

Abstract
Gene expression levels vary heritably, with ∼25–35% of the loci affecting expression acting in cis. We characterized standing cis-regulatory variation among 16 wild-derived strains of Drosophila melanogaster. Our experiment's robust biological and technical replication enabled precise estimates of variation in allelic expression on a high-throughput SNP genotyping platform. We observed concordant, significant differential allelic expression (DAE) in 7/10 genes queried with multiple SNPs, and every member of a set of eight additional, one-assay genes suggest significant DAE. Four of the high-confidence, multiple-assay genes harbor three or more statistically distinguishable allelic classes, often at intermediate frequency. Numerous intermediate-frequency, detectable regulatory polymorphisms cast doubt on a model in which cis-acting variation is a product of deleterious mutations of large effect. Comparing our data to predictions of population genetics theory using coalescent simulations, we estimate that a typical gene harbors 7–15 cis-regulatory sites (nucleotides) at which a selectively neutral mutation would elicit an observable expression phenotype. If standing cis-regulatory variation is actually slightly deleterious, the true mutational target size is larger.
VARIOUS strategies have been used to identify putative cis-regulatory differences between strains or alleles [e.g., microarrays and linkage mapping (Brem et al. 2002) and allelic imbalance/allelic expression (AE) (Yan et al. 2002)], but estimates of the numbers of segregating, functional alleles and their frequencies have not yet been made. Given recent attention paid to the question of whether the evolution of form proceeds largely by changes in cis-regulation (Hoekstra and Coyne 2007; Wray 2007; Stern and Orgogozo 2008), this is a critical gap in knowledge. Like any trait, the evolutionary trajectory of a gene's expression is affected by standing genetic variation and the pressures of mutation, selection, and drift. Mutation-accumulation experiments have indicated that natural selection does curtail mutational variance for expression (Denver et al. 2005; Rifkin et al. 2005), but nevertheless some cis-acting variation in gene expression is readily observed between species (Wittkopp et al. 2004, 2008b; de Meaux et al. 2006; Zhuang and Adams 2007; Genissel et al. 2008). Inbred strains of Drosophila melanogaster afford us the opportunity to address the strength, frequency, and number of distinguishable cis-regulatory alleles in a panel of wild-derived chromosomes.
Screens for differential allelic expression (DAE)—differences in the relative expression of the two gene copies possessed by a diploid individual—have been used to identify genes affected by cis-acting alleles in humans (Yan et al. 2002; Bray et al. 2003; Lo et al. 2003; Pastinen et al. 2004; Pant et al. 2006), mice (Cowles et al. 2002; Campbell et al. 2008), Arabidopsis (de Meaux et al. 2005), and Drosophila (Wittkopp et al. 2004, 2008b; Wittkopp 2005). We adapted the high-throughput oligo ligation assay (OLA) (Macdonald et al. 2005) to quantify allelic expression in 16 D. melanogaster strains over 39 SNP assays querying 18 genes.
MATERIALS AND METHODS
Strains:
Wild-type and mutant-marked strains of D. melanogaster were obtained from stock centers, and most were inbred by brother–sister mating for 18 generations (supplemental Table 1). In this study, there were two distinct functions that an allele inherited from one of the strains could serve: as a “reference” or as a “tester.” The reference allele is always SS (Bloomington 2057) or BWST (Bloomington 686). In any block of experimental individuals, every individual shares its reference allele in common; the other allele is the basis for comparison among individuals and is designated the tester allele.
Gene/assay selection:
Genes were selected irrespective of whether they had been previously shown to be subject to regulatory variation of any kind. To facilitate future genetic dissection, we selected genes that are located on the X chromosome. Biological relevance was assessed from microarray data (Pletcher et al. 2002): X chromosome genes were ranked by average expression level in 3-day-old adult females. After amplification of ∼1-kb PCR products, we sequenced ∼100 highly expressed X-linked genes to discover SNPs. PCR template was cDNA from adult males of three wild-type strains and the BWST reference strain. We used primer3 (Rozen and Skaletsky 2000) to design PCR/sequencing primers (supplemental Table 2) that amplified ∼1000 bp of mRNA sequence from each gene, focusing on 3′-UTR regions. Polymorphisms were discovered by aligning all four sequences to the reference genome of D. melanogaster (Berkeley Drosophila Genome Project Release 3.1, SS). Only polymorphisms for which one or both reference alleles were the minor genotype were selected for assay design; consequently, only genes harboring at least one such SNP received further consideration.
Crossing design:
In conjunction with our SNP selection, we devised a crossing scheme that ensures that at least half of the experimental individuals are informative heterozygotes at every interrogated SNP. In this “two-reference” design (Figure 1a), every tester strain is crossed to each reference strain, yielding 30 unique trans-heterozygous genotypes (or 29 if progeny from reciprocal SS × BWST crosses are considered identical). Under the rule for SNP selection described above, the structure of crosses that result in heterozygous individuals must take one of two forms. Usually, only one reference strain is the rare genotype. Therefore, the testers that are informative in a cross to SS will exactly complement the testers that are informative in a cross to BWST; every tester can be found in heterozygous form once (depicted in Figure 1, c and e). Less commonly, if both reference strains are the same rare genotype, a majority of testers will be heterozygous in the crosses to both reference strains (although some testers will be missed completely); consequently, more than half of the experimental individuals are informative.
Figure 1.—

A high-throughput structure for measuring AE (a–e, clockwise). (a) SNPs are identified by comparing three wild-type sequences with the SS and BWST reference sequences. A subset of SNPs—those for which SS or BWST or both is the rare allele—is selected for assay design. In our “two-reference” cross, each reference strain is crossed to all tester strains, most of which have unknown genotypes. RNA is collected from offspring. (b) AE SNP assays (here, multiplex oligo ligation assays) are performed. The two axes represent log-transformed expression of the two SNP alleles. (c) AE data are first used to ascertain the genotypes of all tester strains so that the genotype of each datum can be inferred. Note that when SS and BWST are of a different genotype, every strain is heterozygous in one of the two crosses, and >50% of experimental individuals are informative. In contrast, any panel of outbred individuals will have a much lower rate of informative samples in a multi-gene, multi-SNP survey (cf. 27.7% of assayed samples that were heterozygous over 193 SNPs surveyed in 63 unrelated human subjects; Pastinen et al. 2004). (d) AE for each datum is calculated as its perpendicular distance from the first principal axis drawn through the heterozygous data, effectively rotating the data clockwise from b. Uninformative samples (gray: homozygotes, controls, and calibration samples) are culled so that heterozygotes (black) can be used to compare testers' AE. (e) Using linear mixed-effects models, the expression of each tester allele is estimated relative to the other strains of the same SNP genotype (left and right plots; unlabeled positions indicate uninformative strains). Separate P-values for the effect of the tester on AE in the two groups are calculated. Error bars indicate 95% confidence intervals.
