

Abstract
Cytochrome P450 46A1 (CYP46A1) and NADPH-cytochrome P450 oxidoreductase (CPR) are the components of the brain microsomal mixed-function monooxygenase system that catalyzes the conversion of cholesterol to 24-hydroxycholesterol. Both CYP46A1 and CPR are monotopic membrane proteins that are anchored to the endoplasmic reticulum via the N-terminal transmembrane domain. The exact mode of peripheral association of CYP46A1 and CPR with the membrane is unknown. Therefore, we studied their membrane topology by using an approach in which solution-exposed portion of heterologously expressed membrane-bound CYP46A1 or CPR was removed by digestion with either trypsin or chymotrypsin followed by extraction of the residual peptides and their identification by mass spectrometry. The identified putative membrane-interacting peptides were mapped onto available crystal structures of CYP46A1 and CPR and the proteins were positioned in the membrane considering spatial location of the missed cleavage sites located within these peptide as well as the flanking residues whose cleavage produced these peptides. Experiments were then carried out to validate the inference from our studies that the substrate, cholesterol, enters CYP46A1 from the membrane. As for CPR, its putative membrane topology indicates that the Q153R and R316W missense mutations found in patients with disordered steroidogenesis are located within the membrane-associated regions. This information may provide insight in the deleterious nature of these mutations.
Keywords: CYP46A1, CPR, Crystal structure, MALDI, Membrane topology
Membrane proteins demonstrate various modes of interaction with the membrane [1-10]. This includes transmembrane segments [7], covalent links to a hydrophobic compound [8], electrostatic binding to phospholipid head groups [9], as well as hydrophobic loops and amphiphilic helices peripherally associated with the membrane [3, 4, 10]. The transmembrane segments are the most studied because they can be easily predicted from the sequence hydropathy plots. Results of such predictions are usually in a good agreement with experimental data. Computing of the membrane interactions other than transmembrane spanning is more difficult because of the diversity of interactions and limited experimental information available [4-6]. The latter hampers our understanding how membrane proteins function and requires development of new approaches to study membrane topology. In the present work, we propose a new approach which combines heterologous expression of a membrane protein of interest in E. coli, proteolytic elimination of the solution-exposed portion of the membrane-bound protein with subsequent extraction of the residual peptides and their identification by mass spectrometry. This approach was used to study the membrane topology of two different monotopic proteins, cytochrome P450 46A1 (CYP46A1; EC 1.14.13.15)1 and NADPH-cytochrome P450 reductase (CPR; NADPH-ferrihemoprotein reductase, EC 1.6.2.4).
CYP46A1 catalyzes cholesterol 24-hydroxylation, the first step in the major pathway of cholesterol elimination from the brain [11, 12]. Medical significance of CYP46A1 in humans is not yet clear. Deficiency of CYP46A1 activity could be one of the risk factors for Alzheimer’s disease [13, 14]. Animal studies also indicate that CYP46A1-mediated cholesterol 24-hydroxylation is an important mechanism that controls cholesterol turnover in the central nervous system [15]. CYP46A1 is a heme-containing monooxygenase that belongs to a group of microsomal cytochrome P450 enzymes (CYP or P450). Microsomal P450s play key roles in the metabolism of drugs, pollutants and other xenobiotics as well as in the biotransformation of some important endogenous compounds [16]. The membrane topology of microsomal P450s in the endoplasmic reticulum and proteoliposomes has been intensively studied by a variety of experimental methods, including hydropathy prediction [17], trypsin digestion [18, 19], immunolocalization with anti-peptide antibodies [20, 21], retaining of hybrid proteins in the endoplasmic reticulum [22, 23], mapping with radioactively labeled photoreactive phospholipids [24], quenching of the tryptophan fluorescence [25], and various combinations of mutagenesis and biochemical and biophysical analyses [26-29]. A notion that emerged from these studies is that microsomal P450s have a mono-facial mode of association with the membrane. A large catalytic domain is anchored to the lipid bilayer through the transmembrane segment of ~20-25 amino acid residues at the N-terminus and may penetrate the membrane to different depths in different P450s. The non-contiguous portions of the polypeptide chain that could provide additional membrane contacts include the sequences before and after the A helix as well as the F-G loop and β1 and β2 sheets [17-19, 25-27). However, it is not currently clear how many segments in the catalytic domain are involved in peripheral association with the membrane and whether they are the same in different microsomal P450s.
Catalytic activity of microsomal P450s requires electrons that are received from NADPH via the P450 redox partner CPR [30]. Although CPR and P450 form a 1:1 complex, there is about 10- to 20-fold excess of P450s over CPR in microsomes [31]. In humans, mutations in the CPR gene lead to disordered steroidogenesis with or without Antley-Bixler skeletal malformation syndrome [32, 33]. CPR is the FAD/FMN-containing protein composed of the N-terminal, 6 kDa hydrophobic domain and the C-terminal, 72 kDa soluble catalytic domain [34]. The hydrophobic domain serves as the membrane anchor and ensures proper spatial interaction of CPR with P450s [35]. P450s do not accept electrons from CPR lacking this hydrophobic domain [34, 35]. The soluble catalytic domain is comprised of the NADPH- and FAD-binding domains, the connecting domain, and the FMN-binding domain that sequentially transfer electrons from NADPH to the redox partner [36]. Structural studies predicted several hydrophobic patches on the surface of the CPR catalytic domain that likely interact with the membrane [36].
By using a new experimental approach, we validated the N-terminal membrane anchoring of CYP46A1 and CPR, identified additional membrane-interacting areas in both proteins, and proposed how CYP46A1 and CPR can be positioned in the membrane. We also obtained experimental evidence in support of our model for the membrane topology of CYP46A1.
Materials and methods
Materials
Sequencing grade chymotrypsin and modified trypsin were from Roche Diagnostics GmbH (Penzberg, Germany) and Promega Corp. (Madison, WI, USA), respectively. All other chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA). The plasmid for bacterial expression of rat CPR was kindly provided by Dr. F.P. Guengerich (Vanderbilt University).
Membrane fractionation
Full-length and truncated forms of recombinant human CYP46A1 were expressed as described [37, 38]. Full-length recombinant rat CPR was also expressed as described [39], except expression time was reduced to 6 hrs. For both proteins, E. coli cells were harvested by centrifugation at 10,000 g for 10 min, resuspended in 10 mM HEPES (pH 7.8) containing 10% sucrose, and treated with 0.2 mg/ml lysozyme on ice for 30 min. Spheroplasts were pelleted at 10,000 g for 10 min, resuspended in 10 mM HEPES (pH 7.8) containing 10% sucrose and sonicated using six 10 seconds pulses at 40% duty cycle (Sonifier 450, Branson Ultrasonics Corp., Dandury, CT). A part of the suspension was subject to 100,000 g centrifugation and the pellet obtained was resuspended in 10 mM potassium phosphate buffer (KPi,), pH 7.2, containing 20% glycerol and used for CYP46A1- and CPR-activity measurements. The other part of the suspension was layered over a cushion of 55% sucrose topped with 10% sucrose in 10 mM HEPES (pH 7.8) and centrifuged in a Ti70 rotor at 35,000 rpm for 60 min. The total membrane fraction recovered from 55% sucrose interface was washed twice with 25 mM NH4HCO3 (pH 7.9) and used for trypsin or chymotrypsin treatment.
