractions, cells were lysed in 10 mM HEPES (pH 7.8), 10 mM KCl, 2 mM MgCl2, 0.1 mM EDTA, and 0.5% NP40, and centrifuged at 16,000 g for 30 s to obtain a nuclei-enriched pellet and cytoplasm/membrane-enriched supernatant. The nuclear pellet was further washed in 50 mM HEPES (pH 7.8), 50 mM KCl, 300 mM NaCl, 0.1 mM EDTA, and 10% glycerol to remove adherent debris before a final resuspension in RIPA buffer. The cell lysates and subcellular fractions were finally sheared by passing several times through a 26 gauge needle and insoluble material subsequently cleared by centrifugation at 16,000 g for 10 min. Protein concentration in the cleared lysates was determined using the BCA assay (Pierce Biotechnology, Rockford, IL). Lysates containing 30 μg total protein were used for immunoblot analysis using standard procedures. Immunoreactivity was assessed using the Odyssey Infrared Imaging System (Li-Cor Biosciences, Lincoln, NE). For the initial characterization of Src expression and cellular phosphotyrosine, subconfluent adherent cells were lysed. The analysis of phosphoERK, β-catenin, and nuclear β-catenin was carried out on lysates prepared from cells growing in suspension on polyHEMA-coated dishes under restrictive conditions.
Cell staining
Cells were cultured under permissive or restrictive conditions, as indicated, for 24-48 hr on 10 μg/ml fibronectin-coated coverslips, then fixed for 20 min in 4% paraformaldehyde in immunostaining buffer (20 mM PIPES (pH 7.1), 127 mM NaCl, 5 mM KCl, 1.1 mM NaH2PO4, 0.4 mM KH2PO4, 2 mM MgCl2, 5.5 mM glucose, 1 mM EGTA), and permeabilized for 30 min in 0.1% Triton-X-100, 1% BSA in PBS. After a 1 hr preincubation in PBS containing 1% BSA to reduce nonspecific binding, the cells were stained by incubation with either Alexa-594-phalloidin (0.4 U/ml, Molecular Probes) to visualize F-actin or β-catenin antibody (1.25 μg/ml) followed by the AlexaFlour 488 secondary antibody. Coverslips were mounted using the Prolong Antifade reagent (Molecular Probes) and imaged on a Nikon Eclipse 80i microscope equipped for fluorescence optics. In studies of the effects of Src inhibition, AZD0530 (or DMSO only control) was added to the media 2 hr after the cells were plated on the coverslips.
FITC-gelatin degradation assay
Cells were cultured under permissive or restrictive conditions, as indicated, for 24 hr on FITC-gelatin coated coverslips [29] and then fixed, permeabilized, stained with Alexa-594-phalloidin, and imaged as described above to visualize both F-actin and degradation of the gelatin matrix. AZD0530 (or DMSO only control) was added to the media 2 hr after the cells were plated on the coverslips. For quantification, the fraction of cells associated with degradation areas were scored (200 total cells) in random fields using 400X magnification.
Cell proliferation assays
For analyses of adherent growth, 3,000 cells in 0.5 ml of growth medium were plated in 24-well plates and maintained under either permissive or restrictive conditions. For each time point, cell number was determined as the average from three independent wells harvested by trypsinization and counted using a Coulter Particle Counter. The entire experiment was repeated three independent times to obtain the final growth curves shown in the Results.
For soft-agar colony formation assays, 20,000 cells were suspended in 1 ml of growth medium and 0.4% SeaPlaque agarose (FMC Bioproducts, Philadelphia, PA), then poured into a 35 mm diameter dish over a base layer of 0.8% agarose in RPMI. For the permissive growth condition, 5 U of γ-interferon was added to the top layer before pouring. After solidification at room temperature, plates were incubated for 2 weeks under permissive or restrictive conditions, with fresh media added daily to the top layer. Colonies above 100 μm diameter were counted in 10 random fields using a Zeiss Axiovert 135 inverted microscope and MetaMorph imaging software (Molecular Devices, Downingtown, PA). Graphs display the average of 5 independent experiments, with 3-4 replicates plates per each cell type. Statistical significance for both adherent and soft-agar growth was determined using the non-paired one-tailed Student's t-test.
Anchorage-independent growth on dishes coated with polyhydroxymethacrylate (poly-HEMA, Sigma) prepared as described previously [28]. Thirty thousand cells were plated initially in replicate 60 mm diameter dishes and incubated under restrictive conditions. Every two days, cells from one dish were harvested, trypsinized to disrupt aggregates, then resuspended in RPMI and counted using a Coulter Particle Counter. AZD0530 (1 μM final concentration), or DMSO vehicle only, was added directly to the plates every 48 hr.