Crosses were performed by mating three to five tester males to four to five virgin reference females in each of seven replicate vials containing standard banana–molasses media. Vials were cleared before the emergence of the first adult progeny. All crosses to a given reference strain were initiated on the same day.
Sampling scheme:
To extend our power to describe the cis-regulatory characteristics of a panel of wild-derived alleles, we collected RNA from 640 experimental individuals, ∼20 individuals per tester × reference cross (with the exception of crosses to tester strain P, from which ∼40 individuals were sampled; see supplemental Table 1). We employed a sampling strategy with the specific goal of minimizing the extent to which differential allelic expression could be confounded with sampling. Experimental samples underwent collection and RNA extraction in 80-fly blocks. In each block, each tester allele was represented by approximately five individuals, and all individuals in a block shared the same reference allele. Four blocks were collected for crosses to each reference strain. For each block, the female experimental flies from each vial (and their male siblings) were transferred, without anesthetic, to a vial of fresh media within their first 24 hr post-eclosion. After 3 days, these flies were transferred to an empty vial without the use of anesthesia. After all strains and replicates had been transferred, the vials were put on ice until flies were motionless. Choosing vials and genotypes in random order, a single female from each vial was quickly transferred to a well of a U-bottom polypropylene 96-well plate (Evergreen Scientific, Los Angeles), which had been prechilled on ice. Although less desirable than flash freezing in liquid nitrogen, collection by this technique allowed us to ensure that every fly in the block experienced ice-anesthesia for the same length of time. Large blocks also ensured that every genotype was multiply represented in each block, reducing the possibility that any sample collection and RNA extraction effects were confounded with genotype.
RNA extraction:
We adapted the manufacturer's standard TRIzol (Invitrogen, Carlsbad, CA) RNA extraction protocol to our unique situation of individual flies extracted in 96-well format. All liquid-handling steps were performed with multichannel pipettors and aerosol-resistant filter tips. Before collecting flies, a stainless-steel 5/32-inch grinding ball (BT&C/OPS Diagnostics, Bridgewater, NJ) was placed in each well of the U-bottom plate. After flies were placed in each well, 150 μl cold TRIzol was added. Plates were sealed with MicroAmp clear adhesive films (Applied Biosystems, Foster City, CA). Homogenization was performed in the Geno/Grinder 2000 (BT&C/OPS Diagnostics, Bridgewater, NJ) for 45 sec at 1500 rpm. After a 5-min room-temperature incubation, the entire 150 μl lysate was transferred to a 96-well PCR-style plate (ABgene, Rochester, NY) containing 30 μl chloroform. After sealing with a new MicroAmp film, plates were shaken in the Geno/Grinder 2000 at 750 rpm for 15 sec. After a 3-min incubation at room temperature, plates were centrifuged for 45 min at 2300 × g. Approximately 65 μl of the top aqueous layer was removed using a multichannel pipettor and mixed with 75 μl isopropanol in another 96-well PCR-style plate. Plates were again centrifuged at 2300 × g for 45 min, after which liquid was removed from the wells by inverting the plate on paper towels and centrifuging at minimal speed for a few seconds. Each pellet was covered in 150 μl ethanol (75%) and samples were centrifuged for 15 min at 2300 × g. Most of this ethanol was removed by inverting plates on paper towels without centrifugation. Pellets were covered in 130 μl ethanol (100%) and stored at −80° until the RNA of all samples had been extracted. Prior to use as template for cDNA synthesis, ethanol was pipetted off. Samples were dried at room temperature and resuspended in 13 μl Molecular Biology Grade water (Eppendorf, Westbury, NY). RNA was dissolved by incubation for 10 min at 55°.
cDNA synthesis:
To synthesize cDNA, we utilized a protocol that allowed us to overcome the hurdle of relatively little RNA starting material yet provided a generic cDNA end product that any assay of any gene could interrogate directly. RNA was used as template for cDNA synthesis using an adapted protocol of the SMART system (Clontech, Mountain View, CA). Briefly, this technology utilizes the terminal transferase activity of Powerscript Moloney murine leukemia virus (MMLV) reverse transcriptase (RT) to incorporate priming sequences at the ends of the first-strand cDNA synthesis product, which can then be used for amplification in a 15- or 25-cycle PCR-like process. At the 5′-end of the first-strand cDNA (3′-end of the gene), the oligo(dT) primer is modified with a tail; at the 3′-end of the first-strand product, an additional SMART oligo has been incorporated by virtue of base pairing with the oligo-C added by MMLV reverse transcriptase. An aliquot of 3.6 μl RNA was used as template for first-strand synthesis by the manufacturer's protocols. Samples were incubated for 90 min at 42°, followed by 15 min at 70° to inactivate the reverse transcriptase. Reverse transcription product was then used as template of a 25-μl second-strand synthesis/amplification reaction according to the manufacturer's protocols, with the following modifications: A substantially larger aliquot of first-strand product, 5.5 μl, was used as template for this reaction; after 15 cycles, 12 μl of the reaction was removed and stored; the remaining mix then underwent 10 additional cycles. Amplified, double-stranded cDNA products (15 and 25 cycles) were stored at −20° until all RNA samples had been processed to cDNA. All samples that share reference genotype and cycle number in common were joined on a single 384-well plate. Samples were then treated with 7.5 units of exonuclease I (Amersham, Piscataway, NJ) by incubating for 1 hr at 37°, followed by 15 min at 80°. Finally, 0.25 μl of each sample's cDNA was aliquoted to 384-well plates and dried; plates were stored at −80° until use in the OLA reaction (below).
Multiplex OLA for allelic expression:
Our multiple-gene, multiple-assay, highly replicated experiment required a low-cost, high-throughput mechanism for quantifying AE. We used a multiplex OLA approach. In OLA, allele-specific upstream oligonucleotides are ligated to a common downstream oligo in proportion to the allele representation among template DNA molecules. Using multiplex OLA for SNP genotyping, Macdonald et al. (2005) showed that under realistic laboratory settings genotype calls are 99.65% accurate, 86% of attempted assays convert, and 93.9% of data can be assigned a genotype. These figures compare favorably to most SNP genotyping methods, including those popularly used to quantify AE (summarized in Bray and O'Donovan 2006). In practice, we experienced a conversion rate of <50% (see supplemental Table 3) when adapting the technology to AE measurement. As the conversion rate is a function of template abundance (not shown), this lower rate is related to our attempts to use OLA on SMART-amplified cDNA as opposed to gene-by-gene reverse-transcription PCR reactions.