Activity of CYP46A1- and CPR-containing E. coli membranes
Catalytic properties of full-length CYP46A1 bound to the E. coli membranes were measured in the in vitro reconstituted system (1 ml). E. coli membrane fraction containing 0.5 nmol of CYP46A1 was mixed with 1 nmol of purified recombinant CPR, 250,000 cpm (11 nM) [3H]cholesterol and 50 mM KPi (pH 7.2) containing 100 mM NaCl. The enzymatic reaction was initiated by the NADPH-regenerating system (1 mM NAPPH, 10 mM glucose-6-phosphate, and 2U of glucose-6-phosphate dehydrogenase), carried out for 1 hr at 37°C and terminated by vortexing with 2 ml of CH2Cl2. The organic phase was isolated, evaporated, dissolved in methanol, and separated by HPLC as described [38]. In control incubations, E. coli membranes were boiled for 1 min prior to reconstitution with CPR and cholesterol.
Electron transfer properties of CPR-containing E. coli membranes were tested using purified CYP46A1 and cytochrome c. In these experiments a truncated Δ(2-50) form of CYP46A1 was used. Unlike the full-length CYP46A1, this form is soluble and does not require the presence of a detergent in the buffer. Thus, the assay mixture did not contain even a trace amount of a detergent that can affect association of CPR with E. coli membranes. The P450 reduction was monitored by recording CO-reduced difference spectra [40] after purified Δ(2-50) CYP46A1 (0.4 μM final) was mixed with CPR-containing E. coli membranes (1.5 mg) and 1 mM NADPH in 1 ml of 50 mM KPi (pH 7.2). Assaying of cytochrome c reduction was similar (50 μM cytochrome c, 1.5 mg of the CPR-containing E. coli membranes and 1 mM NAPDH), except monitored at 550 nm and CO was omitted. In a separate experiment, Δ(2-50) CYP46A1 was reduced chemically with sodium dithionite to assess the extent of the enzymatic reduction with CPR-containing membranes. Reconstitution of Δ(2-50)CYP46A1 and cytochrome c with empty E. coli membranes (containing no CPR) was also carried out to serve as a negative control.
Activity of purified Δ(2-50)CYP46A1 with cholesterol-enriched E. coli membranes
Empty E. coli membranes (600 mg) were incubated with 3,700,000 cpm or 22 nM [3H]cholesterol for 30 min on ice in 3 ml of 50 mM KPi (pH 7.2). The membrane suspension was then subjected to centrifugation at 400,000 g for 10 min. The pellet obtained was re-suspended in 3 ml of 50 mM KPi and sedimented at 400,000 g for 10 min again. The radioactivity was determined in an aliquot (50 μl) by scintillation counting. Since small amounts of [3H]cholesterol were present in the supernatant, the membrane fraction was resuspended in 3 ml of 50 mM KPi and span down at 100,000 g for 60 min 3 more times until no radioactive cholesterol could be detected in the supernatant. The membrane fraction was then re-suspended in 0.5 ml of 50 mM KPi and used for measurements of CYP46A1 activity in the in vitro reconstituted system. The assay mixture (1 ml) comprised 0.1 mg of the E. coli membranes containing 500,000 cpm [3H]cholesterol, 0.1 nmol purified Δ(2-50)CYP46A1, 0.2 nmol purified CPR, and 1 mM NADPH-regenerating system. The assay buffer was 50 mM KPi, and reaction time was 40 min. Steroids were extracted and analyzed by HPLC as described [38]. In control incubations the assay mixture was boiled prior to addition of NADPH-regenerating system.
Control experiments evaluating possible “leakage” of cholesterol from cholesterol-enriched E. coli membranes were performed with and without NADPH-regenerating system on the sample containing [3H]cholesterol-enriched membranes, CYP46A1 and CPR. After a 40-min reaction, the incubations were subjected to the 1st centrifugation at 100,000 g for 60 min and the radioactivity in the supernatant was determined by scintillation counting. The samples were then subjected to the 2nd centrifugation at 100,000 g for 60 min and the radioactivity in supernatant and membrane pellet was determined for a second time.
Treatment with chymotrypsin and trypsin
The membrane pellet obtained from 55% sucrose interface (5 mg, ≈35 μg of total protein) was resuspended in 1 ml of 25 mM NH4HCO3 (pH 7.9) followed by addition of either 10 μg of sequencing grade chymotrypsin or 10 μg of sequencing grade modified trypsin. The suspension was sonicated as described above and left for proteolysis for 15 hrs. Digestion with chymotrypsin was carried out at room temperature, while that with trypsin was at 37 °C. The samples were then subjected to 106,000 g centrifugation for 20 min in Optima TLX ultracentrifuge (Beckman Instruments, Inc., Fullerton, CA, USA). Pellets were collected and washed sequentially with 100 mM Na2CO3 (pH 11.5) containing 0.3 M NaCl (5 times) and with water (1 time). Traces of water from the wet pellets were removed by adding 0.2 ml of methanol/chloroform mixture (4/1, v/v) and drying the sample in Vacufuge (Eppendorf AG, Hamburg, Germany). Dried pellet was then vortexed with 0.2 ml of chloroform, organic phase discarded, and the membranes dried again. Extraction of peptides from the dried membranes was carried out with 100 μl of 50% acetonitrile/0.1% trifluoroacetic acid (TFA). One half of the obtained sample was left untreated while another half reduced with 10 mM DTT for 60 min and alkylated with 50 mM iodoacetamide for 60 min. Reduction/alkylation was performed in 25 mM NH4HCO3 (pH 7.9) at room temperature. Both, non-alkylated and alkylated samples were dried and further purified using ZipTip C18 tips (Millipore Corporation, Bedford, MA, USA).
Mass spectrometry and data analysis
Peptide samples after ZipTip purification were dried, dissolved in 5 mg/ml α-cyano-4-hydroxycinnamic acid in 50% acetonitrile containing 0.1% TFA, and manually spotted onto the ABI 01-192-6-AB target plate. Mass spectrometry (MS) analyses were performed using an AB4700 Proteomics Analyzer (Applied Biosystems, Framingham, MA, USA). MS-mode acquisitions consisted of 1,000 laser shots averaged from 20 sample positions. The collected MS spectra were manually analyzed based on matching of the observed masses with the theoretical masses of CYP46A1 and CPR peptides generated by FindPept software (http://us.expasy.org/tools/findpept.html) for trypsin or chymotrypsin (C-terminal to F/Y/W, not before P) digestions with 4 missed cleavages. Each of the identified CYP46A1 or CPR peptides was systematically interrogated with the MS/MS analysis. In the MS/MS-mode acquisitions, 6,000 laser shots were averaged from 60 sample positions for post-source decay fragments. The signal intensity threshold was set to a signal-to-noise ratio of 10. The precursor ion selection window was set at a resolution of 150, which is a typical setting for this instrument. The analysis of the MS/MS data was performed with GPS Explorer software 3.5 utilizing Mascot 2.0 (MatrixScience, London, UK) as the search engine. The collected spectra were searched against NCBInr database allowing 1 missed cleavage and variable carbamidomethyl modification of cysteines. The mass tolerance was 0.08 Da on the precursor masses and 0.3 Da on the fragment ions.