Genotyping of APC in cells selected for anchorage-independent growth
IMCE-Src cells were cultivated under the restrictive condition at low density in polyHEMA dishes (3,000 cells per 60 mm dish) for 10 days, then collected and plated on a standard 100 mm dish. After cell attachment, the plate was washed with PBS to remove debris and dead cells and adherent colonies were isolated using cloning disks and expanded to confluence in wells of a 24-well plate. Genomic DNA from expanded colonies, as well as from a pool of polyHEMA-selected cells, was isolated using DNeasy Tissue Kit (QIAGEN, Valencia, CA), and the APC gene was amplified by PCR using primers described by Luongo et al. [30]. The PCR product was purified, digested with HindIII restriction enzyme, and the digestion products were separated on a 4% agarose gel (MetaPhor agarose, BMA Products, Rockland, ME). Restriction products from APCWT and APCmin alleles correspond to 123bp and 144bp, respectively.
TOP-Flash assay
To determine β-catenin transcriptional activity, cells were transiently transfected with Super8XTOPFlash reporter plasmid [31] (provided by Randall Moon) using lipofectamine. This reporter expresses firefly luciferase under control of 8 copies of the β-catenin/TCF binding site. To normalize for transfection efficiencies, cells were co-transfected with plasmid pRL-TK (Promega) that constitutively expresses Renilla luciferase. Briefly, 3.5 x 105 cells were plated in 6-well plates in duplicate and co-transfected with 3.2 μg Super8XTOPFlash and 0.8 μg pRL-TK. Cells were cultured for 48 hr under the permissive condition, then held in suspension on polyHEMA-coated dishes for 3 hr under the restrictive condition before lysis in Passive Lysis Buffer (Promega). Luciferase activities in the lysates were measured using a luminometer and Dual-Glo (Promega) reagents. Firefly luciferase activity was normalized to Renilla activity and the mean normalized values from three independent experiments were determined.
Results
Oncogenic Src promotes similar morphologic transformation in YAMC and IMCE cells
Conditionally immortalized colon epithelial cell lines IMCE and YAMC were employed in this study to investigate the potential for APC haploinsufficiency in combination with elevated Src signaling in the neoplastic transformation of colon epithelial cells. Under standard “permissive” culture conditions, these cells are immortalized by expression of a temperature-sensitive SV40 large T antigen (tsA58) under control of a γ-interferon inducible promoter.
An activated form of mouse c-Src (Src-Y529F) was stably expressed in the normal YAMC and preneoplastic IMCE cell lines using a bicistronic retroviral vector that co-expresses GFP. As controls, cells carrying the empty vector (express GFP only) were similarly prepared. Sorting for equivalent GFP levels yielded “YAMC-SrcF” and “IMCE-SrcF” cell populations that express equivalent levels of the activated Src-Y529F as demonstrated by immunoblot analysis for total Src protein, activated Src (activation loop Tyr-418 phosphorylation site), and total phosphotyrosine (Figure 1). Treatment of YAMC-SrcF and IMCE-SrcF cells with the Src-selective inhibitor AZD0530 (1 μM) substantially reduced the phosphorylation of the Src activation loop site as well as the total cellular phosphotyrosine (Figure 1).
Fig. 1.

Stable expression of Src-Y529F to equivalent levels in IMCE (APC+/min) and YAMC (APC+/+) colonic epithelial cell lines. Following infection with the bicistronic retroviral vector and sorting for equivalent GFP levels, subconfluent cultures of the resulting IMCE-SrcF and YAMC-SrcF cell populations and respective vector-only control cells (grown under permissive conditions) were assessed by immunoblot analysis of total cell lysates (30 μg protein/lane). Replicate blots were used to detect either total cellular phosphotyrosine (pTyr, 4G10 antibody), total Src protein, activated Src with phosphorylated kinase domain activation loop (Src pTyr-418), or actin as a loading control. Eight hr prior to lysis, the IMCE-SrcF and YAMC-SrcF cells were either treated with Src-selective inhibitor AZD0530 (1 μM added in 1 μl DMSO to 1 ml culture medium) or given the DMSO vehicle only. Numbers at left indicate positions of molecular mass markers (in kDa).
Src-Y529F expression resulted in a similar morphologic transformation of YAMC and IMCE cells. Cells expressing the empty vector grew as epithelial sheets associated with prominent cell-cell junctions, as indicated by immunostaining for β-catenin (Figure 2A) or E-cadherin (not shown). In contrast, YAMC-SrcF and IMCE-SrcF cells lacked apparent cell-cell junctions and had a mesenchymal appearance (Figure 2B).
Fig. 2.

Similar morphology and cytoskeletal architecture in IMCE-SrcF and YAMC-SrcF cells. (A,B) Vector-only control and Src-Y529F-expressing (SrcF) cells, as indicated, were fixed 24 hr after plating on fibronectin-coated coverslips and immunostained to detect β-catenin. (C-F) Vector-only control and SrcF cells, as indicated, were fixed 48 hr after plating on fibronectin and F-actin was detected using Alexa-594-phalloidin. For C-F, two hr after plating the cells were either treated with either 1 μM AZD0530 (panel F) or the DMSO vehicle only (panels C-E). Cells were cultured under permissive conditions. The scale bar represents 20 μm.
