

Abstract
Estimates of gene flow between subpopulations based on FST (or NST) are shown to be confounded by the reproduction parameters of a model of skewed offspring distribution. Genetic evidence of population subdivision can be observed even when gene flow is very high, if the offspring distribution is skewed. A skewed offspring distribution arises when individuals can have very many offspring with some probability. This leads to high probability of identity by descent within subpopulations and results in genetic heterogeneity between subpopulations even when Nm is very large. Thus, we consider a limiting model in which the rates of coalescence and migration can be much higher than for a Wright–Fisher population. We derive the densities of pairwise coalescence times and expressions for FST and other statistics under both the finite island model and a many-demes limit model. The results can explain the observed genetic heterogeneity among subpopulations of certain marine organisms despite substantial gene flow.
NATURAL populations of organisms are often subdivided by geography. Individuals may or may not migrate between these subpopulations. Modeling gene flow between subpopulations can be traced back to Wright (1931), whose island model describes a population subdivided into discrete, local subpopulations by geography, with limited migration between individual subpopulations. With the advent of techniques to characterize genetic variation, several measures of population subdivision have been proposed on the basis of probabilities of identity (see Rousset 2002 for a review). These include Wright's (1951) FST, Nei's (1982) γST, and Lynch and Crease's (1990) NST.
The quantity FST can, under the assumption of equilibrium, be used to estimate levels of gene flow from allozyme data (Wright 1951). The quantity γST can be calculated from DNA sequence data and is equivalent to FST if the mutation rate is very low (Slatkin 1991). Levels of gene flow between subpopulations can thus also be estimated from DNA sequence data. However, Slatkin (1991) argues that FST is appropriate for allozyme data, whereas the gene genealogy-based method of Slatkin and Maddison (1989) is appropriate for DNA sequence data. As FST continues to be used in investigations of population structure and, recently, as a tool for identifying loci under selection (e.g., Murray and Hare 2006), we are concerned with FST and related measures below.
We derive expressions for FST and NST under the island model of population subdivision with symmetric migration (Nagylaki 1980; Strobeck 1987) and skewed offspring distribution among individuals in a population. When the offspring distribution is skewed, individuals have some nonnegligible probability of having very many offspring. The population model of skewed offspring distribution we adopt in this work can result in an ancestral process with asynchronous multiple mergers (Eldon and Wakeley 2006). An ancestral process with asynchronous multiple mergers, or Λ-coalescent, was introduced by Pitman (1999) and also derived by Sagitov (1999) from a Cannings (1974) model. In a Λ-coalescent, any number of ancestral lines can coalesce at once to a single ancestor. In contrast, the Kingman coalescent (Kingman 1982a,b) allows only two lines to coalesce each time. For a single population, the ancestral process obtained from the population model of Eldon and Wakeley (2006), and employed in this work, is a special case of the Λ-coalescent of Pitman (1999) and Sagitov (1999).
Type III survivorship curve, and high fecundity, characterize a diverse group of organisms (e.g., many plants and marine animals). A prime example are marine species with broadcast spawning, including Atlantic cod (Gadus morhua; Árnason 2004), Pacific oysters (Crassostrea gigas; Beckenbach 1994; Hedgecock 1994a), and red drum (Sciaenops ocellatus; Turner et al. 2002). A model of skewed offspring distribution, in which individuals can have very many offspring with a nonnegligible probability, may therefore better apply in such cases than the Wright–Fisher (Fisher 1930; Wright 1931) or the Moran (Moran 1958, 1962) models.
Genetic observations from these species also argue against the standard population models. Genetic diversity is observed to be much lower than expected on the basis of population size for some marine populations (Hedgecock et al. 1982; Nei and Graur 1984; Avise et al. 1988; Avise 1994). In particular, low effective to actual population size ratios have been reported for Atlantic cod (Árnason 2004), red drum (Turner et al. 2002), and the Pacific oyster (Hedgecock 1994a), and this has been explained by high variance in offspring distribution (Crow and Kimura 1970; Hedrick 2005). Second, models of skewed offspring distribution predict a large number of singleton variants (Eldon and Wakeley 2006; Sargsyan and Wakeley 2008), a feature observed, for example, in Pacific oysters (Boom et al. 1994), Atlantic cod (Árnason 2004), and some hydrothermal vent taxa (Won et al. 2003; Hurtado et al. 2004; Johnson et al. 2006; Young et al. 2008).
Genetic heterogeneity on a small spatial scale has been observed for many marine populations, including the purple sea urchin (Strongylocentrotus purpuratus; Edmands et al. 1996), even though planktonic larvae disperse over wide-ranging habitats (Johnson and Black 1984; Watts et al. 1990; Hedgecock 1994b; David et al. 1997). A range of explanations has been proposed for the observed heterogeneity (see Burton 1983; Palumbi 1994). Our aim is to address, by analytic methods, the problem concerning the genetic population structure of a highly fecund species with potentially highly skewed offspring distribution, like the Atlantic cod (Árnason et al. 2000).
We obtain the probability distributions of pairwise coalescence times, and expressions for FST, for both the finite island and a many-demes limit model. Our main result is that evidence of population subdivision can be observed in genetic data even if the usual migration rate Nm is very large. In essence, a skewed offspring distribution leads to high probabilities of identity by descent within subpopulations and thus high FST. Therefore, patterns in genetic data indicating population subdivision cannot be taken to indicate low levels of gene flow in a population with a skewed offspring distribution. In fact, estimates of migration rate based on FST (or NST) are confounded by the reproduction parameters of our model of skewed offspring distribution. These results may explain the genetic heterogeneity among subpopulations of some marine species like the purple sea urchin (S. purpuratus; Edmands et al. 1996), despite the potential for wide dispersal of long-lived planktotrophic larvae (Burton 1983; Palumbi 1994).
METHODS AND RESULTS
Throughout we are concerned with neutral genetic diversity at a single nonrecombining locus in a haploid population. As usual, N is the population size. The results should hold for a diploid population with gametic migration if we replace N with 2N. The population model we consider is a modification of the well-known Moran model of reproduction (Moran 1958, 1962). In the Moran model, a single randomly chosen individual reproduces each time step. To keep the population size constant a randomly chosen individual, but not the offspring, dies to make room for the offspring.
In our model, which was first presented in Eldon and Wakeley (2006), a single randomly chosen individual (the parent) reproduces each time step. With probability 1 – ɛ the parent has one offspring. Alternatively, with probability ɛ the parent has ψN – 1 offspring (a large reproduction event) with 0 < ψ < 1. To keep population size constant when a large reproduction event occurs, a total of ψN – 1 individuals die to make room for the new offspring. In our model the parent always persists. The parameter ψ represents the fraction of the population that is replaced by the offspring of the parent. Eldon and Wakeley (2006) show that this modified Moran model of overlapping generations gives rise to a coalescent process that allows for asynchronous multiple mergers of ancestral lines, i.e., is of the same type as the ancestral process considered by Pitman (1999) and Sagitov (1999).
For ease of presentation, we define the following quantities: Nγ, cN, λγ, and IA. The quantity Nγ is the coalescence timescale in our model. The coalescence timescale is proportional to the number of time steps, on average, it takes for two individuals to coalesce (in a single population). It depends on the value of ɛ that we assume has the form ɛ ≡ 2φ/Nγ for some constants φ and γ with 0 < φ, γ < ∞. In our model, the coalescence timescale is Nγ/2 time steps when 0 < γ < 2. In the usual Moran model, the timescale is N2/2 time steps, which is also the value of Nγ when γ ≥ 2.
