

SUMMARY
Mice lacking HIP/RPL29, a ribosomal modulator of protein synthesis rate, display a short stature phenotype. To understand the contribution of HIP/RPL29 to bone formation and adult whole bone mechanical properties, we examined both developing and adult bone in our knockout mice. Results indicated that bone shortening in HIP/RPL29-null mice is due to delayed entry of chondro-osteoprogenitors into the cell cycle. Structural properties of adult null bones were analyzed by micro-computed tomography. Interestingly, partial preservation of cortical thickness was observed in null males indicating a gender-specific effect of the genotype on cortical bone parameters. Null males, and to a lower extent null females, displayed increased bone material toughness to counteract decreased bone size. This elevation in a bone material property was associated with increased bone mineral density only in null males. Neither male nor female null animals could withstand the same maximum load as gender-matched controls in three point-bending tests, and smaller post-yield displacements (and thus increased bone brittleness) were found for null animals. These results suggest that HIP/RPL29-deficient mice exhibit increased bone fragility due to altered matrix protein synthesis rates as a consequence of ribosomal insufficiency. Thus, sub-efficient protein translation increased fracture risk in HIP/RPL29-null animals. Taken together, these studies provide strong genetic evidence that the ability to regulate and amplify protein synthesis rates, including those proteins that regulate the cell cycle entry during skeletal development, are important determinants for establishment of normal bone mass and quality.
Keywords: ribosomal protein, protein translation, RPL29 knockout, bone growth delay, skeletal fragility
INTRODUCTION
Although indirectly involved in the process of peptidyl transfer, ribosomal proteins (RPs) play important regulatory functions in ribosome biogenesis, peptide bond formation, and protein synthesis rate.1, 2 Recently, RPs have been proposed to be downstream effectors of pathways integrating environmental cues and coordinators of cell cycle progression in the nucleoli.3–5 Additionally, during bone development, high volume protein synthesis requiring ribosomal activity is essential for growth and bone matrix production.
Several chromosomal mutations in RPs or proteins involved in ribosome biogenesis have been associated recently with human birth defects with clinical features that include skeletal growth deficiencies and deformities and bone marrow failure.6 These observations strongly link the fundamental processes of efficient protein synthesis and skeletogenesis. Because of profound effects on embryonic viability, only a few RP mutations have been characterized in postnatal mouse growth.7–9 Recently, our group generated the first viable null mouse mutant model lacking an individual ribosomal protein.10 In these mutants, the loss of the eukaryote-specific ribosomal protein of the 60S large ribosomal subunit, HIP/RPL29, produced a global skeletal growth defect that persisted into adulthood. Interestingly, the HIP/RPL29-null phenotype in mice is phenotypically similar to that in yeast (S. cerevisiae), where the deletion of the RPL29 gene is accompanied by delayed growth, reduced protein biosynthesis rates, and abnormal assembly of the two ribosomal subunits.11 Because null mutants are viable, HIP/RPL29 must play a role in increasing protein translation rates under high demand rather than as a core component of the basic ribosome translational machinery.
During bone growth, rapid increase of translation capacity provides a considerable advantage for increasing tissue quantity by the addition of both cells and matrix. However, bone quality is controlled by the regulated production of matrix proteins and is determined by the relative proportion of organic versus inorganic phases. Imperfections occurring during skeletal formation ultimately disrupt not only bone element organization and size, but also impact bone microstructure affecting the bone’s ability to resist fracture and deformation under high mechanical stress conditions.12, 13 Dietary protein restriction during early life reduces the mechanical competence of cortical bone because of impaired architectural distribution.14 Whereas deficiencies in RPs or ribosome biogenesis affect bone growth patterns, little is known regarding the role of the protein synthesis machinery in determining bone mechanical behavior and material properties. In the present study, we examined the consequences of HIP/RPL29 deficiency on bone formation and geometry and tested the hypothesis that the cell proliferation defects observed in developing HIP/RPL29 null bones result in altered mechanical properties.
MATERIALS AND METHODS
Animals and Statistics
D18.5 (plug=d0.5) embryos and 24 week-old Hip/Rpl29tm2Udel null and control mice (n≥6) of the C57BL6/J background were obtained.10 Wild type (WT) and heterozygous animals did not exhibit distinct phenotypes, and both were used as controls. Statistical analysis was performed using a two-tailed student’s t-test. All animal handling experiments were in accordance with the University of Delaware IACUC approved guidelines.