We pooled oligos and performed the oligo ligation assay under conditions that were nearly identical to previous SNP genotyping projects. The oligos used to query each SNP by OLA are listed in supplemental Table 2. OLA has been shown to readily tolerate multiplexing for SNP genotyping (Macdonald et al. 2005); expression data from test assays were indistinguishable in 6-plex or 12-plex (not shown). Assay oligos for allelic expression were pooled so that each multiplex had a maximum of 12 assays. Furthermore, none of the queried SNPs in an assay pool were located within 50 bp of one another. Samples were processed essentially as previously described (Macdonald et al. 2005). The 0.25-μl cDNA was used as template for a multiplex ligation reaction, which introduces an allele-specific DNA “barcode.” The ligated product is subsequently amplified by PCR and affixed to nylon membrane arrays. The relative positions of samples on the array are the same as in the 384-well source plate, and the source plate was rotated 180° between the printing of the first spot and the printing of a second, replicate spot to mitigate any plate/array position effects. Allelic expression was quantified by hybridization of radioactively labeled probes that are complementary to the allele-specific tags. It is important to note that in the OLA assay, the two SNP alleles are distinguished by using two 16-bp non-cross-hybridizing bar-code sequences. Spot intensities were quantified with ArrayVision (Imaging Research, St. Catharines, ON, Canada).
Allele quantification by Pyrosequencing:
For one assay at each of three genes (CG2247 assay j07, CG6398 assay b05, CG11674 assay c12), we performed allelic expression quantification by an alternative technology on a subset of samples for comparison to OLA. For each sample, a 4-μl RNA aliquot (∼400 ng) was incubated for 30 min at 37° with 0.6 units DNAse I (Thermo Fisher Scientific, Waltham, MA), followed by the addition of 0.5 μl Stop Solution (20 mm EGTA, pH 8.0) and incubation at 65° for 10 min. Gene-specific reverse transcription was performed according to the manufacturer's protocol using 50 units Moloney murine leukemia virus reverse transcriptase (NEB, Ipswich, MA), 2 μl DNAse-treated RNA template, and 0.5 pmol primer in a 5-μl reaction. The resulting RT product was diluted with 10 μl water; 0.5–1.5 μl diluted RT product served as template for a 30–50 μl PCR. Each 10-μl PCR mix contained 1× ExTaq buffer, 2 nmol each dNTP, 1.25 pmol each primer, 0.25 units ExTaq (Takara, Otsu, Japan). We incubated the samples at 94° for 4 min with 45 cycles of 94° for 30 sec, 58° for 30 sec, 72° for 30 sec, and 72° for 10 min with a 4° hold. Pyrosequencing was performed according to the manufacturer's instructions. Reverse transcription, PCR, and sequencing primers are available in supplemental Table 2. After plotting the log-transformed signal intensity of the two alleles, AE was calculated from the minimum deviation of each datum from the first principal axis through all heterozygous samples. Technical replication and blocking did not pertain to the Pyrosequencing experiment, so we determined the significance of tester as a predictor of AE in a one-way ANOVA.
Sample genotyping:
Because most testers' genotypes at query SNPs were unknown prior to our performing of the allelic expression assays, our first use of “expression” data was to infer the genotypes of the parental strains on the basis of the clustering patterns of each strain's offspring. For each tester allele, we plotted the spot intensities of all of its samples (i.e., all samples from flies that were fathered by the strain) on top of the clusters formed by plotting the intensities of the remaining samples. Assays for which genotypes could not be readily ascertained were excluded (supplemental Table 3). We note that for many assays, genomic DNA from each inbred, homozygous tester strain was also used to amplify the locus in question by standard PCR. OLA genotypes from these amplicons were used as a check against cDNA-inferred genotypes. Additionally, specific cDNA samples whose genotypes were inconsistent with their parents' consensus genotypes were excluded. Finally, data points of abnormally low signal or abnormal distance from the main cluster were excluded on an assay-by-assay basis.
Quantifying allelic expression with principal components analysis:
We devised a single measure to capture the allelic expression described by each data point. After selecting heterozygous individuals for a given SNP, we plotted the log intensities of the two probes (i.e., bar codes that distinguish between alleles) against one another (Figure 1b). Plots of all typable assays are available in supplemental Figure 3. To utilize principal components analysis (PCA), we calculated a first principal axis (Sokal and Rohlf 1995, pp. 586–593), which averages the first principal components, in this situation representing overall expression (the diagonal characteristic of the data cloud in Figure 1b). When the two reference strains had alternative alleles, this axis was defined using solely SS/BWST reciprocal heterozygotes; otherwise, all heterozygous samples were used. We define AE for any datum as its perpendicular distance to the axis (i.e., parallel to the second principal axis). These distances encapsulate the expression of the tester allele normalized to the expression of the reference allele; as described below, significant differences in this metric, not in the value of the metric itself, indicate DAE.
Estimating within-assay tester means and significance:
We used our measure of AE to compare the expression of tester alleles. Under our experimental structure, the AE of a strain is directly comparable to the alleles of all other strains that have the same genotype at the assay SNP. For each such data set, we fit a linear mixed-effects model in R using the lme() function 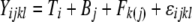 , where
, where 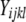 is the lth measurement (random) of the allelic expression of the kth fly (random) nested in the jth block (random) of the ith tester (fixed). In all, 74 models were fit (∼2/assay); therefore, the α = 0.05 Bonferroni correction for the significance of tester is P < 6.76 × 10−4. Separate analysis of data conditional on cycle number (15 or 25) results in nearly identical findings; thus, the
is the lth measurement (random) of the allelic expression of the kth fly (random) nested in the jth block (random) of the ith tester (fixed). In all, 74 models were fit (∼2/assay); therefore, the α = 0.05 Bonferroni correction for the significance of tester is P < 6.76 × 10−4. Separate analysis of data conditional on cycle number (15 or 25) results in nearly identical findings; thus, the 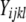 were first determined by extracting the residuals from a linear model fitting cycles as a categorical predictor of AE. For every SNP assay/reference cross combination, an average AE estimate for each tester allele was obtained using the “$coefficients$fixed” function in R. When necessary, these average tester estimates were multiplied by −1 so that the relative sign of AE measures always had a consistent biological syntax.
were first determined by extracting the residuals from a linear model fitting cycles as a categorical predictor of AE. For every SNP assay/reference cross combination, an average AE estimate for each tester allele was obtained using the “$coefficients$fixed” function in R. When necessary, these average tester estimates were multiplied by −1 so that the relative sign of AE measures always had a consistent biological syntax.