For a single population, Eldon and Wakeley (2006) show that different coalescent processes result depending on γ. Multiple mergers of ancestral lines are allowed in the coalescent process when 0 < γ ≤ 2, while Kingman's coalescent (Kingman 1982a,b) results when γ > 2. The probability that two individuals do coalesce in a single time step is denoted by cN and depends on ɛ. The rate λγ of coalescence of two individuals is obtained from cN by “speeding up” time by a factor of Nγ. When 0 < γ ≤ 2, λγ depends on the reproduction parameters φ and ψ. In mathematical notation, Nγ is expressed as 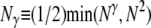 , and the coalescence probability cN is
, and the coalescence probability cN is
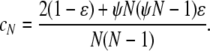 |
(1) |
For notational convenience, we also define the indicator function IA as
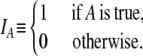 |
For example, Iγ<2 = 1 if γ < 2, and zero otherwise. In our model a large reproduction event occurs when the number of offspring of the parent equals ψN – 1. These events occur with probability ɛ. Our choice of ɛ = 2φ/Nγ results in the coalescence timescale being Nγ. The rate λγ of coalescence is then
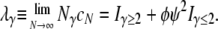 |
(2) |
The coalescence rate λγ is a key quantity in nearly all of our results below.
Model of subdivision:
We now consider the finite island model of population subdivision with the simplifying assumption that migration does not change the sizes of the subpopulations (Nagylaki 1980; Strobeck 1987; Herbots 1997). Reproduction in all the subpopulations follows the modified Moran model described above. The discrete-time ancestral process for a sample of size 2 is a Markov chain with transition probabilities given in Equation A1 in the appendix.
We are concerned with small migration rates, specifically those on the order of 1/Nγ time steps. This means that a single individual resides in the same subpopulation for 2Nγ time steps, on average, before migrating to a different subpopulation. When 0 < γ < 2, each individual resides in the same subpopulation for only Nγ time steps, on average. This time can be much shorter (when 0 < γ < 1) than the usual average of N time steps assumed in Wright–Fisher populations. In other words, a large number of individuals migrate during N time steps when 0 < γ < 1. We let m denote the probability that a single individual resided in a different subpopulation in the previous time step and model m as m = mγ ≡ κ/(2Nγ) in which κ is a finite constant (0 < κ < ∞).
To illustrate the difference between our migration rate κ and the usual migration rate Nm let M* ≡ N2mγ denote a migration rate scaled in units of N2 time steps (or N generations). This corresponds to the usual “Nm” in the Wright–Fisher model. Substituting for mγ gives 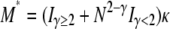 . If, for example, γ =
. If, for example, γ = 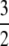 , then
, then 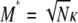 . When γ < 2 the migration rate M* is very high; i.e.,
. When γ < 2 the migration rate M* is very high; i.e., 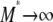 as
as 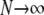 since κ is finite. However, in our modified model of reproduction coalescence also occurs on the timescale of N3/2 time steps (or
since κ is finite. However, in our modified model of reproduction coalescence also occurs on the timescale of N3/2 time steps (or 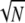 generations when γ =
generations when γ = 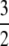 ) and thus “counteracts” the effects of high migration rate.
) and thus “counteracts” the effects of high migration rate.
The main results of this work concern expected coalescence times (Equations 3 and 5) and FST-like measures (Equations 10–12). We also derive the densities of the coalescence times (see appendix). The densities are used to derive distribution functions for the number of segregating sites between two sequences (see the appendix), which in turn yield expressions for FST-like measures including mutation (Equations 13 and 14).
The distributions of the coalescence times are functions of λγ:
DNA sequences differ because they have accumulated mutations from the time of their most recent common ancestor until they are sampled. By assuming a very low mutation rate, Slatkin (1991) derived an expression for FST in terms of expected values of coalescence times. The time until two genes coalesce is therefore a fundamental quantity in theoretical work on structured populations. Given two genes sampled from a structured population, two different coalescence times arise that are of interest: the time T0 until two genes sampled from the same subpopulation coalesce and time T1 until two genes sampled from different subpopulations reach a common ancestor. The densities of T0 and T1 were previously derived under the structured coalescent by Takahata (1988) and Nath and Griffiths (1993) in the case of two subpopulations and by Herbots (1997) for any finite number of subpopulations.
Given the transition rates in Equation A2, we can obtain the distributions of the coalescence times T0 and T1 (see the appendix). Figure 1 shows the distributions of T0 and T1, respectively, as functions of time for different values of ψ (the fraction of the population replaced by the offspring of a single individual). As ψ increases (i.e., tends to 1), the coalescence times T0 and T1 become very short.
Figure 1.—

The densities 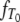 and
and 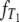 of times to coalescence for two genes sampled from the same (T0), or different (T1), subpopulations as functions of time for different values of ψ when the number of subpopulations D = 3 and φ = κ = 1. The coalescence timescale is N2/2 in a and c and Nγ/2 with 0 < γ < 2 in b and d. The solid lines in a and c are the densities obtained under the standard coalescent (i.e., γ > 2).
of times to coalescence for two genes sampled from the same (T0), or different (T1), subpopulations as functions of time for different values of ψ when the number of subpopulations D = 3 and φ = κ = 1. The coalescence timescale is N2/2 in a and c and Nγ/2 with 0 < γ < 2 in b and d. The solid lines in a and c are the densities obtained under the standard coalescent (i.e., γ > 2).
The expected value and variance of T0 are both less than the corresponding quantities for T1. Specifically,
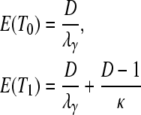 |
(3) |
and
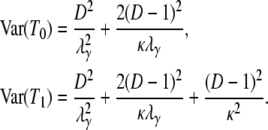 |
(4) |
Equation 3 holds a key result, namely that E(T0) is always less than E(T1).
The significance of the result in Equation 3 is best understood by an example. When γ < 2, say 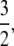 then the timescale is Nγ = N3/2, and λγ = ψ2 (assuming φ = 1). Our migration parameter is then κ = mγNγ = mγN3/2. Migration is scaled in units of N2 time steps in a standard Moran population. If we let M* ≡ N2mγ be a scaled migration rate in units of N2 time steps, then if mγ is of order 1/N3/2 as above, M* becomes very high in a large population. Specifically, since γ =
then the timescale is Nγ = N3/2, and λγ = ψ2 (assuming φ = 1). Our migration parameter is then κ = mγNγ = mγN3/2. Migration is scaled in units of N2 time steps in a standard Moran population. If we let M* ≡ N2mγ be a scaled migration rate in units of N2 time steps, then if mγ is of order 1/N3/2 as above, M* becomes very high in a large population. Specifically, since γ = 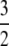 , we have
, we have 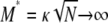 (as
(as 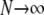 ), since κ is a constant. The result in Equation 3 says that even when
), since κ is a constant. The result in Equation 3 says that even when 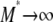 one will still see evidence of population structure in DNA sequence data, since coalescence occurs on a timescale of
one will still see evidence of population structure in DNA sequence data, since coalescence occurs on a timescale of 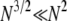 time steps (in a large population) when γ =
time steps (in a large population) when γ = 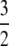 . In fact,
. In fact, 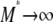 as
as 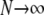 whenever 0 < γ < 2.
whenever 0 < γ < 2.
Similarly, 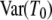 is always less than
is always less than 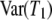 . In addition, the expected value and variance of T0 are inversely proportional to λγ and thus will be small when the probability of large reproduction events is close to one. The expressions for E(T0) and E(T1) (Equation 3) obtained under the usual reproduction models (Nei and Feldman 1972; Li 1976; Griffiths 1981) can be recovered by assuming that large reproduction events occur on a longer timescale (γ > 2) than usual (e.g., Wright–Fisher) sampling, in which case λγ = 1. The variances of T0 and T1 were first derived by Hey (1991) under the structured coalescent and can be recovered in the same way from Equation 4.
. In addition, the expected value and variance of T0 are inversely proportional to λγ and thus will be small when the probability of large reproduction events is close to one. The expressions for E(T0) and E(T1) (Equation 3) obtained under the usual reproduction models (Nei and Feldman 1972; Li 1976; Griffiths 1981) can be recovered by assuming that large reproduction events occur on a longer timescale (γ > 2) than usual (e.g., Wright–Fisher) sampling, in which case λγ = 1. The variances of T0 and T1 were first derived by Hey (1991) under the structured coalescent and can be recovered in the same way from Equation 4.
