

Abstract
Botulinum neurotoxins (BoNTs), etiological agents of the deadly food poisoning disease botulism, are the most toxic proteins currently known. Although only a few hundred cases of botulism are reported in the United States annually, there is growing interest in BoNTs attributable to their potential use as biological warfare agents. Neurotoxicity results from cleavage of the SNARE complex proteins of the presynaptic vesicles by the BoNT light chain subunit, a Zn(II) endopeptidase. Few effective inhibitors of BoNT/A LC activity are known and the discovery process is hampered by the lack of an efficient high throughput assay for screening compound libraries. To alleviate this bottleneck we have synthesized the peptide SNAPtide (FITC-T(D)RIDQANQ(Ψ)RATK(εDABCYL)Nle, and have developed a robust assay for the high throughput evaluation of BoNT/A LC inhibitors. Key aspects for the development of this optimized assay include; the addition of a series of detergents, co-solvents and salts including 0.01% w/v Tween 20 to increase BoNT/A LC catalysis, stability and ease of small molecule screening. To evaluate the effectiveness of the assay a series of hydroxamate-based small molecules were synthesized and examined with BoNT/A LC. The methodology described is superior to other assays reported to date for the high throughput identification of BoNT/A inhibitors.
Introduction
Botulinum neurotoxin (BoNT), an agent responsible for the deadly food poisoning disease botulism and a dreaded biological weapon, is one of the most toxic proteins currently known (~100 billion times more toxic than cyanide).1 Clostridium botulinum is classified into seven strains (A–G) each of which can cause flaccid muscle paralysis and subsequent death by blocking the release of a neurotransmitter, acetylcholine, at neuromuscular junctions. Structurally, BoNT consists of three functional domains; catalytic, translocation, and binding;2 BoNT toxicity results from the catalytic activity of its light chain, a Zn(II) endopeptidase.
The catalytic domain of BoNT is a compact globule consisting of a mixture of α-helices, β-sheets and strands with a gorge-like zinc containing metalloprotease active site (15–20Å deep depending on serotype).3 The metalloprotease activity is responsible for BoNT’s neurotoxicity through the hydrolytic cleavage of one of three SNARE (soluble NSF-attachment protein receptor) proteins that are involved in neuronal synaptic vesicle function. Moreover, the hydrolytic cleavage sites of these SNARE proteins (SNAP-25, VAMP, Sb-1) differ across the BoNT serotypes; however, any degradation of these SNARE proteins disables the exocytosis of acetylcholine, resulting in paralysis and potentially death. Current therapy for BoNT intoxication involves “passive immunization” with equine antitoxin.4 Unfortunately, treatment must start shortly after intoxication, and several safety concerns exist over the use of antitoxins in the general population.5 Therefore, inhibition of the catalytic light chain protease with a small molecule inhibitor may provide an attractive approach to counter the effects of botulism poisoning.
BoNT serotype A (BoNT/A) is the most toxic of the BoNT serotypes and it is considered the most threatening due to a prolonged half-life in vivo and ease of its production.5 While there are reports of success treating BoNT toxicity with multiple monoclonal antibodies as antitoxins,6,7 this is of limited therapeutic utility since the antibodies must be administered prior to, or shortly after, toxin exposure (<12 hrs).
Presently, there are only modest protease inhibitors for BoNT/A with IC50 values in the range of > 20 μM.8–13 Possibly, the lack of potent inhibitors for BoNT/A may be attributed to not only the deficiency but also the reliability of readily available high-throughput assays suitable to screen libraries of compounds. A reliable high-throughput assay is required to identify small molecule inhibitors for BoNT/A LC. To our knowledge, few protease assays,14,15 particularly high-throughput,16 are reported. Schmidt and Stafford report an assay employing a fluorescence resonance energy transfer (FRET) substrate for BoNT/A LC;16 however, we observe that this substrate has low excitation and emission wavelengths generating false hits as a consequence of spectral overlap with the inhibitors being screened. In addition, this substrate degrades when exposed to ambient lighting over the course of several hours, further limiting its utility. Herein, we present the synthesis of the FRET peptide, SNAPtide, its kinetic evaluation and utility in a high-throughput assay.17 Furthermore, we have validated this assay through a screen of a small series of inhibitors we synthesized based on the cleavage site of SNAP-25.
Results and Discussion
SNAPtide sysnthesis
SNAPtide is a truncated sequence of the native BoNT/A light chain (LC) substrate, SNAP-25, and was developed by List biologics as a tool for the detection of botulinum neurotoxin/A.17 While commercially available, its costs are substantial, its synthesis has not been described, nor has any kinetic parameters of this substrate or the evaluation of its ability to serve as a robust substrate for high-throughput screens (HTS). Therefore, we synthesized SNAPtide (Scheme 1.) using custom-modified in situ neutralization protocols for automated solid-phase peptide synthesis via Boc/Benzyl chemistry as previously described.18,19 Specifically, MBHA resin was subjected to Boc/Benzyl solid-phase peptide chemistry employing the Boc-Lys(Alloc)-OH amino acid to give the resin bound peptide 1. Overnight treatment of the resin with Pd(PPh3)4 and 1,3-dimethylbarbituric acid in 1:1 DMF:CH2Cl2 successfully deprotected the lysine side chain. Subsequent coupling of 4-dimethylaminoazobenzene-4′-carboxlic acid (DABCYL) with HBTU produced the resin bound DABCYL peptide 2. Cleavage of the peptide from the resin was accomplished by treatment with anhydrous HF containing 5% p-cresol. The resulting lyophilized peptide was dissolved in 500 mM MOPS (morpholinopropanesulfonic acid), 6 M guanidinium hydrochloride pH = 7.0, to which 3 equivalents of fluorescein thioisocyanite isomer 1 (FITC) was added for the N-terminal conjugation reaction. Reaction progress was determined by use of analytical RP-HPLC. Upon completion, excess FITC was scavenged with the addition of amino methyl resin and the mixture was filtered, then purified via preparative RP-HPLC affording SNAPtide 3. The peptide content was found to be 59.8% as determined by amino acid analysis. Stock solutions for use in enzyme assays were prepared in DMSO and stored protected from light at 4° C.
Scheme 1.

Synthesis of SNAPtide.