In vivo proliferation assay and immunodetection
For in vivo proliferation, d18.5 pregnant females were injected intraperitoneally with 100μg bromodeoxyuridine (BrdU)/per gram body weight (Sigma-Aldrich). Two hours later, embryonic hindlimbs were fixed in 10% (w/v) neutral buffered formalin and processed for paraffin sectioning. Sections (5μm) were immunostained using ZYMED BrdU Staining Kit® (Invitrogen, Carlsbad, CA). Distal femoral epiphyses/growth plates were analyzed for the number of BrdU-positive cells/40 μm2 area using Image J® software (NIH, Bethesda, MD). Adjacent sections were stained using a standard Safranin O/Fast Green procedure. For immunostaining, d18.5 hindlimbs were cryosectioned (10 μm), fixed in 4% (v/v) paraformaldehyde in PBS, and stained using polyclonal antibodies against type II collagen (Abcam, Cambridge, MA) or type X collagen (provided by Dr. W. Horton, Shriners Hospital for Children, Portland, OR) at 1:75 and 1:200 dilutions, respectively. Data for multilabel experiments were acquired by confocal microcopy.10, 15
Micro-computed (μCT) tomography
Femurs were scanned at a resolution of 27μm using a GE Healthcare eXplore Locus Pre-Clinical μCT Scanner® (GE Healthcare Bio-Sciences Corp., Piscataway, NJ), and reconstructed images were analyzed using GE’s Microview® software. For cortical measurements, a 3mm×3mm×2mm region of interest (ROI) was segmented at the mid-diaphysis of the femur by using an average threshold value of 2850 that allowed for the separation of mineralized and non-mineralized voxels. Measurements obtained were total cortical and bone marrow areas and volumetric bone mineral density (BMD). For cortical bone, tissue-level mineral density (TMD) values were found to be equivalent to BMD, and thus were not reported. To obtain bending moment of inertia, the cross-section image at the middle plane of the ROI was converted to a binary image, and the center of mass was identified. A Cartesian coordinate system x’oy’ was chosen with the origin at the center of mass so that the moments of inertia, Ix’x’, Iy’y’, and the product of inertia, Ix’y’, could be calculated. Finally, the principal moments of inertia, Ixx, Iyy, were determined with the following formulas:
The mean cortical width (CTW) was determined by measuring the distance between eight pairs of points that were evenly distributed on the periosteal and endosteal surfaces of a cortical slice at mid-shaft. Trabecular data were collected from a free-form ROI generated 250μm below the distal growth plate using the stereology feature of the Microview® software.
Biomechanical testing
Bending tests were performed using a three-point fixture (with a 4.5 mm lower span) on a mechanical MicroTester (Instron 5848, Norwood, MA). The bones were bent in the anterior-posterior (A/P) direction monotonically with a cross head speed of 0.01mm/s until failure.16 Load-displacement curves were constructed and analyzed for stiffness, ultimate load, and post-yield displacement.16 Stiffness, which corresponds to the slope of the linear section of the loading-displacement curve (S), was used to calculate EI (bending rigidity) by means of the simply-supported beam equation that accounts for the error due to shearing displacement.17 The smaller principle moment of inertia, (Ixx, also known as IML) was used in the calculation of the elastic modulus (E) using the equation: E = EI/IML.
RESULTS
Effects of HIP/RPL29 null mutation on developing growth plates
At d18.5, the distal epiphyses/growth plates of HIP/RPL29−/− femora were reduced in length and width compared with those of control littermates (Fig. 1 A, B). The layer of proliferating chondrocytes (P) was shortened when compared to control samples, whereas the reserve (R) and hypertrophic (H) zones remained normal length (Fig. 1 A, B). Although no difference in cellular proliferation was seen in the P zone between HIP/RPL29 null mice and controls, a small but significant decrease was noted in the R zone using in vivo BrdU-labeling (Fig. 1D, p < 0.05). In addition, the layer of proliferating (P) chondrocytes was shortened, and a significant increase in cellular density was seen in the R zone of null embryos compared to control littermates (Fig. 1C, p < 0.001).
Figure 1.