A many-demes limit:
The structured coalescent simplifies under certain migration mechanisms when the number of subpopulations is taken to be much greater than the sample size of DNA sequences (Wakeley 1998). The convergence of the ancestral process under a many-demes limit (i.e., when 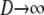 ) follows from the work of Möhle (1998), which shows how events in a stochastic process that occur on different timescales can be separated (see the appendix for a more detailed description). We consider the ancestral process in the limit
) follows from the work of Möhle (1998), which shows how events in a stochastic process that occur on different timescales can be separated (see the appendix for a more detailed description). We consider the ancestral process in the limit 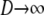 and
and 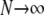 . Switching the order of the limits leads to the same coalescent process (see the appendix).
. Switching the order of the limits leads to the same coalescent process (see the appendix).
The limit process of two genes sampled from a population subdivided into very many subpopulations (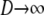 ), each of which is very large (
), each of which is very large (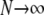 ), is of the form P*etG* in which P* and G* are given by Equations A16 and A19, respectively. The form of P* tells us that the ancestral process immediately enters the continuous-time process if the two genes are sampled from two different demes. If the two genes are sampled from the same subpopulation, they coalesce with probability
), is of the form P*etG* in which P* and G* are given by Equations A16 and A19, respectively. The form of P* tells us that the ancestral process immediately enters the continuous-time process if the two genes are sampled from two different demes. If the two genes are sampled from the same subpopulation, they coalesce with probability 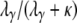 or enter the continuous-time process by moving to different subpopulations with probability
or enter the continuous-time process by moving to different subpopulations with probability 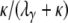 . In the continuous-time process the two lines wait with exponential time with rate
. In the continuous-time process the two lines wait with exponential time with rate 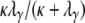 on a timescale of DNγ time steps until they coalesce. The ancestral process under the many-demes limit model (Equation A19) differs from the limit process obtained when the number of subpopulations is finite (Equation A2), in that G* has a zero entry for the transition where the two alleles enter the same subpopulation, after having been separated. When D < ∞, the corresponding rate is κ/(D – 1) (Equation A2). Ancestral lines can coalesce, however, only if they reside in the same subpopulation. The matrix B* (Equation A18) ensures that the two lines do arrive in the same subpopulation.
on a timescale of DNγ time steps until they coalesce. The ancestral process under the many-demes limit model (Equation A19) differs from the limit process obtained when the number of subpopulations is finite (Equation A2), in that G* has a zero entry for the transition where the two alleles enter the same subpopulation, after having been separated. When D < ∞, the corresponding rate is κ/(D – 1) (Equation A2). Ancestral lines can coalesce, however, only if they reside in the same subpopulation. The matrix B* (Equation A18) ensures that the two lines do arrive in the same subpopulation.
Again we are interested in the coalescence times T0 and T1 of two genes sampled from the same, or different, subpopulations, respectively. The distribution of T0 is a mixture distribution (see appendix), and we obtain
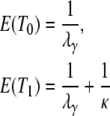 |
(5) |
and
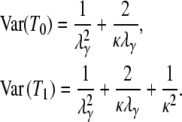 |
(6) |
The expressions for the expected value and variance of T0 and T1 obtained under the many-demes limit model (Equations 5 and 6) are functions of λγ and κ in the same way as the corresponding expected values and variances (Equations 3 and 4) obtained for a finite number of subpopulations. In particular, we always expect a shorter coalescence time for two ancestral lines sampled from the same subpopulation than if they were sampled from different subpopulations.
Deriving FST and NST:
The quantity FST is commonly used to assess levels of population subdivision. The inbreeding coefficient of an individual relative to a collection of subpopulations, FIT, can be attributed to nonrandom mating within a subpopulation (FIS) and to differences among subpopulations (FST; Wright 1951). Two sequences are identical by descent if they have not experienced mutation from the time of their most recent common ancestral sequence until they are sampled. If we let f0 and f denote the probability of identity by descent of two genes sampled from the same subpopulation (f0) and at random from the collection of subpopulations (f), we can express FST as
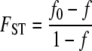 |
(7) |
(Nei 1973). By the definition of FST in terms of inbreeding coefficients (as in Equation 7), FST depends on the mutation rate (μ). By forcing μ to be very low Slatkin (1991) derived an approximation of FST that is a function of expectation of coalescence times and is given by
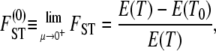 |
(8) |
in which T is the coalescence time of two lines randomly sampled from the collection of subpopulations, T0 is the time to coalescence of two lines from the same subpopulation, and μ is the mutation rate.
To obtain an expression of 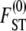 in terms of coalescence times under skewed offspring distribution, we can proceed by first obtaining the expected coalescence time E(T) of two genes randomly sampled from the collection of subpopulations, which is readily obtained from Equations 3 and A10 and is given by
in terms of coalescence times under skewed offspring distribution, we can proceed by first obtaining the expected coalescence time E(T) of two genes randomly sampled from the collection of subpopulations, which is readily obtained from Equations 3 and A10 and is given by
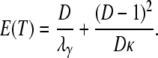 |
(9) |
When the number of subpopulations D is finite, the general form of 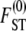 is
is
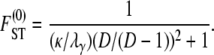 |
(10) |
For example, when 0 < γ < 2, the rate of coalescence is λγ = ψ2 (with φ = 1) and Equation 10 gives 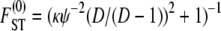 . The expression for
. The expression for 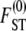 in Equation 10 has the same form as the one derived by Slatkin (1991) under the standard coalescent. The key difference is that, under skewed offspring distribution, FST is a function of the rate λγ (Equation 2) of coalescence and thus a function of the reproduction parameters φ and ψ. The result that Slatkin (1991) obtained can be recovered from Equation 10 by taking γ > 2, in which case λγ = 1 (recall that the probability of large reproduction events ∝ 1/Nγ).
in Equation 10 has the same form as the one derived by Slatkin (1991) under the standard coalescent. The key difference is that, under skewed offspring distribution, FST is a function of the rate λγ (Equation 2) of coalescence and thus a function of the reproduction parameters φ and ψ. The result that Slatkin (1991) obtained can be recovered from Equation 10 by taking γ > 2, in which case λγ = 1 (recall that the probability of large reproduction events ∝ 1/Nγ).
When the number of subpopulations 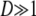 , we obtain from Equation 10
, we obtain from Equation 10
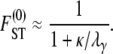 |
(11) |
In Equation 11 we have taken two limits: 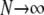 and
and 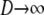 . Switching the order of the limits gives the same limit result for FST in Equation 11.
. Switching the order of the limits gives the same limit result for FST in Equation 11.
Following Wright (1951), the value of FST has often been used to estimate levels of gene flow. Figure 2 shows 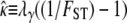 , obtained from Equation 11, as a function of ψ for different values of FST (Figure 2a) and φ (Figure 2b) and for two different values of λγ. Since FST is a function of ψ and φ, so is any estimate of gene flow based on FST, as Figure 2 clearly shows.
, obtained from Equation 11, as a function of ψ for different values of FST (Figure 2a) and φ (Figure 2b) and for two different values of λγ. Since FST is a function of ψ and φ, so is any estimate of gene flow based on FST, as Figure 2 clearly shows.
Figure 2.—

The estimate 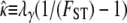 of migration rate from Equation 11 as a function of ψ. (a)
of migration rate from Equation 11 as a function of ψ. (a) 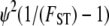 when FST = 0.1 (solid line), FST = 0.2 (dashed line), and FST = 0.5 (dotted line). (b)
when FST = 0.1 (solid line), FST = 0.2 (dashed line), and FST = 0.5 (dotted line). (b) 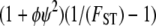 with FST = 0.1 and φ = 1 (solid line), φ = 2 (dashed line), and φ = 5 (dotted line).
with FST = 0.1 and φ = 1 (solid line), φ = 2 (dashed line), and φ = 5 (dotted line).