Kinetic analysis of the SNAPtide substrate
BoNT/A LC (1–425)20 dependent hydrolysis of SNAPtide in 40 mM HEPES pH 7.4 produced a linear increase in fluorescence when monitored at excitation and emission wavelengths of 490 and 523 nm respectively. Product analysis by ESI-MS revealed masses consistent with cleavage between residues Gln-7 and Arg-8 identical to the scissile bond of SNAP-25. As followed by fluorescence spectroscopy, apparent saturation kinetics was observed when the SNAPtide concentration was varied from 1 to 50 μM. However, upon correction for the inner filter effect,21 and as confirmed by endpoint HPLC analysis, BoNT/A LC catalytic activity was linear with respect to SNAPtide concentration up to 80 μM. Over the range of SNAPtide solubility, BoNT/A LC is operating under substrate limiting conditions; as such, we have obtained a second order rate constant (kcat/Km) of 4,300 ± 100 M−1 s−1 for SNAPtide. A Km in excess of 100 μM such as we observe with SNAPtide is not unexpected, Schmidt and Bostian have characterized the truncated SNAP-25 sequence SNKTRIDEANQ(Ψ)RATKML and report a Km of 5 mM for that 17 amino acid sequence.14 As a substrate for high throughput inhibitor screening, SNAPtide will produce an IC50 value for a competitive inhibitor that is essentially the Ki of the inhibitor.22 Characterization of the kinetic mechanism of inhibition (competitive, mixed/noncompetitive or uncompetitive) requires a follow up HPLC-based assay with a larger peptide substrate such as SNAP-25 (141–206) so that the inhibitor may be evaluated at substrate concentrations bracketing the Km. We optimized the assay conditions so as to be amenable to high-throughput screening. In addition, we synthesized several rationally designed competitive inhibitors to validate the SNAPtide assay.
Enzyme activity optimization
Many enzymes are stabilized by, and/or operate more efficiently in the presence of nondenaturating detergents.23 Furthermore, as reported recently by Shoichet24,25 the phenomenon of promiscuous aggregating inhibitors is mitigated by the presence of detergents below their critical micelle concentration (CMC). To optimize fluorescence assay conditions a series of detergents including Polyoxyethylenesorbitan monolaurate (Tween® 20), N-Lauroylsarcosine (NLS), 3-[(3-Cholamidopropyl)dimethylammonio]-1-propanesulfonate (CHAPS) and deoxycholate were evaluated at varied wt percent for their influence on enzyme activity (Figure 1).
Figure 1. Influence of Several Commonly used Detergents on BoNT/A LC catalytic activity.a.

aAssays were conducted at 22.5 oC, pH 7.4 in 40 mM HEPES, 5 μM SNAPtide substrate and 50 nM enzyme. Values are the mean of duplicate measurements.
Three of the detergents Tween® 20, CHAPS and NLS produced bell-shaped activity vs concentration profiles. At very low detergent concentrations BoNT/A LC catalysis of SNAPtide is approximately 20% greater than in their absence. Incremental addition of detergent concentration resulted in an increase in enzymatic activity to roughly 2-fold over the detergent absent rate. On the other hand, we note that the increase in rate is accompanied by a large increase in the standard error. Increasing the detergent concentration further produces an abrupt reduction in enzyme activity presumably because under these reaction conditions the CMC is attained. At detergent concentrations greater than CMC, BoNT/A LC activity was lost as seen in Figure 1. The detergent deoxycholate was inhibitory at all concentrations examined. From this study, detergents present 10-fold below their CMC appear to give slightly elevated activity and cleaner results.
In addition to employing detergents in an in vitro metalloprotease assay, DTT is commonly added to prevent disulfide formation. Since BoNT/A LC (1–425)20 is a truncated construct, with the wild type C-terminal cysteines removed the addition of 1.25 μM DTT had no influence on catalytic activity (data not shown).
The necessity of adding zinc salts to increase the protease activity was evaluated. Up to a concentration of 100 μM, no increase in catalytic activity was observed. Previously, the addition of zinc salts above 100 μM was shown to have an inhibitory effect.26 Furthermore, it has been observed previously that adding zinc salts to in vitro assays leads to non-physiologically relevant inhibitors.27
Co-solvent stability studies
We assessed several solvents commonly used to solublize test compounds these included methanol, ethanol, glycerol and DMSO at various volume fractions for their influence on BoNT/A LC catalytic activity. In Table 1 the relative activity of BoNT/A LC at the highest percent co-solvent examined is presented. Methanol and especially ethanol are inhibitory; therefore, BoNT/A LC assays should contain not more than 3% by volume of either alcohol co-solvent. Glycerol at concentrations from 5 to 25% appears stimulatory but reproducibility became problematic. Specifically, at glycerol concentrations greater than 10% by volume the standard error of triplicate measurements approached 20%, thus, glycerol should be avoided. Conversely, the catalytic activity of BoNT/A LC is largely insensitive to the presence of DMSO making this an ideal co-solvent.
Table 1.
Influence of various Co-Solvents on the catalytic activity of BoNT/A LCa
| Methanol(25%) | Ethanol (25%) | Glycerol (25%) | DMSO (25%) | |
|---|---|---|---|---|
| Vcosol./Vo | 0.83 ± 0.1 | 0.41 ± 0.06 | 1.6 ± 0.6 | 1.2 ± 0.2 |
Assays were conducted at 22.5 °C, pH 7.4 in 40 mM HEPES 0.01% (W/V) Tween® 20, 5 μM SNAPtide substrate and 200 nM enzyme. Values are the mean ± SEM of triplicate measurements.
Prior assays of BoNT/A LC activity determined with the Schmidt16 FRET substrate, termed Fl-A, were problematic and suggestive of photo-degradation. Exposure of Fl-A stock solutions to ambient lighting over the course of several hours of experimentation resulted in diminished fluorescence which was not due to enzyme degradation. This was not observed with SNAPtide. In view of these results, the photostability of both substrates were examined by a 1 hour incubation of each substrate at its excitation wavelength in a spectrophotometer (Figure 2).
Figure 2. Photostability of the BoNT/A LC substrates Fl-A and SNAPtide at their excitation wavelengths.a.

aEach Y-axis scaled to 7% of the substrate’s initial absorbance. 20 μM of substrate was incubated at 20° C in 40 μM HEPES pH 7.4 in a Shimadizu UV2100U spectrophotometer. Data were collected at 12 second intervals for 1 hour. High frequency noise was reduced by subjecting the digitized signal to a 3 point rolling average.
Fl-A displays a biphasic change in absorbance over the course of one hour exposure to its excitation wavelength. Significantly, the greatest change occurs within the initial 10 minutes of exposure corresponding to the time of interest during an enzyme assay. On the other hand, no detectable change in absorbance is observed for SNAPtide upon exposure to its excitation wavelength over the course of an hour. These data suggest the instability of Fl-A is attributable to bleaching of the chromophore, resulting in an un-hydrolyzable or otherwise undetectable substrate for BoNT/A LC.