Calibration to estimate fold differences:
For 30/39 assays, we were able to include calibration samples that enabled us to calibrate our average AE estimates, which are quantified in relative, arbitrary units, to fold differences between tester alleles. To generate a set of calibration samples, RNA was extracted from homozygous reference strain flies and resuspended to 2× standard concentration. We pooled these pure samples of reference alleles in known proportions to construct 12 RNA calibration samples, with relative volumetric fold difference between alleles ranging 10-fold from ∼3.2-fold to its inverse, ∼0.31-fold. This pooling procedure was performed for three different pairs of RNA samples (each pair known as a “pool”); the calibration samples were then randomly positioned on an 8 × 6 PCR plate and used as template for reverse transcription and cDNA amplification, as above. Each calibration sample cDNA was used as template for four separate OLA reactions: one sample from a 15-cycle SMART amplification and three from a 25-cycle amplification.
Although the exact molar ratio of any calibration sample was unknown, the molar fold differences between calibration samples are expected to reflect the known volumetric fold differences. Separately for each pool, we used R's smooth.spline() function to fit a spline to a plot of calibration samples' measure of AE vs. log2(volume B/volume A) (supplemental Figure 1). The equation of this spline was used to convert each experimental AE datum into theoretical log-transformed volumetric ratios (log2V.R.). We analyzed log2V.R. values by our standard procedures to estimate the mean log2V.R. of each tester. The difference between log2V.R. of each possible pair of testers was calculated and averaged across pools. Thus, 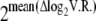 gives the average fold difference between any two testers. For two reasons—(1) because an absolute molar ratio of each individual's two alleles cannot be calculated, even with the calibration data, and (2) because calibration was not available for all assays—all further analysis was conducted on the relative AE estimates described in Quantifying allelic expression with principal components analysis (above).
gives the average fold difference between any two testers. For two reasons—(1) because an absolute molar ratio of each individual's two alleles cannot be calculated, even with the calibration data, and (2) because calibration was not available for all assays—all further analysis was conducted on the relative AE estimates described in Quantifying allelic expression with principal components analysis (above).
Estimating within-gene tester means and significance:
For 10 genes, we obtained multiple working SNP assays, which were then combined to create a single estimate of AE for that gene. For each gene, estimates of each tester strain's average relative AE (obtained from different assays and/or reference alleles) were treated as a separate observation of the true AE. An initial model 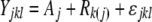 was fit, where
was fit, where 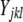 was the lth observation of a block effect applied to any strain crossed to the kth reference strain nested within the jth assay. The residual error (
was the lth observation of a block effect applied to any strain crossed to the kth reference strain nested within the jth assay. The residual error (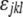 ) from this initial “nuisance model” was used as the response variable for a second model. In the second model,
) from this initial “nuisance model” was used as the response variable for a second model. In the second model, 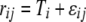 , the ith tester allele is the sole predictor of allelic expression. The P-value associated with this predictor was determined in a one-way ANOVA. Because the foundation of the model is the mean of the AE measures assessed over multiple assays and crosses, significance indicates that the allelic differences observed in multiple assays are more concordant than expected by chance. Moreover, for each gene examined, the use of this linear model results in a single measure (
, the ith tester allele is the sole predictor of allelic expression. The P-value associated with this predictor was determined in a one-way ANOVA. Because the foundation of the model is the mean of the AE measures assessed over multiple assays and crosses, significance indicates that the allelic differences observed in multiple assays are more concordant than expected by chance. Moreover, for each gene examined, the use of this linear model results in a single measure (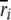 ) of the cis-regulatory propensity of each tester allele.
) of the cis-regulatory propensity of each tester allele.
Estimating the number of allelic classes represented in the tester panel:
We compared our results from gene-level analysis (the model  , as above) to null expectations to estimate the number of distinguishable allelic classes (ACs) likely to exist in the tester panel. The general principle of this comparison is the following: if a statistic can be calculated from a group of testers' data that significantly exceeds the expectations of a reasonable null model (H0, no significant difference among testers), it is likely that the group of testers is composed of at least two true alleles. The power of this approach is dependent on properties of the data such as the magnitude of the difference between putative ACs and the number of observations of each. Starting with a single class encompassing all testers, we progressively defined ACs by accepting the most significant bifurcation of any existing class, with significance assessed by permutation testing. The heuristic terminated when no prospective division of a class achieved significance. A full description of the heuristic can be found in the supplemental materials. There, we demonstrate that (1) the heuristic overestimates the number of alleles ∼5% of the time or less and (2) the heuristic's power is dependent upon strong penetrance of allelic effects and the size of the data set.
, as above) to null expectations to estimate the number of distinguishable allelic classes (ACs) likely to exist in the tester panel. The general principle of this comparison is the following: if a statistic can be calculated from a group of testers' data that significantly exceeds the expectations of a reasonable null model (H0, no significant difference among testers), it is likely that the group of testers is composed of at least two true alleles. The power of this approach is dependent on properties of the data such as the magnitude of the difference between putative ACs and the number of observations of each. Starting with a single class encompassing all testers, we progressively defined ACs by accepting the most significant bifurcation of any existing class, with significance assessed by permutation testing. The heuristic terminated when no prospective division of a class achieved significance. A full description of the heuristic can be found in the supplemental materials. There, we demonstrate that (1) the heuristic overestimates the number of alleles ∼5% of the time or less and (2) the heuristic's power is dependent upon strong penetrance of allelic effects and the size of the data set.