Results
Recombinant CYP46A1 and CPR are catalytically active when associated with E. coli membranes
Previously we expressed human CYP46A1 in E. coli and established that, like other bacterially expressed microsomal P450s, full-length CYP46A1 is localized exclusively to the E. coli membrane fraction [37, 38]. The level of heterologously expressed full-length CYP46A1 in E. coli is relatively low, ~60 nmol/l of culture, therefore, in the present study no precautions were taken to prevent protein overexpression, which may lead to non-specific aggregation and non-functional association with the membrane. For measurements of the enzyme activity, CYP46A1-containing E. coli membranes were isolated and used for reconstitution with exogenous cholesterol, CPR and NADPH-regenerating system. A product peak corresponding to the retention time of 24-hydroxycholesterol was seen in the incubations with CYP46A1-containing E. coli membranes (Fig. 1a) but not in control incubations with boiled membranes (Fig. 1b). Thus, CYP46A1 is catalytically active when associated with E. coli membranes.
Fig. 1.
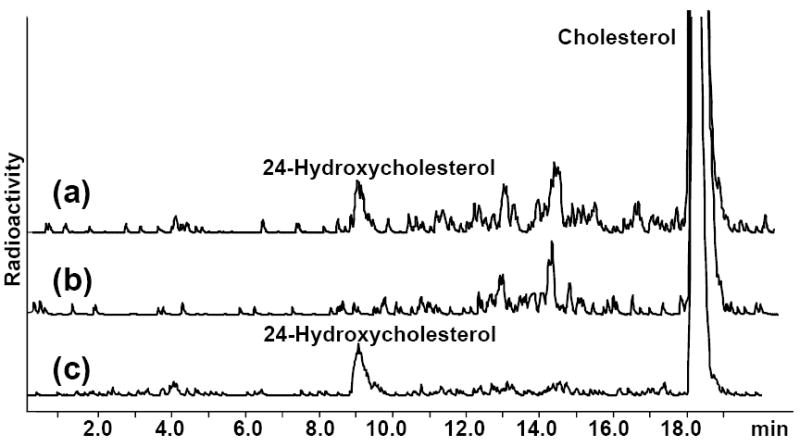
The HPLC product profiles of the extracts from the enzyme assays carried out with (a) CYP46A1-containing E. coli membranes; (b) boiled CYP46A1-containing E. coli membranes; and (c) purified recombinant CYP46A1. Assay conditions were as described in Materials and Methods.
Compared to CYP46A1, recombinant CPR is expressed at higher levels in E. coli [39]. To avoid overexpression, the E. coli cells were harvested at 6 hrs after the induction of CPR expression. The amount of CPR expressed at this time point was about two times less as compared to the 20 hr-expression used routinely for isolation of recombinant CPR. The CPR containing E. coli membranes were isolated and assayed for the ability to transfer electrons from NADPH to CYP46A1 and non-physiological redox partner cytochrome c. Both redox partners can be reduced by the CPR-containing E. coli membranes. The P450 reduction was ~55% of that observed with the sodium dithionite (Fig. 2), and cytochrome c reduction was 0.086 μM cytochrome c/mg of membrane protein (not shown). The empty E. coli membranes (contained no CPR) did not reduce CYP46A1 but were able to reduce cytochrome c, although at a 10-fold lower rate equal to 0.008 μM cytochrome c/mg of membrane protein.
Fig. 2.
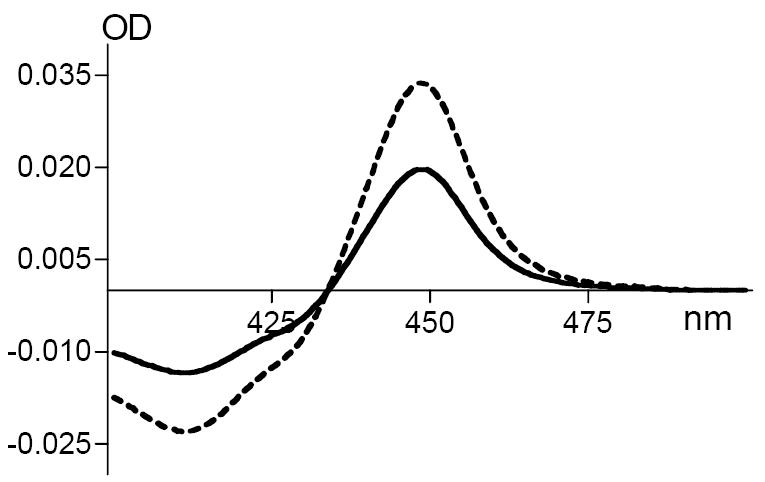
Enzymatic reduction of purified recombinant CYP46A1 with NADPH and CPR-containing E. coli membranes (solid line) and with sodium dithionite (dashed line). Assay conditions were as described in Materials and Methods.
Identification of the putative membrane-interacting peptides in CYP46A1 and CPR
CYP46A1- and CPR-containing E. coli membranes were each treated separately with trypsin and chymotrypsin and washed extensively with 0.1 M Na2CO3/0.3 M NaCl to remove hydrophobic peptides from the interior of the proteins that may bind non-specifically to the membranes. Lipids were then extracted from the membranes followed by the extraction of the peptide material and its MS analysis.
Representative MS spectra of the extracts derived from the non-alkylated CYP46A1 samples treated with trypsin and chymotrypsin are shown in Fig. 3a and Fig. 3c, respectively. Manual analysis revealed 3 peptides with m/z [M+H]+ values expected for the CYP46A1 hydrolysis with trypsin (Fig. 3a). In addition, the fourth tryptic peptide with m/z [M+H]+ value 2716.41 was recovered from the alkylated CYP46A1 sample. The enlarged spectrum for this peptide is shown in Fig. 3b since the intensity of signal was very low. Chymotrypsin treatment led to identification of two additional peptides in CYP46A1 (Fig. 3c, m/z [M+H]+ 972.56 and 1119.51). All of CYP46A1 peptides obtained after tryptic and chymotryptic digestion are summarized in Table 1 and clustered into two groups on the basis of their location in the domains of CYP46A1. The expected m/z [M+H]+ values were calculated using MASCOT software and rounded to two decimals. The observed m/z [M+H]+ values deviated from the expected m/z [M+H]+ values no more than 0.01-0.04 Da indicating the high accuracy of mass measurements. For three peptides (m/z [M+H]+ 2716.41, 1333.70, and 1119.51), the signal intensity was too low to acquire MS/MS spectra with confident ions score. For three other peptides (m/z [M+H]+ 1971.10, 1617.90, and 972.56), the identity was confirmed by MS/MS analysis (Table 1) and the MS/MS spectrum for the chymotryptic peptide ion with m/z [M+H]+ value 972.56 is shown in Fig. 3d for illustration. This spectrum has b1 (R), b3 (RLV), b7 (RLVPGQR), y2 (RF), y5 (PGQRF), y5-NH3 (loss of ammonia from y5), and internal fragment (PGQ) ions that match the RLVPGQRF sequence. In addition, all identified CYP46A1 peptides were searched against E. coli genome using blastp (http://blast.ncbi.nlm.nih.gov/Blast.cgi). No matches corresponding to the E.coli proteins were found in the non-redundant database.
Fig. 3.