Inhibitor synthesis
BoNT/A LC cleaves the protein SNAP-25 between Gln 197 and Arg 198. In an effort to discover possible small molecule inhibitor leads we evaluated these two amino acids coupled with a zinc-binding function (ZBF). The hydroxamate functionality was chosen as the ZBF due to prior success with other zinc metalloprotease enzymes.28 Specifically, L-arginine hydroxamate and L-glutamine hydroxamate were screened in the optimized SNAPtide assay. Gratifyingly, L-arginine hydroxamate (NHR) produced 75% inhibition at 50 μM while L-glutamine hydroxamate showed only 5% inhibition at 50 μM. In total, these results follow the cleavage pattern seen for BoNT/A.14,15
With this promising result we synthesized a small series of arginine hydroxamates to crudely evaluate structure activity relationships (SAR) of NHR of the α-nitrogen. To accomplish this task, we developed a general route to enable easy diversification from the α-nitrogen outlined as shown in scheme 2. The route (scheme 2) utilizes commercially available H-Arg-OMe 2HCl that was deprotonated with 1 equiv of DIPEA in DMF followed by the addition of either sulfonyl chloride, isocyanate, or aldehyde at 0° C overnight. In the case of an aldehyde, formation of the alkyl imine was monitored by mass spectrometry before reduction with sodium cyanoborohydride. The corresponding arginine N-alpha sulfonamide, urea, or alkyl methyl esters (5a–j) were then concentrated in vacuo by freezing with liquid N2 followed by lyophilization overnight. It is important to note that removal of the solvent by lyophilization is critical due to an observed side reaction which ensues readily upon heating >30 °C on rotary evaporation29 where the guanidinium group undergoes a cyclization with displacement of the methyl ester. The resulting oils (5a–j) were used without further purification. Conversion to the hydroxamate was with 50% aqueous hydroxylamine and catalytic potassium cyanide.30 Reaction progress was monitored by mass spectrometry and terminated upon consumption of methyl ester. Next, the reaction mixture was concentrated in vacuo and purified by reverse phase preparative HPLC affording the corresponding arginine hydroxamates (6a–j).
Scheme 2. Synthesis of arginine hydroxamate derivatives.a.

aConditions: (a) DIPEA, R1-Cl, R1-NCO, DMF 0°C; (b) 50% H2NOH soln., cat. KCN, 1:1 THF:MeOH, 40–80% yield over 2 steps.
To analyze further the SAR of substituents on the NHR α-nitrogen we synthesized the constrained lactams 10a and 10b. The synthesis is shown in scheme 3. Commercially available Boc arginine (diZ) 7, was converted to the methyl ester followed by TFA deprotection of the N-Boc. The arginine (diZ) methyl ester was then coupled to Boc-methionine using BOP as a coupling reagent, producing dipeptide 8. Dipeptide 8 was treated with neat methyl iodide followed by sodium hydride to give lactam 9a. Lactam methyl ester 9c was converted into hydroxamic acid 10a by hydrogenolysis of the arginine side chain protecting group followed by treatment with a hydroxylamine solution and catalytic potassium cyanide. While these two steps can be carried out in the reverse order it is preferable to form the hydroxamic last to avoid unwanted reduction of the N-O bond during the hydrogenation step; in addition, at this stage it was necessary to purify 10a by HPLC. Finally, deprotection of the N-terminus with TFA gave the desired hydroxamic acid 10b.
Scheme 3. Synthesis of Lactam arginine hydroxamate.a.

aConditions: (a) MeI, K2CO3, DMF, 96 %; (b) TFA:CH2Cl2 (1:3), quant; (c)BocMet-OH, BOP, NEt3, CH2Cl2, 71 %; (d) MeI (neat); (e) NaH, DMF, 58 % over 2 steps; (f) LiOH, THF:H2O; (g) H2, Pd/C, MeOH, 25 % over 2 steps; (h) TFA:CH2Cl2 (1:1), 95 %(i) H2, Pd/C, MeOH; (j) 50% NH2OH soln., cat. KCN, 1:1 THF:MeOH, 53 % over 2 steps; (k) TFA:CH2Cl2 (1:1), 80 %;
Inhibitor evaluation with SNAPtide
The compounds were tested in the optimized SNAPtide assay to validate whether the assay would reliably identify inhibitors of BoNT/A LC. Reactions were performed in 40 mM HEPES, 2% DMSO, 0.01% Tween® 20 and 100 nM BoNT/A LC with a final volume of 100 μL. Reactions were initiated by the addition of SNAPtide at a final concentration of 4 μM. As seen in table 2, little difference was observed for D-NHR and L-NHR suggesting stereochemistry is not important for this class of inhibitors. Not surprisingly, when the amino acid L-arginine was tested in the SNAPtide assay no inhibition was observed indicative of the hydroxamte being required for inhibition. Furthermore, when electron withdrawing groups were introduced, inhibition was significantly improved suggesting the pKa of the α-nitrogen is important for inhibition.
Table 2.
Inhibitor activity with SNAPtide.a
| Compound | R | % inhibition of hydrolysis rate |
|---|---|---|
| L-NHR | – | 75% |
| L-Glutamine hydroxamate | – | 5% |
| D-NHR | – | 73% |
| 6a |
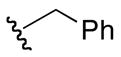
|
55% |
| 6b |
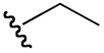
|
53% |
| 6c |
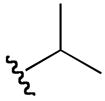
|
82% |
| 6d |
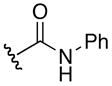
|
44% |
| 6e |
|
23% |
| 6f |
|
39% |
| 6g |
|
68% |
| 6h |
|
68% |
| 6i |
|
50% |
| 6j |
|
58% |
| 10a | – | 15% |
| 10b | – | 12% |
Assays were conducted with 50 μM inhibitor at 22.5 °C, pH 7.4 in 40 mM HEPES 0.01% (W/V) Tween® 20, 5 μM SNAPtide substrate and 200 nM enzyme.
In order to validate further the integrity of our high-throughput assay, we also synthesized the native sequence SNAP-25 (141–206) and evaluated this native substrate in an end-point HPLC-base assay. We observed a Km and kcat of 10.0 μM and 0.49 sec−1 respectively. These kinetic parameters are in excellent agreement with the reported native substrate and enzyme.9 Although an HPLC-based assay is not amenable to high-throughput screening, the low Km of the SNAP-25 (141–206) substrate allows for the determination of the kinetic mechanism of inhibition. Furthermore, by employing the native substrate SNAP-25 (141–206) all of the unique binding features such as the α- and β-exosites of the BoNT/A LC and SNAP-25 complex are engaged.3
On the other hand, an important attribute of a high-throughput assay is the rapid and reliable identification of lead compounds. To validate SNAPtide as a substrate for the identification of inhibitors of BoNT/A LC, we followed up L-NHR with our HPLC assay employing SNAP-25 (141–206) as a substrate. Gratifyingly, L-NHR competitively inhibited BoNT/A LC hydrolysis of SNAP-25 (141–206) with a Ki = 60 μM. These data are in excellent agreement with the observed inhibition profile of L-NHR with SNAPtide validating SNAPtide as a robust high-throughput substrate for BoNT/A LC.
Conclusion
In summary, we have developed a robust assay for the high-throughput screening of BoNT/A LC inhibitors. We have provided an efficient synthesis of SNAPtide via an automated peptide synthesizer, allowing us to prepare multi-milligram to gram quantities of the peptide. We have developed high throughput assay conditions for a SNAPtide-based assay to screen potential inhibitors of BoNT/A LC protease. The inclusion of a detergent such as Tween® 20 or CHAPS at 0.01 wt% increases catalytic activity, reproducibility, and DMSO is well tolerated as a co-solvent. Additionally, a small series of rationally designed inhibitors validated the utility of this assay for high throughput screening. Moreover, the inhibitors identified using SNAPtide as a substrate were confirmed and kinetically characterized by use of the native SNAP-25 (141–206) substrate. Further application of this assay for the identification of protease inhibitors via HTS of the BoNT/A LC will be reported in due course.
