

Abstract
The zebra finch has long been an important model system for the study of vocal learning, vocal production, and behavior. With the imminent sequencing of its genome, the zebra finch is now poised to become a model system for population genetics. Using a panel of 30 noncoding loci, we characterized patterns of polymorphism and divergence among wild zebra finch populations. Continental Australian populations displayed little population structure, exceptionally high levels of nucleotide diversity (π = 0.010), a rapid decay of linkage disequilibrium (LD), and a high population recombination rate (ρ ≈ 0.05), all of which suggest an open and fluid genomic background that could facilitate adaptive variation. By contrast, substantial divergence between the Australian and Lesser Sunda Island populations (KST = 0.193), reduced genetic diversity (π = 0.002), and higher levels of LD in the island population suggest a strong but relatively recent founder event, which may have contributed to speciation between these populations as envisioned under founder-effect speciation models. Consistent with this hypothesis, we find that under a simple quantitative genetic model both drift and selection could have contributed to the observed divergence in six quantitative traits. In both Australian and Lesser Sundas populations, diversity in Z-linked loci was significantly lower than in autosomal loci. Our analysis provides a quantitative framework for studying the role of selection and drift in shaping patterns of molecular evolution in the zebra finch genome.
THE zebra finch Taeniopygia guttata has long been a model system for studies of avian behavior and neurobiology (reviewed in Slater et al. 1988; Zann 1996). As an oscine passerine, or songbird, the zebra finch is part of a diverse clade composed of >4000 species (Raikow 1986; Edwards 1998; Barker et al. 2004) and is a member of the family Estrildidae, which itself includes 140 finch species distributed across Africa and Australasia (Goodwin 1982; Sorenson et al. 2004). The zebra finch has been of particular interest because songbirds, like humans, learn their vocalizations by imprinting on their parents (reviewed in Jarvis 2004). A number of parallels have already been discovered between the genetic underpinnings of vocal learning in humans and songbirds (e.g., Haesler et al. 2004; Teramitsu et al. 2004), and as genomic resources continue to develop, the zebra finch will only increase its importance as a model system for studies of neurobiology. With the production of bacterial artificial chromosome (BAC) libraries, cDNA microarrays (Naurin et al. 2008; Replogle et al. 2008), and the forthcoming complete genome sequence (Clayton et al. 2005), the zebra finch is now also a model system for genomics (Clayton 2004). Indeed, the first large-scale comparisons of orthologous genes in birds have been made possible through analysis of the chicken and zebra finch genomes (Ellegren 2007; Mank et al. 2007; Axelsson et al. 2008). The zebra finch genome will provide valuable insights into whether patterns observed in the chicken can be generalized across all birds or whether there are important differences among avian lineages, such as those that learn songs and those that do not.
Zebra finches are extremely common in the wild, frequenting habitats such as the cattle pastures, small towns, and homesteads of inland Australia. They are thus distributed across all of Australia with the exception of the extreme north and south of the continent (Figure 1). A second zebra finch subspecies, the Timor zebra finch T. guttata guttata (hereafter the “island” population), occurs on the Lesser Sunda Islands of southeast Asia, just north of Australia (Zann 1996). While the subspecies are well characterized behaviorally (Clayton 1990; Clayton et al. 1991), the history of the divergence between them is not well understood. Mayr (1944) analyzed a group of >40 bird species, including zebra finches, with ranges spanning Australia and the Lesser Sunda Islands. He proposed that faunal exchange occurred during the Pleistocene at which point reduced sea levels may have facilitated the crossing between the Lesser Sundas and Australia. Zebra finch populations therefore allow for a test of whether Pleistocene environmental change and the colonization of the islands contributed to population divergence and have led to speciation.
Figure 1.—

Range map and sampling localities of two zebra finch subspecies.
In birds, as in all lineages, a number of factors interact to shape the physical structure and pattern of variation in the genome and, crucially, these factors can be studied reliably only by analysis of natural populations or strains derived from nature. Such factors include demographic events such as population subdivision, life history variation, natural selection, and genetic drift, as well as genetic processes such as mutation and recombination (Reich et al. 2002). The efficiency with which natural selection can act to shape the genome is dependent in part on the effective population size of the species and on the genomic recombination rate (e.g., Bachtrog and Charlesworth 2002; Marais and Charlesworth 2003; Charlesworth and Eyre-Walker 2006; Maside and Charlesworth 2007). Large effective population sizes provide favorable conditions for the spread of adaptive mutations, while small effective population sizes and strong population substructure allow genetic drift to influence the fate of non-neutral mutations more strongly (Wright 1931; Kimura 1983). High recombination rates in turn allow favorable gene combinations to be brought together and deleterious combinations to be broken apart more quickly.
The rate of recombination is also a key factor influencing the extent of linkage disequilibrium (LD) in the genome and therefore has important consequences for genetic mapping studies for which zebra finches could be useful. In chickens, however, genomic data has indicated higher levels of recombination that would tend to break up blocks of LD and require a higher density of markers for linkage mapping (Wong et al. 2004). Higher recombination rates in chickens may in part be due to the occurrence of microchromosomes in the avian karyotype, resulting in a higher frequency of crossing over, but it is not clear if recombination rates are consistently high across all birds and avian chromosomes (Edwards and Dillon 2004; Backström et al. 2006a; Stapley et al. 2008). Stapley et al. (2008) recently published the first genomewide estimates of recombination based on a pedigree of captive zebra finches. They found that the zebra finch map length was only one-quarter that of the chicken map and that the estimated rates of genomewide recombination were substantially lower than in chicken. The role of domestication or even short-term captivity in modulating evolutionarily recent rates of recombination (in either the chicken or the zebra finch) is unclear; hence it is of interest to also estimate long-term rates of recombination and patterns of LD in natural populations of birds. It also is highly likely that estimates of recombination as measured from pedigrees vs. natural populations will differ due to methodological differences. Although our study does not attempt to measure genomewide rates of recombination, we have nonetheless accumulated multiple estimates of recombination and LD in several distinct regions of the zebra finch genome.
In view of the fact that little is known of the population genetics in this emerging model species, we set out to describe the basic features of population variation and history using a semi-targeted locus sampling approach. We developed 30 genomic loci occurring in seven “locus trios” of clustered loci distributed across several noncoding regions of autosomes, autosomal introns, and the Z chromosome. By sampling the diversity of trios of loci separated by known physical distances, we have been able to study the interacting effects of population history, genetic drift, and recombination in shaping the genomic context in which we must interpret variation in protein coding and other functional regions of the genome.
MATERIALS AND METHODS
Zebra finch samples:
We analyzed samples of 44 wild zebra finches from six populations spanning much of the natural range of zebra finches in Australia and the Lesser Sunda Islands (Figure 1, supplemental Table 2). The birds from the two northern populations, near Fitzroy Crossing in Western Australia (n = 12), and Longreach, Queensland (n =12), were collected using shotguns or mist nets and were prepared as morphological voucher specimens under appropriate permits (Queensland: WISP02899905; Western Australia: SF5943). Heart, liver, muscle tissue, and gonads were frozen in liquid nitrogen in the field. We also collected specimens of the double-barred finch Taeniopygia bichenovii (n = 4) as an outgroup to root gene trees and to estimate mutation rates for the genetic loci in this study. All specimens and tissues have been deposited in the collections of Harvard University's Museum of Comparative Zoology and/or the Philadelphia Academy of Natural Sciences. DNA was extracted from 25 mg of tissue using a QIAamp tissue kit (Qiagen). DNA samples from Shark Bay (n = 12), Flinder's Ranges (n = 12), and two populations in the Lesser Sunda Islands [West Timor (n = 6) and Lombok (n = 6)] were kindly provided by David Runciman (LaTrobe University).
Laboratory methods:
We studied nucleotide variation and the decay of linkage disequilibrium using resequencing at loci within seven “locus trios” (for a similar approach, see Frisse et al. 2001). Within each locus trio, the loci were separated by ∼2, 8, and 10 kb, as judged by distances derived from sequenced contigs within BAC clones published online (see supplemental material). We designed primers using PRIMER3 (Rozen and Skaletesky 2000). These whole BACs were BLASTed against the nr database in GenBank to ensure that they did not contain known coding regions. We also sequenced four nuclear introns using previously published primers: α-enolase intron 8 (Sorenson et al. 2004), ornithine decarboxylase intron 6 (Muñoz-Fuentes et al. 2007), transforming growth factor-β2 intron 5 (Sorenson et al. 2004), and phosphoenolpyruvate carboxykinase inton 9 (Muñoz-Fuentes et al. 2007). Finally, we designed a set of primers for sex-linked genetic markers. Four of these were based on Z-linked sequences from the pied flycatcher (Ficedula albicollis; Backström et al. 2006a). Published sequences were BLASTed against the zebra finch trace archive (http://www.ncbi.nlm.nih.gov/Traces/trace.cgi) and primers were targeted by eye to conserved domains. One additional Z-linked locus was designed by BLASTing sequences from a genomic library for the Cameroon indigobird (Vidua camerunensis) against zebra finch and chicken databases (H. Schull, personal communication). We confirmed that the selected loci were located on the Z chromosome in the zebra finch by comparing PCR band intensity in male and female samples and by checking that no females showed evidence of heterozygosity in chromatograms for any of these loci. For anonymous loci, we ensured that we were not sequencing multiple paralogous loci by confirming that no PCR products generated multiple bands and that BLAST searches against whole-genome sequencing reads in the zebra finch GenBank trace archive produced only a few hits for any one query, none of which suggested the presence of divergent paralogous sequences.