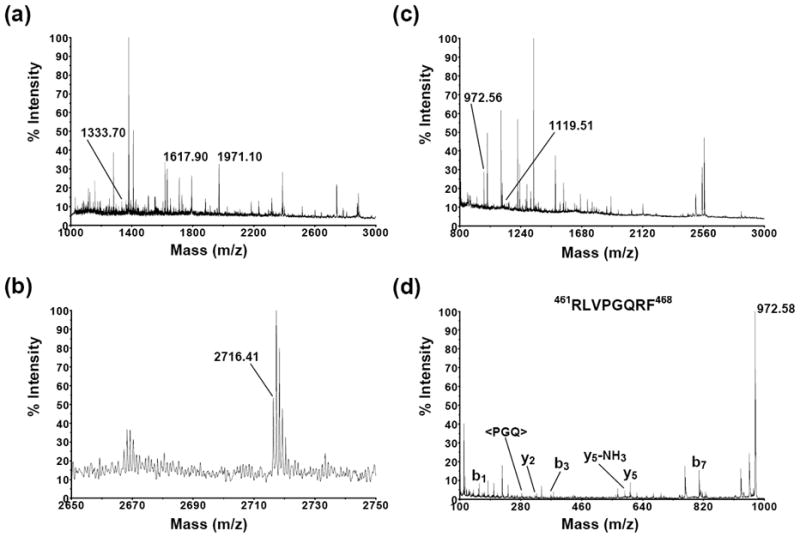
MS spectra of the extracts from the trypsin- (a) and chymotrypsin-treated (c) CYP46A1-containing E. coli membrane. Peptides that correspond to CYP46A1 are labeled and those from E. coli are not. Enlarged MS spectrum of the tryptic peptide ion with m/z [M+H]+ value 2716.41 is shown in panel (b). The MS/MS spectrum of the chymotryptic peptide ion with m/z [M+H]+ value 972.56 is shown in panel (d) and has b1 (R), b3 (RLV), b7 (RLVPGQR), y2 (RF), y5 (PGQRF), y5-NH3 (loss of ammonia from y5), and internal fragment (PGQ) ions that match the RLVPGQRF sequence.
Table 1.
Putative membrane-interacting peptides in CYP46A1. Missed cleavage sites are underlined.
| Peptide | m/z [M+H]+ expected/observed | Ions scorea | Protease |
|---|---|---|---|
| N-terminal membrane anchor | |||
| 1MAPGLLLLGSAVLLAFGLCCTFVHR25 | 2716.45/2716.41 | - | trypsin |
| Catalytic domain | |||
| 59VLQDVFLDWAK69 | 1333.72/1333.70 | - | trypsin |
| 77VNVFHKTSVIVTSPESVK94 | 1971.09/1971.10 | 59(>40) | trypsin |
| 217LMLEGITASRNTLAK231 | 1617.90/1617.90 | 77(>40) | trypsin |
| 397VMGRMDTYF405 | 1119.50/1119.51 | - | chymotrypsin |
| 461RLVPGQRF468 | 972.57/972.56 | 62(>40) | chymotrypsin |
Ions scores from Mascot software are shown for peptides with available MS/MS spectra. The number in parentheses is the threshold ions score that indicates identity or extensive homology (p < 0.05) for that particular search.
The MS analysis of the CPR samples was performed similarly. The representative spectra for non-alkylated CPR samples are shown in Fig. 4. A total of 5 peptides from CPR were found after trypsin treatment. These peptides are labeled in Fig. 4a. Seven more CPR peptides were found in the chymotrypsin-treated sample. Five of these peptides are labeled in Fig. 4c. Intensity of the other two peptides was low and enlarging of the spectrum is necessary to observe them. Peptides having the m/z [M+H]+ values matching those expected for the CPR peptides after tryptic or chymotryptic digestion are summarized in Table 2. They are clustered into four groups on the basis of their location in the domains of CPR as well as aligned on the basis of their primary sequence. The sequence identity for seven peptides out of twelve was confirmed by MS/MS analysis (Table 2). The representative MS/MS spectra for the tryptic peptide ion with m/z [M+H]+ value 1637.85 and for the chymotryptic peptide ion with m/z [M+H]+ value 1137.55 are shown in Fig. 4b and Fig. 4d, respectively. The spectrum for m/z [M+H]+ 1637.85 has b2 (TA), b3 (TAL), b8 (TALTYYLD), y1 (R), y3 (PPR), y4 (NPPR), y5 (TNPPR), y6 (ITNPPR), y7 (DITNPPR), y8 (LDITNPPR), y9 (YLDITNPPR), and y10 (YYLDITNPPR) fragment ions that match the TALTYYLDITNPPR sequence. The spectrum for m/z [M+H]+ 1137.55 has b2 (MG), b3 (MGF), b4 (MGFI), b8 (MGFIQERA), y1 (W), y2 (AW), y3 (RAW), y4 (ERAW), y5 (QERAW), and y6 (IQERAW) fragment ions that match the MGFIQERAW sequence. All identified CPR peptides were also searched against E. coli genome using blastp (http://blast.ncbi.nlm.nih.gov/Blast.cgi). No matches corresponding to the E.coli proteins were found in the non-redundant database.
Fig. 4.
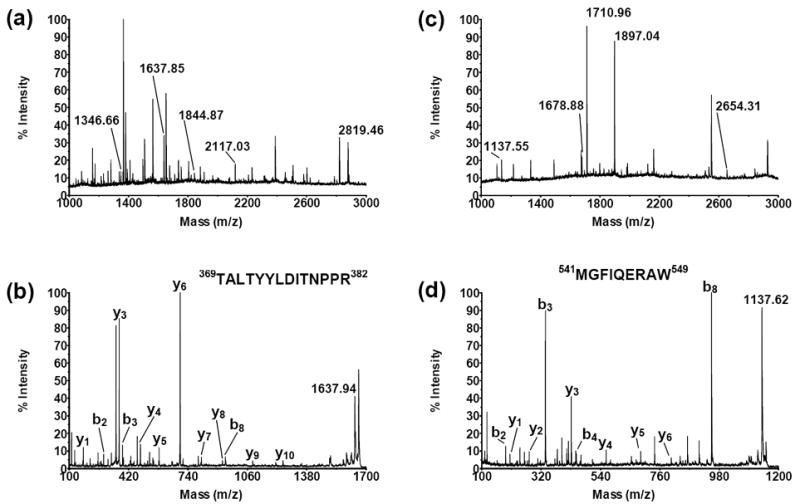
MS spectra of the extracts from the trypsin- (a) and chymotrypsin-treated (c) CPR-containing E. coli membrane. Peptides that correspond to CPR are labeled and those from E. coli are not. The MS/MS spectrum of the tryptic peptide ion with m/z [M+H]+ value 1637.85 is shown in panel (b) and has b2 (TA), b3 (TAL), b8 (TALTYYLD), y1 (R), y3 (PPR), y4 (NPPR), y5 (TNPPR), y6 (ITNPPR), y7 (DITNPPR), y8 (LDITNPPR), y9 (YLDITNPPR), and y10 (YYLDITNPPR) fragment ions that match the TALTYYLDITNPPR sequence. The MS/MS spectrum of the chymotryptic peptide ion with m/z [M+H]+ value 1137.55 is shown in panel (d) and has b2 (MG), b3 (MGF), b4 (MGFI), b8 (MGFIQERA) y1 (W), y2 (AW), y3 (RAW), y4 (ERAW), y5 (QERAW), and y6 (IQERAW) fragment ions that match the MGFIQERAW sequence.
Table 2.