Experimental Procedures
General Procedures
All reactions were carried out under an argon atmosphere with dry solvents under anhydrous conditions, unless otherwise noted. Yields refer to chromatographically and spectroscopically (1H NMR) homogeneous materials, unless otherwise stated. Reagents were purchased at the highest commercial quality and used without further purification, unless otherwise stated. Methylene chloride and chloroform were distilled from calcium hydride. Tetrahydrofuran (THF) was distilled from sodium/benzophenone. Methanol was distilled from magnesium. Analytical thin-layer chromatography (TLC) was performed using 0.25 mm pre-coated silica gel Kieselgel 60 F254 plates. TLC visualization was by UV absorbance, iodine, dinitrophenylhydrazine, ceric ammonium molybdate, ninhydrin or potassium permanganate, as appropriate. Preparative and semi-preparative TLC was performed using Merck 1 mm or 0.5 mm coated silica gel Kieselgel 60 F254 plates respectively. Merck silica gel (60, particle size 0.040–0.063 mm) was used for flash column chromatography). RP-HPLC employed binary gradients of solvents A and B, where A is 0.1% TFA in water and B is 0.09% TFA in acetonitrile. NMR spectra were recorded on either a Varian 300MHz or a 400MHz instrument and calibrated using residual undeuterated solvent as an internal reference. The following abbreviations were used to explain the multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, quin = quintuplet, sext = sextet, sep = septet, br = broad, AB = AB quartet, app = apparent. Electrospray ionization (ESI) time-of-flight reflection experiments were performed on an Agilent ESI-TOF mass spectrometer. Samples are electrosprayed into the TOF reflection analyzer at an ESI voltage of 4000V and a flow rate of 200 microliters/minute.
General procedure for N-alpha-derivatized-Arg-OMe (5)
To a 20 mL scintillation vial charged with H-Arg-OMe 2HCl (4) (250 mg, 0.957 mmol) was added anhydrous DMF (5 mL), and DIPEA (167 mL, 0.957 mmol), and the reaction was cooled to 0° C. Sulfonyl chloride, aldehyde, or isocyanate (1.00 mmol) was then added and stirred at 0° C for 10 min. The cooling bath was then removed and the reaction was stirred at r.t. overnight. After the disappearance of starting material as judged by mass spectrometry the reaction was transferred to a separatory funnel containing water with 1%TFA (10 mL) and washed with CH2Cl2 (3X, 20 mL). The water layer was then collected, frozen with N2, and concentrated on a lyophilizer overnight. The crude Arginine Methyl ester (5) was used without further purification.
N-alpha-derivatized-Arg-hydroxamic acid (6a–j)
To a scintillation vial containing crude arginine methyl ester (100 mg, ~300 mmol) was added 1:1:0.5 THF:MeOH:50% hydroxylamine in water (10 mL) along with KCN (5 mg). The reaction was stirred overnight. Following completion of the reaction as confirmed by mass spectrometry, the crude products (6a–j) were dried in vacuo, and purified via reverse phase preparative HPLC. Yield 30–80%.
2-(benzylamino)-5-guanidino-N-hydroxypentanamide (6a)
1H NMR (400 MHz, DMSO-d6, δ): 10.80 (s, 1H), 7.37 (m, 1H), 7.36 (d, J = 4 Hz, 2H), 7.21 (t, J = 8 Hz, 2H), 7.10 (broad m, 4H), 6.89 (t, J = 8 Hz, 1H), 6.54 (d, J = 4 Hz, 1H), 4.11 (q, J = 8.0, 16.0 Hz, 1H), 3.12 (m, 2H), 1.51 (m, 4H). 13C NMR (100 MHz, DMSO-d6, δ): 168.5, 156.8, 140.2, 128.7, 126.7, 121.2, 117.5, 50.0, 40.3, 30.5, 25.1. ESI-TOF calc. C8H19N5O2 [M + H+]: 280.1695, found: 280.1770. 40% yield.
2-(ethylamino)-5-guanidino-N-hydroxypentanamide (6b)
1H NMR (400 MHz, DMSO-d6, δ): 10.80 (s, 1H), 8.07 (t, J = 4 Hz, 1H), 7.10 (broad m, 4H), 3.59 (m, 2H), 3.12 (q, J = 8.0, 16.0 Hz, 1H), 2.83 (m, 2H), 1.42 (m, 4H), 1.17 (t, J = 8 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ): 168.8, 158.4, 119.0, 23.0, 22.1, 17.0, 14.5, 11.2. ESI-TOF calc. C8H19N5O2 [M + H+]: 218.1611, found: 218.1611. 67% yield.
5-guanidino-N-hydroxy-2-(isopropylamino)pentanamide (6c)
1H NMR (400 MHz, DMSO-d6, δ): 10.34 (s, 1H), 8.90 (m, 1H), 7.78 (t, J = 6.0 Hz, 1H), 7.10 (broad m, 4H), 3.59 (m, 1H), 3.12 (m, 2H), 1.72 (q, J = 8.4, 15.6 Hz, 2H), 1.44 (m, 2H), 1.21 (t, J = 6.0 Hz, 6H). 13C-NMR (100 MHz, DMSO-d6, δ): 163.2, 156.7, 54.6, 48.4, 26.8, 24.5, 19.3, 18.0. ESI-TOF calc. C9H21N5O2 [M + H+]: 232.1768, found: 232.1774. 47% yield.
5-guanidino-N-hydroxy-2-(3-phenylureido)pentanamide (6d)
1H NMR (400 MHz, DMSO-d6, δ): 10.79 (s, 1H), 8.65 (s, 1H), 7.58 (d, J = 1.2 Hz, 1H), 7.51 (t, J = 6.0 Hz, 2H) 7.36 (m, 2H), 7.22 (t, J = 7.6 Hz, 2H),7.10 (broad m, 4H), 6.90 (m, 1H), 4.11 (q, J = 8.0, 15.3 Hz, 1H), 3.11 (q, J = 6.4, 12.2 Hz, 2H), 1.49 (m, 4H). 13C NMR (100 MHz, DMSO-d6, δ): 168.9, 157.4, 156.8, 128.6, 128.2, 126.8, 49.9, 42.5, 30.5, 25.1. ESI-TOF calc. C13H20N6O3 [M + H+]: 309.1670, found: 309.1675. 30% yield.