PCR products were amplified in 25-μl reactions with 0.2 units of EconoTaq DNA polymerase (Lucigen), 1 μm of each primer, and 0.25 mm of each dNTP. Thermal cycling was generally done using an annealing temperature of 55°. PCR products were purified using Millipore Montage μ96 plates, and cleaned products were directly sequenced using forward primers, Big Dye version 3.1 (Applied Biosystems), and an ABI 3100 or 3730 capillary sequencer. Raw sequence data were assembled into contigs using Sequencher (Gene Codes), and alignments and base calls were checked by eye. In cases where heterozygous length polymorphisms were discovered, reverse primers were used in an additional sequencing reaction to obtain sequence reads on either side of the indels. The length of indels was determined by visual inspection of the chromatograms. Because the software packages used in this study generally do not make use of indel data, these portions of the sequences were trimmed before analyses.
Diploid sequences (all those in addition to Z-linked loci amplified from females) were resolved into haplotypes using PHASE (Stephens et al. 2001; Stephens and Scheet 2005), which has been shown to perform well even when sample sizes are relatively small and diversity is high (Harrigan et al. 2008). Loci were phased individually and as trios of linked loci. In the former case, both variable and invariant sites were included to allow subsequent estimation of genetic variation statistics. For locus trios, PHASE was run using only variable sites to calculate linkage and recombination parameters across linked loci and between variable sites. For the seven linked trios, sequences were concatenated and gapped and constant sites were removed prior to running PHASE (further details below). Due to the high diversity of the loci studied here, alleles were sometimes resolved with less than perfect certainty by PHASE. Although incorrectly resolved haplotypes will not influence estimates of nucleotide diversity, they may subtly influence coalescent analyses, estimates of LD, and recombination analyses in LDhat.
Polymorphism and population structure:
Basic population genetic statistics were estimated using DNAsp (Rozas et al. 2003) and tested with relevant statistics (Nei 1987; Hudson et al. 1992). We also used neighbor-joining gene trees for each locus, generated in PAUP* (Swofford 2002), and the Mesquite software package (Maddison and Maddison 2007) to calculate the S statistic (Slatkin and Maddison 1989). We tested for population structure by comparing empirical estimates of S with values calculated from 1000 gene trees simulated under a coalescent model again using Mesquite. Finally, we used the program Structure (Pritchard et al. 2000; Falush et al. 2003), which uses a model-based clustering approach to infer population structure on the basis of multilocus genotypes. We tested alternative models of population structure ranging from K = 1, or no population structure, to K = 6, where each sampled population is genetically differentiated from each other. Because our data consisted of linked sequence loci, they were entered as such in the Structure input file with genetic distances between loci specified. The data set therefore consisted of 16 independent loci (7 locus trios, 4 nuclear introns, and 5 Z-linked introns), each consisting of multiple linked SNPs. The model of population structure that best fit the data was determined by examining changes in likelihood scores across runs with different K. Structure was run with a burn-in period of 10,000 cycles followed by another 500,000 cycles. Three replicate runs for each value of K were made to test for convergence.
We used the isolation-with-migration model implemented in the software IM (Hey and Nielsen 2004) to reconstruct the history of the divergence between mainland and Lesser Sundas zebra finches. This model is particularly appropriate for our analysis, given its constraint of analyzing two populations within which there is random mating, yet allowing for different population sizes, population size change, and potential gene flow between them. We ran IM numerous times with varying priors and heating schemes to optimize priors and to test for convergence among analyses. Two final runs presented here were conducted with 10,000 generations of burn-in followed by 25 million cycles. We conducted these runs assuming the Hasegawa–Kishino–Yano (HKY) model of sequence evolution and identical priors (θ1, 0–15; θ2, 0–0.5; θA, 0–1; t, 0–3; m1, 0–0.5; m2, 0–15). We also placed a minimum bound on our estimate of s of 0.5. We used the HKY model because the assumption of infinite sites was violated in our data in the form of sites with more than two character states. We did not use Markov coupling of multiple chains in the final analyses because doing so did not greatly improve effective sample sizes (ESS) estimates for difficult parameters (e.g., t), but rather vastly increased computation time. Because the results presented below are based on runs with the same priors, we were able to simply sum both distributions to determine point estimates for parameters.
As an additional test of alternative demographic scenarios, we modeled the divergence of the two subspecies in a coalescent framework using Serial SIMCOAL (Excoffier et al. 2000; Anderson et al. 2005). We modeled two isolated populations (i.e., no migration) that merged into a single ancestral population 1.5 MYA. To test for population growth and to determine the severity of the founder event, we modeled the populations under histories of constant population size and under exponential population decline from the present back to the time of population splitting (in other words, exponential growth since the time of splitting). Summary statistics from simulated data sets were compared with empirical results using Kolmogorov–Smirnov tests to assess the fit of the models to the empirical data. Current population size and growth rate parameters used in simulations were based on results from IM but were varied to generate simulations that more closely approximated empirical results. Because our approximation of the mutation rate (see details on calculation below) was also uncertain, we varied this parameter among runs. Given the population size, growth rate, and divergence time parameters for each simulation, we used the exponential growth equation (NT = N0ert, where NT is the current population size, N0 is the ancestral population size, r is the growth rate, and t is the divergence time) to determine the ancestral population size and the proportion of individuals founding the island population.
Morphological divergence and speciation history:
To test whether genetic drift could explain the morphological divergence observed between zebra finch subspecies, we used Lande's Ne* statistic and six morphological measures derived from Clayton et al. (1991). Clayton et al. (1991) provide a thorough description of morphological differences between the two zebra finch subspecies, including differences in body size (wing length, weight, bill length, bill depth) and coloration (bill color, breast-band size). Raw data from these analyses were no longer available so we digitally measured figures from Clayton et al. (1991) using the X–Y coordinate scale in Adobe Photoshop version 7.0. Such analyses provided proportional estimates of means and standard deviations for the six morphological measures.
Ne* is an estimate of the effective population size that would be required for drift alone to explain the observed morphological divergence and is estimated assuming a multigenic, additive model of trait divergence, given known trait heritabilities. In these analyses, we assumed a range of heritabilities (0.1–0.5) although those presented here are based on previous studies of birds (Price and Burley 1993; Merila et al. 2001; Hadfield et al. 2006; Frentiu et al. 2007). Only the heritability estimate for bill color (Price and Burley 1993) is based on data from the zebra finch. We also assumed two possible divergence times based on IM results and that the current effective population size estimated for the island population is a reasonable approximation of the historic size. Estimates of Ne* were compared with the estimated Ne based on sequence data and IM analyses. We also estimated the proportion of the observed divergence in phenotypic traits, possibly explained by drift, by assuming the island Ne suggested by IM and then calculating the expected divergence (z) under drift.
Linkage disequilibrium and recombination:
Haplotypes estimated by PHASE were used to estimate levels of linkage disequilibrium using Haploview (Barrett et al. 2005). Only sites that were resolved at ≥70% confidence were included. Using Haploview, we calculated r2 and D′, two commonly used measures of linkage between pairs of linked sites. High-frequency polymorphisms are preferable for accurate estimation of LD (Reich et al. 2001; Backström et al. 2006b) so we restricted our analysis to sites where the frequency of the rare allele was at least 10%. Because pairwise estimates of D′ and r2 are non-independent, we used the permutation test implemented in LDhat (McVean et al. 2002) to test for a significant decline of the two parameters with genetic distance.
We estimated the population recombination parameter ρ = 4Nec using both PHASE and LDhat. We ran PHASE four times per locus, using 10,000 iterations, a random number used to check for convergence, and two different priors ρ [0.0004 from humans and 0.0588 from a previous study of birds (Edwards and Dillon 2004)]. We also used the PAIRWISE module in LDhat to estimate ρ per locus while relaxing the infinite-sites assumption (McVean et al. 2002). In these analyses, θ for each locus was determined using Watterson's estimator as calculated in DNAsp (Rozas et al. 2003). Confidence levels were assessed by Monte Carlo coalescent simulation in LDhat, conditioned on the estimated recombination rate and θ. These simulations were used to generate the sampling distribution around the point estimate of ρ. To test for significant evidence of recombination, we used the likelihood permutation test implemented in LDhat (McVean et al. 2002). We used LAMARC (Kuhner 2006) to estimate the per site recombination rate r = ρ/θ for each of 21 anonymous loci and to generate a multilocus estimate across loci.
To assess the genealogical consequences of recombination, we quantified topological similarity among gene trees within and between locus trios. We surmised that if loci are in complete LD, we expect their gene trees to be similar in structure, even in a large randomly mating population. To study this effect fully, we also examined the topological similarity of gene trees for the two adjacent halves of individual loci within locus trios. Topological similarity of neighbor-joining gene trees based on different portions of the data set was assessed using PAUP* version 4.0b10 (Swofford 2002). The similarity of trees was measured using the symmetric length difference (SLD) measure.
RESULTS
Nucleotide polymorphism:
In total, we sequenced 8061 bases spanning 30 loci in each of 44 individuals (88 chromosomes) from six populations (Figure 1). A total of 4781 bp were from autosomal noncoding anonymous regions (21 loci), 1327 were from nuclear introns (4 loci), and 1953 were from Z-linked loci (5 loci). Overall, the loci in the study showed high levels of polymorphism (Table 1, Figure 2). Among the four Australian populations, we discovered 566 SNPs, yielding an average nucleotide diversity (π) of 0.010, whereas only 63 sites were polymorphic in the Lesser Sundas population (average π = 0.002), revealing a statistically significant difference in diversity among populations (two-tailed t-test: t = 7.97, P < 1.00 × 10−8). Levels of diversity in autosomal introns were very similar to anonymous regions (Table 1) but, as expected, the five Z-chromosome-linked markers showed much lower levels of polymorphism than autosomal loci (introns and anonymous loci, Table 1). Among Australian populations, this difference in diversity was approximately threefold and was statistically significant (one-tailed t-test: t = 2.40, P < 0.01) while, among Lesser Sundas populations, this difference was roughly sixfold and was also statistically significant, assuming unequal variances among populations (one tailed t-test: t = 2.43, P = 0.01). The site frequency distribution of haplotypes for most loci is characterized by an excess of rare polymorphisms, as evidenced by negative and generally nonsignificant values of Tajima's D across loci (Table 1). A total of 11 sites distributed among the 30 loci had more than two nucleotide states.