Lynch and Crease (1990) used the number of pairwise sequence differences of DNA sequences to estimate levels of genetic heterogeneity. In that context, Lynch and Crease (1990) introduced the quantity NST that has the form 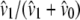 in which
in which 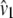 and
and 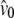 are the average number of pairwise differences between sequences sampled from different, or the same, subpopulations, respectively. If mutation rate is constant and mutations occur according to the infinite-sites model (Watterson 1975), then NST estimates
are the average number of pairwise differences between sequences sampled from different, or the same, subpopulations, respectively. If mutation rate is constant and mutations occur according to the infinite-sites model (Watterson 1975), then NST estimates 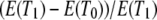 (Slatkin 1993). Using the results obtained for expected coalescence times (Equation 3), we obtain
(Slatkin 1993). Using the results obtained for expected coalescence times (Equation 3), we obtain  as in Equation 11 for the many-demes limit model of population subdivision and
as in Equation 11 for the many-demes limit model of population subdivision and
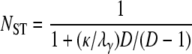 |
(12) |
when D < ∞. The effect of skewed offspring distribution is the same on NST as it is on FST. Under the infinite-sites mutation model we do not need an assumption of small mutation rate to obtain an expression of NST in terms of coalescence times, unlike the case for FST. As NST is defined, the mutation parameter cancels out (Slatkin 1993).
Number of segregating sites between pairs of sequences:
By the definition of FST in terms of probabilities of identity by descent (Equation 7), FST depends on mutation. Eldon and Wakeley (2006) show that the limit process (as 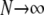 ) of our model of skewed offspring distribution predicts nonzero levels of genetic variation only when γ > 1. If we (as in Eldon and Wakeley 2006) let μ denote the probability of mutation for each offspring in a single time step, we define the mutation rate θ as
) of our model of skewed offspring distribution predicts nonzero levels of genetic variation only when γ > 1. If we (as in Eldon and Wakeley 2006) let μ denote the probability of mutation for each offspring in a single time step, we define the mutation rate θ as 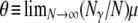 (and γ > 1). We can include mutation in an expression for FST by first obtaining the probability distributions of the number of segregating sites, under the infinite-sites model (Watterson 1975), between two genes given a model of population subdivision with migration. Let K0 denote the number of segregating sites between two genes sampled from the same subpopulation and K denote the number of segregating sites between two genes sampled randomly from the collection of subpopulations. The distributions of K0 and K are derived in the appendix, along with the distribution of the number of segregating sites K1 between two genes sampled from different subpopulations. Then by the definition of FST given in Equation 7 we obtain
(and γ > 1). We can include mutation in an expression for FST by first obtaining the probability distributions of the number of segregating sites, under the infinite-sites model (Watterson 1975), between two genes given a model of population subdivision with migration. Let K0 denote the number of segregating sites between two genes sampled from the same subpopulation and K denote the number of segregating sites between two genes sampled randomly from the collection of subpopulations. The distributions of K0 and K are derived in the appendix, along with the distribution of the number of segregating sites K1 between two genes sampled from different subpopulations. Then by the definition of FST given in Equation 7 we obtain
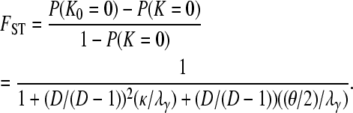 |
(13) |
When 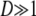 ,
,
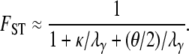 |
(14) |
From Equation 14 we conclude that mutation can affect FST only if θ is large relative to λγ. The expression for FST in Equation 14 has the same form as the one derived by Wilkinson-Herbots (1998) and by Nei (1975) and Takahata (1983) by other methods, under the Wright–Fisher model, including mutation. In Figure 3, FST from Equation 14 is graphed as a function of ψ for different values of θ and κ. The interpretation of Figure 3 is that FST, as a function of ψ, can vary considerably when the timescale of coalescence (and migration) is in units of Nγ/2 generations with 1 < γ < 2 (Figure 3, b and d).
Figure 3.—

The quantity FST from Equation 14 as a function of ψ (with φ = 1) for different values of θ, κ, and rate of coalescence (λγ). Solid lines, θ = 10; dashed lines, θ = 1; dotted lines, θ = 0.1.
Nei's genetic distance d:
Not all indicators of separation between populations depend on λγ. Nei's (1972) genetic distance is more appropriate for estimating divergence time between species, and FST-like quantities are more suitable for inferring population structure within species (Slatkin 1991). Nei's (1972) genetic distance measure is given by 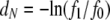 in which f0 and f1 are the probabilities of identity by descent of two genes sampled from the same or different subpopulations, respectively, and we add the subscript N to remind us that time is discrete. If we now assume that 0 < μE(ti) < 1 for i = 0, 1, then using the Maclaurin series expansion of the logarithmic function
in which f0 and f1 are the probabilities of identity by descent of two genes sampled from the same or different subpopulations, respectively, and we add the subscript N to remind us that time is discrete. If we now assume that 0 < μE(ti) < 1 for i = 0, 1, then using the Maclaurin series expansion of the logarithmic function 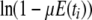 we obtain
we obtain 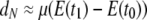 (previously obtained by Slatkin 1991) in which t0 and t1 are the coalescence times for two genes sampled from the same, or different, subpopulations, respectively. To obtain an expression of d for continuous time, we assume that the product
(previously obtained by Slatkin 1991) in which t0 and t1 are the coalescence times for two genes sampled from the same, or different, subpopulations, respectively. To obtain an expression of d for continuous time, we assume that the product 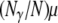 converges to a constant θ as
converges to a constant θ as 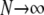 (and γ > 1). Rewriting the approximation for dN gives
(and γ > 1). Rewriting the approximation for dN gives
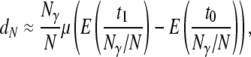 |
(15) |
which has the continuous-time limit
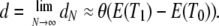 |
(16) |
However, using the expressions for E(T1) and E(T0) (Equation 3), we obtain 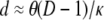 and so Nei's (1972) genetic distance is independent of λγ. Another way of deriving an expression for d is to note that the probability of identity by descent of two genes is the same as the probability that no mutations occur from the time they are sampled until they reach a common ancestor. Thus fi = P(Ki = 0) for i = 0, 1. We can therefore write
and so Nei's (1972) genetic distance is independent of λγ. Another way of deriving an expression for d is to note that the probability of identity by descent of two genes is the same as the probability that no mutations occur from the time they are sampled until they reach a common ancestor. Thus fi = P(Ki = 0) for i = 0, 1. We can therefore write
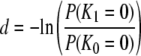 |
(17) |
for any model of population subdivision. For the many-demes limit model under consideration,
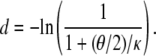 |
Using either the limit approach (Equation 16) or the substitution approach (Equation 17) in the many-demes limit model, and assuming small θ/κ (i.e., 0 < θ/κ < 1), d is of the form θ/κ. The same result is obtained for a finite number of subpopulations. Indeed, when D is finite, we obtain from Equations A28 and A29
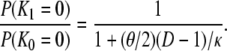 |
(18) |
Thus, if 0 < (θ/2)(D – 1)/κ < 1, we have from Equations 17 and 18
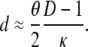 |
(19) |
Even if (θ/2)(D – 1)/κ > 1, we have from Equations 17 and 18 that d is not a function of λγ. Thus Nei's (1972) genetic distance can be used to estimate divergence times of species even if one or both species have skewed offspring distribution, since d is proportional to the time of separation of two populations (Nei 1972; Slatkin 1991).
DISCUSSION
Some organisms, for example Pacific oysters (Beckenbach 1994; Hedgecock 1994a) and Atlantic cod (Bekkevold et al. 2002; Árnason 2004), may exhibit skewed offspring distribution among individuals in a population. Both Beckenbach (1994) and Hedgecock (1994a) describe the reproductive mode of oysters, for example, as a lottery, in which only the offspring of a few lucky females survive. Oyster and cod females have very high reproductive potential, as they may produce millions of eggs in a single spawning (May 1967; Strathmann 1987; Chambers and Waiwood 1996; Kjesbu et al. 1996). The Wright–Fisher model does not capture the skewed offspring distribution possibly exhibited by organisms with high fecundities and high early mortality. The models of Pitman (1999) and Sagitov (1999), and later of Eldon and Wakeley (2006) and Sargsyan and Wakeley (2008) for overlapping generations in a single population, incorporate the skewness and may thus better apply to organisms with highly fecund individuals and sweepstakes-style recruitment. By deriving distributions of coalescence times for two genes sampled from a subdivided population, we show how skewed offspring distribution confounds estimates of migration rate between subpopulations when based on FST-like measures of population subdivision.