Estimating the size of the neutral mutational target with the coalescent:
We simulated the process of selecting 16 chromosomes at random from a population evolving with neutral Wright–Fisher dynamics using the program MS (Hudson 2002). Specifically, we compared the average number of unique haplotypes obtained ( ) when considering only polymorphisms that occur at i = {2, 3, 4 … 26} regulatory sites. Each set of 1000 simulations was defined by a value of θ and ρ. We utilized the locus θ = 3Nμ (all genes studied are X linked) as a compound parameter equal to the number of consequential regulatory sites (i) multiplied by a recent estimate of per-site θ (Andolfatto 2007; θ = 0.031 obtained from selection models using 12 Zimbabwe alleles of 137 X-linked loci). The population recombination rate ρ = 2Nc was set arbitrarily at values between 0 and 2000, inclusive, a reasonable range for a 10-kb locus (Haddrill et al. 2005). Simulations were conducted for each value of ρ paired with each value of θ (and therefore i). Custom perlscripts were used to determine the number of unique haplotypes (k) represented in the output of each run. To place our empirical data in the context of the simulation results, we used bootstrapping to obtain an 80% confidence interval on our observed
) when considering only polymorphisms that occur at i = {2, 3, 4 … 26} regulatory sites. Each set of 1000 simulations was defined by a value of θ and ρ. We utilized the locus θ = 3Nμ (all genes studied are X linked) as a compound parameter equal to the number of consequential regulatory sites (i) multiplied by a recent estimate of per-site θ (Andolfatto 2007; θ = 0.031 obtained from selection models using 12 Zimbabwe alleles of 137 X-linked loci). The population recombination rate ρ = 2Nc was set arbitrarily at values between 0 and 2000, inclusive, a reasonable range for a 10-kb locus (Haddrill et al. 2005). Simulations were conducted for each value of ρ paired with each value of θ (and therefore i). Custom perlscripts were used to determine the number of unique haplotypes (k) represented in the output of each run. To place our empirical data in the context of the simulation results, we used bootstrapping to obtain an 80% confidence interval on our observed  (the average number of ACs estimated by the heuristic over genes).
(the average number of ACs estimated by the heuristic over genes).
RESULTS
Analysis of calibration samples showed that the properties of the high-throughput OLA platform are appropriate for quantifying AE. Like all surveys of AE, the signal intensities of two transcribed alleles assessed on our SNP genotyping platform serve as a proxy for the relative expression of each diploid's homologous gene copies. To evaluate OLA, we included samples of known allelic input in 30/39 SNP assays. Overwhelmingly, these showed an appropriately linear relationship over a 10-fold range of variation of relative transcript abundance (supplemental Figure 1), demonstrating that our metric for AE effectively accounts for experimental differences between alleles (e.g., differential probe hybridization efficiency). Unlike other SNP genotyping methods, our multiplex OLA assay is intrinsically high throughput, allowing economical expansion of the set of experimental samples and the battery of assays performed. Enabled in part by a novel crossing design (Figure 1; materials and methods), a typical SNP assay in our experiment manifested as ∼1100 informative AE measures comprising ∼300 heterozygous individuals representing the 16 different genotypes.
These high levels of replication are necessary for precise estimates of AE and for detection of small differences across a panel of alleles. We found that significant DAE is detectable even when the estimated range of variation over alleles was as subtle as 1.14-fold. For comparison, we assayed three SNPs on a subset of RNA samples using Pyrosequencing (Biotage AB, Uppsala, Sweden), quantifying AE once in four to five individuals per genotype. For two such SNPs, the significant results of the full experiment could not be recapitulated by Pyrosequencing, and the same subset of samples showed significant results by OLA data only when technical replicates were considered. Results from the Pyrosequencing assay of the third SNP were not concordant with the comparable OLA assay, although we detected significant DAE in the subset using either platform. This observed discrepancy may be associated with the different methodologies used to amplify template: Pyrosequencing employs a SNP-specific 45-cycle RT–PCR, whereas OLA reactions were carried out on a universal SMART-amplified cDNA template (materials and methods).
In our experiment, we detected significant DAE among the panel of 16 paternally transmitted tester alleles for 17/18 genes, although two genes failed to remain significant when discordance between multiple assays was considered (see below). We first used a linear mixed-effects model to estimate the significance of the tester allele as a predictor of observed AE. This model was applied after grouping samples according to their maternal “reference” allele because the heterozygous pairing of the same reference with different informative testers serves as a normalizing factor. At first we judged that a gene was subject to DAE if there was a significant difference in relative AE in either of the two groups of informative testers for at least one SNP assay. By these initial criteria, 34/39 Bonferroni-corrected SNP assays showed DAE. The significant assays comprised all 18 genes with at least one assayable SNP except CG9012, for which 0/3 SNPs showed significant DAE.
Relative AE estimates and confidence intervals (example in Figure 1e) and the estimated ranges of the fold differences observed (when applicable) are provided in supplemental Figure 2. For assays that showed significant DAE and for which calibration samples were available, our estimates of the expression fold difference between extreme alleles were restricted to <2-fold for seven genes, while some alleles differed by 2.01- to 3.55-fold in assays of the four remaining calibrated genes. For the SNP assays with calibration curves, the largest fold difference in AE observed (i.e., <4-fold) is much smaller than the range over which fold differences appear to be linear (10-fold).
For genes with multiple SNP assays interrogating AE, examining all the data in concert for that gene leads to a more conservative measure of DAE than simply assessing DAE on a SNP-by-SNP basis. We use linear models to combine AE estimates across SNP assays and reference crosses for the 10 multiple-SNP genes; by this approach, we detected significant DAE in 7 genes (Figure 2). Such combined estimates of AE greatly reduce the danger of concluding the existence of DAE due to an assay-specific artifact. In this regard, two (CG2913 and CG11674) of the 10 genes with multiple assays showed incongruent results across SNPs, a phenomenon that has been previously reported (Pastinen et al. 2005; Campbell et al. 2008). Whether the confounding factors are biological (e.g., alternative splicing) or technical is unclear, but in such cases at least one SNP assay must be influenced by factors other than canonical cis-regulatory variation. In the combined analyses, one of these genes, CG2913, as well as CG1372 did not show significant overall DAE despite having had highly significant tester effects for 5/5 and 1/2 assayed SNPs, respectively. Moreover, CG2913 clearly illustrated the value of multiple assays: when heterozygous with the SS reference allele, we estimate that the tester allele F was 3.35-fold more abundant than allele B in SNP assay f16, 2.48-fold less abundant in SNP assay f10, and showed no significant difference in SNP assay f06 (see supplemental Figure 2). Overall, we detected high rates of DAE in both SNP-level and gene-level analyses. Because the gene-level analyses are more robust against assay artifacts, our further efforts to elucidate the number and frequency of regulatory alleles were performed solely on the 10 multiple-assay genes (Figure 2).