TABLE 1.
Polymorphism and divergence statistics for zebra finch subspecies
|
T. guttata castanotis (mainland)
|
T. guttata guttata (island)
|
Divergence
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Locus | Length (bp) | Indel (bp) | n | S | π | D | H | 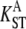 |
n | S | π | D | H | 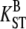 |
S-S |
| Anonymous | |||||||||||||||
| 005.01 | 200 | ND | 64 | 3 | 0.0024 | −0.50 | −0.58 | −0.007 | 24 | 0 | 0.0000 | — | — | 0.065* | 3 |
| 005.02 | 236 | 3, 7 | 64 | 15 | 0.0096 | −0.85 | 0.11 | 0.008 | 24 | 0 | 0.0000 | — | — | 0.227* | 3 |
| 005.10 | 218 | 1 | 64 | 8 | 0.0085 | −1.04 | −0.09 | 0.038 | 24 | 0 | 0.0000 | — | — | 0.094* | 3 |
| 035.01 | 224 | ND | 64 | 12 | 0.0046 | −1.70 | 0.93 | 0.008 | 24 | 2 | 0.0007 | −1.51 | −1.75 | 0.467* | 2 |
| 035.02 | 204 | — | 62 | 27 | 0.0201 | −1.08 | −0.97 | −0.009 | 24 | 3 | 0.0076 | 2.35 | −0.28 | 0.100* | 3 |
| 035.10 | 231 | 1, 1 | 62 | 25 | 0.0120 | −1.53 | −0.55 | 0.012 | 22 | 8 | 0.0033 | −2.19 | −3.12 | 0.314* | 2 |
| 175.01 | 267 | 1 | 64 | 18 | 0.0066 | −1.89 | −0.67 | 0.016 | 24 | 1 | 0.0003 | −1.15 | 0.08 | 0.236* | 3 |
| 175.02 | 272 | 1 | 56 | 11 | 0.0042 | −1.50 | −3.04 | 0.098* | 24 | 0 | 0.0000 | — | — | 0.055 | 4 |
| 175.10 | 204 | 1 | 62 | 19 | 0.0200 | −1.43 | ND | 0.024 | 24 | 4 | 0.0045 | −0.41 | ND | 0.289* | 2 |
| 276.01 | 150 | 7 | 50 | 7 | 0.0051 | −1.35 | −0.89 | 0.004 | 22 | 0 | 0.0000 | — | — | 0.029 | 4 |
| 276.02 | 218 | — | 64 | 25 | 0.0247 | −0.06 | 2.07 | 0.039 | 24 | 14 | 0.0123 | −1.00 | −1.87 | 0.091* | 6 |
| 276.10 | 241 | — | 64 | 21 | 0.0177 | −0.26 | 1.30 | 0.004 | 22 | 9 | 0.0031 | −2.26 | −8.93 | 0.181* | 5 |
| 319.01 | 244 | 8 | 64 | 29 | 0.0184 | −1.02 | −1.52 | −0.008 | 24 | 11 | 0.0076 | −1.25 | −4.02 | 0.090* | 8 |
| 319.02 | 286 | — | 64 | 11 | 0.0044 | −1.29 | −1.10 | 0.013 | 24 | 0 | 0.0000 | — | — | 0.040 | 3 |
| 319.10 | 196 | ND | 60 | 7 | 0.0068 | −0.27 | −3.08 | 0.096* | 22 | 0 | 0.0000 | — | — | 0.665* | 1 |
| 359.01 | 216 | ND | 60 | 24 | 0.0124 | −1.60 | 0.91 | 0.007 | 24 | 0 | 0.0000 | — | — | 0.281* | 1 |
| 359.02 | 284 | 1 | 56 | 42 | 0.0173 | −1.60 | 1.61 | 0.023 | 18 | 2 | 0.0028 | 0.88 | 1.60 | 0.129* | 3 |
| 359.10 | 212 | 3 | 64 | 18 | 0.0075 | −1.84 | 1.48 | 0.015 | 24 | 2 | 0.0043 | 1.53 | 0.17 | 0.086* | 6 |
| 365.01 | 238 | 7 | 64 | 21 | 0.0160 | −0.57 | 1.19 | 0.035 | 24 | 4 | 0.0017 | −1.69 | −3.27 | 0.223* | 3 |
| 365.02 | 211 | — | 64 | 7 | 0.0074 | 0.14 | 0.46 | −0.022 | 24 | 0 | 0.0000 | — | — | 0.412* | 1 |
| 365.10 | 228 | 1 | 58 | 7 | 0.0047 | −0.76 | ND | 0.004 | 24 | 0 | 0.0000 | — | — | 0.060 | 3 |
| Mean (SE) | 227.7 (1.5) | 17.0 (0.5) | 0.0110 (0.0014) | −1.03 (0.03) | −0.12 (0.07) | 0.018 (0.001) | 2.9 (0.2) | 0.0023 (0.0010) | −0.61 (0.14) | −2.13 (0.27) | 0.197 (0.008) | 3.29 (0.08) | |||
| Introns | |||||||||||||||
| TGF2B | 616 | — | 54 | 62 | 0.0099 | −2.04 | 3.18 | 0.003 | 8 | 0 | 0.0000 | — | — | 0.081 | 1 |
| OD | 415 | — | 58 | 36 | 0.0039 | −2.14 | ND | −0.010 | 8 | 0 | 0.0000 | — | — | 0.048 | 2 |
| Enol | 289 | — | 34 | 48 | 0.0242 | −1.00 | 1.03 | 0.031 | 24 | 0 | 0.0000 | — | — | 0.327* | 1 |
| PepCK9 | 633 | — | 60 | 21 | 0.0060 | −2.18 | 2.81 | −0.002 | 6 | 1 | 0.0005 | −0.93 | 0.27 | 0.011 | 2 |
| Mean (SE) | 488.3 (41.4) | 41.8 (4.4) | 0.0109 (0.0046) | −1.84 (0.14) | 2.34 (0.38) | 0.006 (0.004) | 0.0001 (0.0001) | −0.93 | 0.27 | 0.117 (0.036) | 1.5 (0.14) | ||||
| Z linked | |||||||||||||||
| GHR | 293 | — | 42 | 1 | 0.0003 | −0.85 | 0.12 | −0.050 | 14 | 0 | 0.0000 | — | — | 0.012 | 3 |
| NNT | 242 | — | 42 | 0 | 0.0000 | — | — | NA | 13 | 0 | 0.0000 | — | — | — | — |
| Z24638 | 259 | — | 35 | 3 | 0.0014 | −1.12 | 0.32 | −0.022 | 14 | 1 | 0.0010 | −0.34 | −0.22 | 0.033 | 3 |
| P35FF4 | 262 | — | 42 | 6 | 0.0030 | −1.16 | −0.14 | 0.022 | 14 | 0 | 0.0000 | — | — | 0.613* | 1 |
| ZFYVE | 271 | 18, 2 | 41 | 32 | 0.0114 | −2.02 | 1.90 | 0.003 | 13 | 1 | 0.0006 | −1.15 | −0.80 | 0.530* | 1 |
| Mean (SE) | 265.4 (3.7) | 8.4 (2.7) | 0.0032 (0.0021) | −1.29 (0.13) | 0.55 (0.23) | −0.009 (0.007) | 0.0003 (0.0002) | −0.75 (0.29) | −0.51 (0.21) | 0.238 (0.055) | 2 (0.29) | ||||
Sample size in alleles (n), the number of segregating sites (S), nucleotide diversity (π), Tajima's (D), and Fay and Wu's (H) are given for mainland and island populations. KST is estimated among four mainland populations (KSTA) and between mainland and island zebra finches (KSTB). Slatkin's S statistic (S-S) is given for neighbor-joining genealogies for each locus. Of note are the difference in diversity of mainland and island populations, the consistently negative Tajima's D, and the strong differentiation between subspecies (high KSTB, low S). Significant chi-square tests of genetic differentiation are indicated by asterisks. Among mainland birds, indel lengths are given unless no indel was present (−) or length could not be determined (ND). In cases where there was no polymorphism (−), Tajima's D, Fay and Wu's H, and Slatkin's S are not calculated.
Figure 2.—

Nucleotide diversity (π) in Australian and Lesser Sundas zebra finch subspecies across 21 anonymous nuclear loci, 4 nuclear introns, and 5 Z-linked introns. Sixteen of the 30 loci are monomorphic in the Timor zebra finch T. guttata gutatta.
Although insertion–deletion (indel) polymorphisms were not used in our population genetics analyses, they were common in the data set (Table 1). Sixteen of 21 anonymous nuclear loci had indels and, among these, 2 loci had indel polymorphisms at two different sites. None of the autosomal introns had indels, but one of the five Z-linked loci had two indels. Where we could clearly characterize the indel in terms of sequence and length (n =16), the size ranged from a single base, which was the most common (n = 8), to an 18-bp indel in the Z-linked locus ZFYVE. The average indel size was 3.94 bases. The ratio of indel mutations to SNPs is therefore ∼20:566, or 3.5%.