Oncogenic Src similarly enhances the invasive properties of YAMC and IMCE cells
In fibroblasts, oncogenic Src expression has been associated with the formation of podosomes, actin-rich ventral structures associated with matrix degradation and invasion [reviewed in 32, 33]. To assess podosome formation in the colon epithelial lines, the cells were cultivated on fibronectin-coated coverslips for 48 hr and stained with fluorescent-tagged phalloidin to detect F-actin. In contrast to the control cells where F-actin staining was prominent at the cell-cell junctions and as internal stress fibers (Figure 2C), IMCE-SrcF and YAMC-SrcF cells were observed to similarly form podosomes evident as large rosettes (Figure 2E) and, more typically, as smaller actin-dense puncta (Figure 2D). The podosomes did not form when the cells were plated and maintained in the presence of 1 μM AZD0530 (Figure 2F).
The invasive capacity of the YAMC-SrcF and IMCE-SrcF cells was examined by cultivating the cells for 24 hr on FITC-gelatin coated coverslips, followed by cell staining with fluorescent-tagged phalloidin and visualization of cells and the underlying gelatin matrix by fluorescence microscopy (Figure 3A). While the vector-only control cells were unable to degrade FITC-gelatin, non-fluorescent regions in the matrix were observed under 40-45% of both IMCE-SrcF and YAMC-SrcF cells and this was sensitive to AZD0530 treatment (Figure 3B).
Fig. 3.

Similar matrix-degrading activity in IMCE-SrcF and YAMC-SrcF cells. (A) Representative micrographs of vector-only control and Src-Y529F-expressing (SrcF) cells after 24 hr cultivation at low density on FITC-gelatin coated coverslips. Using two-channel fluorescence microscopy, fixed cells were visualized by Alexa-594-phalloidin staining of F-actin while FITC-gelatin in the same field was also revealed. Matrix degradation is evident as dark spots or patches in the FITC-gelatin layer (arrows) typically observed directly under a cell. The scale bar represents 20 μm. (B) Graph displaying the cell fraction (from 200 scored) associated with matrix degradation. Included in the analysis are IMCE-SrcF and YAMC-SrcF cells treated with either 1 μM AZD0530 or the DMSO vehicle only, all cultured under the permissive condition.
While the above results were obtained from analyzing cells maintained under the permissive growth condition, podosome formation, ECM degradation, and AZD0530 inhibition of these properties were all similarly observed under restrictive growth (data not shown).
IMCE-SrcF cells exhibit enhanced anchorage-independent growth, compared to YAMC-SrcF cells
The proliferative capacity of YAMC-SrcF and IMCE-SrcF cells under adherent and nonadherent conditions was investigated. Adherent growth was measured by growth curves after plating the cells at low density. Adherent growth was minimal under the restrictive condition, with both YAMC-SrcF and IMCE-SrcF cells showing no growth advantage over the control cells during a 12 day period after plating (Figure 4A). Under the permissive condition, after a lag period, the cell lines expanded 2-4 fold faster than under the restrictive condition, as expected (Figure 4B). Under the permissive condition, adherent growth of YAMC-SrcF cells was not significantly enhanced relative to their vector-only control cells. However, the IMCE-SrcF cell population expanded significantly faster than either their control cells or the YAMC-SrcF cells under the permissive condition.
Fig. 4.

Enhanced adherent growth of IMCE-SrcF cells, but not YAMC-SrcF cells, under the permissive growth condition. Growth curves of IMCE-SrcF and YAMC-SrcF cell populations, and respective vector-only control cells, were obtained under the restrictive (A) or permissive (B) culture conditions. Each time point represents the mean cell number from three independent experiments, with bars indicating standard error. At the later time points under the permissive condition the IMCE-SrcF cells reached a significantly higher number in comparison to either the vector-only control cells (p = 0.038 for day 12) or the YAMC-SrcF cells (p = 0.046 for day 10 and p = 0.010 for day 12), as determined by the Student's t-test.
Nonadherent (anchorage-independent) growth was determined using the soft agar assay. Under the permissive condition (Figure 5A) the number of colonies >100 μm diameter, detected after 2 weeks, was significantly higher (∼7.5-fold) in IMCE-SrcF cells compared to their vector-only control cells. In contrast YAMC-SrcF cells formed far fewer colonies, with no significant increase compared to their control cells. Under the restrictive condition, the ability of IMCE-SrcF cells to form large colonies was greatly impaired (Figure 5A, compare open and solid bars), indicating the importance of p53 inactivation in the nonadherent growth. Nevertheless, a small but significant number of colonies were formed by IMCE-SrcF cells maintained under the restrictive condition (Figure 5B), suggesting some circumvention of the growth-suppressive activity of p53.
Fig. 5.