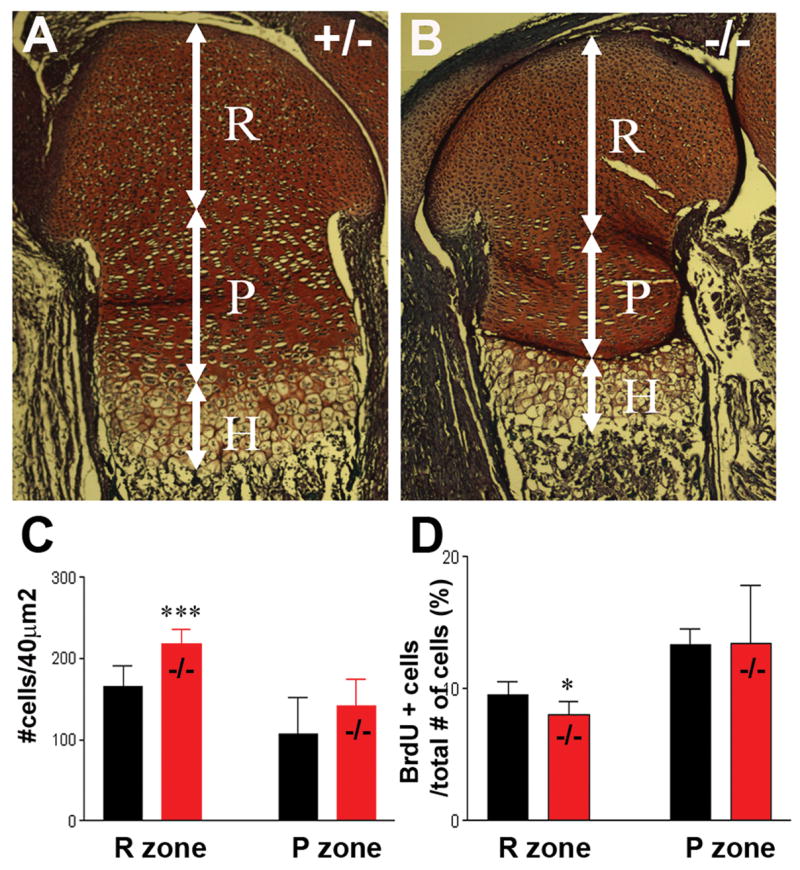
Cell density is increased in the reserve zone of HIP/RPL29 null epiphyses of distal embryonic femurs. Although the epiphysis/growth plates of d18.5 embryos (B) were shorter (white arrows) compared with controls (A), no overall alteration of cartilage organization was observed. However, the layer of proliferating (P) chondrocytes was shortened, and a significant increase in cellular density was seen in the reserve zone (R) of null embryos (C). BrdU-labeling revealed a small but significant decrease in chondroprogenitor proliferation in the R zone of developing nulls bones compared to controls (D). *** and * denote statistical significance differences (p < 0.001 and p < 0.05, respectively).
We examined the expression of cartilage matrix markers in distal femoral growth plates from HIP/RPL29 null embryos and control littermates. Although the total area of type II and X collagen-expressing cells was reduced in HIP/RPL29 nulls due to global reduction of bone size (Fig. 2A–D), the length of the hypertrophic layer was unaffected (arrows in Fig. 2 C–D). Additionally, the in vitro differentiation potential of HIP/RPL29 null progenitors was severely reduced compared to WT controls (Supplementary Fig. 1).
Figure 2.
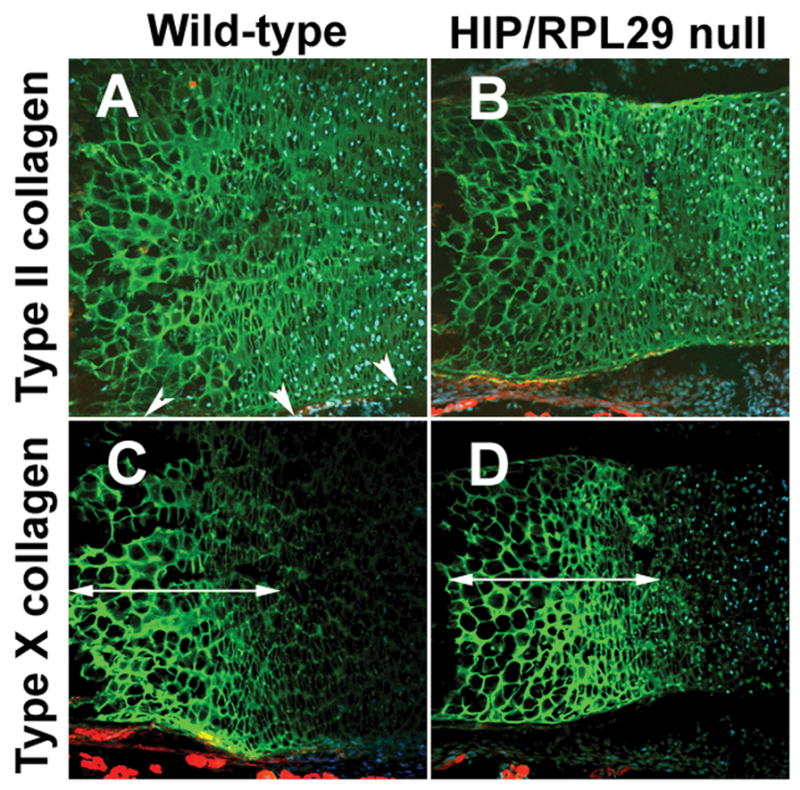
Immunostaining for early and late markers of chondrocyte differentiation on consecutive sections: type II (A, B) and type X collagen (C, D). Arrows (C, D) delineate the length of hypertrophic zones. Arrowheads in A indicate the periphery of developing bone.