Putative membrane-interacting peptides in CPR. Missed cleavage sites are underlined.
| Peptide | m/z [M+H]+ expected/observed | Ions scorea | protease |
|---|---|---|---|
| N-terminal membrane anchor | |||
| 41WFIFRKKKEEIPEF54 | 1897.04/1897.04 | 104(>30) | chymotrypsin |
| 42FIFRKKKEEIPEF54 | 1710.96/1710.96 | 129(>30) | chymotrypsin |
| FMN-binding domain | |||
| 70VEKMKKTGRNIIVFY84 | 1826.04/1826.03 | - | chymotrypsin |
| 79NIIVFYGSQTGTAEEFANR97 | 2117.03/2117.03 | 134(>30) | trypsin |
| Connecting and FAD-binding domains | |||
| 260TGEMGRLKSYENQKPPFDAKNPF282 | 2654.30/2654.31 | - | chymotrypsin |
| 266LKSYENQKPPFDAKNPFLAAVTANR290 | 2819.48/2819.46 | 71(>40) | trypsin |
| 358KHPFPCPTTYR368 | 1346.67/1346.66 | - | trypsin |
| 369TALTYYLDITNPPR382 | 1637.85/1637.85 | 98(>32) | trypsin |
| 374YLDITNPPRTNVLY387 | 1678.88/1678.88 | 53(>40) | chymotrypsin |
| NADPH-binding domain | |||
| 541MGFIQERAW549 | 1137.55/1137.55 | 64(>40) | chymotrypsin |
| 550LREQGKEVGETLLY563 | 1634.87/1634.86 | - | chymotrypsin |
| 552EQGKEVGETLLYYGCR567 | 1844.88/1844.87 | - | trypsin |
Ions scores from Mascot software are shown for peptides with available MS/MS spectra. The number in parentheses is the threshold ions score that indicates identity or extensive homology (p < 0.05) for that particular search.
Positioning of CYP46A1 and CPR in the membrane
At first, we examined the putative membrane-interacting peptides in CYP46A1 and CPR for the presence of the missed cleavage sites. In our experiments, high 1 to 4 ratio (μg of protease/μg of total protein) of trypsin and chymotrypsin was used to achieve complete hydrolysis of accessible bonds. Under this protease/protein ratio, the purified CYP46A1 and CPR were completely hydrolyzed in solution in control experiments (not shown). However, hydrolysis of membrane-associated CYP46A1 and CPR generated peptides with missed cleavage sites. Trypsin and chymotrypsin are not reported to cleave amino acid residues inside the membrane. Consequently, the missed cleavage sites could indicate the regions in CYP46A1 and CPR that are located either inside or close to the membrane and are not accessible for proteolysis. Likewise, the amino acid residues that were proteolysed should be located outside the membrane. To position CYP46A1 and CPR in the membrane, we first mapped the identified missed cleavage site on the available crystal structures of CYP46A1 and CPR (PDB ID codes 2Q9F and 1amo, respectively) (Fig. 5a and Fig. 6a). Then, we mapped the putative membrane-interacting peptides and their flanking residues whose cleavage produced these peptides. While positioning CYP46A1 and CPR in the membrane, we kept in mind the following. Trypsin and chymotrypsin are known to digest only solution-exposed amino acid residues, therefore peptides extracted from the membrane contain both membrane-bound and soluble portions. Also, certain classes of long peptides with high hydrophobicity can escape MS analysis [41], therefore a set of identified peptides may not represent all membrane-interacting areas. Finally, the first available N-terminal amino acid residue in the crystal structures, which is next to the membrane anchor in the full-length proteins, should be spatially close to the membrane.
Fig. 5.
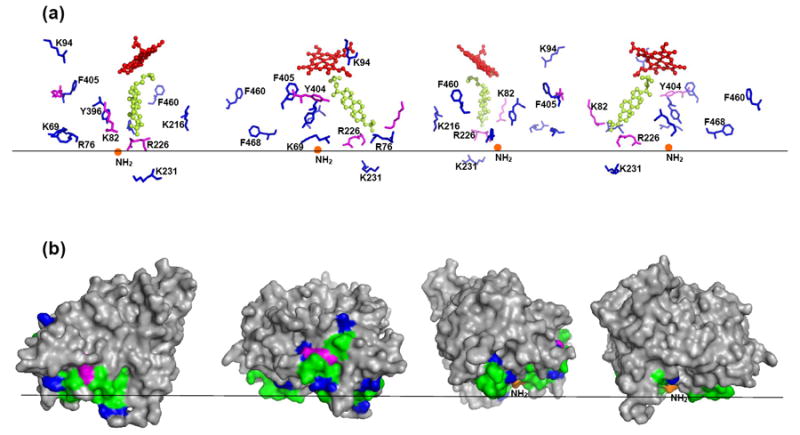
The proposed membrane orientation of CYP46A1. Black horizontal line separates the cytosol (above) and the lipid bilayer (below). The CYP46A1 molecule (PDB ID 2Q9F) is shown in four different views rotated counterclockwise by 90°. (a) Spatial position of the heme (shown as sticks and balls in red), substrate (cholesterol sulfate, shown as sticks and balls in lime), the missed cleavage sites (shown as sticks in magenta), and amino acid residues flanking the putative membrane-interacting peptides (shown as sticks in blue). The N-terminus, which connects to the transmembrane anchor, is shown as orange ball. (b) The surface representation of CYP46A1. The views and coloring are the same as in (a); the putative membrane-interacting peptides are in green.
Fig. 6.
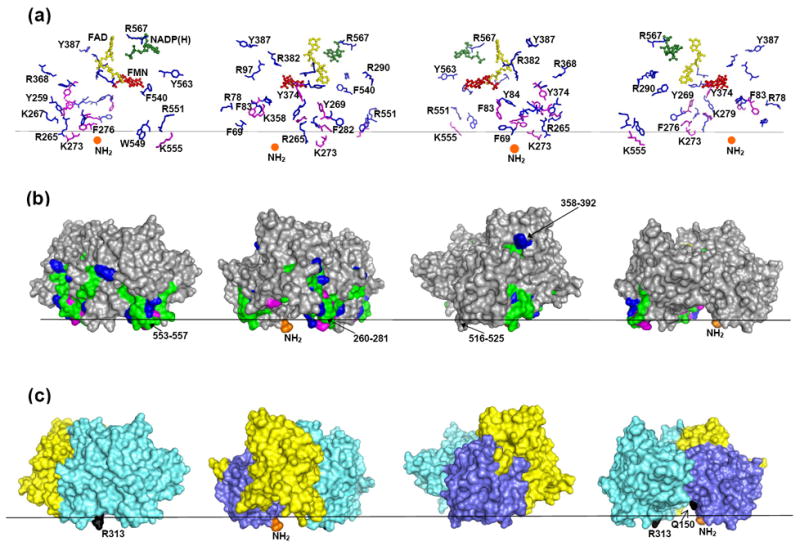
The proposed membrane orientation of CPR. Black horizontal line separates the cytosol (above) and the lipid bilayer (below). The CPR molecule (PDB ID 1AMO) is shown in four different views rotated counterclockwise by 90°. (a) Spatial position of NADP(H) (shown as sticks and balls in green), FAD (shown as sticks and balls in yellow), FMN (shown as sticks and balls in red), the missed cleavage sites (shown as sticks in magenta), and amino acid residues flanking the putative membrane-interacting peptides (shown as sticks in blue). The N-terminus, which connects to the transmembrane segment, is shown as orange ball. (b) The surface representation of CPR. The views and coloring are the same as in (a); the putative membrane-interacting peptides are in green. (c) The surface representation of CPR showing the domain structure; the FAD- and NADPH-binding domains are in aquamarine, the connecting domain is in yellow, and the FMN-binding domain is in violet. Q150 and R313 (Q153 and R316 in human CPR) whose mutations result in disordered steroidogenesis are in black.