An important result of this work is that FST depends not only on the migration rate κ but also on the parameters (ψ and φ) of our model of large offspring numbers. Demographic processes such as population size fluctuations, founder effects, or skewed offspring distribution have been thought to increase genetic differentiation among subpopulations. As defined and calculated from genetic data, common indicators of population subdivision then take on high values, thus suggesting low levels of migration (Boileau et al. 1992; Whitlock 1992; Slatkin 1993; Hedgecock 1994a). Our main conclusions are twofold. First, FST is shown to depend on the parameters controlling the size (ψ) and frequency (φ) of large reproduction events (the probability that the offspring of a single individual replace a fraction ψ of the population is ɛ = 2φ/Nγ) and can thus indicate high or low levels of genetic heterogeneity depending on ψ and φ. To illustrate, consider the expression for FST derived under the many-demes limit model without mutation (Equation 11), and let the timescale of coalescence occur on Nγ/2 time steps (i.e., 0 < γ < 2). In that case the rate of coalescence λγ = ψ2 (by taking φ = 1), and the rate of large reproduction events is high. By fixing κ, we see that FST ranges from very low (when ψ is low), to ≈1/(1 + κ), when ψ ≈ 1. Second, to the extent that FST (or NST) is used in estimating levels of gene flow, these estimates are confounded by λγ and thus by φ and ψ. Also, migration in our model is not the usual Nm quantity, but is given by κ = mγNγ. This means that even when Nm is very large, we may still observe genetic heterogeneity, since the rate of large reproduction events is also large. In a population where individuals can have very many offspring, gene flow is not the only demographic force that influences genetic heterogeneity.
The coalescence times T0 and T1 (for genes sampled from the same or different subpopulations, respectively) are fundamental quantities of the ancestral process of genes in subdivided populations. The time during which DNA sequences accumulate mutations is determined by T0 and T1. As we have shown, the coalescence times depend on the skewness of the offspring distribution through the rate λγ (Equation 2) of coalescence. By deriving the distributions of T0 and T1 for two genes in a structured population, we have obtained insight into how skewed offspring distribution shapes the genetics of structured populations. Since T0 and T1 are functions of φ and ψ through the rate of coalescence λγ, all the quantities of interest in regard to investigation of the genetics of structured populations, including expected values, number of segregating sites, and indicators of population subdivision, are functions of λγ.
One such insight is that genetic heterogeneity can be observed in genetic data even if gene flow is very high by the usual standard (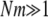 ). Edmands et al. (1996) found significant genetic heterogeneity among subpopulations of the purple sea urchin S. purpuratus sampled along the coast of California and Baja California. The ecology and physiology of S. purpuratus indicate the capacity for highly skewed offspring distribution: external fertilization and very high fecundity. Despite a planktonic larval period of several weeks (Strathmann 1978), and thus a potential for high dispersal, both allozyme and mtDNA sequence data revealed genetic differentiation, even over short distances (Edmands et al. 1996). We have shown that, regardless of the timescale of migration, E(T0) < E(T1). Genetic heterogeneity can, therefore, be observed in DNA sequence data even if gene flow is very high, in a population with skewed offspring distribution. Population turnover in a metapopulation model when demes that become extinct are recolonized by one or a few individuals can also lead to increased FST (Wade and McCauley 1988; Whitlock and McCauley 1990; Pannell 2003). Indeed, a model of metapopulation structure that allows only one founder for every deme that is recolonized necessarily results in a coalescent process with multiple mergers, if the founder can have many offspring.
). Edmands et al. (1996) found significant genetic heterogeneity among subpopulations of the purple sea urchin S. purpuratus sampled along the coast of California and Baja California. The ecology and physiology of S. purpuratus indicate the capacity for highly skewed offspring distribution: external fertilization and very high fecundity. Despite a planktonic larval period of several weeks (Strathmann 1978), and thus a potential for high dispersal, both allozyme and mtDNA sequence data revealed genetic differentiation, even over short distances (Edmands et al. 1996). We have shown that, regardless of the timescale of migration, E(T0) < E(T1). Genetic heterogeneity can, therefore, be observed in DNA sequence data even if gene flow is very high, in a population with skewed offspring distribution. Population turnover in a metapopulation model when demes that become extinct are recolonized by one or a few individuals can also lead to increased FST (Wade and McCauley 1988; Whitlock and McCauley 1990; Pannell 2003). Indeed, a model of metapopulation structure that allows only one founder for every deme that is recolonized necessarily results in a coalescent process with multiple mergers, if the founder can have many offspring.
In summary, we consider the coalescence times of a subdivided population following a sweepstakes-style recruitment. The expected coalescence time for two genes sampled from the same subpopulation is always less than the expected coalescence time for two genes sampled from different subpopulations, even when migration occurs on a very short timescale. Estimates of migration rate based on FST are confounded by the rate λγ of coalescence, since FST-like measures of genetic heterogeneity are a function of the reproduction parameters of our model of skewed offspring distribution. These results underscore the importance of choosing an appropriate limit process for the population under consideration.
Acknowledgments
We thank two anonymous reviewers for helpful comments and suggestions.
APPENDIX
The discrete-time transition matrix:
The probability transition matrix ΠN (Equation A1) for the ancestral process over one time step for the case of arbitrary fixed number D ≥ 2 of subpopulations has three states: (1) two lines in the same deme but not coalesced, (2) two lines in different demes, and (3) the two lines have coalesced. We do not distinguish between subpopulations. The transition probabilities in ΠN are derived under the assumption that migration does not alter the subpopulation sizes (Nagylaki 1980; Strobeck 1987; Herbots 1997). We let m denote the single backward migration fraction. The matrix ΠN is
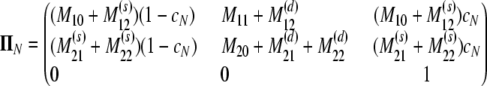 |
(A1) |
in which cN is the coalescence probability and
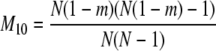 |
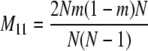 |
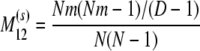 |
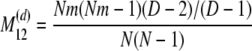 |
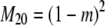 |
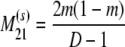 |
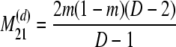 |
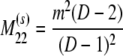 |
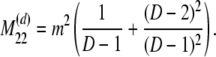 |
The matrix in Equation A1 is a generalization of the matrix for the same migration mechanism (cf. Wakeley 2008) obtained under the usual Wright–Fisher model of reproduction. In Equation A1, the probability of coalescence is cN, instead of the usual 1/N, as is the case for the haploid Wright–Fisher model. The corresponding continuous-time process has rate matrix G given by
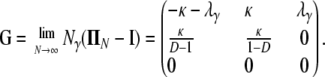 |
(A2) |
Distribution functions of the coalescence times when D is finite:
In this section we derive the distribution functions for the coalescence times T0 and T1 when the number of subpopulations is finite. Given the distributions of T0 and T1 we can determine the distribution of the coalescence time T of two genes sampled at random from the collection of subpopulations. The distributions of these coalescence times allow us to derive expressions for FST with or without mutation.