Skeletal phenotype in adult HIP/RPL29−/− mice
A significant decrease in total length of both HIP/RPL29-deficient males’ and females’ tibias and femurs (Fig. 3A, −7%, p < 0.05 null vs. control) was accompanied by a 30% decrease in wet weight (Fig. 3B, p < 0.001 null vs. control).
Figure 3.
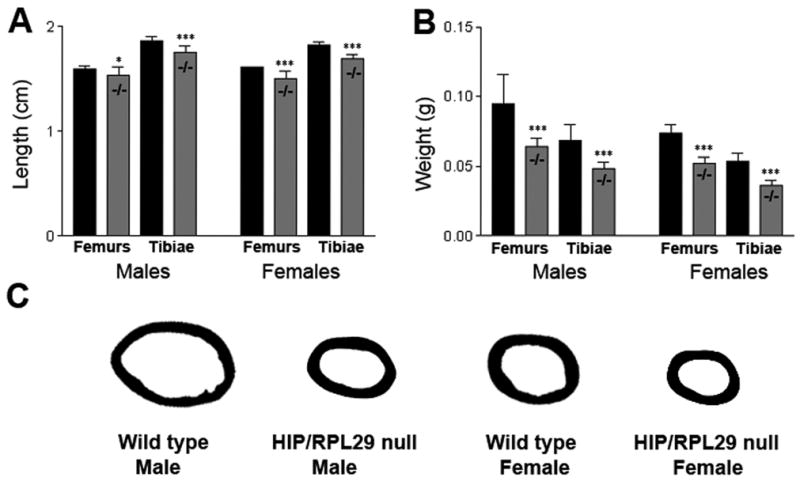
Individual bone measurements showed a 30% decrease in wet bone weight (B) in HIP/RPL29 null (−/−). Also, overall bone length 7% less in HIP/RPL29-deficient bones (A). Representative mid-diaphyseal cross-sections showed a decrease in bone area of mutant animals compared to gender-matched controls (C). Cortical thickness was maintained in null males relative to controls. *** and * denote significant differences (p < 0.001 and p < 0.05, respectively).
Analysis of male femoral mid-shaft cross-sections (Fig. 3C) revealed a severe decrease in bone marrow area in HIP/RPL29 null mutant when compared to controls. A similar observation was made, but to a lesser extent, in age-matched null and control female femurs (Fig. 3C). Accordingly, the inner (−21.3% in males and −9.6% in females, p < 0.05) and outer (−13.9% in males and −9.2% in females, p < 0.05) perimeters of HIP/RPL29 null also were significantly reduced compared with control bones with a more pronounced decrease observed in null males (Table 1A). Consistent with these data, the decrease in bone marrow area was higher in males than in females (Table 1A, −38.1% in males vs. only −17.2% in females) and was accompanied by a non-significant decrease in cortical area in males (Table 1A, −11% in males, p = 0.298 vs. −19% in females, p < 0.05). Interestingly, HIP/RPL29 mutant females had smaller reductions in their external medial-lateral (M/L) and A/P bone diameters compared to males (Table 1A, M/L:−9.9% and A/P:−9.7% in females and M/L:−13.2% and A/P:−14.8% in males). In contrast to null females in which cortical width was decreased (−7.6%, p < 0.05), no significant difference in cortical thickness was found in null males when compared to controls (Table 1A and Fig. 3C). The M/L and A/P moments of inertia (IML and IAP), which indicate bending resistance, were severely decreased in both female (IML,−36.5% and IAP,−36.6%) and male (IML,−49.5% and IAP,−55%) null bones when compared to appropriate control bones (p < 0.05). μCT analysis indicated that BMD of cortical bone was increased in null males (Table 1A, +4.3%, p < 0.05) but not significantly changed in null females when compared to gender-matched controls.
TABLE 1.