2-(3-benzylureido)-5-guanidino-N-hydroxypentanamide (6e)
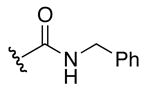
1H NMR (400 MHz, DMSO-d6, δ): 10.70 (s, 1H), 10.19 (s, 1H), 7.68 (t, J = 6.0 Hz, 2H), 7.31 (m, 2H), 7.23 (m, 3H), 7.10 (broad m, 4H), 6.57 (t, J = 6.0 Hz, 1H), 6.27 (d, J = 8.4 Hz, 1H), 4.52 (dd, J = 4.0, 19.6 Hz, 1H), 4.206 (s, 2H), 4.07 (q, J = 7.6, 14.8 Hz, 2H), 3.11 (m, 2H), 1.48 (m, 4H). 13C-NMR (100 MHz, DMSO-d6, δ): 168.8, 157.4, 156.7, 140.6, 128.2, 126.9, 126.5, 50.1, 42.7, 30.7, 25.1. ESI-TOF calc. C14H22N6O3 [M + H+]: 323.1826, found: 323.1826. 75% yield.
2-(3-ethylureido)-5-guanidino-N-hydroxypentanamide (6f)
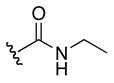
1H NMR (400 MHz, DMSO-d6, δ): 10.66 (s, 1H), 10.192 (s, 1H), 7.68 (m, 1H), 6.07 (m, 2H), 4.03 (m, 2H), 3.36 (q, J = 9.6, 14.0 Hz, 1H), 1.47 (m, 4H), 0.96 (t, J = 7.2 Hz, 3H). 13C NMR (100 MHz, DMSO-d6, δ): 169.9 157.3, 156.8, 49.9, 33.9, 32.5, 30.7, 25.1, 15.5. ESI-TOF calc. C9H20N6O3 [M + H+]: 261.1670, found: 261.1670. 56% yield.
2-(N,N-dimethylsulfamoylamino)-5-guanidino-N-hydroxypentanamide (6g)
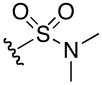
1H NMR (400 MHz, DMSO-d6, δ): 10.67 (s, 1H), 7.54 (m, 3H), 7.10 (broad m, 4H), 3.54 (q, J = 9.2, 15.6 Hz, 1H), 3.09 (q, J = 6.0, 11.6 Hz, 2H), 2.60 (s, 6H), 1.53 (m, 4H). 13C NMR (100 MHz, DMSO-d6, δ): 167.9, 156.6, 53.9, 40.1, 37.4, 30.1, 25.2. ESI-TOF calc. C8H20N6O4S [M + H+]: 297.1339, found: 297.1340. 52% yield.
2-(5-(dimethylamino)naphthalene-1-sulfonamido)-5-guanidino-N-hydroxypentanamide (6h)
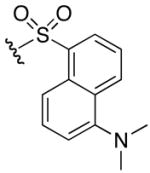
1H NMR (400 MHz, DMSO-d6, δ): 10.55 (s, 1H), 8.42 (dd, J = 6.0, 14.8 Hz, 2H), 8.30 (d, J = 8.8 Hz, 1H), 8.09 (dd, J = 6.0, 7.2 Hz, 1H), 7.58 (m, 2H), 7.43 (t, J = 6.0 Hz, 1H), 7.26 (d, J = 7.6 Hz, 2H), 7.10 (broad m, 4H), 3.58 (q, J = 8.4, 15.6 Hz, 1H), 2.92 (q, J = 6.4, 13.2 Hz, 2H), 1.47 (m, 2H), 1.32 (m, 1H), 1.18 (m, 1H). 13C NMR (100 MHz, DMSO-d6, δ): 167.0, 156.5, 136.6, 129.2, 128.9, 128.7, 127.8, 126.6, 123.5, 123.5, 119.6, 117.6, 115.1, 53.5, 45.0, 30.0, 24.9. ESI-TOF calc. C18H26N6O4S [M + H+]: 423.1809, found: 423.1809. 60% yield.
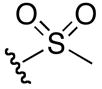
5-guanidino-N-hydroxy-2-(methylsulfonamido)pentanamide (6i)
1H NMR (400 MHz, DMSO-d6, δ): 10.78 (s, 1H), 7.61 (t, J = 6.0 Hz, 1H), 7.52 (d, J = 8.8 Hz, 1H), 7.10 (broad m, 4H), 6.62 (q, J = 8.8, 16 Hz, 1H), 3.09 (q, J = 6.0, 12.4 Hz, 2H), 2.85 (s, 3H), 1.55 (m, 2H), 1.41 (m, 2H). 13C NMR (100 MHz, DMSO-d6, δ): 167.4, 158.6, 156.6, 53.6, 40.8, 29.8, 25.0. ESI-TOF calc. C7H17N5O4S [M + H+]: 268.1074, found: 268.1075. 55% yield.
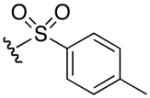
5-guanidino-N-hydroxy-2-(4-methylphenylsulfonamido)pentanamide (6j)
1H NMR (400 MHz, DMSO-d6, δ): 10.60 (s, 1H), 7.97 (d, J = 8.4 Hz, 1H), 7.65 (d, J = 8.4 Hz, 2H), 7.58 (t, J = 5.6 Hz, 1H), 7.34 (d, J = 8.0 Hz, 2H), 7.10 (broad m, 4H), 3.53 (q, J = 8.0, 15.2 Hz, 1H), 2.97 (q, J = 6.0, 12.8 Hz, 2H), 2.37 (s, 3H) 1.48 (m, 1H), 1.36 (m, 2H), 1.24 (m, 1H). 13C NMR (100 MHz, DMSO-d6, δ): 166.9, 158.7, 156.6, 142.4, 138.4, 129.3, 126.2, 53.5, 29.9, 24.8, 20.9. ESI-TOF calc. C13H21N5O4S [M + H+]: 344.1387, found: 344.1387. 80% yield
methyl 5-(2,3-bis(benzyloxycarbonyl)guanidino)-2-(2-(tert-butoxycarbonyl)-4-(methylthio)butanamido)pentanoate (8)
To a solution of (7) (8.39 g, 18.3 mmol, 1 eq) in CH2Cl2 (250 mL) was added Boc-Met-OH (5.47 g, 21.96 mmol, 1.2 eq), BOP (9.7 g, 21.96 mmol, 1.2 eq), NEt3 (7.7 ml, 54.9 mmol, 3 eq) and the reaction allowed to stir at rt for 2 h. The reaction mixture was washed with brine then dried (MgSO4) and concentrated in vacuo. The crude product mixture was then purified by flash chromatography using 40 % EtOAc in hexane as the eluant to give (8) (8.92 g, 12.98 mmol) as a white solid. 1H NMR (400 MHz, CDCl3, δ): 9.47 (broad s, 1H), 9.28 (broad s, 1H), 7.36 (m, 10H), 6.91 (d, J = 7.6 Hz, 1H), 5.25 (s, 2H), 4,26 (m, 1H), 4.57 (m, 1H), 5.17 (s, 2H,), 3.96 (m, 2H), 2.50 (t, J = 7.2 Hz, 2H), 3.64 (s, 3H), 2.04 (m and s, 5H), 1.84 (m, 2H), 1.65 (m, 2H), 1.43 (s, 9H). 13C NMR (100 MHz, CDCl3, δ): 171.8, 171.0, 163.4, 160.2, 155.4, 155.0, 136.5, 134.3, 128.2, 128.1, 127.8, 127.6, 127.3, 127.2, 79.6, 68.6, 66.7, 52.9, 52.0, 51.7, 43.8, 31.2, 29.6, 28.2, 27.8, 24.4, 14.49. ESI-TOF calc. C33H46N5O9S [M+H+]: 688.3011, found: 688.3012. 71 % yield.