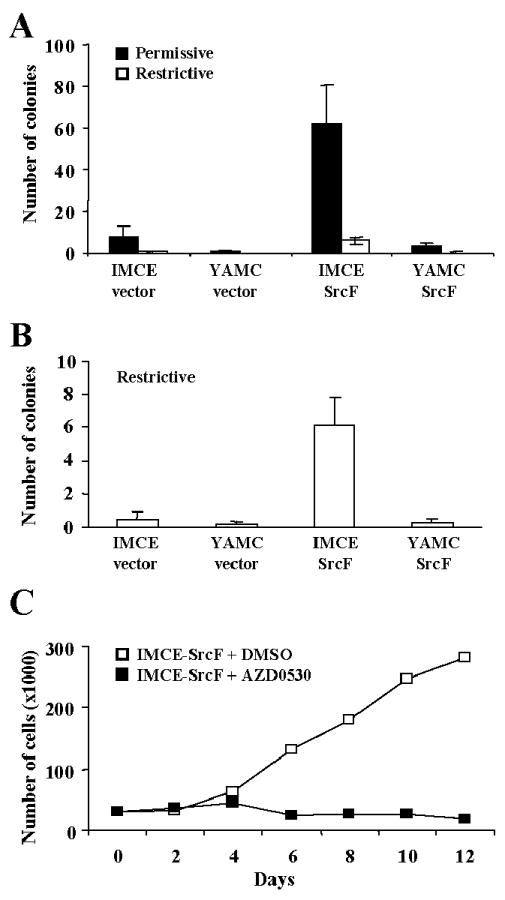
Enhanced anchorage-independent growth of IMCE-SrcF cells, but not YAMC-SrcF cells. The soft agar colony formation assay was carried out under either permissive (A, solid bars) or restrictive (A and B, open bars) culture conditions. Histogram bars represent the average number of large colonies (> 100 μm) observed in five independent experiments, with error bars representing the standard error. The IMCE-SrcF cells formed a significantly higher number of large colonies in comparison to either the vector-only control cells (p = 0.014 for permissive growth, p = 0.002 for restrictive growth) or the YAMC-SrcF cells (p = 0.008 for permissive growth, p = 0.001 for restrictive growth), as determined by the Student's t-test. (C) Anchorage-independent growth of IMCE-SrcF cells on polyHEMA plates is blocked by the Src-selective inhibitor AZD0530. A representative experiment under the restrictive growth condition is shown.
To test whether the anchorage-independent growth of IMCE-SrcF cells was dependent on Src kinase activity, the cells were plated on polyHEMA-coated (non-adherent) dishes and growth curves were determined under restrictive conditions in the presence or absence of AZD0530. IMCE-SrcF cells proliferated in suspension on the polyHEMA dishes, and this was blocked by treatment with 1 μM AZD0530 (Figure 5C).
To rule out the possibility that the capacity for IMCE-SrcF cell anchorage-independent growth was due to loss of the WT APC allele, cells selected through growth on polyHEMA were genotyped using a PCR-based assay. DNA isolated from parental IMCE cells was included as a control. As shown in Figure 6A, the WT APC allele is similarly detected in the polyHEMA-selected IMCE-SrcF and parental IMCE cells, indicating that there was no loss-of-heterozygosity. Immunoblot analysis showed that the expression level of the WT APC protein was also unaffected by SrcF expression (Figure 6B), indicating there is no Src-mediated silencing of the WT APC allele.
Fig. 6.

The WT APC allele is retained in IMCE-SrcF cells selected for anchorage-independent growth. (A) Using a PCR-based strategy, the APC genotype was determined from two different clonal expansions of large cell aggregates harvested after culturing 10 days under the restrictive growth condition on polyHEMA-coated dishes, as well as a larger pool of cell aggregates. A standard adherent culture of parental IMCE cells was genotyped for comparison. Hind III restriction products from the PCR-amplified APCWT and APCmin alleles migrate in the agarose gel according to expected sizes of 123 and 144 bp, respectively. (B) Immunoblot detection of WT APC protein in IMCE-SrcF and YAMC-SrcF cell populations, and respective vector-only control cells. Total cell lysates (100 μg protein/lane) were analyzed. The number at left indicates the position of a 250 kDa molecular mass marker protein.
Distinct signaling pathways are implicated in the anchorage-independent growth capacity of IMCE-Src and IMCE-Ras cells
Reminiscent of our findings with oncogenic Src-Y529F, D'Abaco [15] reported that stable expression of oncogenic H-Ras in IMCE cells was associated with enhanced capacity for anchorage-independent growth as compared to YAMC-Ras cells. The major mitogenic pathway acting downstream of Ras is the Raf > MEK > ERK cascade [reviewed in 34]. Since oncogenic Src may exert its proliferative effect, at least in part, through stimulating Ras and the ERK pathway, it was of interest to determine if the anchorage-independent growth capacity of IMCE-SrcF cells was associated with ERK activation. Immunoblotting with a phosphospecific antibody that recognizes the activated forms of ERK1 and ERK2 revealed that ERK activity is elevated in suspension cultures of IMCE-Ras cells, but not IMCE-SrcF cells, relative to control cells (Figure 7). The basal level of ERK phosphorylation was slightly higher in the IMCE cells in comparison to the YAMC cells, but for both cell types the signal was unchanged by Src-Y529F expression.