The resulting model of CYP46A1 is shown in Fig. 5b. All five putative membrane-interacting peptides in the soluble domain are located on the same side of the protein molecule and could be placed into a plane of membrane with the missed cleavage sites being close to the membrane surface and all flanking residues except one (K231) located outside the membrane. K231 resides on the F-G loop, a flexible region in the CYP46A1 structure, which undergoes significant conformational movement upon substrate binding [42] and, therefore, could be outside the membrane in substrate-free P450 that was used in the present work. Unfortunately, this region is disordered in the crystal structure of substrate-free CYP46A1 [42]. According to the model obtained, the entrance to the substrate access channel in CYP46A1 is in the membrane and the heme group is outside the membrane at ~45° angle relative to the plane of the membrane.
The same rules were used for positioning CPR in the membrane and the resulting model is shown in Fig. 6b. All missed cleavage sites found in the membrane-interacting peptides of the soluble domain are located on the same side of the protein molecule and in close proximity to the first N-terminal residue available in the crystal structure. All three cofactors in CPR are located above the membrane and the NADP(H) can enter/exit the protein from above.
Studies of cholesterol access to the CYP46A1 active site
The proposed membrane topology of CYP46A1 suggests that cholesterol enters the enzyme through the membrane. Since E. coli membranes do not contain cholesterol, we tested this inference by conducting the following experiments. At first, membrane fraction was isolated from the E. coli cells transformed with the expression vector that did not contain the coding sequence for either CYP46A1 or CPR (empty membranes). These empty membranes were then incubated with exogenous radioactive cholesterol followed by extensive washes of the membranes with the buffer to remove cholesterol that did not incorporate into the membranes. The cholesterol-enriched membranes were reconstituted with purified substrate-free CYP46A1, CPR, and the enzyme reaction initiated by addition of NADPH-regenerating system. Upon completion of the reaction, steroids were extracted and analyzed by HPLC for the presence of 24-hydroxycholesterol and 24,25- and 24,27-dihydroxycholesterols, the products of CYP46A1 activities. Two peaks, one corresponding to the retention time of 24-hydroxycholesterol, and the other one to the retention time of dihydroxycholesterol (24,25- and 24, 27-dihydroxycholesterols are not separated under the HPLC conditions used) were seen in the HPLC profiles of the extracts from the incubations of CYP46A1 with cholesterol-enriched membranes (Fig. 7). No products, however, were seen in the control incubations in which the reaction mixture was boiled prior to addition of NADPH.
Fig. 7.
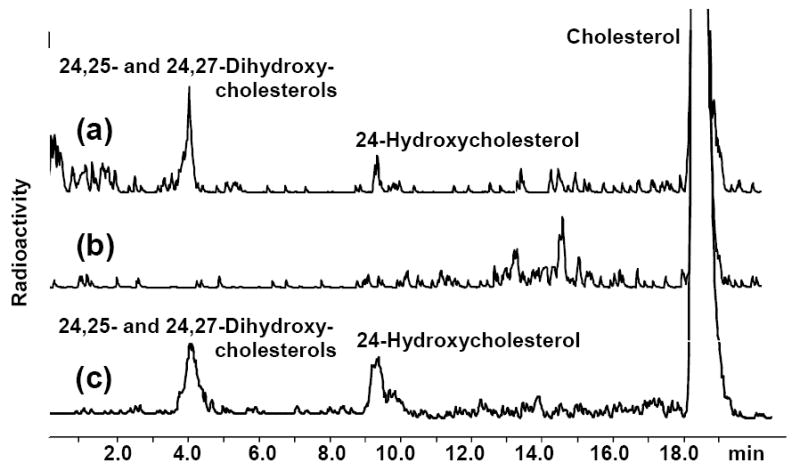
The HPLC product profiles of the extracts from the enzyme assays carried out with (a) purified CYP46A1 and cholesterol-enriched E. coli membranes; (b) control incubation with boiled CYP46A1; and (c) purified recombinant CYP46A1 and cholesterol. Assay conditions were as described in Materials and Methods.
To establish whether membrane-incorporated cholesterol is the only substrate for CYP46A1 in these incubations, we evaluated a possible partitioning of [3H]cholesterol into aqueous phase during the enzyme assay. After a 40-min reaction, a set of incubations with and without NADPH-regenerating system was subjected to two sequential ultracentrifugations and distribution of radioactivity was determined in the supernatant and pellet (membrane fraction). Incubations containing [3H]cholesterol-enriched E. coli membranes, CYP46A1, CPR and NADPH-regenerating system, had 13,748 cpm of the radioactivity in the supernatant after 1st ultracentrifugation and 1,323 less cpm in the supernatant (12,425 cpm) after 2nd ultracentrifugation (Table 3). Omitting NADPH from the incubations resulted in a much less radioactivity in the supernatant (2,198 cpm) after 1st ultracentrifugation but to a similar 1,409 cpm decrease in radioactivity after 2nd ultracentrifugation. Although these control experiments cannot absolutely exclude the partitioning of membrane-bound [3H]cholesterol into the aqueous solution, the similar decrease of the supernatant radioactivity in the NADPH-containing and NAPDH-lacking incubations after 2nd ultracentrifugation indicates that a source of small radioactivity found in the supernatant of NADPH-lacking incubation is rather due to incomplete precipitation of [3H]cholesterol-enriched membranes after the 1st centrifugation. A significant difference in the supernatant radioactivity in the NADPH-containing and NAPDH-lacking incubations (6.3-fold and 15.7-fold after the 1st and 2nd ultracentrifugations, respectively) also suggested that products of CYP46A1 activities are released in the supernatant during the enzyme reaction. To test this, we compared propensity of cholesterol and 24-hydroxycholesterol toward E. coli membranes. The incubation and washing procedures were exactly the same as those used for saturation of the membranes with cholesterol for enzyme activity measurements. Of a total amount of steroid taken for saturation, 85% of cholesterol and 34% of 24-hydroxycholesterol were found in the supernatant after the incubation step. Subsequent washes of the membranes with the buffer did not affect cholesterol retention in the membrane but led to a constant leakage of 24-hydroxycholesterol from the membranes in the supernatant. Only 2.6% of 24-hydroxycholesterol was found in the membranes after the last washing step. These experiments suggest that most of the radioactivity detected in the supernatant in the incubations containing NADPH-regenerating system in Table 3 is likely due to partitioning of the cholesterol hydroxylated products from the CYP46A1 active site into the aqueous phase.
Table 3.
Distribution of radioactivity between the supernatant and pellet (membrane fraction) in the incubations of [3H]cholesterol-enriched E. coli membranes with CYP46A1 and CPR
| Composition of the assay mixture | [3H]cholesterol taken for saturation of the membranes, cpm | Radioactivity in the supernatant after 1st ultracentrifugation, cpm | Radioactivity after 2nd ultracentrifugation, cpm | |
|---|---|---|---|---|
| supernatant | pellet | |||
| [3H]cholesterol-enriched membranes, CYP46A1, CPR, and NADPH-regenerating system | 444,984 | 13,748 | 12,425 | 432,504 |
| [3H]cholesterol-enriched membranes, CYP46A1, and CPR | 444,697 | 2,198 | 789 | 443,698 |
Discussion
Levels of protein expression and complexity of the native biological membranes limit analysis of membrane binding of microsomal P450s to the parental endoplasmic reticulum. The majority of current information on P450-membrane interactions was obtained in various model system, such as proteoliposomes or expression of P450s in non-parental cell lines [19, 22-25, 28, 29]. In the present work we used E. coli membranes containing heterologously expressed CYP46A1 and CPR as a model system to study membrane topology of these proteins. As any model, the E. coli system also has its limitation. The lipid composition of E. coli membrane differs from that of human membranes, therefore positioning of the heterologously expressed proteins may not be exactly the same as in parental endoplasmic reticulum. Recombinant P450s embedded in the E. coli membranes are usually catalytically active and efficiently catalyze the oxidation of representative substrates at efficient rates [16]. Thus, despite differences in lipid composition of mammalian and E. coli membranes, E. coli membranes containing a heterologously expressed P450 seem to represent a suitable model to study structure/function relationships in P450s. In the present work, we capitalized on the fact that E. coli expression systems were previously developed for CPR and CYP46A1 [37, 39]. Further, both CYP46A1 and CPR stay associated with the membrane fraction when expressed in E. coli and are catalytically active. Finally, crystal structures of CYP46A1 and CPR are available [36, 42].