methyl 5-(2,3-bis(benzyloxycarbonyl)guanidino)-2-(3-(tert-butoxycarbonyl)-2-oxopyrrolidin-1-yl)pentanoate (9a)
Compound (8) (0.66 g, 0.97 mmol, 1 eq) was allowed to stir in neat MeI (10 mL) for 2 d. The reaction mixture was then concentrated in vacuo to give the intermediate sulfonium salt (803 mg, 0.97 mmol) as a yellow solid which was used without further purification. Sulfonium salt (803 mg, 0.97 mmol, 1 eq) was dissolved in DMF (20 mL). NaH (46 mg of 60 % in mineral oil, 1.16 mmol, 1.2 eq) was then added and the reaction was allowed to stir at rt for 1 h. The reaction was quenched with aq. sat. NH4Cl and the aqueous layer extracted three times with EtOAc. The combined organic layers were dried (MgSO4) and concentrated in vacuo. The crude product mixture was purified by flash chromatography using 50% EtOAc in hexane as the eluant to give (9a) (356 mg, 0.56 mmol) as a clear oil. 1H NMR (400 MHz, CDCl3, δ): 9.43 (broad s, 1H), 9.24 (broad s, 1H), 7.36 (m, 10H), 5.28 (d, J = 12.0 Hz, 1H), 5.22 (d, J = 12.0 Hz, 1H), 5.14 (s, 2H,), 5.14 (s, 2H), 5.03 (broad m, 1H), 4.67 (m, 1H), 4.18 (m, 1H), 3.96 (m, 2H), 3.66 (s, 3H), 3.09 (m, 1H), 3.33 (m, 1H), 2.47 (m, 1H), 1.72 (m, 5H), 1.49 (s, 9H). 13C NMR (100 MHz, CDCl3, δ): 173.0, 170.8, 163.8, 160.3, 155.7, 129.0, 128.9, 128.5, 128.1, 129.9, 79.8, 68.9, 66.9, 54.0, 52.0, 44.1, 40.9, 28.6, 28.2, 26.0, 25.4. ESI-TOF calc. C32H42N5O9 [M+H+]: 640.2977, found: 640.2975. 58 % yield.
5-(2,3-bis(benzyloxycarbonyl)guanidino)-2-(3-(tert-butoxycarbonyl)-2-oxopyrrolidin-1-yl)pentanoic acid (9b)
To a solution of (9a) (247 mg, 0.39 mmol, 1 eq) in 1:1 THF: H2O (14 mL) was added LiOH.H2O (130 mg, 3.12 mmol, 8 eq) and the reaction allowed to stir at rt for 1 h. H2O was added and the aqueous layer was washed with CH2Cl2. The aqueous layer was acidified with 1 M HCl and extracted three times with EtOAc. The combined EtOAc extracts were dried (MgSO4) and concentrated in vacuo. The product was dissolved in MeOH (40 mL) under argon. 10% Pd/C (20 mg, cat) was added. The argon atmosphere was replaced with hydrogen and the reaction was allowed to stir under hydrogen (balloon pressure) at rt for 18 h. The reaction was then filtered through celite and concentrated in vacuo to give (9b) (35 mg, 0.099 mmol) as a white solid which was used without further purification. 1H NMR (400 MHz, MeOD-d3, δ): 4.50 (dd, J = 11.0, 4.0 Hz, 1H), 4.32 (apparent t, J = 9.5 Hz, 1H) 3.47 (apparent t, J = 9.0 Hz, 1H), 3.29 (m, 1H), 3.16 (m, 2H), 2.39 (m, 1H), 1.74 (m, 5H), 1.41 (s, 9H). 13C NMR (100 MHz, MeOD-d3, δ): 175.6, 158.7, 158.2, 80.6, 56.6, 53.3, 42.2, 41.8, 28.8, 27.8, 27.4, 26.7. ESI-TOF calc. C15H28N5O5 [M+H+]: 358.2085, found: 358.2087. 25 % yield over 2 steps.
1-(1-carboxy-4-guanidinobutyl)-2-oxopyrrolidin-3-aminium 2,2,2-trifluoroacetate (9c)
To a suspension of (9b) (15 mg, 0.04 mmol, 1 eq) in CH2Cl2 (1.5 mL) was added TFA (1.5 mL). The reaction was allowed to stir at rt for 2 h. The reaction was concentrated in vacuo to give (9c) (14 mg, 0.038 mmol). 1H NMR (400 MHz MeOD-d3, δ): 4.57 (dd, J = 10.8, 4.8 Hz, 1H), 4.10 (dd, J = 10.5, 8.7 Hz, 1H), 3.55 (t, J = 9.3 Hz, 1H), 3.42 (m, 1H), 3.15 (t, 2H), 2.56 (m, 1H), 2.06–1.44 (m, 5H). 13C NMR (100 MHz, MeOD-d3, δ): 173.0, 171.5, 158.8, 55.7, 51.8, 42.6, 41.7, 27.1, 26.8, 25.7. ESI-TOF calc. C10H20N5O3 [M+H+]: 258.1561, found 258.1566. 95 % yield.
tert-butyl 1-(5-guanidino-1-(hydroxyamino)-1-oxopentan-2-yl)-2-oxopyrrolidin-3-ylcarbamate (10a)
To a solution of (9a) (350 mg, 0.55 mmol, 1 eq) in MeOH (40 mL) under argon was added 10% Pd/C (20 mg, cat). The argon atmosphere was replaced with hydrogen and the reaction was allowed to stir under hydrogen (balloon pressure) at rt for 18 h. The reaction was then filtered through celite and concentrated in vacuo (177 mg, 0.48 mmol, 87 %) and used immediately. To a solution of the hydrogenation product (166 mg, 0.45 mmol, 1 eq) in THF: MeOH (4 mL) was added 50 % aq. NH2OH sol (1 mL) and KCN (5 mg, cat) and the reaction allowed to stir at rt for 2 h. The reaction was then concentrated in vacuo. The crude product mixture was purified using HPLC (0–40 %B over 30 mins, 16.7 mins) to give (10a) (88 mg, 0.23 mmol) as a clear oil. 1H NMR (400 MHz, MeOD-d3, δ): 4.42 (m, 1H) 4.48 (m, 1H), 4.05–4.18 (m, 1H), 4.00 (m, 1H), 3.36 (m, 1H), 3.23 (m, 2H), 2.40 (m,1H), 1.75 (m, 5H) 1.43 (s, 9H). 13C NMR (100 MHz, MeOD-d3, δ): 176.4, 175.4, 168.1, 167.9, 162.2, 161.8, 158.4, 157.8, 80.7, 53.9, 53.1, 41.8, 41.6, 28.6, 26.6, 26.2, 26.1. ESI-TOF calc. C15H29N6O5 [M+H+]: 373.2194, found: 373.2192. 52 % yield.