We can use the rate matrix (Equation A2) to obtain the density functions for T0 and T1. Using Laplace transforms (see Herbots 1997) we obtain
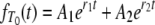 |
(A3) |
in which
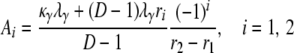 |
(A4) |
and
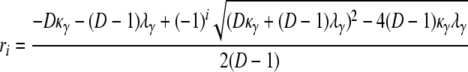 |
(A5) |
for i = 1, 2. To obtain the density function of T1, we note that T1 can be represented as a sum of two independent random variables, T1 = Y1 + T0, where Y1 is an exponential random variable with rate κγ/(D – 1). By direct calculation,
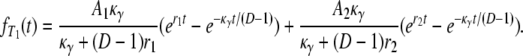 |
(A6) |
The form of the continuous functions 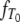 and
and 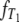 (Equations A3 and A6) immediately yields the cumulative densities
(Equations A3 and A6) immediately yields the cumulative densities 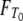 and
and 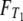 for T0 and T1, respectively. Namely, writing λi = –ri for i = 1, 2,
for T0 and T1, respectively. Namely, writing λi = –ri for i = 1, 2,
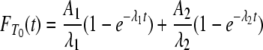 |
(A7) |
and
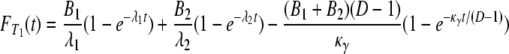 |
(A8) |
in which
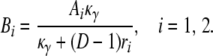 |
Let T denote the time to coalescence for two genes sampled at random from the collection of subpopulations. Then, with probability 1/D the two genes are sampled from the same subpopulation, and with probability 1 – 1/D they are sampled from different subpopulations. The cumulative density function (c.d.f.) FT of T is then given by
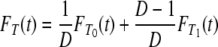 |
(A9) |
in which  denotes the c.d.f. of Ti for i = 0, 1. The expected value of T is
denotes the c.d.f. of Ti for i = 0, 1. The expected value of T is
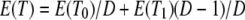 |
(A10) |
in which E(T0) and E(T1) are given by Equation 3 and the variance of T is
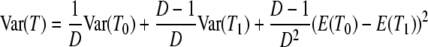 |
(A11) |
in which Var(T0) and Var(T1) are given by Equation 4. Note that although the expected value of T lies between E(T0) and E(T1), Equation A11 tells us that the variance of T may not lie between the variance of T0 and T1. If 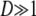 , then Var(T) > Var(T1).
, then Var(T) > Var(T1).
A many-demes limit model:
In this section we derive the ancestral process for two genes in the many-demes limit (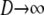 ) and as
) and as 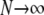 . Since now
. Since now 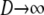 , the single-generation backward transition matrix, following Möhle (1998), can be written
, the single-generation backward transition matrix, following Möhle (1998), can be written
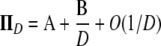 |
(A12) |
in which the matrices A and B are given below. The matrix A describes the probabilities of the transitions that occur on a timescale of time steps. The matrix B/D describes transitions that occur on a timescale of D time steps, thus forming the continuous-time part of the ancestral process. The limit process (as 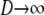 ) is then given by Π(t) = PetPBP in which
) is then given by Π(t) = PetPBP in which 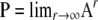 describes the equilibrium process of the events that occur on the timescale of time steps (Möhle 1998). The ancestral history of a sample is first adjusted by an instantaneous process described by P and then enters a continuous-time process described by the rate matrix PBP. Given the ancestral process, we can derive the distributions of T0 and T1.
describes the equilibrium process of the events that occur on the timescale of time steps (Möhle 1998). The ancestral history of a sample is first adjusted by an instantaneous process described by P and then enters a continuous-time process described by the rate matrix PBP. Given the ancestral process, we can derive the distributions of T0 and T1.
The instantaneous matrix A is
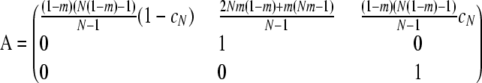 |
(A13) |
with eigenvalues λ1 = λ2 = 1 and
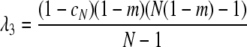 |
and we observe that 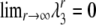 . By calculating the corresponding eigenvectors (not shown) we obtain the limit matrix P by diagonalizing A,
. By calculating the corresponding eigenvectors (not shown) we obtain the limit matrix P by diagonalizing A,
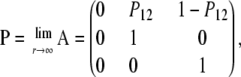 |
(A14) |
in which
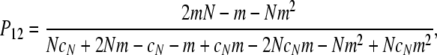 |
(A15) |
and in the limit  we obtain
we obtain
 |
(A16) |
When 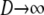 but holding N finite, the ancestral process is given by PetPBP, in which the matrix B is given by
but holding N finite, the ancestral process is given by PetPBP, in which the matrix B is given by
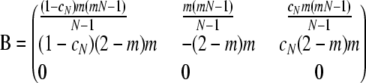 |
(A17) |
and we obtain
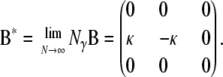 |
(A18) |
The rate matrix G* = P*B*P* then takes the general format
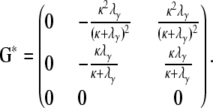 |
(A19) |
The ancestral process in the limit  and
and 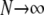 is P*etG* and immediately yields the density functions for T0 and T1 as follows. The time T1 to coalescence for two lines sampled from different demes follows in each case (γ > 2, γ = 2, and 0 < γ < 2) an exponential distribution with rate
is P*etG* and immediately yields the density functions for T0 and T1 as follows. The time T1 to coalescence for two lines sampled from different demes follows in each case (γ > 2, γ = 2, and 0 < γ < 2) an exponential distribution with rate 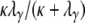 . Now consider the time T0 to coalescence for two lines sampled from the same subpopulation. Going back in time, two lines in the same subpopulation can either coalesce with probability
. Now consider the time T0 to coalescence for two lines sampled from the same subpopulation. Going back in time, two lines in the same subpopulation can either coalesce with probability 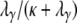 or they enter the continuous-time process with probability
or they enter the continuous-time process with probability 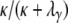 , in which case one of the two lines migrates to a different subpopulation. Thus T0 follows a mixture distribution with cumulative density function
, in which case one of the two lines migrates to a different subpopulation. Thus T0 follows a mixture distribution with cumulative density function
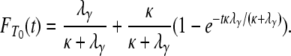 |
(A20) |
Order of limits irrelevant in the many-demes limit model:
In this section we show that the same ancestral process is obtained irrespective of the order of the limits 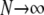 and
and 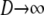 . We have already derived the process when first
. We have already derived the process when first 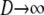 and then
and then 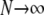 . Now we show that the same ancestral process is obtained when first
. Now we show that the same ancestral process is obtained when first 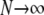 and then
and then 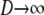 .
.
As 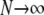 , we obtain the ancestral process described by the rate matrix
, we obtain the ancestral process described by the rate matrix
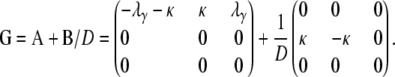 |
(A21) |
We remark that An = (–κ – λγ)n−1A for n ≥ 1. Since A is a rate matrix, we have, with a = κ + λγ,
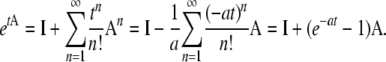 |
(A22) |
Equation A22 immediately gives us the instantaneous matrix
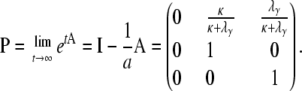 |
(A23) |
Equations A21 and A23 then give us the rate matrix G = PBP after first taking the limit 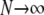 and then
and then 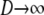 .
.
By similar arguments we can show that the ancestral process does indeed result in coalescence regardless of initial state. Indeed,
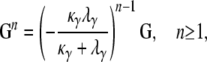 |
(A24) |
which gives, writing b = κλγ/(κ + λγ),
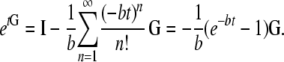 |
(A25) |
The equilibrium distribution (as 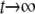 ) is then
) is then
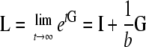 |
(A26) |
and we obtain that two genes do reach a common ancestor regardless of initial state—i.e.,
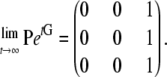 |
(A27) |
Number of segregating sites:
We now consider the number of segregating sites, under the infinite-sites model (Watterson 1975), between two genes in the two models of population subdivision discussed previously. First, consider the model of finite number of subpopulations. Let K0 and K1 denote the number of segregating sites for two genes sampled from the same or different subpopulations, respectively. We let θ denote the scaled mutation rate and note that, given a specific length of time t, the number of segregating sites is Poisson distributed with rate θt/2. The probability distribution for the number of segregating sites for two genes sampled from the same subpopulation is
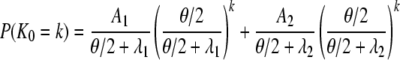 |
(A28) |
in which A1 and A2 are given in Equation A4, and λi = –ri from Equation A5.