Figure 2.—

A permutation-based heuristic assigns testers to allelic classes according to estimates of AE combined across SNP assays. Combined estimates of AE for the 10 genes with at least two working SNP assays are shown, with strains ordered consistently across plots (labeled below CG11129 and CG11674). For each gene, bar plots are color coded according to the allelic classes identified by the heuristic. Error lines indicate 95% confidence intervals for relative AE. The table summarizes the data for all genes with at least one working assay: Assays, assays attempted (assays showing significant DAE); Combined P, the P-value for a significant difference among testers in the linear model fitted to all relevant assays AE estimates; n, number of observations per tester, as a harmonic mean; k, number of allelic classes identified by the permutation-testing heuristic; Membership, number of testers assigned to each allelic class.
Inspection of the relative AE estimates of each gene (Figure 2) suggested that the tester alleles do not often cluster into visually obvious groupings. To estimate the number of allelic classes represented, we developed a permutation-based heuristic to systematically partition the tester alleles into statistically distinguishable ACs (materials and methods; supplemental materials). Applying our heuristic enabled us to determine the groups of tester strains that are likely to be different from each other, to the resolution limits imposed by the number of observations and the magnitudes of underlying allelic differences relative to technical precision. Our analysis indicated that the tester panel was composed of at least three unique alleles (k > 2) for four genes and was at least biallelic (k = 2) for three genes. As expected, the heuristic did not support any division of the panel for the three genes in which we detected no significant difference among tester genotypes. We also challenged the heuristic with simulated data sets to verify the procedure's power and accuracy (supplemental materials). The simulations showed that we overestimated the true number of ACs by one class ∼4.5% of the time and by two classes ∼0.5% of the time. They also showed that the heuristic's power to increment up to the correct number of ACs decreases drastically as the true number of alleles increases. For example, in simulations with only four true ACs and a highly penetrant AE phenotype, we underestimated the true number of alleles ∼50% of the time. Underestimates of the true number of ACs are even more common in simulations of less penetrant AE phenotypes because a consequence of weakly replicated and/or noisy data is a failure to subdivide an AC that is actually composed of two or more alleles. Nonetheless, applying our heuristic to the 10 multi-assay genes gave us preliminary insight into the population genetics architecture of each gene's AE diversity.
Under neutral dynamics, population genetics theory offers predictions for the number of unique alleles (k) in a sample of n chromosomes as a function of the mutational target size (c.f. Ewens 1972 for a nonrecombining region), leading us to investigate the number of consequential regulatory sites consistent with our data. For each unique set of mutation and recombination parameters, we performed 1000 coalescent simulations and calculated the average k (per 16 samples), considering only the mutations allowed to occur at a prescribed number of regulatory sites (Figure 3). To compare simulation results to our data, we bootstrapped our 10 multiple-SNP estimates of k to establish the 10% lower and 90% upper bounds on 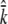 of 1.8 and 2.8 (of 16), respectively. At these bounds (Figure 3, boxed area), our simulations suggest that the number of sites at which individual mutations will result in selectively neutral yet observable cis-regulatory differences is 9–23 or 7–15 sites (under zero or high recombination, respectively).
of 1.8 and 2.8 (of 16), respectively. At these bounds (Figure 3, boxed area), our simulations suggest that the number of sites at which individual mutations will result in selectively neutral yet observable cis-regulatory differences is 9–23 or 7–15 sites (under zero or high recombination, respectively).
Figure 3.—

The effect of recombination on the number of haplotype classes in a 16-chromosome sample. Dashed lines indicate the mean number of unique haplotypes (conservatively assuming each class is phenotypically distinguishable) calculated from 1000 simulations of i = 2, 3, 4 … 26 mutable sites; corresponding solid lines indicate splines fit to these means. The rectangle indicates the 80% confidence interval calculated from bootstrapping the number of haplotype classes observed in our original 10 genes.
DISCUSSION
Our experiments profile cis-regulatory variation in a panel of 16 genotypes, and the structure of our work emphasizes strong biological and technical replication of samples and multiple SNPs within genes. This experimental structure enabled us to regularly detect variation, particularly effects as subtle as 1.14-fold, and to examine the concordance of different SNPs that should function as proxies for the same gene's expression. We often, but not always, observed concordance between AE assays. As occasional discrepancies have also been observed in different organisms and assay technologies (Pastinen et al. 2005; Campbell et al. 2008), we suggest that, in general, AE data from single SNPs may be prone to misinterpretation and ought to be complemented with additional markers. It should also be noted that experiments on “natural” heterozygotes will face difficulties because the set of informative samples will differ between any two such SNPs. This highlights an advantage to the crossing employed in our experiment: as long as SNPs are chosen such that the two reference strains always differ in genotype, all the tester genotypes are informative at all assays. Moreover, the ability to systematically compare cis-regulatory properties within a large panel will add an important hypothesis-testing utility to future AE studies. For example, one could construct the panel to ask whether the strains' phenotypic or geographic parameters covary with the AE of a candidate gene.
Using differential allelic expression as an indicator of cis-regulatory variation, in this study we find that a panel of 16 D. melanogaster alleles harbors cis-acting genetic diversity at a majority of surveyed genes. Although direct comparisons to other studies are confounded by a number of experimental variables, our observation of a large fraction of genes showing DAE is consistent with other claims in the literature (Yan et al. 2002; Bray et al. 2003; Lo et al. 2003; Pastinen et al. 2004; Pant et al. 2006; Campbell et al. 2008). In 4/10 high-confidence genes we detect more than two alleles, implying that either multiple biallelic sites or polyallelic features such as microsatellites affect cis-regulation. Previously, cis-regulatory variation affected by multiple sites has been documented for only a single human gene, and in that instance only two population alleles were observable owing to linked transmission as a haplotype (Tao et al. 2006).
Unique to our study is a rudimentary view of the frequencies of cis-regulatory haplotypes, a modest step toward understanding the population genetics architecture of standing cis-regulatory variation. Many outstanding questions exist for this field. How many sites, when mutated, affect cis-regulatory properties? What fraction of such sites is polymorphic in populations? What is the distribution of allelic effects and the selection coefficients for cis-regulatory variants? Unfortunately, direct answers to such questions may require large, potentially unwieldy experiments. Our study is suitable for asking whether patterns of detected cis-regulatory variation are consistent with large-effect mutations that are strongly deleterious (i.e., |2Ns| > 100), which would likely be unique in a sample of 16 chromosomes. Under such a model (not dissimilar to classical Mendelian phenotypes such as Drosophila eye and body color mutants), mutations that change cis-regulation would be detected at low frequency, and many genes would harbor no variation. In our data, we examined the frequency of different ACs in search of this pattern. Of our 10 multiple-assay, high-confidence genes, three (CG1372, CG2913, CG9012) show no significant evidence of variation and three (CG4070, CG6398, CG8408) have variation in the form of one very common and one very rare AC. These genes are consistent with a large-effect mutation-selection balance model, but four other genes (CG2221, CG2247, CG11129, CG11674) do not fit this pattern.