Fig. 7.

Increased ERK activation is not associated with the nonadherent growth of IMCE-SrcF cells. IMCE-SrcF, YAMC-SrcF, their vector-only control cells, and IMCE-Ras cells were held in suspension on polyHEMA plates for 24 hr under the restrictive growth condition and evaluated by immunoblotting analysis of whole cell lysates (30 μg protein/lane). ERK1/ERK2 activation was assessed using a phosphospecific antibody recognizing the activation loop phosphorylation sites (pERK, top), with control duplicate blots used to detect total ERK1/ERK2 proteins (middle, note that ERK2 is detected more efficiently by the antibody employed) or actin (bottom).
We also investigated whether the differential capacity of oncogenic Src to promote anchorage-independent growth of IMCE and YAMC cells could be associated with increased β-catenin signaling. This might be anticipated if β-catenin is displaced from the cell-cell junctions as a result of oncogenic Src activity and is more stable in IMCE cells due to the reduced APC function. As expected, β-catenin tyrosine phosphorylation was elevated in both YAMC-SrcF and IMCE-Src cells relative to their vector-only controls and IMCE-Ras cells (data not shown). However, total and nuclear β-catenin levels are clearly elevated in IMCE-Src cells compared to YAMC-SrcF, IMCE-vector, and IMCE-Ras cells (Figure 8 A-C). Moreover, the TOPFlash reporter assay indicated that β-catenin transcriptional activity is significantly elevated in IMCE-SrcF cells compared to the other cell types (Figure 8D).
Fig. 8.

Nonadherent growth of IMCE-SrcF cells is associated with enhanced β-catenin signaling. (A-C) Immunoblot analysis of β-catenin in IMCE-SrcF, YAMC-SrcF, their vector-only control cells, and IMCE-Ras cells after holding in suspension on polyHEMA plates for 24 hr under the restrictive growth condition. (A) Representative blot detecting β-catenin from total cell lysates (30 μg protein/lane) (top) with control duplicate blot detecting actin (bottom). (B) Histogram plot representing the average β-catenin band intensity from total cell lysates, normalized to the actin loading control, from five independent experiments. IMCE-SrcF cells have a significantly higher total β-catenin level in comparison to either vector-only control cells (p = 0.042) or YAMC-SrcF cells (p = 0.001), as determined by the Student's t-test. (C) Blot detecting β-catenin in nuclear and cytoplasmic/membrane fractions, with control duplicate blots used to detect actin (middle) and histone H1 (bottom). (D) Assay of β-catenin transcriptional activity using the TOPFlash reporter system. Cells were held in suspension for 3 hr under the restrictive culture condition prior to analysis. Histogram bars represent the mean normalized β-catenin/TCF activity from three independent experiments, with bars indicating standard error. IMCE-SrcF cells have significantly higher β-catenin transcriptional activity in comparison to either vector-only control cells (p = 0.0005), YAMC-SrcF cells (p = 0.0048) or IMCE-Ras cells (p = 0.0014), as determined by the Student's t-test.
Discussion
Among the prominent molecular lesions associated with colon cancer development and progression are loss of function of the APC tumor suppressor and elevation of Src activity. To gain possible new insight into the effects of elevated Src activity on the development and progression of colorectal cancer, we investigated the capacity of oncogenic Src to confer morphologic and proliferative transformation to two mouse colonic epithelial cell lines that differ with regard to APC genotype. We found that for both normal YAMC (APC+/+) and preneoplastic IMCE (APC+/min) cell lines, expression of activated Src-Y529F equally gave rise to morphologic transformation associated with loss of cell-cell adhesion, formation of invasion-associated podosomes, and acquisition of the capacity to degrade the ECM. The presence or absence of functional p53, regulated through culture under restrictive or permission conditions, respectively, also had no apparent effect on these properties. However Src-transformed IMCE cells much more efficiently formed colonies in soft agar, as compared to Src-transformed YAMC cells, and this capacity for anchorage-independent growth was further enhanced by p53 inactivation. Oncogenic Src expression and p53 inactivation in the APC+/min background also afforded a proliferative advantage under adherent growth conditions.
While the ability of constitutively active Src to confer morphologic and proliferative transformation to epithelial cells has been well established, the mechanism(s) involved are not well understood, likely to be complex, and may differ in different epithelial cell types. In studies of RIE-1 rat intestinal epithelial cells, oncogenic Src-mediated anchorage-independent growth appeared to be largely independent of Ras (in contrast to studies of Src-transformed fibroblasts), MEK, or PI3K signaling [35, 36]. Another study, however, indicated a more critical role for Ras signaling in Src-induced anchorage-independent growth of a human gallbladder epithelial cell line [37]. We examined the possible role of the Ras/ERK pathway in the anchorage-independent growth capacity of Src-transformed YAMC and IMCE cells by assessing ERK activation in suspension cultures. In contrast to Ras-transformed IMCE cells, ERK activation was not apparent in either Src-transformed population. Thus elevated ERK signaling does not appear to be responsible for the observed Src-mediated anchorage-independent growth of these cells.