The probability distribution for the number of segregating sites between two genes sampled from different subpopulations is
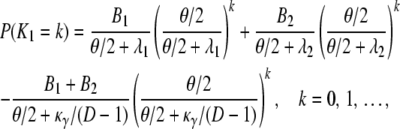 |
(A29) |
in which Bi = Aiκ/(κ + (D – 1)ri) for i = 1, 2. The expected value and variance of K0 are both less than the corresponding quantities for K1. Indeed,
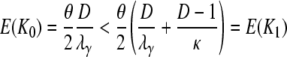 |
(A30) |
and
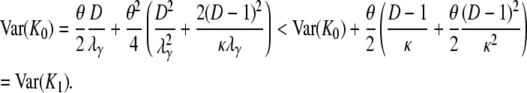 |
(A31) |
The probability mass distribution of the number of segregating sites K for two genes sampled at random from the collection of subpopulations is
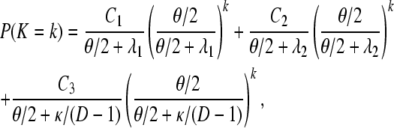 |
(A32) |
in which Ci = Ai/D + (D – 1)Bi/D for i = 1, 2 and 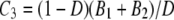 . One can write K ∼ (1/D)K0 + (1 – 1/D)K1; i.e., K is a mixture distribution. In fact, K is distributed as K0 with probability 1/D and as K1 with probability 1 – 1/D. Hence,
. One can write K ∼ (1/D)K0 + (1 – 1/D)K1; i.e., K is a mixture distribution. In fact, K is distributed as K0 with probability 1/D and as K1 with probability 1 – 1/D. Hence,
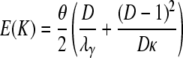 |
(A33) |
and so E(K0) < E(K) < E(K1). However, the variance of K (Equation A34), which can be obtained in the same way as the variance of the time to coalescence of two genes sampled at random from the collection of subpopulations (Equation A11), may not lie between the variance of K0 and K1. The variance of K is
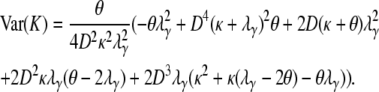 |
(A34) |
Number of segregating sites under the many-demes limit model:
Under the many-demes limit model of population subdivision, the probability mass distribution for the number of segregating sites K1 between two genes sampled from different subpopulations is
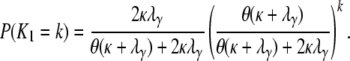 |
(A35) |
The expected number and the variance of the number of segregating sites between two genes sampled from different subpopulations is then
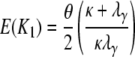 |
(A36) |
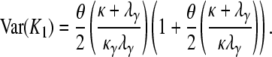 |
(A37) |
The probability mass function for K0, the number of segregating sites between two genes sampled from the same subpopulation, is (k = 0, 1, …)
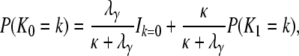 |
(A38) |
which gives expected value
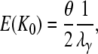 |
(A39) |
and the variance of K0 is
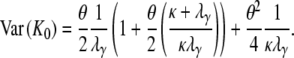 |
(A40) |
References
- Árnason, E., 2004. Mitochondrial cytochrome b variation in the high-fecundity Atlantic cod: trans-Atlantic clines and shallow gene genealogy. Genetics 166 1871–1885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Árnason, E., P. H. Petersen, K. Kristinsson, H. Sigurgíslason and S. Pálsson, 2000. Mitochondrial cytochrome b DNA sequence variation of Atlantic cod from Iceland and Greenland. J. Fish Biol. 56 409–430. [Google Scholar]
- Avise, J. C., 1994. Molecular Markers, Natural History and Evolution. Chapman & Hall, New York.
- Avise, J. C., R. M. Ball and J. Arnold, 1988. Current versus historical population sizes in vertebrate species with high gene flow: a comparison based on mitochondrial DNA lineages and inbreeding theory for neutral mutations. Mol. Biol. Evol. 5 331–344. [DOI] [PubMed] [Google Scholar]
- Beckenbach, A. T., 1994. Mitochondrial haplotype frequencies in oysters: neutral alternatives to selection models, pp. 188–198 in Non-Neutral Evolution, edited by B. Golding. Chapman & Hall, New York.
- Bekkevold, D. M., M. Hansen and V. Loeschcke, 2002. Male reproductive competition in spawning aggregations of cod (Gadus morhua l.). Mol. Ecol. 11 91–102. [DOI] [PubMed] [Google Scholar]
- Boileau, M. G., P. D. N. Hebert and S. S. Schwartz, 1992. Non-equilibrium gene frequency divergence: persistent founder effects in natural populations. J. Evol. Biol. 5 25–39. [Google Scholar]
- Boom, J. D. G., E. G. Boulding and A. T. Beckenbach, 1994. Mitochondrial DNA variation in introduced populations of Pacific oyster, Crassostra gigas, in British Columbia. Can. J. Fish. Aquat. Sci. 51 1608–1614. [Google Scholar]
- Burton, R. S., 1983. Protein polymorphisms and genetic differentiation of marine invertebrate populations. Mar. Biol. Lett. 4 193–206. [Google Scholar]
- Cannings, C., 1974. The latent roots of certain Markov chains arising in genetics: a new approach. Adv. Appl. Probab. 6 260–290. [Google Scholar]
- Chambers, R. C., and K. G. Waiwood, 1996. Maternal and seasonal differences in egg sizes and spawning characteristics of captive Atlantic cod, Gadus morhua. Can. J. Fish. Aquat. Sci. 53 1986–2003. [Google Scholar]
- Crow, J. F., and M. Kimura, 1970. Introduction to Population Genetics Theory. Harper & Row, New York.
- David, P., M. Perdieu, A. Pernot and P. J. Jarne, 1997. Fine-grained spatial and temporal population genetic structure in the marine bivalve Spisula ovalis l. Evolution 51 1318–1322. [DOI] [PubMed] [Google Scholar]
- Edmands, S., P. E. Moberg and R. S. Burton, 1996. Allozyme and mitochondrial DNA evidence of population subdivision in the purple sea urchin Strongylocentrotus purpuratus. Mar. Biol. 126 443–450. [Google Scholar]
- Eldon, B., and J. Wakeley, 2006. Coalescent processes when the distribution of offspring number among individuals is highly skewed. Genetics 172 2621–2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Griffiths, R. C., 1981. The number of heterozygous loci between two randomly chosen completely linked sequences of loci in two subdivided population models. J. Math. Biol. 12 251–261. [Google Scholar]
- Hedgecock, D., 1994. a Does variance in reproductive success limit effective population sizes of marine organisms?, pp. 1222–1344 in Genetics and Evolution of Aquatic Organisms, edited by A. Beaumont. Chapman & Hall, London.
- Hedgecock, D., 1994. b Temporal and spatial genetic structure of marine animal populations in the California Current. Calif. Coop. Ocean Fish. Invest. Rep. 35 73–81. [Google Scholar]
- Hedgecock, D., M. Tracey and K. Nelson, 1982. Genetics, pp. 297–403 in The Biology of Crustacea, Vol. 2, edited by L. G. Abele. Academic Press, New York.
- Hedrick, P. W., 2005. Large variance in reproductive success and the Ne/N ratio. Evolution 59 1596–1599. [PubMed] [Google Scholar]
- Herbots, H. M., 1997. The structured coalescent, pp. 231–255 in Progress of Population Genetics and Human Evolution, edited by P. Donnelly and S. Tavaré. Springer, New York.