Approximation of the genomic mutation rate:
A mutation rate is required to convert estimates of scaled population genetics parameters into demographic units. We used mitochondrial DNA (mtDNA) sequences to estimate the divergence time between the zebra finch and the double-barred finch T. bichenovii. These two species are 10% divergent in mtDNA sequences from the NADH dehydrogenase subunit 2 gene (ND2) (Sorenson et al. 2004). Using an approximate rate calibration for mtDNA coding genes of 2% divergence/million years (reviewed in Lovette 2004), 10% divergence in ND2 suggests a divergence time of ∼5 million years for zebra and double-barred finches. We used this divergence time and sequence data from the loci in this study to estimate the mutation rate for the loci in this study. This calibration and the harmonic mean of the estimated rate for each locus results in an average rate of 7.38 × 10−7 substitutions/locus per year. Given the lengths of the loci in our study, our per-locus estimate translates into a rate of 2.95 × 10−9 substitutions/site/year, similar to a previous estimate of 1.5 × 10−9 substitutions/site/year on the basis of the divergence of galliformes chicken and turkey in autosomal introns (Ellegren 2007).
Divergence and population growth in Australian and Lesser Sundas zebra finches:
We find no evidence of population differentiation among the four Australian populations studied here. KST (Hudson et al. 1992), a measure of differentiation related to FST, indicated little genetic substructure within Australia; chi-square tests suggested significant differentiation at only 2 of the 30 loci and, for both loci, different populations appeared to be genetic outliers in pairwise comparisons (Table 1). Even in the two cases where chi-square tests were statistically significant, estimates of KST were still relatively low (∼0.09). In contrast, KST estimates of divergence between Australian and island populations were generally high (mean = 0.19, range = 0.01–0.66), indicating substantial genetic substructure between subspecies (Table 1). In no case were individual gene trees of island and mainland populations reciprocally monophyletic even though all showed a departure from random mixing. Empirical estimates of the S statistic (Slatkin and Maddison 1989), which is a measure of the degree to which a gene tree tracks geographic populations, were in all cases significantly lower than estimates based on 1000 simulated gene trees that had the same geographic sampling at each locus (Table 1). In fact, the empirical estimate was lower than the distribution of simulated values in all cases (P = 0), supporting the hypothesis of strong geographic structuring of gene trees among island and mainland populations.
The clustering approach Structure (Pritchard et al. 2000) indicated that a model in which the zebra finch was composed of three populations (K) had the highest likelihood (Figure 3). Falush et al. (2003), however, suggest choosing the K at which the likelihood reaches a plateau, which is essentially the point following the greatest change in likelihood and after which the likelihood remains relatively constant. By this criterion, a two-population model best fits the data (Figure 3). Results from Structure therefore suggest that differentiation occurs only significantly between the two subspecies and that no significant substructure exists within Australia.
Figure 3.—

Results from clustering analysis in Structure. (A and B) Probabilistic assignments of individual genotypes to either three or two populations, respectively. (C) The mean and standard error around likelihoods from three replicate runs testing models of one to six populations. The likelihood estimate clearly plateaus at K = 2, suggesting a two-population model best fits the data (despite a slightly higher likelihood for K = 3 and 4).
IM analyses using different priors, heating schemes, and subsets of the data were generally consistent across runs. Hey (2005) advocates optimizing run settings until the ESS for each parameter reaches a minimum of 50. Possibly due to the complexity of our data set, composed of one very diverse and one nearly monomorphic population, we were unable to attain such ESS values in some cases. The divergence time parameter t (ESS = 27 in each run) and θA (ESS = 28 and 30 in each run) were consistently lower than those of the other parameters (ESS: θ1 = 438, 486; θ2 = 38, 46; s = 225, 738; m1= 57, 67; m2 = 43, 53). Nevertheless, results from replicate runs were generally very consistent (Figure 4).
Figure 4.—

Posterior probability distributions for seven parameters estimated using IM. Depicted are results from replicate runs of 25 million post-burn-in iterations. Each run was conducted using the same priors and thus can be combined. Point estimates for each parameter for each run are given with 95% quantiles in parentheses.
The most striking result of the IM analysis was the dramatic bottleneck that is suggested in the founding of the Lesser Sundas subspecies (Figure 4). This is implied by a large estimate of s (0.9995), the proportion of the ancestral population founding the mainland population, which in turn indicates a small fraction of founders for the Lesser Sundas subspecies (1 − s, or 0.0005). On the basis of our estimated ancestral θA of 0.06 from IM and our mutation rates (Figure 4), we infer the ancestral Ne of the two forms to be ∼18,760 individuals. This would suggest that only about 9 individuals colonized the Lesser Sunda Islands, although this estimate should be interpreted cautiously, given the lack of a right tail to the posterior distribution of s. We estimate a current Ne for Australia at 7 million individuals (θ = 220.70) and the current Ne of the Lesser Sundas population at ∼26,750 (θ = 0.08), or just slightly larger than our estimated ancestral Ne. By contrast, estimating current Ne for Australia using our mutation rates and Watterson's θ across loci (0.015), a conversion that assumes demographic equilibrium, suggests an effective population size of only ∼1.3 million. In fact, judging from casual observation of birds in the field and from estimates of abundance of other continental songbirds, both numbers are likely drastically smaller than the census size of Australian zebra finches, a discordance that can arise due to a number of departures from demographic equilibrium. Nonetheless, the lower estimates of ancestral as compared with current Ne from IM imply that populations in Australia have experienced population growth (r = 2.9 × 10−6, where r is the growth rate in the exponential growth equation) and are not at demographic equilibrium.
We compared empirical estimates with summary statistics based on data sets from coalescent simulations and were able to reject a model in which there was no population growth in island and mainland populations (Table 2). Models in which the current Ne for the island population was comparable to those estimated in IM (25,000) generated less genetic diversity than was observed in the empirical data and were also rejected (Table 2). Simulations incorporating a larger Ne for the island (1 × 105), a higher mutation rate (2 × 10−6 substitutions/locus/year), and population growth, however, produced distributions of summary statistics that were not significantly different from empirical values (Table 2). This model suggests a founding population size of ∼5000 individuals for the island, or 1.4% of the ancestral population (Ne ancestral ∼350,000). It is also possible to generate similar summary statistics by further raising the current Ne of the island population without increasing the mutation rate (data not shown). Because our estimates of mutation rate are derived indirectly, we view a potential bias in mutation rate estimation as more likely than a very large Ne (1 × 106) for the island. We note, however, that the mutation rate that we have estimated for these noncoding loci in zebra finches is very similar to that estimated by slightly different methods for anonymous loci in another Australian bird species, the red-backed fairy wren (Malurus melanocephalus; Lee and Edwards 2008).
TABLE 2.
Means and 95% confidence intervals for data sets simulated in Serial SIMCOAL
| Mainland
|
Island
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| μ | Ne | r | S | π | D | Ne | r | S | π | D |
| 7 × 10−7 | 7 × 106 | 0 | 34.25 (31.94, 36.57) | 0.029* (0.026, 0.032) | 0.01* (−0.16, 0.19) | 25,000 | 0 | 0.12* (0.05, 0.20) | 5.81 × 10−5* (9.12 × 10−6, 1.07 × 10−4) | 0.4* (−0.25, 1.09) |
| 7 ×10−7 | 7 × 106 | 1 × 10−8 | 27.77* (25.94, 29.59) | 0.020* (0.018, 0.023) | −0.45* (−0.70, −0.20) | 25,000 | 1 × 10−8 | 0.02* (−0.02, 0.07) | 3.17 × 10* (−3.23 × 10−5, 9.57 × 10−5) | 0.054a |
| 7 ×10−7 | 7 × 106 | 1 × 10−6 | 15.90 (14.81, 16.98) | 0.010 (0.009, 0.011) | −0.86* (−0.98, −0.73) | 25,000 | 1 × 10−6 | 0.13* (0.07, 0.20) | 1.32 × 10−4* (5.62 × 10−5, 2.08 × 10−4) | −0.24* (−0.84, 0.35) |
| 7 × 10−7 | 7 × 106 | 1 × 10−6 | 15.46* (14.52, 16.44) | 0.010 (.009, 011) | −082* (−0.97, −0.67) | 100,000 | 1 × 10−6 | 0.41 (0.28, 0.54) | 4.03 × 10−4* (2.57 × 10−4, 5.49 × 10−4) | −0.32* (−0.63, −0.01) |
| 1 × 10−6 | 7 × 106 | 2 × 10−6 | 14.77* (13.94, 15.60) | 0.008 (0.072, 0.084) | −1.19 (−1.30, −1.09) | 100,000 | 2 × 10−6 | 0.63 (0.47, 0.79) | 6.29 × 10−4* (4.60 × 10−4, 7.98 × 10−4) | −0.22* (−0.47, 0.03) |
| 2 × 10−6 | 7 × 106 | 2 × 10−6 | 30.20* (29.02, 31.38) | 0.016 (0.015, 0.17) | −1.29 (−1.38, −1.21) | 100,000 | 2 × 10−6 | 1.230 (0.98, 1.48) | 0.001 (9.41 × 10−4, 0.01) | −0.26 (−0.50, −0.02) |
| Empirical values (95% CI): | 18.87 (13.40, 24.33) | 0.010 (0.074, 0.13) | −1.18 (−1.42, −0.94) | 2.10 (0.74, 3.64) | 0.002 (5.68 × 10−4, 2.78 × 10−3) | −0.65 (−1.44, 0.13) | ||||
Mutation rate per locus (μ), effective population sizes (Ne), and population growth rate (r) were varied for the mainland and island populations. A model with no growth (r = 0) for the mainland population produced summary statistics (S, number of segregating sites; π, nucleotide diversity; and Tajima's D) that were significantly different from empirical distributions (*Kolmogorov–Smirnov test, P < 0.05). Although none of the scenarios we tested were perfectly consistent with our data (K–S test, P > 0.05 across all parameters and populations), models incorporating population growth, higher mutation rates, and Ne = 100,000 for the island population values yielded summary statistics comparable with the observed data.