Two proteases with different selectivity, trypsin and chymotrypsin, were used to digest the solution-exposed portions of CYP46A1 and CPR. Peptides remaining in the membrane were extracted and identified by MS. The use of two proteases allowed mapping more putative membrane-associated regions in each protein than would have been found if only one type of proteolysis was used. Also, the two proteases produced overlapping peptides in CPR (Table 2). It is the presence of the missed cleavage sites in both tryptic and chymotryptic Y269-K279 segment of CPR that made us decide to place this segment in the membrane (Fig. 6).
In both CYP46A1 and CPR, we identified the peptides originating from the N-terminal regions known to anchor these proteins to the membrane. This result lends a support to the validity of our approach. Peptides other than the N-terminal membrane anchor were found in the extracts from the protease-treated CYP46A1- and CPR-containing membranes. In CYP46A1, the three putative membrane-interacting peptides in the soluble domain, 59V-K69, 77V-K94, and 217L-K231 (Table 1), form the entrance to the substrate access channel and represent the secondary structural elements (the A helix and β1 sheet and F-G loop regions) that are suggested to be associated with the membrane in some microsomal P450s [43, 44]. The identification of these peptides in CYP46A1 is, thus, consistent with the notion in the field. The fourth and fifth putative membrane-interacting peptides in Table 1, 397V-F405 and 461R-F468, are the part of the K’ helix and β3 sheet, respectively. These topological elements were not previously implicated in membrane binding and could be specific to CYP46A1. Positioning of the five putative membrane-interacting peptides in the membrane placed the entrance to the substrate access channel into the membrane and suggested that cholesterol enters CYP46A1 directly from the membrane. Our experiments with cholesterol-enriched E. coli membranes supported this suggestion, although interpretation of this data should be cautious as cholesterol is known to be not absolutely insoluble in water. Trace partitioning of membrane-bound cholesterol into the aqueous solution cannot be ruled out. Knowledge of the membrane topology of CYP46A1 and the route of cholesterol access is important in a view of recent findings that some marketed drugs inhibit CYP46A1 activity in vitro, and the enzyme contains several channels connecting the protein surface and active site [42, 45]. It is conceivable that highly hydrophobic cholesterol and less hydrophobic therapeutic agents may enter the CYP46A1 active site via different paths. While cholesterol has a preferable channel that opens into the membrane, xenobiotics, depending on their hydrophobicity, may reach the active site from the channel having the entrance above the membrane surface [45].
Unlike P450 enzymes, CPR is a multidomain protein suggested to undergo domain motions upon interaction with P450 [36]. A total of 10 putative membrane-interacting peptides were identified in the soluble domain of CPR (Table 2). These peptides originate from 4 regions in the protein: 70V-R97, 260T-R290, 358K-Y387, and 541M-R567. Two of these regions, 260T-R290 and 541M-R567, overlap with the segments, 250-281 and 553-557, that were previously suggested to bind to the membrane based on the analysis of the CPR crystal structure [36]. These segments are in the membrane in our model as well (Fig. 6). The third membrane-binding segment indicated by the crystal structure comprises residues 516-525. We did not identify the peptides from this segment in our membrane extracts but this segment turned out to be in the membrane in our model as well. Such good correlation between the predictions based on the analysis of the CPR crystal structure and model of membrane topology obtained in our study, made us superimpose the two suggested models. We found them to be similar (not shown), despite our model being constructed based on experimental data and independent of the previously reported model [36].
Recently, a number of mutations have been found in CPR in patients with Antley-Bixer Syndrome and disordered steroidogenesis [32, 33]. Two of them, R316W and Q153R, are located in the regions associated with/close to the membrane in our model, in the FMN- and FAD-binding domains, respectively (Fig. 6c). Huang et al. recreated these mutations in vitro by site-directed mutagenesis and showed that, depending on the redox partner, the R316W mutant had either moderately reduced or increased catalytic activity (up to 40%), whereas the catalytic properties of the Q153R mutant are significantly impaired in all types of assays [33]. Our data raise a possibility that the R316W and Q153R mutations affect correct interaction of CPR with the membrane. These mutations may also have other catalytic or conformational effects.
In summary, we propose to use E.coli expression of membrane proteins as a model to study their membrane topology. It cannot be ruled out that expression of human proteins in non-parental E.coli membranes may lead to false positive identifications. However, when applied to two different proteins, CYP46A1 and CPR, our new approach generated the data that are in a good agreement with previous studies in the field. We also obtained peptides pointing to the regions that were not previously predicted as membrane-bound based on the analysis of the hydrophobicity and secondary and tertiary structures. Lastly, this study provided important insights pertinent to the function of CYP46A1 and CPR.
Acknowledgments
Certain commercial materials, instruments, and equipment are identified in this manuscript in order to specify the experimental procedure as completely as possible. In no case does such identification imply a recommendation or endorsement by the National Institute of Standards and Technology (NIST) nor does it imply that the materials, instruments, or equipment identified is necessarily the best available for the purpose.
This work was supported in part by National Institutes of Health Grants GM062882 and AG024336.