2-(3-amino-2-oxopyrrolidin-1-yl)-5-guanidino-N-hydroxypentanamide (10b)
To a suspension of (10a) (17 mg, 0.046 mmol, 1 eq) in CH2Cl2 (2 mL) was added TFA (2mL). The reaction was allowed to stir at rt for 3.5 h. The reaction was then concentrated in vacuo to give (10b) (10 mg, 0.038 mmol) as a clear oil. The crude product was purified by HPLC (100 % H2O, isocratic, 5.87 min). 1H NMR (400 MHz, MeOD-d3, δ): 4.42 (m, 1H), 4.06 (m, 1H), 3.75 (apparent t, J = 9.4 Hz, 1H), 3.46 (m, 1H), 3.15 (m, 2H), 2.52 (m, 1H), 1.87 (m, 3H), 1.50 (m, 2H). 13C NMR (100 MHz, MeOD-d3, δ): 169.9, 169.7, 157.0, 53.0, 50.9, 41.5, 40.8, 26.2, 25.4, 24.4. ESI-TOF calc. C10H21N6O3 [M+H+]: 273.1675, found: 273.1667. 80 % yield.
Peptide Synthesis
Fl-A was prepared by published procedures.16 The SNAPtide and SNAP-25 (141–206) were prepared by stepwise solid phase peptide synthesis using in situ neutralization protocols for Boc chemistry as previously described.18,19 Automated chain assembly was performed on a CSBio Model 136 peptide synthesizer using custom protocols (available upon request). All Boc amino acids were obtained from Senn Chemicals (Dielsdorf, Switzerland). p-Methylbenzhydrylamine (MBHA) resin was prepared by Advanced Chemtech at a loading of 0.64 mmol NH2/g (100–200 mesh). Side chain protecting groups were as follows: Ser, Thr (Bzl); Asp, Glu (OcHex); Asn (Xan); Trp (formyl); Met (sulfoxide); Cys (4-MeBzl); Tyr (2-BrZ); Lys (Alloc); all other amino acids were incorporated without side chain protection. Trifluoroacetic acid was from Halocarbon (River Edge, NJ), N,N-dimethylformamide (BioAnalyzed) was from J.T. Baker (St. Louis, MO), N,N-diisopropylethylamine, and Pd(PPh3)4 were from Aldrich (Milwaukee, WI), and anhydrous hydrogen fluoride (UHP) was from Matheson Gas (Cucamonga, CA). HF cleavage was performed in a Type II vacuum-driven HF apparatus from The Peptide Institute (Minoh, Osaka, Japan). All other reagents, solvents, and chemicals were of the highest purity commercially available and used as received. RP-HPLC employed binary gradients of solvents A and B, where A is 0.1% TFA in water and B is 0.09% TFA in acetonitrile. Analytical RP-HPLC was performed using a Vydac 218TP5415 column at a flow rate of 1 mL/min, with detection at 214 nm during a linear gradient of 10–50 over 30 min. Preparative RP-HPLC was performed using a Vydac 218TP101522 column at a flow rate of 10 mL/min, with detection at 220 nm during a linear gradient of 25–55 over 40 min. In all cases, fractions were analyzed off-line using an ABI/Sciex 150EX single quadrupole mass spectrometer and judged for purity after a consistent summing of 50 scans in multichannel analysis (MCA) mode. For preparative purification purposes, fractions containing no single charged species accounting for more than 10% of the total ion intensity were designated “pure” and pooled; the homogeneity of this pool was >95% as verified by analytical RP-HPLC.
SNAP-25 (141–206) was prepared as a C-terminal free acid and the N-terminal was acylated with acetic anhydride at the conclusion of chain elongation.
SNAPtide (3)
At the conclusion of chain assembly, the N-terminus was protected with Z-OSu, after which the Lys- alloc side chain protection was removed by overnight treatment with Pd(PPh3)4 (0.3 eq.) and N,N-dimethylbarbituric acid (10 eq.) in 1:1 CH2Cl2/DMF with exclusion of light. The completeness of this deprotection step was verified by small scale (20 mg) HF cleavage followed by ESI analysis of the crude product. SNAPtide was then coupled to the newly-unmasked Lys side chain in DMF with excess DIEA. The completed peptide-resin was then washed successively with DMF and CH2Cl2, dried in vacuo overnight, and global side-chain deprotection/cleavage was performed with anhydrous HF (1hr, 0°C) containing 5 wt% p-cresol as a carbocation scavenger. After evaporation of HF, the crude peptide was extracted from the resin with TFA; following evaporation and trituration with diethyl ether (3x), the crude peptide was solubilized in aqueous acetonitrile and lyophilized with exclusion of light for solution-phase N-terminal FITC conjugation. The resulting lyophilized peptide was dissolved in 500 mM MOPS, 6 M guanidinium hydrochloride pH = 7.0, (2 mL), then 3 equivalents of fluorescein thioisocyanite isomer 1 (FITC) was added for the N-terminal conjugation reaction. The progress of the reaction was monitored by analytical RP-HPLC and ES-MS. The reaction was deemed complete when no starting material was observed and excess FITC was scavenged with the addition of amino methyl resin. The mixture was filtered and purified via preparative RP-HPLC (as described above) affording SNAPtide 3.
Assays for BoNT/A LC activity with SNAPtide
BoNT/A LC at 100 nM was assayed at 22.5 °C, pH 7.4 in 40 mM HEPES in 100 μL volumes by use of a Molecular Devices SpectraMax GeminiEM plate reader. Fluorimeter parameters were as follows, excitation wavelength 490 nm, emission wavelength 532 nm with 2 nm slit widths for both excitation and emission and a cutoff filter at 495 nm. Enzyme velocities were determined from within the linear window of the assay typically from data collected over the range of 100 to 300 seconds. Detergents at varied concentrations were diluted from stocks prepared as 4% weight-to-volume in 40 mM HEPES pH 7.4. SNAPtide substrate was prepared as a concentrated stock in DMSO and protected from exposure to light.
Assays for BoNT/A LC activity with SNAP-25 (141–206)
BoNT/A LC at 5 nM was assayed at 22.5 °C, pH 7.4 in 40 mM HEPES in 500 μL volumes with a total DMSO concentration not exceeding 4.0%. 110 μL aliquot’s were quenched with 10 μL of AcOH and analyzed using HPLC with a Hitachi elite LaChrom on an agilent 300SB-C18, 5 mm pore size, 2.1 × 75 mm column.