Estimates of nucleotide diversity (π) island population were often 0, making Tajima's D undefined. Averages of Tajima's D for the island population therefore represent only cases where π > 0. Where <5 simulations of 100 yielded π > 0, we did not calculate confidence intervals.
Our estimate of divergence time, t, between Australian and island zebra finches from IM is ∼1.9 MYA, with 95% confidence limits at 1.2 and 2.8 MYA, placing divergence in the early Pleistocene or the late Pliocene. Estimates of gene flow between subspecies from IM were very low, but clearly nonzero, with gene flow from the mainland to the island (m2) estimated at 2.94 × 10−6 migrants/generation and gene flow from the island to the mainland (m1) of ∼2.05 × 10−8 migrants/generation. These estimates may also reflect a departure from the model assumed by IM, given that there was probably a single founder event leading to the surviving island populations, with little evidence from the field or phenotypic traits for ongoing gene flow between the two forms. We cannot, however, rule out the possibility that cycles of sea level change during the Pleistocene have allowed for occasional dispersal between Australia and the Lesser Sundas.
Morphological divergence and speciation:
Despite the fact that only a small number of individuals were estimated to have founded the Lesser Sundas population, estimates of Ne* are generally smaller than the current Ne estimated for the island or for the ancestral species (Table 3). This suggests that drift alone may not be a sufficient mechanism for explaining the divergence in most quantitative characters. Two possible exceptions to this are bill color and depth, traits with least divergence (z) and lowest variance. Even if heritabilities are relatively high (h2 = 0.5) and divergence times are closer to the upper confidence bound of our estimates (2.8 MYA), divergence in only five of the six characters can be fully explained by drift. Our estimate of the predicted morphological divergence under drift alone also emphasizes that while divergence in bill length and depth may be explained largely by drift, only between 27% and 67% of the divergence in the other four traits may be explained by drift (Table 3).
TABLE 3.
Estimates of Ne* for six morphological traits
| Trait | z | σ | h2 | Ne* (t = 1.2 MY) | Ne* (t = 2.8 MY) | % drift (t = 1.2 MY) |
|---|---|---|---|---|---|---|
| Wing length | 0.58 | 0.04 | 0.3 | 2,005 | 4,678 | 27 |
| Weight | 0.43 | 0.05 | 0.3 | 5,699 | 13,297 | 46 |
| Bill length | 0.52 | 0.07 | 0.3 | 7,638 | 17,821 | 53 |
| Bill depth | 0.29 | 0.07 | 0.3 | 24,557 | 57,299 | 96 |
| Bill color | 0.29 | 0.09 | 0.5 | 112,761 | 263,110 | 100 |
| Breast band | 0.67 | 0.11 | 0.1 | 1,262 | 2,945 | 21 |
z is the mean morphological shift observed between birds from Timor and birds from Australia and σ is the phenotypic standard deviation of the colonized population (see materials and methods and results for details). Two estimates of Ne* are provided using the 95% high and low bounds of the estimate of divergence time (t) from IM. Heritability estimates (h2) are based on previous studies (Price and Burley 1993; Merila et al. 2001; Hadfield et al. 2006; Frentiu et al. 2007). Ne* estimates in italics represent cases where Ne* > 26,750, the Ne estimated in IM for the island population. These represent the traits for which drift alone may explain the observed divergence. The rightmost column is an estimate of the proportion of divergence potentially explained by drift.
Linkage disequilibrium and recombination:
Linkage disequilibrium was observed to decay rapidly with physical distance in the genome (Figure 5, Figure 6). Many pairwise site comparisons in our locus trios, particularly in the Australian population, showed low levels of LD even within 300 bp, and very few strong LD signals were evident across loci separated by 10 kb. Point estimates of the population recombination rate ρ = 4Nec, where c is the intersite recombination rate, were only partly consistent using the estimation approaches available in LDhat and PHASE (Table 4). Confidence intervals surrounding point estimates and the average across loci, however, were quite similar and suggest relatively high values for this parameter (mean PHASE = 0.05/site/generation, LDhat = 0.08; see Table 4). Consistent with the low overall levels of LD, likelihood permutation tests in LDhat suggest significant evidence of recombination within each of the 10-kb regions analyzed. While estimates of ρ necessarily confound the effects of recombination and effective population size, we were able to assess the relative influences of recombination and population size by estimating the ratio ρ/θ using LAMARC. The multilocus estimate of this ratio across each of 21 autosomal, anonymous loci (ρ/θ = 0.14, 95% CI = 0.09–0.19) emphasizes the large value of θ relative to ρ, although there was tremendous variation among loci (range ρ/θ = 1.75 × 10−5–0.64). A multilocus estimate of θ across the 21 loci using LAMARC of 0.031 (95% CI = 0.028–0.034) can be used with r estimates to calculate a ρ of ∼0.004. As expected, this reflects a lower recombination rate across individual loci than across locus trios. Elevated levels of LD in the smaller island population also highlight the role of Ne in shaping patterns of LD and indicate a shift in LD following the population bottleneck (Figure 6).
Figure 5.—

Rapid decay of LD in Australian zebra finches. Point estimates represent empirical r-squared values from pairwise comparisons among sites. Curves represent predictions of the decay of LD based on equation 3 from Weir and Hill (1986). The top solid line is based on ρ estimated from humans (0.0004), the bottom line is the multilocus average estimated using PHASE (ρ = 0.051), and the middle line is based on the minimum point estimate of ρ estimated by PHASE (ρ = 0.006).
Figure 6.—

Enhanced LD measured as r2 across 10-kb trios in Timor vs. Australian zebra finches. Solid squares indicate r2 values of 1, or perfect linkage. Shaded squares indicate r2 between zero and 1. Open squares indicate r2 values of zero. Although Lesser Sundas populations show greater LD, most significant LD is restricted to intralocus comparisons. High LD, therefore, is rarely detected at scales of even 1 kb.
TABLE 4.
Linkage disequilibrium and recombination statistics for mainland zebra finches
| Locus | D′ × d | r2 × d | ρ per site PHASEa | ρ per site PHASEb | ρ per site LDhat |
|---|---|---|---|---|---|
| 005 | −0.18** | −0.16** | 0.009 (0.003–0.022) | 0.011 (0.004–0.024) | 0.098 (0.035–0.147) |
| 035 | −0.09** | −0.06* | 0.028 (0.016–0.049) | 0.030 (0.018–0.051) | 0.127 (0.039–0.150) |
| 175 | −0.07* | −0.12** | 0.006 (0.002–0.013) | 0.007 (0.003–0.017) | 0.005 (0.002–0.007) |
| 276 | −0.19** | −0.12** | 0.058 (0.035–0.098) | 0.058 (0.036–0.094) | 0.108 (0.056–0.130) |
| 319 | −0.15** | −0.06* | 0.212 (0.109–0.384) | 0.199 (0.103–0.344) | 0.193 (0.085–0.223) |
| 359 | −0.06* | −0.12** | 0.050 (0.028–0.087) | 0.051 (0.028–0.087) | 0.029 (0.008–0.029) |
| 365 | −0.15** | −0.10* | 0.012 (0.004–0.027) | 0.014 (0.006–0.028) | 0.003 (0.001–0.005) |
| Mean (SE): | 0.053 (0.010) | 0.051 (0.010) | 0.08 (0.010) |
D′ × d and r2 × d are correlation coefficients of two measures of LD with genetic distance. Significance values are the proportion of data sets simulated in LDhat that show correlation coefficients that are less than or equal to the observed values (*P < 0.05, **P < 0.01). In parentheses are the 10% and 90% quantiles from the posterior distribution. For LDhat, we assessed confidence by running simulations conditioned on the point estimates of ρ. In parentheses are 10% and 90% quantiles of the distribution of these conditioned estimates.
Estimates of the recombination parameter ρ = 4Nec per site are given from PHASE using priors for ρ from humans (0.0004).
Estimates of the recombination parameter ρ = 4Nec per site are given from PHASE using priors for ρ from blackbirds (0.0588).
As expected, gene tree topologies based on the 21 pairs of adjacent halves within loci were the most similar (mean SLD = 107.14), but were not significantly more similar than SLDs among different loci (mean = 111.14; t-test, P = 0.38). Comparisons among loci separated by 2, 8, and 10 kb were only slightly less similar and were not significantly different from each other (SLD = 110.00, 111.71, and 111.71, respectively), further suggesting high levels of recombination even at this small genomic scale.
DISCUSSION
Nucleotide polymorphism, population structure, linkage disequilibrium, and recombination rate are four fundamental parameters that characterize the genetic architecture of a species. We have provided here a first glimpse of these parameters among wild zebra finch populations. While the avifauna of northern and coastal Australia often show striking patterns of population structure (e.g., Cracraft 1986; Jennings and Edwards 2005), broadly distributed bird species in the arid zone of Australia often lack such structure (e.g., Joseph and Wilke 2007). The Australian zebra finch populations analyzed here fall into this latter category, showing no evidence of population structure despite a very large geographic range spanning several potential biogeographic barriers (Cracraft 1986). Zebra finch colonies are nomadic and frequently exchange members (Zann and Runciman 2008), two factors that could contribute to the lack of phylogeographic structure within Australia. A recent, smaller study of two other zebra finch populations also suggests a lack of genetic differentiation among Australian zebra finches (Forstmeier et al. 2007).