Footnotes
Abbreviations used: CYP46A1, cytochrome P450 46A1; CPR, NADPH-cytochrome P450 oxidoreductase; TFA, trifluoroacetic acid; MALDI MS, matrix assisted laser desorption ionization mass spectrometry; TOF, time-of-flight.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Granseth E, von Heijne G, Elofsson A. J Mol Biol. 2005;346:377–385. doi: 10.1016/j.jmb.2004.11.036. [DOI] [PubMed] [Google Scholar]
- 2.Fernandez-Vidal M, Jayasinghe S, Ladokhin AS, White SH. J Mol Biol. 2007;370:459–470. doi: 10.1016/j.jmb.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dubovskii PV, Dementieva DV, Bocharov EV, Utkin YN, Arseniev AS. J Mol Biol. 2001;305:137–149. doi: 10.1006/jmbi.2000.4283. [DOI] [PubMed] [Google Scholar]
- 4.Sapay N, Guermeur Y, Deleage G. BMC Bioinformatics. 2006;7:255. doi: 10.1186/1471-2105-7-255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lomize AL, Pogozheva ID, Lomize MA, Mosberg HI. BMC Struct Biol. 2007;7:44. doi: 10.1186/1472-6807-7-44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roberts MG, Phoenix DA, Pewsey AR. Comput Appl Biosci. 1997;13:99–106. doi: 10.1093/bioinformatics/13.1.99. [DOI] [PubMed] [Google Scholar]
- 7.Jayasinghe S, Hristova K, White SH. Protein Sci. 2001;10:455–458. doi: 10.1110/ps.43501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eisenhaber B, Bork P, Eisenhaber F. Protein Eng. 1998;11:1155–1161. doi: 10.1093/protein/11.12.1155. [DOI] [PubMed] [Google Scholar]
- 9.Mukhopadhyay S, Cho W. Biochim Biophys Acta. 1996;1279:58–62. doi: 10.1016/0005-2736(95)00201-4. [DOI] [PubMed] [Google Scholar]
- 10.Pratt JM, Jackson ME, Holland IB. EMBO J. 1986;5:2399–2405. doi: 10.1002/j.1460-2075.1986.tb04510.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bjorkhem I, Lutjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J. J Lipid Res. 1998;39:1594–1600. [PubMed] [Google Scholar]
- 12.Lund EG, Guileyardo JM, Russell DW. Proc Natl Acad Sci U S A. 1999;96:7238–7243. doi: 10.1073/pnas.96.13.7238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wellington CL. Clin Genet. 2004;66:1–16. doi: 10.1111/j.0009-9163.2004.00280.x. [DOI] [PubMed] [Google Scholar]
- 14.Carter CJ. Neurochem Int. 2007;50:12–38. doi: 10.1016/j.neuint.2006.07.007. [DOI] [PubMed] [Google Scholar]
- 15.Kotti TJ, Ramirez DM, Pfeiffer BE, Huber KM, Russell DW. Proc Natl Acad Sci U S A. 2006;103:3869–3874. doi: 10.1073/pnas.0600316103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guengerich FP. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. Ortiz de Montellano PR, editor. Kluwer Academic/Plenum Publishers; New York: 2005. pp. 377–530. [Google Scholar]
- 17.Nelson DR, Strobel HW. J Biol Chem. 1988;263:6038–6050. [PubMed] [Google Scholar]
- 18.Brown CA, Black SD. J Biol Chem. 1989;264:4442–4449. [PubMed] [Google Scholar]
- 19.Vergeres G, Winterhalter KH, Richter C. Biochemistry. 1989;28:3650–3655. doi: 10.1021/bi00435a005. [DOI] [PubMed] [Google Scholar]
- 20.De Lemos-Chiarandini C, Frey AB, Sabatini DD, Kreibich G. J Cell Biol. 1987;104:209–219. doi: 10.1083/jcb.104.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monier S, Van Luc P, Kreibich G, Sabatini DD, Adesnik M. J Cell Biol. 1988;107:457–470. doi: 10.1083/jcb.107.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ahn K, Szczesna-Skorupa E, Kemper B. J Biol Chem. 1993;268:18726–18733. [PubMed] [Google Scholar]
- 23.Szczesna-Skorupa E, Ahn K, Chen C-D, Doray B, Kemper B. J Biol Chem. 1995;270:24327–24333. doi: 10.1074/jbc.270.41.24327. [DOI] [PubMed] [Google Scholar]
- 24.Uvarov VYu, Sotnichenko AI, Vodovozova EL, Molotkovsky JG, Kolesanova EF, Lyulkin YuA, Stier A, Krueger V, Archakov AI. Eur J Biochem. 1994;222:483–489. doi: 10.1111/j.1432-1033.1994.tb18889.x. [DOI] [PubMed] [Google Scholar]
- 25.Ozalp C, Szczesna-Skorupa E, Kemper B. Biochemistry. 2006;45:4629–4637. doi: 10.1021/bi051372t. [DOI] [PubMed] [Google Scholar]
- 26.von Wachenfeldt C, Johnson EF. In: Cytochrome P450: Structure, Mechanism, and Biochemistry. Ortiz de Montellano PR, editor. Plenum Press; New York: 1995. pp. 183–223. [Google Scholar]
- 27.Williams PA, Cosme J, Sridhar V, Johnson EF, McRee DE. Mol Cell. 2000;5:121–131. doi: 10.1016/s1097-2765(00)80408-6. [DOI] [PubMed] [Google Scholar]
- 28.Cosme J, Johnson EF. J Biol Chem. 2000;275:2545–2553. doi: 10.1074/jbc.275.4.2545. [DOI] [PubMed] [Google Scholar]
- 29.Nakayama K, Puchkaev A, Pikuleva IA. J Biol Chem. 2001;276:31459–31465. doi: 10.1074/jbc.M103943200. [DOI] [PubMed] [Google Scholar]
- 30.Lu AY, Coon MJ. J Biol Chem. 1968;243:1331–1332. [PubMed] [Google Scholar]
- 31.Estabrook RW, Franklin MR, Cohen B, Shigamatzu A, Hildebrandt AG. Metabolism. 1971;20:187–199. doi: 10.1016/0026-0495(71)90091-6. [DOI] [PubMed] [Google Scholar]
- 32.Fluck CE, Tajima T, Pandey AV, Arlt W, Okuhara K, Verge CF, Jabs EW, Mendonca BB, Fujieda K, Miller WL. Nat Genet. 2004;36:228–230. doi: 10.1038/ng1300. [DOI] [PubMed] [Google Scholar]
- 33.Huang N, Pandey AV, Agrawal V, Reardon W, Lapunzina PD, Mowat D, Jabs EW, Van Vliet G, Sack J, Fluck CE, Miller WL. Am J Hum Genet. 2005;76:729–749. doi: 10.1086/429417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Black SD, Coon MJ. J Biol Chem. 1982;257:5929–5938. [PubMed] [Google Scholar]
- 35.Black SD, French JS, Williams CH, Jr, Coon MJ. Biochem Biophys Res Commun. 1979;91:1528–1535. doi: 10.1016/0006-291x(79)91238-5. [DOI] [PubMed] [Google Scholar]
- 36.Wang M, Roberts DL, Paschke R, Shea TM, Masters BS, Kim JJ. Proc Natl Acad Sci U S A. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mast N, Norcross R, Andersson U, Shou M, Nakayama K, Bjorkhem I, Pikuleva IA. Biochemistry. 2003;42:14284–14292. doi: 10.1021/bi035512f. [DOI] [PubMed] [Google Scholar]
- 38.Mast N, Andersson U, Nakayama K, Bjorkhem I, Pikuleva IA. Arch Biochem Biophys. 2004;428:99–108. doi: 10.1016/j.abb.2004.05.012. [DOI] [PubMed] [Google Scholar]
- 39.Hanna IH, Teiber JF, Kokones KL, Hollenberg PF. Arch Biochem Biophys. 1998;350:324–332. doi: 10.1006/abbi.1997.0534. [DOI] [PubMed] [Google Scholar]
- 40.Omura R, Sato R. J Biol Chem. 1964;239:2370–2378. [PubMed] [Google Scholar]
- 41.Eichacker LA, Granvogl B, Mirus O, Muller BC, Miess C, Schleiff E. J Biol Chem. 2004;279:50915–50922. doi: 10.1074/jbc.M405875200. [DOI] [PubMed] [Google Scholar]
- 42.Mast N, White MA, Bjorkhem I, Johnson EF, Stout CD, Pikuleva IA. Proc Natl Acad Sci U S A. 2008;105:9546–9551. doi: 10.1073/pnas.0803717105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graham-Lorence S, Amarneh B, White RE, Peterson JA, Simpson ER. Protein Sci. 1995;4:1065–1080. doi: 10.1002/pro.5560040605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Peterson JA, Graham SE. Structure. 1998;6:1079–1085. doi: 10.1016/s0969-2126(98)00109-9. [DOI] [PubMed] [Google Scholar]
- 45.Pikuleva IA. Expert Opin Drug Metab Toxicol. 2008;4:1403–1414. doi: 10.1517/17425255.4.11.1403. [DOI] [PMC free article] [PubMed] [Google Scholar]