Comparative μCT analysis
| A. Cross-sectional cortical properties at femoral mid-shaft | ||||
|---|---|---|---|---|
| Females | Males | |||
| Control | Hip/Rpl29−/− % reduction vs. control | Control | Hip/Rpl29−/− % change vs. control | |
| Outer Perimeter (mm) | 4.67 (+/−0.144) | 4.24 (+/−0.258) −9.2% | 5.53 (+/−0.506) | 4.76 (+/−0.188) −13.9% |
| Inner Perimeter (mm) | 3.03 (+/−0.145) | 2.74 (+/−0.293) −9.6% | 4.08 (+/−0.568) | 3.21 (+/−0.191) −21.3% |
| Marrow Area (mm2) | 0.65 (+/−0.06) | 0.54 (+/−0.12) −17.2% | 1.13 (+/−0.31) | 0.70 (+/−0.07) −38.1% |
| Cortical Area (mm2) | 0.99 (+/−0.05) | 0.80 (+/−0.07) −19.0% | 1.08 (+/−0.22) | 0.96 (+/−0.06) −11.0% n.s. |
| A/P bone diameter (Depth, mm) | 1.556 (+/−0.07) | 1.40 (+/−0.06) −9.7% | 1.76(+/−0.21) | 1.50(+/−0.07) −14.8% |
| M/L bone diameter (Width, mm) | 2.20 (+/−0.06) | 1.98(+/−0.16) −9.9% | 2.68(+/−0.22) | 2.33(+/−0.14) −13.2% |
| Cortical width, (CTW, mm) | 0.30 (+/−0.02) | 0.28 (+/−0.02) −7.6% | 0.30(+/−0.03) | 0.30(+/−0.01) No change. |
| IML(=Ixx, mm4) | 0.277 (+/−0.035) | 0.176 (+/−0.042) −36.5% | 0.463(+/−0.068) | 0.234 (+/−0.048) −49.5% |
| IAP (=Iyy, mm4) | 0.566 (+/−0.068) | 0.359 (+/−0.103) −36.6% | 0.947(+/−0.204) | 0.521(+/−0.074) −55% |
| BMD (mg/cm3) | 883 (+/−21) | 861 (+/−26) −2.5% | 976 (+/−22) | 1018 (+/−19) +4.3% |
| Total tissue area (mm2) | 1.64(+/−0.1) | 1.34(+/0.15) −18.3% | 2.21(+/−0.34) | 1.66(+/−0.12) −24.9% |
| B. Trabecular properties of distal femora | ||||
|---|---|---|---|---|
| Females | Males | |||
| Control | Hip/Rpl29−/− (%change vs. Control) | Control | Hip/Rpl29−/− (%change vs. Control) | |
| BV/TV | 0.177(+/− 0.029) | 0.158(0.028) −10.7% | 0.254(+/−0.029) | 0.302 (+/− 0.041) |
| Trabecular thickness (Tb. Th, μm) | 68 (+/− 9.8) | 59 (+/− 9.9) −12.4% | 71 (+/− 5.8) | 76 (+/−6.2) +7.2% |
| Trabecular number (Tb. N/mm) | 2.65 (+/−0.21) | 2.66 (+/−0.24) No change | 3.62 (+/− 0.48) | 3.96 (+/−0.37) +9.6% |
| Trabecular Separation (Tb. Sp, μm) | 312 (+/− 32.1) | 319 (+/− 35.9) No change | 210 (+/−39.5) | 178 (+/−28.8) −15.1% |
| BMD (mg/cm3) | 247 (+/−23) | 214 (+/−21) −13.4% | 347 (+/−13) | 363 (+/−35) +4.6% n.s. |
| TMD (mg/cm3) | 582 (+/−40) | 529 (+/−40) −9.1% | 533 (+/−71) | 578 (+/−60) +8.4 n.s. |
| Total volume (TV, mm3) | 2.34 (+/−0.12) | 1.96 (+/− 0.19) −16.6% | 3.32 (+/− 0.5) | 2.49 (+/− 0.19) −25% |
change in bold are significant p < 0.05. n.s, non-significant. Data are mean ±std dev.
Compared to controls, HIP/RPL29 null males had a significant increase in trabecular volume (Table 1B, +19%, p < 0.05) with moderate increases in trabecular thickness and number, and a slight decrease in trabecular spacing, while trabecular BMD remained unchanged (Table 1B). In contrast, null females, which did not show dramatic changes in their trabecular bone volume, demonstrated a significant decrease in their trabecular BMD and TMD compared to age and gender-matched control mice (Table 1B, −13.4% and −9.1%, respectively; p < 0.05).
Mechanical testing of adult HIP/RPL29−/− mice
Although HIP/RPL29 null male femora displayed bending rigidity (EI) comparable to controls, HIP/RPL29 null female femurs exhibited a significant decrease of approximately 17% in EI relative to controls (Fig. 4A, p < 0.05). The same observations were made for whole bone stiffness (data not shown). These data are consistent with the absence of change in cortical width between null and control males. However, maintenance in geometrical properties at mid-shaft in null males did not compensate for withstanding comparable forces at failure. Thus, as expected from structural properties, both males and females demonstrated a significant decrease in maximum force (Fig. 4B, −17.5%, p < 0.05 in males and −14.7%, p < 0.05 in females). Because of reduced bone size and concomitant decrease in IML in mutants versus controls (Fig. 4E, −49.4% in males and −36.6% in females, p < 0.05), HIP/RPL29-deficient femora would be weaker and lower energy would be required to bend them to failure if the material properties remained the same. In null males femurs, cortical BMD was increased (Fig. 4D), and post yield displacement (PYD) was significantly smaller (Fig. 4C). Both HIP/RPL29 null males and females exhibited a higher elastic modulus compared to controls (Fig. 4F, +82.9% in males and +36.1% in females, p < 0.05).