Nucleotide diversity among 25 autosomal loci was remarkably high (π = 0.01), >10 times the level observed in the human species, and comparable to levels found in natural populations of some Drosophila species (e.g., Andolfatto 2001). Levels of diversity in Z-linked markers were significantly lower, and this difference may be attributed to the difference in effective population size among sex-linked and autosomal markers. When populations are expanding, we expect to see a deviation from the 1:0.75 ratio of diversity expected for autosomal and Z-linked loci solely on the basis of their equilibrium population sizes (Pool and Nielsen 2007). Coalescent analyses of both gene genealogies and summary statistics suggest a history of population expansion among mainland birds, apparently driven by their adaptation to the arid environment of inland Australia (Zann 1996), which has been expanding from its former rainforest state for the past 15 million years (White 1996). The observed allele-frequency spectrum, however, is also consistent with the expectation under recurrent selective sweeps (e.g., Przeworski 2002; Kim 2006). Although this possibility warrants further exploration, given the high rates of recombination estimated for this and other avian genomes (e.g., Edwards and Dillon 2004), it would be surprising to find that 16 randomly chosen noncoding loci consistently occurred in close enough proximity to sites experiencing selection to undergo frequent hitchhiking.
Analyses of divergence between the two zebra finch subspecies suggest substantial genetic differentiation and imply that the zebra finch colonized the Lesser Sunda Islands between 1.2 and 2.8 MYA, with little subsequent gene flow. These multilocus estimates coincide with ∼2% divergence at the mitochondrial ND2 gene (M. D. Sorenson, personal communication), which suggests a divergence time of ∼1 million years. In accord with Mayr's (1944) hypothesis, these divergence times date to the Pleistocene (although confidence bounds extend into the Pliocene) during which cycles of glaciation reduced the water barrier between the Lesser Sundas and Australia (Yokoyama et al. 2001; Lambeck et al. 2002). While glaciation-driven oscillations in sea level are relatively well characterized for the late Pleistocene (e.g., Lambeck and Chappel 2001), earlier cycles are less well understood. Lambeck et al. (2002) suggest that early glaciations also lowered sea levels, but perhaps to a lesser extent than during the last glacial maximum. It appears, however, that such reductions in sea level were sufficient to allow the colonization of the Lesser Sundas by zebra finches. It will be of great interest to test whether other bird species with similar distributions colonized the Lesser Sundas at similar times or whether repeated cycles of colonization occurred during the Pleistocene (Mayr 1944).
Although the 30 loci that we analyzed were linked into locus trios, the levels of LD detected among loci within these trios suggested that we were justified in treating each locus as an independent coalescent sample in analyses of population divergence. Given potential intralocus recombination, it is also possible that we should have further split our loci into recombination-free blocks to accommodate the assumptions of the IM model. The failure to account for recombination has been shown to bias parameter estimates in IM (see Becquet and Przeworski 2007). Given the difficulty of distinguishing homoplasy and recombination, particularly given the high levels of polymorphism in our study, we did not feel that such a procedure was warranted. Indeed, homoplasy may contribute in part to our low estimates of LD.
The colonization of islands by continental birds has played a critical role in the development of speciation theory (Mayr 1942, 1963; MacArthur and Wilson 1967). In highly vagile birds in particular, dispersal to new habitats may be accomplished readily and may lead to speciation either by genetic drift or by adaptation to the new habitat. One of the most controversial mechanisms of speciation is speciation by founder effect (reviewed in Templeton 2008) and there has been little empirical support for founder-effect speciation in birds (Clegg et al. 2002; Walsh et al. 2005; Price 2007; but see Friesen et al. 2007). Founder-induced divergence in birds has been rejected either due to the maintenance of relatively high levels of genetic variation (e.g., Walsh et al. 2005) or due to evidence that genetic drift could not fully explain the observed phenotypic divergence (e.g., Clegg et al. 2002). Natural selection, however, acts in concert with genetic drift in many founder-effect speciation models, and genetic diversity can be maintained following the founder event (Templeton 2008).
Previous studies suggest that the two zebra finch subspecies differ in morphology and song, with both characteristics influencing patterns of mate choice in captivity (Clayton 1990). Despite this, the two forms are clearly not completely reproductively isolated and can produce viable offspring when mating is constrained. By the criterion of the biological species concept, they are therefore not separate species. The two forms, however, are diagnosable morphologically, and although their gene trees are not reciprocally monophyletic, they are also diagnosable genetically by the multilocus Structure approach that we have employed (see Edwards et al. 2005; Knowles and Carstens 2007 for a discussion of diagnosability without gene tree monophyly). Thus, the use of other species concepts and criteria would likely favor recognition as full species (reviewed in de Queiroz 1998).
Our results suggest that the divergence of two zebra finch subspecies involved a founder event that is reflected in the reduced levels of genetic diversity in the Timor zebra finch (π = 0.002) relative to Australian zebra finches (π = 0.01). Results from both IM and coalescent simulations suggest that only a small proportion of the ancestral population founded the island population and that both populations have experienced a history of population growth. The details of the demographic history, however, differ among approaches with IM analyses, suggesting a much stronger founder event. Differences between these results may be due in part to the fact that the IM model included migration among populations. We believe that the broad details of the demographic history that we have estimated are accurate and that such details will be essential for quantifying the pattern of protein-coding evolution in zebra finches. We note that the particular demographic history that we have described makes the zebra finch particularly appropriate for methods of estimating adaptive evolution in proteins that require populations to differ strongly in Ne (e.g., Loewe et al. 2006; Maside and Charlesworth 2007).
Given our IM estimates of divergence time and Ne for the island population, application of a simple quantitative genetics model suggested that genetic drift alone cannot account for all of the observed morphological divergence between island and mainland subspecies of zebra finch. To the contrary, analyses using Lande's (1976) Ne* indicate that unless the traits in question are highly heritable (h2 = 0.5), divergence times are relatively deep (∼2.8 MYA), or we have overestimated the Ne for the island subspecies, drift alone is not a sufficient explanation for the observed divergence in most traits. This last situation is likely since the long-term Ne of the island population probably resides somewhere between the founder size and the current size. Because the Ne* estimates are relatively small and because we assumed a relatively large island Ne, these conclusions would be similar even if the demographic history was more similar to that suggested by our simulations (Ne island between 5000 and 100,000) than that suggested by IM. This contrast between fixed differences at phenotypic traits and lack of complete differentiation at neutral nuclear loci has been found between other Australian grassfinches (Jennings and Edwards 2005) and suggests an important role for natural and/or sexual selection in the divergence of phenotypic traits in this clade (Edwards et al. 2005). Given the evidence for a founder event, however, it is unlikely that speciation has been driven solely by natural or sexual selection. In fact, our data suggest that drift can explain anywhere from 21 to 100% of the morphological divergence in specific traits that show subspecific differentiation. For this reason, we suggest that both drift and selection have been important for speciation in this clade and that the founding of the island population may have facilitated speciation in so far as it caused shifts in levels of LD, bringing together and fixing novel gene combinations in the Lesser Sundas population.
Variation in the extent of LD is caused in part by the interacting influences of effective population size and recombination rate, which in birds is expected to be quite high (Smith and Burt 1998; but see Backström et al. 2006b; Dawson et al. 2007; Stapley et al. 2008). Estimates of the ratio ρ/θ and the elevated level of LD following the colonization of the islands demonstrate the contribution of Ne to the low levels of LD in the mainland populations. On the other hand, the fact that LD among the island population has apparently decayed almost back to the level seen in the mainland, confirms that recombination rates and an open population structure cause rapid decay of LD over time (Anderson and Slatkin 2007; Leblois and Slatkin 2007). The rapid decay of LD in zebra finches is similar to what has been observed in a number of studies of wild, outbred organisms (e.g., Conway et al. 1999; Remington et al. 2001; Morrell et al. 2005; Cutter 2006; Cutter et al. 2006; but see Laurie et al. 2007). In studies of wild birds, however, results have been varied. Edwards and Dillon (2004) found quite low levels of LD at small genomic scales on autosomes while Backström et al. (2006b) found relatively high levels of LD extending across hundreds of kilobases on the Z chromosome.
The average recombination rate, ρ, for zebra finches is similar to the only previous estimate for a songbird based on SNPs, the red-winged blackbird Agelaeius pheonicus (Edwards and Dillon 2004; see Table 3), another species with a relatively large Ne. Estimates of effective population size for zebra finches, however, are over two orders of magnitude greater than for red-winged blackbirds, suggesting a lower per-base-pair recombination rate (c) in the zebra finch. Linkage mapping studies have also demonstrated that recombination rates may vary greatly among birds (Backström et al. 2006a; Dawson et al. 2007) and a recent estimate based on a pedigree of captive zebra finches suggests that recombination rates in zebra finches are lower than in chickens (Stapley et al. 2008). Until the zebra finch genome project progresses, we will be unable to map our locus trios directly to the genome or to correlate our estimates of recombination with those found for similar regions using the pedigree approach. However, Ne estimates for Australian zebra finches, which are at a minimum 500 times greater than those for humans (Takahata 1993; Tenesa et al. 2007), are more than large enough to explain the disparity in estimates of ρ between humans (∼0.0004/site) and zebra finches (∼0.05/site). All of these parameters—large effective population size, high recombination rate, population growth, and a founder event in the Lesser Sundas subspecies—will be crucial for informing our understanding and analysis of the evolution of protein-coding sequences as we begin to unravel the myriad links between molecular, morphological, and behavioral variation in this emerging model species.