The key indictment of the deleterious, large-effect model observed in the four other genes is the apparent presence of more than one intermediate-frequency AC, leaving several plausible interpretations for the existence of this pattern. One interpretation is that AC assignments are essentially correct (i.e., to assume that all strains in a given AC really do have the same cis-regulatory allele). In this case the detection of multiple intermediate frequency classes suggests that no allele has a large selection coefficient. Or it is formally possible that detected classes represent alternative states subject to balancing selection or reflect local adaptation (progenitors of the tester strains were collected on several continents). But the claim of pervasive balancing selection or local adaptation for cis-regulation throughout the genome seems incredible, and the latter would predict that AC groupings would be highly similar between genes, which is not observed (Figure 2).
A second broad interpretation of multiple intermediate-frequency ACs must also be considered; each AC is underclassified and actually reflects a functional grouping of haplotypes whose similar regulatory properties belie genetic differences. A number of lines of reasoning support the “underclassified” interpretation. First, as noted by Ewens (1972), even under neutral dynamics it is unusual for several haplotypes to segregate at intermediate frequency. Second, testing of the heuristic showed that it is indeed likely to undercount the number of true alleles, presumably by failing to distinguish between strains of functionally similar, nonidentical genotypes. Finally, a greater number of undetected unique ACs implies a moderately greater number of consequential segregating sites.
Such a model is appealingly unremarkable, plausible, and explanatory. Although selection continuously acts to reduce moderately deleterious variation, a large number of segregating non-neutral sites can explain the same sampling properties as a small number of neutral sites. And if the architecture of cis-regulatory diversity is one in which a moderate or a large number of sites can harbor alleles of small average effect, neither selective neutrality nor balancing selection nor local adaptation needs to be invoked to explain our results. This model is in line with a recent analysis showing that 40–70% of the D. melanogaster noncoding genome is evolving under positive or negative selection (Andolfatto 2005). We must also consider that a portion of the standing variation may be due to sites where allelic effects are sufficiently subtle to escape selection altogether. Such variation has been observed in the human ENCODE project, which estimated that ∼50% of biochemically functional noncoding sequence is not evolutionarily constrained (Encode Consortium 2007). Our experiments have shown that cis-regulatory diversity is not wholly caused by deleterious alleles of large effect, and it appears that regulatory haplotypes represented in the panel are not typically subject to strong selection. However, until the actual AC assignments can be made with less ambiguity (perhaps with a larger set of strains) or until the regulatory haplotypes can be otherwise distinguished, it will remain difficult to demonstrate the superiority of one of the remaining explanations.
Comparing our data to coalescent simulations offers a conservative view of the cis-regulatory mutational target size in terms of the number of potentially biallelic sites consistent with the data. Given the limits of our experiment, it is reasonable to question the value of the simulations, which assume selective neutrality. We see two points of utility to this exercise. First, the simulations show that, under neutrality, recombination has a small effect (∼20%) on the lower limit and a slightly higher impact (∼35%) on the upper limit of our target size estimate. Diminishing returns were observed for additional recombination, as ρ = 1000 and ρ = 2000 yield essentially identical estimates of the mutational target size (data not shown). As the actual genetic distance across the relevant cis-regulatory region is essentially unknown for most genes, a sense of the magnitude of potential impacts from recombination will be critical for future work on mutational targets. Second, almost all the potential biases of inputs to our analysis would result in underestimation of the mutational target size, so our conclusions provide a conservative baseline. These biases include practical limits on our ability to assign strains to ACs (the real number of alleles sampled in the panel may be higher) compared to inerrant counting of alleles in the simulations, the use of an estimate of θ from the highly polymorphic “Zimbabwe” D. melanogaster (the tester strains are the less-diverse “Cosmopolitan”), and the assumption of neutrality (to assume that mutations at all cis-regulatory nucleotides have neutral selection coefficients is certainly erroneous). However, it is reasonable to expect that much of this selection is negative and is likely to reduce the diversity of the pool of unique haplotypes in the population, thereby decreasing the average number of observed alleles and contributing to the underestimation of the target size. In a pragmatic sense, it is a simple exercise to calculate θ on the basis of the number of observed alleles if one cares to ignore recombination and assume neutrality (Ewens 1972). Given the complexity of describing the mutational target more realistically, the effective neutral mutational target size may be a useful descriptor for each gene's functional diversity.
Interpretation of our data depends on the verisimilitude of a reasonable but only modestly examined relationship between AE and cis-regulatory variation. Although each is limited in its scope, a particular subset of our experiments and a previously published study suggest that observed variation in AE is not influenced by trans-acting effects or cis × trans interactions. An idealized implementation of AE surveys would contrast the cis-regulatory properties of a given gene's alleles in a standard genetic background, but this is highly impractical. In our experiment, the individuals that shared a unique cis-regulatory heterozygous genotype also shared a private paternal haploid genome. If it affects AE quantification, non-cis-acting variation in each background would indeed be confounded with cis-regulatory diversity. However, we find no evidence to support this particular concern. Our data include six assays (five genes) in which the same subset of testers is interrogated in the different haploid backgrounds of the two reference strains. In each of these assays, the reference × tester interaction term is not significant (at P < 0.05) in a linear mixed-effects model predicting AE (data not shown), suggesting that the ratio of differentially expressed alleles may be invariant with respect to genetic background or at least that cis × trans effects are subtle compared to cis effects. This supports other work that also failed to document AE × trans interactions, even when trans-regulatory effects on total gene expression are significant (Wittkopp et al. 2008a). Combining the two studies, the number of genes (seven) and particularly the number of genetic backgrounds compared (two per investigation) are both extremely limited and explicit study is warranted. But there is currently no empirical evidence suggesting that cis–trans interactions are likely to complicate the interpretation of AE as a proxy of cis-regulatory variation.