Figure 4.

EI, ultimate force, and PYD of HIP/RPL29-deficient and control femurs (A–C). BMD (D) measurement IML were obtained from μCT. The elastic modulus (F) was calculated from EI and IML. *, p < 0.05; n.s., non-significant. Data are mean ±std dev.
DISCUSSION
Disruption of the coordinated processes that link protein synthesis with entry and withdrawal from the cell cycle, hypertrophy, and eventual apoptosis of chondrocytes that occur during endochondral bone growth18 lies at the heart of the skeletal phenotype seen in HIP/RPL29 null mice. When comparing growth plate morphology of HIP/RPL29 null embryos to controls, a reduction in the length of the chondrocytic proliferative zone was seen in null animals. This phenotype agrees with previous observations that HIP/RPL29 is abundant in proliferating chondrocytes of WT growth plate.15 A decrease in the size of the proliferative zone results either from a delay in transitioning from reserve into proliferating chondrocytes or from accelerated terminal differentiation into hypertrophic chondrocytes.19 Because HIP/RPL29 null mice display an increase of cell density in the reserve zone of the growth plate and no change in the size of the hypertrophic zone, we believe that reduction in the overall volume occupied by dividing chondrocytes is due to a slower entry of prechondrocytes into the cell cycle leading to a decrease in the pool of differentiating chondrocytes. Consistent with these in vivo results, previous in vitro work using MEFs demonstrated defects in cell cycle progression and a significant decrease in the percentage of cells transitioning into S phase in null compared to WT cells.10 Recently, the impaired transition of precursor cells into the erythroid lineage in patients with RP gene mutations was attributed to temporal compression of the cellular division cycle.5 Because the nucleoli contains cell cycle machinery components in close vicinity to nascent ribosomes20, we hypothesize that the HIP/RPL29 null mutation induces defects in ribosomal assembly and destabilization of cell cycle machinery co-resident. Ultimately, these early changes in the kinetics of osteogenesis can lead to global bone shortening. Sub-efficient biogenesis of ribosomes also can have secondary effects on ECM production as shown herein using a primary MEF culture system. For these reasons, the HIP/RPL29 null phenotype could be the consequence of a direct negative effect on cell cycle progression and/or sub-optimal protein biosynthesis due to misshapen nascent ribosomes.
Interestingly, the delay in HIP/RPL29-deficient developing growth plate development is not compensated for during post-natal life, and adult HIP/RPL29 null mice display a short stature phenotype.10 Skeletal growth abnormalities were observed in another RP mouse mutant model that displays kinetic defects in ribosome biogenesis.7 Furthermore, genetic variations in RP genes are associated with human syndromes displaying skeletal growth defects.6 These observations support the concept that a strict regulation of RP gene expression is an important determinant for skeletogenesis and the establishment of normal adult bone mass.
Here we demonstrated that the major defects observed in adult HIP/RPL29 null bones were diminished wet weight and concomitant decrease in global geometrical properties. Thus, both null male and female long bones were significantly shorter and displayed smaller cross-sectional parameters when compared to controls. Interestingly, although HIP/RPL29-deficient bones were globally smaller, the cortical bone width of HIP/RPL29 null males was about the same as measured in controls. Thus, gender modulated the effect of the genotype on cortical bone parameters. At six months of age, gender specific effects are usually visible, and male bones are generally considered more robust with better adaptation capabilities than female bones.21 For example, exercise-induced anabolic changes in cortical bone are gender-specific, with males demonstrating the greater response.22 In our study, the trabecular bone region below the growth plate of null females displayed decreased BMD. This observation suggests that a higher bone turnover occurs in null females when compared to gender-matched controls.
As expected, the smaller null bones failed under lower maximum loads compared with control bones. The reciprocal relationship between geometry and material stiffness was documented by others.16 However, a dramatic gender difference was seen in the elastic and post-yield phases of deformation for null bones. Compared to controls, null male bones exhibited compensable bending rigidity and stiffness during elastic deformation and displayed a significantly reduced PYD. In contrast, null female bones showed significant reduction in both bending rigidity and stiffness when compared to controls.