Acknowledgments
We thank A. DiRienzo and two anonymous reviewers for their feedback on this manuscript. We especially thank Dave Runciman for providing DNA samples, particularly those from the Lesser Sundas, that were crucial to the completion of this project. Michaela Hau and James Adelman also provided samples that were used in the development of markers. Angus Emmott, June Yong Lee, Jeremiah Trimble, and Nate Rice provided assistance in the field, while Charles Chapus, Tim Gernat, Dan Janes, June Yong Lee, Liang Liu, Patricia Brito, and Nancy Rotzel provided assistance with laboratory work and/or data analysis. We thank the property manager and owner of Ellendale Station, Western Australia, and Angus Emmott for granting us permission to work on their properties. Walter Boles provided invaluable help with logistics in Australia. Gil McVean provided guidance in the use of LDhat and Christian Anderson helped with Serial SIMCOAL analyses. This manuscript benefited greatly from discussions with Kurt Lambeck, Trevor Price, John Todd, and Monty Slatkin. C.N.B. and the laboratory research were supported by funds from Harvard University and the Nuttall Ornithological Club.
References
- Anderson, C. N. K., U. Ramakrishnan, Y. L. Chan and E. A. Hadly, 2005. Serial SimCoal: a population genetics model for data from multiple populations and points in time. Bioinformatics 21 1733–1734. [DOI] [PubMed] [Google Scholar]
- Anderson, E. C., and M. Slatkin, 2007. Estimation of the number of individuals founding colonized populations. Evolution 61 972–983. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., 2001. Contrasting patterns of X-linked and autosomal nucleotide variation in Drosophila melanogaster and Drosophila simulans. Mol. Biol. Evol. 18 279–290. [DOI] [PubMed] [Google Scholar]
- Axelsson, E., L. Hultin-Rosenberg, M. Brandstrom, M. Zwahlen, D. F. Clayton et al., 2008. Natural selection in avian protein-coding genes expressed in brain. Mol. Ecol. 17 3008–3017. [DOI] [PubMed] [Google Scholar]
- Bachtrog, D., and B. Charlesworth, 2002. Reduced adaptation of a non-recombining neo-Y chromosome. Nature 416 323–326. [DOI] [PubMed] [Google Scholar]
- Backström, N., A. Qvarnström, L. Gustafsson and H. Ellegren, 2006. a Levels of linkage disequilibrium in a wild bird population. Biol. Lett. 2 435–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backström, N., M. Brandström, L. Gustafsson, A. Qvarnström, H. Cheng et al., 2006. b Genetic mapping in a natural population of collared flycatchers (Ficedula albicollis): conserved synteny but gene order rearrangements on the avian Z chromosome. Genetics 174 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker, F. K., A. Cibois, P. Schikler, J. Feinstein and J. Cracraft, 2004. Phylogeny and diversification of the largest avian radiation. Proc. Natl. Acad. Sci. USA 101 11040–11045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett, J. C., B. Fry, J. Maller and M. J. Daly, 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21 263–265. [DOI] [PubMed] [Google Scholar]
- Becquet, C., and M. Przeworski, 2007. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 17 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth, J., and A. Eyre-Walker, 2006. The rate of adaptive evolution in enteric bacteria. Mol. Biol. Evol. 23 1348–1356. [DOI] [PubMed] [Google Scholar]
- Clayton, D. F., 2004. Songbird genomics: methods, mechanisms, opportunities, and pitfalls. Ann. NY Acad. Sci. 1016 45–60. [DOI] [PubMed] [Google Scholar]
- Clayton, D. F., A. P. Arnold, W. Warren, E. Jarvis, C. Mello, et al., 2005. Proposal to sequence the genome of the zebra finch (Taeniopygia guttata). National Institutes of Health. http://www.genome.gov.
- Clayton, N. S., 1990. Assortative mating in zebra finch subspecies, Taeniopygia guttata guttata and T.g. castanotis. Philos. Trans. R. Soc. Lond. B 330 351–370. [Google Scholar]
- Clayton, N. S., D. Hodson and R. Zann, 1991. Geographic variation in zebra finch subspecies. Emu 91 2–11. [Google Scholar]
- Clegg, S. M., S. M. Degnan, C. Moritz, A. Estoup, J. Kikkawa et al., 2002. Microevolution in island forms: the roles of drift and directional selection in morphological divergence of a passerine bird. Evolution 56 2090–2099. [DOI] [PubMed] [Google Scholar]
- Conway, D. J., C. Roper, A. M. J Oduola, D. E. Arnot, P. G. Kremsner et al., 1999. High recombination rate in wild populations of Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 96 4506–4511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cracraft, J., 1986. Origin and evolution of continental biotas: speciation and historical congruence within the Australian avifauna. Evolution 40 977–996. [DOI] [PubMed] [Google Scholar]
- Cutter, A. D., 2006. Nucleotide polymorphism and linkage disequilibrium in wild populations of the partial selfer Caenorhabditis elegans. Genetics 172 171–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cutter, A. D., S. E. Baird and D. Charlesworth, 2006. High nucleotide polymorphism and rapid decay of linkage disequilibrium in wild populations of Caenorhabditis remanei. Genetics 174 901–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, D. A., M. Akesson, T. Burke, J. M. Pemberton, J. Slate et al., 2007. Gene order and recombination rate in homologous chromosome regions of the chicken and a passerine bird. Mol. Biol. Evol. 24 1537–1552. [DOI] [PubMed] [Google Scholar]
- de Queiroz, K., 1998. The general lineage concept of species, species criteria, and the process of speciation: a conceptual unification and terminological recommendations, pp. 57–75 in Endless Forms: Species and Speciation, edited by D. J. Howard and S. H. Berlocher. Oxford University Press, Oxford.
- Edwards, S. V., 1998. Diversity of birds, pp. 358–369 in Encyclopedia of Reproduction, edited by E. Knobil and J. D. Neill. Academic Press, San Diego.
- Edwards, S. V., and M. Dillon, 2004. Hitchhiking and recombination in birds: evidence from Mhc-linked and unlinked loci in red-winged blackbirds (Agelaius phoeniceus). Genet. Res. 84 175–192. [DOI] [PubMed] [Google Scholar]
- Edwards, S. V., S. B. Kingan, J. D. Calkins, C. N. Balakrishnan, W. B. Jennings et al., 2005. Speciation in birds: genes, geography, and sexual selection. Proc. Natl. Acad. Sci. USA 102 6550–6557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellegren, H., 2007. Molecular evolutionary genomics of birds. Cytogenet. Genome Res. 117 120–130. [DOI] [PubMed] [Google Scholar]
- Excoffier, L., J. Novembre and S. Schneider, 2000. SIMCOAL: a general coalescent program for the simulation of molecular data in interconnected populations with arbitrary demography. J. Hered. 91 506–509. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstmeier, W., G. Segelbacher, J. C. Mueller and B. Kempenaers, 2007. Genetic variation and differentiation in captive and wild zebra finches (Taeniopygia guttata). Mol. Ecol. 16 4039–4050. [DOI] [PubMed] [Google Scholar]
- Frentiu, F. D., S. M. Clegg, M. W. Blows and I. P. F. Owens, 2007. Large body size in an island-dwelling bird: a microevolutionary analysis. J. Evol. Biol. 20 639–649. [DOI] [PubMed] [Google Scholar]
- Friesen, V. L., T. M. Burg and K. D. McCoy, 2007. Mechanisms of population differentiation in seabirds. Mol. Ecol. 16 1765–1785. [DOI] [PubMed] [Google Scholar]
- Frisse, L., R. R. Hudson, A. Bartozewicz, J. D. Wall, J. Donfrack et al., 2001. Gene conversion and different population histories may explain the contrast between polymorphism and linkage disequilibrium. Am. J. Hum. Genet. 69 831–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodwin, D., 1982. Estrildid Finches of the World. Cornell University Press, Ithaca, NY.
- Hadfield, J. D., M. D. Burgess, A. Lord, A. B. Phillimore, S. M. Clegg et al., 2006. Direct versus indirect sexual selection: genetic basis of colour, size and recruitment in a wild bird. Proc. R. Soc. Lond. B 273 1347–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haesler, S., K. Wada, A. Nshdejan, E. Morrisey, T. Lints et al., 2004. FoxP2 expression in avian vocal learners and non-learners. J. Neurol. 24 3164–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrigan, R. J., M. E. Mazza and M. D. Sorenson, 2008. Computation versus cloning: evaluation of two methods for haplotype determination. Mol. Ecol. Res. 8 1239–1248. [DOI] [PubMed] [Google Scholar]
- Hey, J., 2005. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 3 965–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J., and R. Nielsen, 2004. Multilocus methods for estimating population sizes, migration rates and divergence time, with applications to the divergence of Drosophila pseudoobscura and D. persimilis. Genetics 167 747–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., D. D. Boos and N. L. Kaplan, 1992. A statistical test for detecting population subdivision. Mol. Biol. Evol. 9 138–151. [DOI] [PubMed] [Google Scholar]
- Jarvis, E. D., 2004. Learned birdsong and the neurobiology of human language. Ann. NY Acad. Sci. 1016 749–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennings, W. B., and S. V. Edwards, 2005. Speciational history of Australian grass finches (Poephila) inferred from thirty gene trees. Evolution 59 2033–2047. [PubMed] [Google Scholar]
- Joseph, L., and T. Wilke, 2007. Lack of phylogeographic structure in three widespread Australian birds reinforces emerging challenges in Australian historical biogeography. J. Biogeogr. 34 612–624. [Google Scholar]
- Kim, Y., 2006. Allele frequency distribution under recurrent selective sweeps. Genetics 172 1967–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura, M., 1983. The Neutral Theory of Molecular Evolution. Cambridge University Press, Cambridge, UK.