- Hey, J., 1991. A multi-dimensional coalescent process applied to multi-allelic selection models and migration. Theor. Popul. Biol. 39 30–48. [DOI] [PubMed] [Google Scholar]
- Hurtado, L. A., R. A. Lutz and R. C. Vrijenhoek, 2004. Distinct patterns of genetic differentation among annelids of eastern Pacific hydrothermal vents. Mol. Ecol. 13 2603–2615. [DOI] [PubMed] [Google Scholar]
- Johnson, M. S., and R. Black, 1984. Pattern beneath the chaos: the effect of recruitment on genetic patchiness in an intertidal limpet. Evolution 38 1371–1383. [DOI] [PubMed] [Google Scholar]
- Johnson, S. B., C. R. Young, W. J. Jones, A. Warén and R. C. Vrijenhoek, 2006. Migration, isolation, and speciation of hydrothermal vent limpets (gastropoda; lepetodrilidae) across the Blanco Transform Fault. Biol. Bull. 210 140–157. [DOI] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. a The coalescent. Stoch. Proc. Appl. 13 235–248. [Google Scholar]
- Kingman, J. F. C., 1982. b On the genealogy of large populations. J. Appl. Probab. 19A 27–43. [Google Scholar]
- Kjesbu, O. S., P. Solemdal, P. Bratland and M. Fonn, 1996. Variation in annual egg production in individual captive Atlantic cod (Gadus morhua). Can. J. Fish. Aquat. Sci. 53 610–620. [Google Scholar]
- Li, W., 1976. Distribution of nucleotide difference between two randomly chosen cistrons in a subdivided population: the finite island model. Theor. Popul. Biol. 10 303–308. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and T. J. Crease, 1990. The analysis of population survey data on DNA sequence variation. Mol. Biol. Evol. 7 377–394. [DOI] [PubMed] [Google Scholar]
- May, A. W., 1967. Fecundity of Atlantic cod. J. Fish. Res. Brd. Can. 24 1531–1551. [Google Scholar]
- Möhle, M., 1998. A convergence theorem for Markov chains arising in population genetics and the coalescent with selfing. Adv. Appl. Probab. 30 493–512. [Google Scholar]
- Moran, P. A. P., 1958. Random processes in genetics. Proc. Camb. Philos. Soc. 54 60–71. [Google Scholar]
- Moran, P. A. P., 1962. Statistical Processes of Evolutionary Theory. Clarendon Press, Oxford.
- Murray, M. C., and M. P. Hare, 2006. A genomic scan for divergent selection in a secondary contact zone between Atlantic and Gulf of Mexico oysters, Crassostrea virginica. Mol. Ecol. 15 4229–4242. [DOI] [PubMed] [Google Scholar]
- Nagylaki, T., 1980. The strong migration limit in geographically structured populations. J. Math. Biol. 9 101–114. [DOI] [PubMed] [Google Scholar]
- Nath, H. B., and R. C. Griffiths, 1993. The coalescent in two colonies with symmetric migration. J. Math. Biol. 31 841–852. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1972. Genetic distance between populations. Am. Nat. 106 283–292. [Google Scholar]
- Nei, M., 1973. Analysis of gene diversity in subdivided populations. Proc. Natl. Acad. Sci. USA 70 3321–3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M., 1975. Molecular Population Genetics and Evolution. Elsevier, New York. [PubMed]
- Nei, M., 1982. Evolution of human races at the gene level, pp. 167–181 in Human Genetics, Part A: The Unfolding Genome, edited by B. Bohhe-Tamir, P. Cohen and R. N. Goodman. Alan R. Liss, New York.
- Nei, M., and M. Feldman, 1972. Identity of genes by descent within and between populations under mutation and migration pressure. Theor. Popul. Biol. 3 460–465. [DOI] [PubMed] [Google Scholar]
- Nei, M., and D. Graur, 1984. Extent of protein polymorphism and the neutral mutation theory. Evol. Biol. 17 73–118. [Google Scholar]
- Palumbi, S. R., 1994. Genetic divergence, reproductive isolation, and marine speciation. Annu. Rev. Ecol. Syst. 25 547–572. [Google Scholar]
- Pannell, J. R., 2003. Coalescence in a metapopulation with recurrent local extinction and recolonization. Evolution 57 949–961. [DOI] [PubMed] [Google Scholar]
- Pitman, J., 1999. Coalescents with multiple collisions. Ann. Probab. 27 1870–1902. [Google Scholar]
- Rousset, F., 2002. Inbreeding and relatedness coefficients: What do they measure? Heredity 88 371–380. [DOI] [PubMed] [Google Scholar]
- Sagitov, S., 1999. The general coalescent with asynchronous mergers of ancestral lines. J. Appl. Probab. 36 1116–1125. [Google Scholar]
- Sargsyan, O., and J. Wakeley, 2008. A coalescent process with simultaneous multiple mergers for approximating the gene genealogies of many marine organisms. Theor. Popul. Biol. 74 104–114. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1991. Inbreeding coefficients and coalescence times. Genet. Res. 58 167–175. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., 1993. Isolation by distance in equilibrium and non-equilibrium populations. Evolution 47 264–279. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and W. P. Maddison, 1989. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strathmann, M. F., 1987. Reproduction and Development of Marine Invertebrates of the Northern Pacific Coast. University of Washington Press, Seattle.
- Strathmann, R. R., 1978. The length of pelagic period in echinoderms with feeding larvae from the northeastern pacific. J. Exp. Mar. Biol. Ecol. 34 23–27. [Google Scholar]
- Strobeck, C., 1987. Average number of nucleotide differences in a sample from a single subpopulation: a test for population subdivision. Genetics 117 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1983. Gene identity and genetic differentiation of populations in the finite island model. Genetics 104 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahata, N., 1988. The coalescent in two partially isolated diffusion populations. Genet. Res. Camb. 52 213–222. [DOI] [PubMed] [Google Scholar]
- Turner, T. F., J. P. Wares and J. R. Gold, 2002. Genetic effective size is three orders of magnitude smaller than adult size census size in an abundant, estuarine-dependent marine fish (Sciaenops ocellatus). Genetics 162 1329–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wade, M. J., and D. E. McCauley, 1988. Extinction and recolonization: their effects on the genetic differentiation of local populations. Evolution 42 995–1005. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 1998. Segregating sites in Wright's island model. Theor. Popul. Biol. 53 166–174. [DOI] [PubMed] [Google Scholar]
- Wakeley, J., 2008. Coalescent Theory: An Introduction. Roberts & Company, Greenwood Village, CO.
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7 256–276. [DOI] [PubMed] [Google Scholar]
- Watts, R. J., M. S. Johnson and R. Black, 1990. Effects of recruitment on genetic patchiness in the urchin Echinometra mathaei in Western Australia. Mar. Biol. 105 145–151. [Google Scholar]
- Whitlock, M. C., 1992. Temporal fluctuations in demographic parameters and the genetic variance among populations. Evolution 46 608–615. [DOI] [PubMed] [Google Scholar]
- Whitlock, M. C., and D. E. McCauley, 1990. Some population genetic consequences of colony formation and extinction: genetic correlations within founding groups. Evolution 44 1717–1724. [DOI] [PubMed] [Google Scholar]
- Wilkinson-Herbots, H. M., 1998. Genealogy and subpopulation differentiation under various models of population structure. J. Math. Biol. 37 535–585. [Google Scholar]
- Won, Y., C. R. Young, R. A. Lutz and R. C. Vrijenhoek, 2003. Dispersal barriers and isolation among deep-sea mussel populations (mytilidae: Bathymodiolus) from eastern Pacific hydrothermal vents. Mol. Ecol. 12 169–184. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1951. The genetical structure of populations. Ann. Eugen. 15 323–354. [DOI] [PubMed] [Google Scholar]
- Young, C. R., S. Fujio and R. C. Vrijenhoek, 2008. Directional dispersal between mid-ocean ridges: deep-ocean circulation and gene flow in Ridgeia piscesae. Mol. Ecol. 17 1718–1731. [DOI] [PubMed] [Google Scholar]