Cell-cell adherens junctions are a major site of Src localization within epithelial cells, and Src-mediated tyrosine phosphorylation can result in cadherin/β-catenin complex dissociation and disruption of cell-cell junctions [24, 38, 39]. Thus it is quite plausible that Src activity promoting release of β-catenin from its complex with E-cadherin, in combination with reduced APC function allowing the free β-catenin to escape the fate of rapid degradation, could contribute to the observed growth cooperativity of oncogenic Src and the APCmin allele. We found that, indeed, β-catenin signaling was elevated in IMCE-SrcF compared to YAMC-SrcF cells as measured by both nuclear localization and a transcriptional reporter assay in experiments carried out under the restrictive condition. Regulation of growth control genes, e.g. MYC (c-Myc) and CCND1 (cyclin D1), by the β-catenin/TCF complex could thus be a contributing factor to the enhanced growth properties of IMCE-SrcF cells, at least in the presence of functional p53. Additional studies involving functional impairment of β-catenin in these cells could reveal the importance of this signaling pathway in tumorigenic growth.
APC has functions in addition to its classical role as a negative regulator of β-catenin, including the regulation of epithelial cell polarity and migration through control of the actin cytoskeleton and microtubule stabilization [reviewed in 40]. Loss of these cytoskeletal functions in APC-deficient cells could further contribute to colon cancer progression towards invasiveness [8, 10]. While it is thus conceivable that the ability of oncogenic Src to disrupt cell-cell adhesion and promote invasive behavior could be impacted by APC deficiency, our study found no apparent difference in these aspects of neoplastic transformation between YAMC and IMCE cells expressing Src-Y529F. The capacity of oncogenic Src to promote tumorigenesis and invasiveness in a human adenoma-derived cell line has been demonstrated previously [41], but this study did not investigate the potential for oncogenic Src to combine with APC deficiency in promoting the neoplastic properties. In our study, the effect of oncogenic Src on the invasiveness of colon epithelial cells lines was also independent of p53 status.
There has been renewed interest in Src as a therapeutic target in the treatment of cancer including colorectal cancer [23, 42]. In the course of our studies, we further evaluated AZD0530, a highly selective inhibitor of Src-family kinases [27]. Our findings that a low dose (1 μM) of AZD0530 effectively reverted the invasive phenotype/behavior of the Src-transformed colon epithelial cells (independent of either APC genotype or the functional state of p53) lend additional support to the notion that pharmacologic Src inhibition may have a therapeutic benefit in treating the metastatic spread of colon cancer. Furthermore, our findings of cooperativity between oncogenic Src activity and APC deficiency in promoting anchorage-independent growth suggest that Src inhibition may also have a benefit in reducing colon tumor growth.
In conclusion, we presented evidence for a cooperative mechanism that may contribute to tumorigenic growth of colon epithelial cells involving the combined effects of three molecular lesions commonly observed in colon cancer: elevated Src signaling, partial-loss of APC function, and p53 inactivation. We also demonstrated the ability of oncogenic Src signaling to confer invasive characteristics to colon epithelial cells, irrespective of the functional states of APC and p53.
Acknowledgments
We are grateful to Bob Whitehead for cell lines and helpful discussions, Bob Coffey for advice and support, Jeff Franklin for help with soft agar colony size measurements, Sergey Ryzhov, Kristi Neufeld and Christine Cartwright for antibody reagents, Ethan Lee and Curtis Thorne for TOP-Flash assay constructs and Larisa Ryzhova for excellent technical assistance. Supported by NIH grant R01 GM-49882, GI-SPORE grant 2P50 CA95103-06, and a contract from Astrazeneca UK, Ltd.