- Knowles, L. L, and B. C. Carstens, 2007. Delimiting species without monophyletic gene trees. Syst. Biol. 56 887–895. [DOI] [PubMed] [Google Scholar]
- Kuhner, M., 2006. LAMARC 2.0: maximum likelihood and Bayesian estimation of population parameters. Bioinformatics 22 768–770. [DOI] [PubMed] [Google Scholar]
- Lambeck, K., and J. Chappel, 2001. Sea level change through the last glacial cycle. Science 292 679–686. [DOI] [PubMed] [Google Scholar]
- Lambeck, K., T. M. Esat and E. K. Potter, 2002. Links between climate and sea levels for the past three million years. Nature 419 199–206. [DOI] [PubMed] [Google Scholar]
- Lande, R., 1976. Natural selection and random genetic drift in phenotypic evolution. Evolution 30 314–334. [DOI] [PubMed] [Google Scholar]
- Laurie, C. C., D. A. Nickerson, A. D. Anderson, B. S. Weir, R. J. Livingston et al., 2007. Linkage disequilibrium in wild mice. PLoS Genet. 3 e144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblois, R., and M. Slatkin, 2007. Estimating the number of founder lineages from haplotypes of closely linked SNPs. Mol. Ecol. 16 2237–2245. [DOI] [PubMed] [Google Scholar]
- Lee, J. Y., and S. V. Edwards, 2008. Divergence across Australia's Carpentarian barrier: statistical phylogeography of the red-backed fairy wren (Malurus melanocephalus). Evolution 62 3117–3134. [DOI] [PubMed] [Google Scholar]
- Loewe, L., B. Charlesworth, C. Bartolome and V. Noel, 2006. Estimating selection on nonsynonymous mutations. Genetics 172 1079–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovette, I. J., 2004. Mitochondrial dating and mixed-support for the “2% rule” in birds. Auk 121 1–6. [Google Scholar]
- MacArthur, R. H., and E. O. Wilson, 1967. The Theory of Island Biogeography. Princeton University Press, Princeton, NJ.
- Maddison, W. P. and D. R. Maddison, 2007. Mesquite: A Modular System for Evolutionary Analysis. http://www.mesquiteproject.org.
- Mank, J. E., E. Axelsson and H. Ellegren, 2007. Fast-X on the Z: rapid evolution of sex-linked genes in birds. Genome Res. 17 618–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marais, G., and B. Charlesworth, 2003. Genome evolution: recombination speeds up adaptive evolution. Curr. Biol. 13 R68–R70. [DOI] [PubMed] [Google Scholar]
- Maside, X., and B. Charlesworth, 2007. Patterns of molecular variation and evolution in Drosophila americana and its relatives. Genetics 176 2293–2305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayr, E., 1942. Systematics and the Origin of Species. Harvard University Press, Cambridge, MA.
- Mayr, E., 1944. Timor and the colonization of Australia by birds. Emu 44 113–130. [Google Scholar]
- Mayr, E., 1963. Animal Species and Evolution. Harvard University Press, Cambridge, MA.
- McVean, G., P. Awadalla and P. Fearnhead, 2002. A coalescent-based method for detecting and estimating recombination from gene sequences. Genetics 160 1231–1241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merila, J., L. E. B. Kruuk and B. C. Sheldon, 2001. Natural selection on the genetical component of variance in body condition in a wild bird population. J. Evol. Biol. 14 918–929. [Google Scholar]
- Morrell, P. L., D. M. Toleno, K. E. Lundy and M. T. Clegg, 2005. Low levels of linkage disequilibrium in wild barley (Hordeum vulgare ssp. spontaneum) despite high rates of self-fertilization. Proc. Natl. Acad. Sci. USA 102 2442–2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz-Fuentes, V., C. Vila, A. J. Green, J. J. Negro and M. D. Sorenson, 2007. Hybridization between white-headed ducks and introduced ruddy ducks in Spain. Mol. Ecol. 16 629–638. [DOI] [PubMed] [Google Scholar]
- Naurin, S., S. Bensch, B. Hansson, T. Johansson, D. F. Clayton et al., 2008. A microarray for large-scale genomic and transcriptional analyses of the zebra finch (Taeniopygia guttata) and other passerines. Mol. Ecol. Res. 8 275–281. [DOI] [PubMed] [Google Scholar]
- Nei, M., 1987. Molecular Evolutionary Genetics. Columbia University Press, New York.
- Pool, J. E., and R. Nielsen, 2007. Population size changes reshape genomic patterns of diversity. Evolution 61 3001–3006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, D. K., and N. T. Burley, 1993. Constraints on the evolution of attractive traits: genetic (co)variance of zebra finch bill color. Heredity 71 405–412. [DOI] [PubMed] [Google Scholar]
- Price, T., 2007. Speciation in Birds. Roberts & Company, Greenwood Village, CO.
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of selection at randomly chosen loci. Genetics 160 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raikow, R. J., 1986. Why are there so many kinds of passerine birds? Syst. Zool. 35 255–259. [Google Scholar]
- Reich, D. E., M. Cargill, S. Bolk, J. Ireland, P. C. Sabeti et al., 2001. Linkage disequilibrium in the human genome. Nature 411 199–204. [DOI] [PubMed] [Google Scholar]
- Reich, D. E., S. F. Schaffner, M. J. Daly, G. McVean, J. C. Mullikin et al., 2002. Human genome sequence variation and the influence of gene history, mutation and recombination. Nat. Genet. 32 135–142. [DOI] [PubMed] [Google Scholar]
- Remington, D. L., J. M. Thornsberry, Y. Matsuoka, L. M. Wilson, S. R. Whitt et al., 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Acad. Natl. Sci. USA 98 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replogle, K. L., A. P. Arnold, G. F. Ball, M. Band, S. Bensch et al., 2008. The songbird neurogenomics (SoNG) initiative: community-based tools for study of brain gene function and evolution. BMC Genomics 9: 131. [DOI] [PMC free article] [PubMed]
- Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP: DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19 2496–2497. [DOI] [PubMed] [Google Scholar]
- Rozen, S., and H. Skaletesky, 2000. Primer3 on the WWW for general users and biologist programmers, pp. 365–386 in Bioinformatics Methods and Protocols: Methods in Molecular Biology, edited by S. Krawetz and S. Misener. Humana Press, Totowa, NY. [DOI] [PubMed]
- Slater, P. J. B., L. A. Eales and N. S. Clayton, 1988. Song learning in zebra finches (Taeniopygia guttata): progress and prospects, pp. 1–34 in Advances in the Studies of Behavior, edited J. S. Rosenblatt, C. Beer, M. C. Busnel and P. J. B. Slater. Harcourt Brace Jovanovich, New York.
- Slatkin, M., and W. P. Maddison, 1989. A cladistic measure of gene flow inferred from the phylogeny of alleles. Genetics 123 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, J., and D. W. Burt, 1998. Parameters of the chicken genome (Gallus gallus). Anim. Genet. 29 290–294. [DOI] [PubMed] [Google Scholar]
- Sorenson, M. D., C. N. Balakrishnan and R. B. Payne, 2004. Clade-limited colonization in brood parasitic finches (Vidua spp.). Syst. Biol. 53 140–153. [DOI] [PubMed] [Google Scholar]
- Stapley, J., T. R. Birkhead, T. Burke and J. Slate, 2008. A linkage map of the zebra finch Taeniopygia guttata provides new insights into avian genome evolution. Genetics 179 651–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., and P. Scheet, 2005. Accounting for decay of linkage disequilibrium in haplotype inference and missing-data imputation. Am. J. Hum. Genet. 76 449–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens, M., N. J. Smith and P. Donnelly, 2001. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 68 978–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L., 2002. PAUP*: Phylogenetic Analysis Using Parsimony (*and Other Methods). Sinauer Associates, Sunderland, MA.
- Takahata, N., 1993. Allergy geneology and human evolution. Mol. Biol. Evol. 2 2–22. [DOI] [PubMed] [Google Scholar]
- Templeton, A.R., 2008. The reality and importance of founder speciation in evolution. BioEssays 30 470–479. [DOI] [PubMed] [Google Scholar]
- Tenesa, A., P. Navarro, B. J. Hayes, D. L. Duffy, G. M. Clarke et al., 2007. Recent human effective population size estimated from linkage disequilibrium. Genome Res. 17 520–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teramitsu, I., L. C. Kudo, S. E. London, D. H. Geschwind and S. A. White, 2004. Parallel FoxP1 and FoxP2 expression in songbird and human brain predicts functional interaction. J. Neurol. 24 3152–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, H. E., I. L. Jones and V. L. Friesen, 2005. A test of founder effect speciation using multiple loci in the auklets (Aethia spp.). Genetics 171 1885–1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir, B. S., and W. G. Hill, 1986. Nonuniform recombination within the human beta-globin gene cluster. Am. J. Hum. Genet. 38 776–778. [PMC free article] [PubMed] [Google Scholar]
- White, M. E., 1996. After the Greening: The Browning of Australia. Kangaroo Press, Kenthurst, NSW, Australia.
- Wong, G. K., B. Liu, J. Wang, Y. Zhang, X. Yang et al., 2004. A genetic variation map for chicken with 2.8 million single nucleotide polymorphisms. Nature 432 717–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama, Y., A. Purcell, K. Lambeck and P. Johnson, 2001. Shoreline reconstruction around Australia during the last glacial maximum and late glacial states. Quat. Int. 83–85: 9–18. [Google Scholar]
- Zann, R. A., 1996. The Zebra Finch: A Synthesis of Field and Laboratory Studies. Oxford University Press, Oxford.
- Zann, R. A., and D. Runciman, 2008. Survivorship, dispersal and sex ratios of zebra finches Taeniopygia guttata in southeast Australia. Ibis 136 136–143. [Google Scholar]