We were surprised to note that HIP/RPL29 deficiency was associated with significantly greater elastic modulus in both males and females. Although the non-uniformed cross-sectional shape may introduce errors in our estimation of E, they are expected to be small. The greater E values for HIP/RPL29 null bones are mainly due to their decreased I. In null male bones, increased toughness compensated for the reduction in bone size and restored overall stiffness to control levels. These observations agree with the finding that HIP/RPL29 null males displayed increased cortical BMD when compared to controls. However, the compensation of E with increasing mineralization usually comes with a price, and null male bones became more brittle.16 In contrast, despite increased E value, female null bones did not display significant changes in BMD and PYD, but nonetheless were weaker than bones of control females.
Bone architecture and tissue composition are genetically co-regulated during growth leading to the construction of sets of bone traits that are well adapted to loading conditions.12 Genetic perturbations that alter these functional interactions during growth can lead to suboptimal adult bone quality.13 Thus, differences in bone growth patterns due to low volume protein synthesis in HIP/RPL29 mutants can explain differences in bone strength and fragility. Elevation of material properties to compensate for decreased geometric properties occurs in other mouse mutant models of molecules involved in endochondral bone growth.23, 24 The underlying mechanisms of the increase in material properties in HIP/RPL29-deficient female femora are unclear. The quantity and quality of hydroxyapatite mineral and type I collagen are controlled by cellular components and non-collagenous proteins that influence their relative contribution to material properties. Strength and stiffness in three-point bending are closely correlated to mineralization, but also collagen density, fiber orientation and cross-linking.25 Thus, differences in the relative distribution of organic versus inorganic phases, rather than absolute quantity of mineral, are likely responsible for the increase in material properties in null females. Changes in mineral crystal size and shape are also important contributors to modulus,26, 27 and these could readily be disrupted by alterations in protein translation.
In summary, our results identify that HIP/RPL29 deficiency, a component of the translational machinery, playing an important role in cell cycle entry and matrix protein synthesis, is associated with elevated bone material properties, but increased fragility. Insights from the HIP/RPL29 mouse model provide new tools for understanding mechanisms underlying gender modulation of bone fragility and fracture risk.
Supplementary Material
Acknowledgments
We give special thanks to Dr. John Novotny for providing access to the Instron mechanical MicroTester. We are grateful to Ms. Julie Mis at the OLAM for her help with animal breeding. This work was supported by the NIH (P20 RR016458-06 and HD25235).
References
- 1.Moritz M, Pulaski BA, Woolford JL., Jr Assembly of 60S ribosomal subunits is perturbed in temperature-sensitive yeast mutants defective in ribosomal protein L16. Molecular and cellular biology. 1991;11:5681–92. doi: 10.1128/mcb.11.11.5681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dresios J, Panopoulos P, Frantziou CP, et al. Yeast ribosomal protein deletion mutants possess altered peptidyltransferase activity and different sensitivity to cycloheximide. Biochemistry. 2001;40:8101–8. doi: 10.1021/bi0025722. [DOI] [PubMed] [Google Scholar]
- 3.Powers T. Ribosome biogenesis: giant steps for a giant problem. Cell. 2004;119:901–2. doi: 10.1016/j.cell.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 4.Pende M. mTOR, Akt, S6 kinases and the control of skeletal muscle growth. Bulletin du cancer. 2006;93:E39–43. [PubMed] [Google Scholar]
- 5.Pederson T. Ribosomal protein mutations in Diamond-Blackfan anemia: might they operate upstream from protein synthesis? Faseb J. 2007;21:3442–5. doi: 10.1096/fj.07-8766hyp. [DOI] [PubMed] [Google Scholar]
- 6.Liu JM, Ellis SR. Ribosomes and marrow failure: coincidental association or molecular paradigm? Blood. 2006;107:4583–8. doi: 10.1182/blood-2005-12-4831. [DOI] [PubMed] [Google Scholar]