References
- 1.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 2.Fodde R, Smits R, Clevers H. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 2001;1:55–67. doi: 10.1038/35094067. [DOI] [PubMed] [Google Scholar]
- 3.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87:159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 4.Groden J, Thliveris A, Samowitz W, et al. Identification and characterization of the familial adenomatous polyposis coli gene. Cell. 1991;66:589–600. doi: 10.1016/0092-8674(81)90021-0. [DOI] [PubMed] [Google Scholar]
- 5.Nishisho I, Nakamura Y, Miyoshi Y, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253:665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 6.Levy DB, Smith KJ, Beazer-Barclay Y, Hamilton SR, Vogelstein B, Kinzler KW. Inactivation of both APC alleles in human and mouse tumors. Cancer Res. 1994;54:5953–5958. [PubMed] [Google Scholar]
- 7.Rowan AJ, Lamlum H, Ilyas M, et al. APC mutations in sporadic colorectal tumors: A mutational “hotspot” and interdependence of the “two hits”. Proc Natl Acad Sci USA. 2000;97:3352–3357. doi: 10.1073/pnas.97.7.3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schneikert J, Behrens J. The canonical Wnt signaling pathway and its APC partner in colon cancer development. Gut. 2007;56:417–425. doi: 10.1136/gut.2006.093310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sparks AB, Morin PJ, Vogelstein B, Kinzler KW. Mutational analysis of the APC/beta-catenin/Tcf pathway in colorectal cancer. Cancer Res. 1998;58:1130–1134. [PubMed] [Google Scholar]
- 10.Nathke I. Cytoskeleton out of the cupboard: colon cancer and cytoskeletal changes induced by loss of APC. Nature Rev Cancer. 2006;6:967–974. doi: 10.1038/nrc2010. [DOI] [PubMed] [Google Scholar]
- 11.Segditsas S, Tomlinson I. Colorectal cancer and genetic alterations in the Wnt pathway. Oncogene. 2006;25:7531–7537. doi: 10.1038/sj.onc.1210059. [DOI] [PubMed] [Google Scholar]
- 12.Venesio T, Balsamo A, Rondo-Spaudo M, Varesco L, Risio M, Ranzani GN. APC haploinsufficiency, but not CTNNB1 or CDH1 gene mutations, accounts for a fraction of familial adenomatous polyposis patients without APC truncating mutations. Lab Invest. 2003;12:1859–1866. doi: 10.1097/01.lab.0000106722.37873.8d. [DOI] [PubMed] [Google Scholar]
- 13.Whitehead RH, VanEeden PE, Noble MD, Ataliotis P, Jat PS. Establishment of conditionally immortalized epithelial cell lines from both colon and small intestine of adult H-2Kb-tsA58 transgenic mice. Proc Natl Acad Sci USA. 1993;90:587–591. doi: 10.1073/pnas.90.2.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whitehead RH, Joseph JL. Derivation of conditionally immortalized cell lines containing the Min mutation from the normal colonic mucosa and other tissues of an “Immortomouse”/Min hybrid. Epithelial Cell Biol. 1994;3:119–125. [PubMed] [Google Scholar]
- 15.D'Abaco GM, Whitehead RH, Burgess AW. Synergy between APC min and an activated ras mutation is sufficient to induce colon carcinomas. Mol Cell Biol. 1996;16:884–891. doi: 10.1128/mcb.16.3.884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wagenaar RA, Crawford HC, Matrisian LM. Stabilized beta-catenin immortalizes colonic epithelial cells. Cancer Res. 2001;61:2097–2104. [PubMed] [Google Scholar]
- 17.Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Canc Metas Rev. 2003;22:337–358. doi: 10.1023/a:1023772912750. [DOI] [PubMed] [Google Scholar]
- 18.Cartwright CA, Meisler AI, Eckhart W. Activation of pp60c-src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci USA. 1990;87:558–562. doi: 10.1073/pnas.87.2.558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Talamonti MS, Roh MS, Curley SA, Gallick GE. Increase in activity and level of pp60c-src in progressive stages of human colorectal cancer. J Clin Invest. 1993;91:53–60. doi: 10.1172/JCI116200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Allgayer H, Boyd DD, Heiss MM, Abdalla EK, Curley SA, Gallick GE. Activation of Src kinase in primary colorectal carcinoma: an indicator of poor clinical prognosis. Cancer. 2002;94:344–351. doi: 10.1002/cncr.10221. [DOI] [PubMed] [Google Scholar]
- 21.Avizienyte E, Frame MC. Src and FAK signalling controls adhesion fate and the epithelial-to-mesenchymal transition. Curr Op Cell Biol. 2005;17:542–547. doi: 10.1016/j.ceb.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 22.Thomas SM, Brugge JS. Cellular functions regulated by Src family kinases. Ann Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 23.Yeatman TJ. A renaissance for SRC. Nature Rev Cancer. 2004;4:470–480. doi: 10.1038/nrc1366. [DOI] [PubMed] [Google Scholar]