The kinetic constants Km and kcat were determined by varying substrate concentration and fitting to the Michaelis-Menton equation using a non-linear fit with KaleidaGraph. The inhibition constant Ki was evaluated by fitting the data of varied substrate and inhibitor concentrations to the general equation for competitive inhibition using GraFit software (Erithacus Software Ltd.).
Acknowledgments
This work was supported by the National Institute of Health BT010-04 and The Skaggs Institute for Chemical Biology.
References
- 1.Singh BR. Nature. 2000;7:617–619. doi: 10.1038/77900. [DOI] [PubMed] [Google Scholar]
- 2.Simpson LL. Annu Rev Pharmacol Toxicol. 2004;44:167–193. doi: 10.1146/annurev.pharmtox.44.101802.121554. [DOI] [PubMed] [Google Scholar]
- 3.Lacy BD, Tepp W, Cohen AC, DasGupta BR, Stevens RC. Nat Struct Biol. 1998;5:898–902. doi: 10.1038/2338. [DOI] [PubMed] [Google Scholar]
- 4.Arnon SS, Schechter R, Inglesby TV, Henderson DA, Bartlett JG, Ascher MS, Eitzen E, Fine AD, Hauer J, Layton M, Lillibridge S, Osterholm MT, O’Toole T, Parker G, Perl TM, Russell PK, Swerdlow DL, Tonat K. JAMA. 2001;285:1059–1070. doi: 10.1001/jama.285.8.1059. [DOI] [PubMed] [Google Scholar]
- 5.Hicks RP, Hartell MG, Nichols DA, Bhattacharjee AK, Von Hamont JE, Skillman DR. Curr Med Chem. 2005;12:667–690. doi: 10.2174/0929867053202223. [DOI] [PubMed] [Google Scholar]
- 6.Amersdorfer P, Wong C, Smith T, Chen S, Deshpande S, Sheridan R, Marks JD. Vaccine. 2002;20:1640–1648. doi: 10.1016/s0264-410x(01)00482-0. [DOI] [PubMed] [Google Scholar]
- 7.Nowakowski A, Wang C, Powers DB, Amersdorfer P, Smith TJ, Montgomery VA, Sheridan R, Blake R, Smith LA, Marks JD. Proc Natl Acad Sci USA. 2002;99:11346–11350. doi: 10.1073/pnas.172229899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Burnett JC, Schmidt JJ, Connor F, McGrath CF, Nguyen TL, Hermone AR, Panchal RG, Vennerstrom JL, Kodukula K, Zaharevitz DW, Gussio R, Bavari S. Bioorg Med Chem. 2005;13:333, 341. doi: 10.1016/j.bmc.2004.10.026. [DOI] [PubMed] [Google Scholar]
- 9.Sukonpan C, Oost T, Goodnough M, Tepp WH, Johnson EA, Rich DH. J Peptide Res. 2004;63:181–193. doi: 10.1111/j.1399-3011.2004.00124.x. [DOI] [PubMed] [Google Scholar]
- 10.Burnett JC, Schmidt JJ, Stafford RG, Panchal RG, Nguyen TL, Hermone AR, Vennerstrom JL, McGrath CF, Lane DJ, Sausville EA, Zaharevitz DW, Gussio R, Bavari S. Biochem Biophys Res Commun. 2003;310:84–93. doi: 10.1016/j.bbrc.2003.08.112. [DOI] [PubMed] [Google Scholar]
- 11.Hayden J, Pires J, Roy S, Hamilton M, Moore GJ. J Appl Toxicol. 2003;23:1–7. doi: 10.1002/jat.870. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt JJ, Stafford RG. FEBS Lett. 2003;532:423–426. doi: 10.1016/s0014-5793(02)03738-9. [DOI] [PubMed] [Google Scholar]
- 13.Schmidt JJ, Stafford RG, Bostian KA. FEBS Lett. 1998;435:61–64. doi: 10.1016/s0014-5793(98)01041-2. [DOI] [PubMed] [Google Scholar]
- 14.Schmidt JJ, Bostian KA. J Protein Chem. 1995;14:703–708. doi: 10.1007/BF01886909. [DOI] [PubMed] [Google Scholar]
- 15.Schmidt JJ, Bostian KA. J Protein Chem. 1997;16:19–26. doi: 10.1023/a:1026386710428. [DOI] [PubMed] [Google Scholar]
- 16.Schmidt JJ, Stafford RG. Appl Environ Microbiol. 2003;69:297–303. doi: 10.1128/AEM.69.1.297-303.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shine NR. US 6,504,006 B1. Patent application. 2003
- 18.Kim YS, Moss JA, Janda KD. J Org Chem. 2004;69:7776–7778. doi: 10.1021/jo048922y. [DOI] [PubMed] [Google Scholar]
- 19.Schnolzer M, Alewood P, Jones A, Alewood D, Kent SBH. Int J Peptide Protein Res. 1992;40:180. doi: 10.1111/j.1399-3011.1992.tb00291.x. [DOI] [PubMed] [Google Scholar]
- 20.Baldwin MR, Bradshaw M, Johnson EA, Barbieri JT. Protein Expression Purif. 2004;37:187–195. doi: 10.1016/j.pep.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 21.Liu Y, Kati W, Chen CH, Tripathi R, Molla A, Kohlbrenner W. Anal Biochem. 1999;267:331. doi: 10.1006/abio.1998.3014. [DOI] [PubMed] [Google Scholar]
- 22.Mertens ML, Kagi JH. Anal Biochem. 1979;96:448–455. doi: 10.1016/0003-2697(79)90605-5. [DOI] [PubMed] [Google Scholar]
- 23.Helenius A, McCaslin DR, Fries E, Tanford C. Methods in Enzymology. 1979;56:734–737. doi: 10.1016/0076-6879(79)56066-2. [DOI] [PubMed] [Google Scholar]
- 24.Seidler J, McGovern SL, Doman TN, Shoichet BK. J Med Chem. 2003;46:4477–4486. doi: 10.1021/jm030191r. [DOI] [PubMed] [Google Scholar]
- 25.McGovern SL, Helfand BT, Feng B, Shoichet BK. J Med Chem. 2003;46:4265–4272. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 26.Schiffmann R, Heine A, Klebe G, Klein CDP. Angew Chem Int Ed. 2005;44:3620–3623. doi: 10.1002/anie.200500592. [DOI] [PubMed] [Google Scholar]
- 27.Katz BA, Clark JM, Finer-Moore JS, Jenkins TE, Johnson CR, Ross MJ, Luong C, Moore WR, Stroud RM. Nature. 1998;391:608–613. doi: 10.1038/35422. [DOI] [PubMed] [Google Scholar]
- 28.Rao BG. Curr Pharm Des. 2005;11:295–322. doi: 10.2174/1381612053382115. [DOI] [PubMed] [Google Scholar]
- 29.KDJ. unpublished results
- 30.Ho CY, Strobel E, Ralbovsky J, Galemmo RA. J Org Chem. 2005;70:4873–4875. doi: 10.1021/jo050036f. [DOI] [PubMed] [Google Scholar]