- 7.Oliver ER, Saunders TL, Tarle SA, et al. Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development. 2004;131:3907–20. doi: 10.1242/dev.01268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Matsson H, Davey EJ, Draptchinskaia N, et al. Targeted disruption of the ribosomal protein S19 gene is lethal prior to implantation. Molecular and cellular biology. 2004;24:4032–7. doi: 10.1128/MCB.24.9.4032-4037.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Panic L, Tamarut S, Sticker-Jantscheff M, et al. Ribosomal protein S6 gene haploinsufficiency is associated with activation of a p53-dependent checkpoint during gastrulation. Molecular and cellular biology. 2006;26:8880–91. doi: 10.1128/MCB.00751-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kirn-Safran CB, Oristian DS, Focht RJ, et al. Global growth deficiencies in mice lacking the ribosomal protein HIP/RPL29. Dev Dyn. 2007;236:447–60. doi: 10.1002/dvdy.21046. [DOI] [PubMed] [Google Scholar]
- 11.DeLabre ML, Kessl J, Karamanou S, et al. RPL29 codes for a non-essential protein of the 60S ribosomal subunit in Saccharomyces cerevisiae and exhibits synthetic lethality with mutations in genes for proteins required for subunit coupling. Biochimica et biophysica acta. 2002;1574:255–61. doi: 10.1016/s0167-4781(01)00372-4. [DOI] [PubMed] [Google Scholar]
- 12.Price C, Herman BC, Lufkin T, et al. Genetic variation in bone growth patterns defines adult mouse bone fragility. J Bone Miner Res. 2005;20:1983–91. doi: 10.1359/JBMR.050707. [DOI] [PubMed] [Google Scholar]
- 13.Jepsen KJ, Hu B, Tommasini SM, et al. Genetic randomization reveals functional relationships among morphologic and tissue-quality traits that contribute to bone strength and fragility. Mamm Genome. 2007;18:492–507. doi: 10.1007/s00335-007-9017-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alippi RM, Meta MD, Bozzini C, et al. Dynamics of recovery of morphometrical variables and pQCT-derived cortical bone properties after a short-term protein restriction in maturing rats. Growth Dev Aging. 2002;65:67–72. [PubMed] [Google Scholar]
- 15.Miller SA, Brown AJ, Farach-Carson MC, et al. HIP/RPL29 down-regulation accompanies terminal chondrocyte differentiation. Differentiation; research in biological diversity. 2003;71:322–36. doi: 10.1046/j.1432-0436.2003.7106002.x. [DOI] [PubMed] [Google Scholar]
- 16.Jepsen KJ, Pennington DE, Lee YL, et al. Bone brittleness varies with genetic background in A/J and C57BL/6J inbred mice. J Bone Miner Res. 2001;16:1854–62. doi: 10.1359/jbmr.2001.16.10.1854. [DOI] [PubMed] [Google Scholar]
- 17.Schriefer JL, Robling AG, Warden SJ, et al. A comparison of mechanical properties derived from multiple skeletal sites in mice. Journal of biomechanics. 2005;38:467–75. doi: 10.1016/j.jbiomech.2004.04.020. [DOI] [PubMed] [Google Scholar]
- 18.Olsen BR, Reginato AM, Wang W. Bone development. Annual review of cell and developmental biology. 2000;16:191–220. doi: 10.1146/annurev.cellbio.16.1.191. [DOI] [PubMed] [Google Scholar]
- 19.Kronenberg HM. Developmental regulation of the growth plate. Nature. 2003;423:332–6. doi: 10.1038/nature01657. [DOI] [PubMed] [Google Scholar]
- 20.Politz JC, Polena I, Trask I, et al. A nonribosomal landscape in the nucleolus revealed by the stem cell protein nucleostemin. Molecular biology of the cell. 2005;16:3401–10. doi: 10.1091/mbc.E05-02-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tommasini SM, Nasser P, Jepsen KJ. Sexual dimorphism affects tibia size and shape but not tissue-level mechanical properties. Bone. 2007;40:498–505. doi: 10.1016/j.bone.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 22.Wallace JM, Rajachar RM, Allen MR, et al. Exercise-induced changes in the cortical bone of growing mice are bone- and gender-specific. Bone. 2007;40:1120–7. doi: 10.1016/j.bone.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akhter MP, Wells DJ, Short SJ, et al. Bone biomechanical properties in LRP5 mutant mice. Bone. 2004;35:162–9. doi: 10.1016/j.bone.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 24.Maloul A, Rossmeier K, Mikic B, et al. Geometric and material contributions to whole bone structural behavior in GDF-7-deficient mice. Connective tissue research. 2006;47:157–62. doi: 10.1080/03008200600719142. [DOI] [PubMed] [Google Scholar]
- 25.Martin RB, Boardman DL. The effects of collagen fiber orientation, porosity, density, and mineralization on bovine cortical bone bending properties. Journal of biomechanics. 1993;26:1047–54. doi: 10.1016/s0021-9290(05)80004-1. [DOI] [PubMed] [Google Scholar]
- 26.Boskey AL. Assessment of bone mineral and matrix using backscatter electron imaging and FTIR imaging. Current osteoporosis reports. 2006;4:71–5. doi: 10.1007/s11914-006-0005-6. [DOI] [PubMed] [Google Scholar]
- 27.Camacho NP, Rinnerthaler S, Paschalis EP, et al. Complementary information on bone ultrastructure from scanning small angle X-ray scattering and Fourier-transform infrared microspectroscopy. Bone. 1999;25:287–93. doi: 10.1016/s8756-3282(99)00165-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.