- 24.Behrens J, Vakaet L, Friis R, et al. Loss of epithelial differentiation and gain of invasiveness correlates with tyrosine phosphorylation of the E-cadherin/beta-catenin complex in cells transformed with a temperature-sensitive v-SRC gene. J Cell Biol. 1993;120:757–766. doi: 10.1083/jcb.120.3.757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Piedra J, Martinez D, Castano J, Miravet S, Dunach M, de Herreros AG. Regulation of beta-catenin structure and activity by tyrosine phosphorylation. J Biol Chem. 2001;276:20436–20443. doi: 10.1074/jbc.M100194200. [DOI] [PubMed] [Google Scholar]
- 26.Roura S, Miravet S, Piedra J, García de Herreros A, Duñach M. Regulation of E-cadherin/Catenin association by tyrosine phosphorylation. J Biol Chem. 1999;274:36734–36740. doi: 10.1074/jbc.274.51.36734. [DOI] [PubMed] [Google Scholar]
- 27.Hennequin LF, Allen J, Breed J, et al. N-(5-chloro-1,3-benzodioxol-4-yl)-7-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(tetrahydro-2H-pyran-4-yloxy)quinazolin-4-amine, a novel, highly selective, orally available, dual-specific c-Src/Abl kinase inhibitor. J Med Chem. 2006;49:6465–6488. doi: 10.1021/jm060434q. [DOI] [PubMed] [Google Scholar]
- 28.Brabek J, Constancio SS, Shin NY, Pozzi A, Weaver AM, Hanks SK. CAS promotes invasiveness of Src-transformed cells. Oncogene. 2004;23:7406–7415. doi: 10.1038/sj.onc.1207965. [DOI] [PubMed] [Google Scholar]
- 29.Bowden ET, Coopman PJ, Mueller SC. Invadopodia: unique methods for measurment of extracellula matrix degradation in vitro. Meth Cell Biol. 2001;63:613–627. doi: 10.1016/s0091-679x(01)63033-4. [DOI] [PubMed] [Google Scholar]
- 30.Luongo C, Moser AR, Gledhill S, Dove WF. Loss of APC+ in intestinal adenomas from Min mice. Cancer Res. 1994;54:5947–5952. [PubMed] [Google Scholar]
- 31.Veeman MT, Slusarski DC, Kaykas A, Louie SH, Moon RT. Zebrafish prickle, a modulator of noncanonical wnt/fz signaling, regulates gastrulation movements. Curr Biol. 2003;13:680–685. doi: 10.1016/s0960-9822(03)00240-9. [DOI] [PubMed] [Google Scholar]
- 32.Buccione R, Orth JD, McNiven MA. Foot and mouth: podosomes, invadopodia and circular dorsal ruffles. Nat Rev Mol Cell Biol. 2004;5:647–657. doi: 10.1038/nrm1436. [DOI] [PubMed] [Google Scholar]
- 33.Linder S, Aepfelbacher M. Podosomes: adhesion hot-spots of invasive cells. Trends Cell Biol. 2003;13:376–385. doi: 10.1016/s0962-8924(03)00128-4. [DOI] [PubMed] [Google Scholar]
- 34.Dhillon AS, Hagan S, Rath O, Kolch W. MAP kinase signalling pathways in cancer. Oncogene. 2007;26:3279–3290. doi: 10.1038/sj.onc.1210421. [DOI] [PubMed] [Google Scholar]
- 35.Nguyen KT, Wang WJ, Chan JLK, Wang LH. Differential requirements of the MAP kinase and PI3 kinase signaling pathways in Src- versus insulin and IGF-1 receptors-induced growth and transformation of rat intestinal epithelial cells. Oncogene. 2000;19:5385–5397. doi: 10.1038/sj.onc.1203911. [DOI] [PubMed] [Google Scholar]
- 36.Oldham SM, Cox AD, Reynolds ER, Sizemore NS, Coffey RJ, Der CJ. Ras, but not Src, transformation of RIE-1 epithelial cells is dependent on activation of the mitogen-activated protein kinase cascade. Oncogene. 1998;16:2565–2573. doi: 10.1038/sj.onc.1201784. [DOI] [PubMed] [Google Scholar]
- 37.Tokumitsu Y, Nakano S, Ueno H, Niho Y. Suppression of malignant growth potentials of v-Src-transformed human gallbladder epithelial cells by adenovirus-mediated dominant negative H-Ras. J Cell Physiol. 2000;183:221–227. doi: 10.1002/(SICI)1097-4652(200005)183:2<221::AID-JCP8>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 38.Irby RB, Yeatman TJ. Increased Src activity disrupts cadherin/catenin-mediated homotypic adhesion in human colon cancer and transformed rodent cells. Cancer Res. 2002;62:2669–2674. [PubMed] [Google Scholar]
- 39.Daugherty RL, Gottardi CJ. Phospho-regulation of β-catenin adhesion and signaling functions. Physiol. 2007;22:303–309. doi: 10.1152/physiol.00020.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Aoki K, Taketo MM. Adenomatous polyposis coli (APC): a multi-functional tumor suppressor gene. J Cell Sci. 2007;120:3327–3335. doi: 10.1242/jcs.03485. [DOI] [PubMed] [Google Scholar]
- 41.Empereur S, Djelloul S, Di Gioia Y, et al. Progression of familial adenomatous polyposis (FAP) colonic cells after transfer of the src or polyoma middle T oncogenes: cooperation between src and HGF/Met in invasion. Br J Cancer. 1997;75:241–250. doi: 10.1038/bjc.1997.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Summy JM, Gallick GE. Treatment for advanced tumors: Src reclaims center stage. Clin Cancer Res. 2006;12:1398–1401. doi: 10.1158/1078-0432.CCR-05-2692. [DOI] [PubMed] [Google Scholar]