Unfortunately, the slow progress that has characterized attempts to dissect standing phenotypic variation (for example, Robin et al. 2002; Macdonald and Long 2004; Gruber et al. 2007) may well apply to cis-regulatory phenotypes. Chief among these concerns is difficulty defining the set of relevant, functional mutations contributing to intraspecific organismal variation, and our observation that cis-regulatory variation is often characterized by several function alleles suggests that this may be an equally formidable challenge with cis-regulatory variation. In general, it also remains to be seen whether standing variation is the general source of evolutionary novelty at all. A hopeful possibility is that the presumed localized nature of cis-regulatory variation may yet propel investigations toward greater success in addressing this fundamental question.
Acknowledgments
We thank S. J. Macdonald for inbred fly strains and for technical advice on OLA typing and analysis. P. J. Wittkopp, B. S. Gaut, and K. J. Hertel offered valuable comments on the project, and we thank two anonymous readers for their detailed reviews of the manuscript. This work was supported by National Science Foundation grant DEB-0614429 to A.D.L., by National Institutes of Health service award 5 T15 LM007443 to J.D.G., and by a fellowship from the Achievement Rewards for College Scientists Foundation of Orange County to J.D.G.
References
- Andolfatto, P., 2005. Adaptive evolution of non-coding DNA in Drosophila. Nature 437 1149–1152. [DOI] [PubMed] [Google Scholar]
- Andolfatto, P., 2007. Hitchhiking effects of recurrent beneficial amino acid substitutions in the Drosophila melanogaster genome. Genome Res. 17 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray, N. J., and M. C. O'Donovan, 2006. Investigating cis-acting regulatory variation using assays of relative allelic expression. Psychiatr. Genet. 16 173–177. [DOI] [PubMed] [Google Scholar]
- Bray, N., P. Buckland, M. Owen and M. O'Donovan, 2003. Cis-acting variation in the expression of a high proportion of genes in human brain. Hum. Genet. 113 149–153. [DOI] [PubMed] [Google Scholar]
- Brem, R., G. Yvert, R. Clinton and L. Kruglyak, 2002. Genetic dissection of transcriptional regulation in budding yeast. Science 296 752–755. [DOI] [PubMed] [Google Scholar]
- Campbell, C., A. Kirby, J. Nemesh, M. Daly and J. Hirschhorn, 2008. A survey of allelic imbalance in F1 mice. Genome Res. 18 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowles, C., J. Hirschhorn, D. Altshuler and E. Lander, 2002. Detection of regulatory variation in mouse genes. Nat. Genet. 32 432–437. [DOI] [PubMed] [Google Scholar]
- de Meaux, J., U. Goebel, A. Pop and T. Mitchell-Olds, 2005. Allele-specific assay reveals functional variation in the chalcone synthase promoter of Arabidopsis thaliana that is compatible with neutral evolution. Plant Cell 17 676–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Meaux, J., A. Pop and T. Mitchell-Olds, 2006. Cis-regulatory evolution of chalcone-synthase expression in the genus Arabidopsis. Genetics 174 2181–2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denver, D., K. Morris, T. Streelman, S. Kim, M. Lynch et al., 2005. The transcriptional consequences of mutation and natural selection in Caenorhabditis elegans. Nat. Genet. 37 544–548. [DOI] [PubMed] [Google Scholar]
- ENCODE Project Consortium, E. Birney, J. A. Stamatoyannopoulos, A. Dutta, R. Guigó et al., 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature 447 799–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewens, W. J., 1972. The sampling theory of selectively neutral alleles. Theor. Popul. Biol. 3 87–112. [DOI] [PubMed] [Google Scholar]
- Genissel, A., L. McIntyre, M. Wayne and S. Nuzhdin, 2008. Cis and trans regulatory effects contribute to natural variation in transcriptome of Drosophila melanogaster. Mol. Biol. Evol. 25 101–110. [DOI] [PubMed] [Google Scholar]
- Gruber, J. D., A. Genissel, S. J. Macdonald and A. D. Long, 2007. How repeatable are associations between polymorphisms in achaete-scute and bristle number variation in Drosophila? Genetics 175 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddrill, P. R., K. R. Thornton, B. Charlesworth and P. Andolfatto, 2005. Multilocus patterns of nucleotide variability and the demographic and selection history of Drosophila melanogaster populations. Genome Res. 15 790–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekstra, H., and J. Coyne, 2007. The locus of evolution: Evo-Devo and the genetics of adaptation. Evolution 61 995–1016. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 2002. Generating samples under a Wright-Fisher neutral model of genetic variation. Bioinformatics 18 337–338. [DOI] [PubMed] [Google Scholar]
- Lo, S., Z. Wang, Y. Hu, H. Yang, S. Gere et al., 2003. Allelic variation in gene expression is common in the human genome. Genome Res. 13 1855–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, S. J., and A. D. Long, 2004. A potential regulatory polymorphism upstream of hairy is not associated with bristle number variation in wild-caught Drosophila. Genetics 167 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald, S., T. Pastinen, A. Genissel, T. Cornforth and A. Long, 2005. A low-cost open-source SNP genotyping platform for association mapping applications. Genome Biol. 6 R105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pant, K., H. Tao, E. Beilharz, D. Ballinger, D. Cox et al., 2006. Analysis of allelic differential expression in human white blood cells. Genome Res. 16 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastinen, T., R. Sladek, S. Gurd, A. A. Sammak, B. Ge et al., 2004. A survey of genetic and epigenetic variation affecting human gene expression. Physiol. Genomics 16 184–193. [DOI] [PubMed] [Google Scholar]
- Pastinen, T., B. Ge, S. Gurd, T. Gaudin, C. Dore et al., 2005. Mapping common regulatory variants to human haplotypes. Hum. Mol. Genet. 14 3963–3971. [DOI] [PubMed] [Google Scholar]
- Pletcher, S., S. Macdonald, R. Marguerie, U. Certa, S. Stearns et al., 2002. Genome-wide transcript profiles in aging and calorically restricted Drosophila melanogaster. Curr. Biol. 12 712–723. [DOI] [PubMed] [Google Scholar]
- Rifkin, S., D. Houle, J. Kim and K. White, 2005. A mutation accumulation assay reveals a broad capacity for rapid evolution of gene expression. Nature 438 220–223. [DOI] [PubMed] [Google Scholar]
- Robin, C., R. F. Lyman, A. D. Long, C. H. Langley and T. F. C. Mackay, 2002. hairy: a quantitative trait locus for Drosophila sensory bristle number. Genetics 162 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozen, S., and H. Skaletsky, 2000. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 132 365–386. [DOI] [PubMed] [Google Scholar]
- Sokal, R., and J. Rohlf, 1995. Biometry. W. H. Freeman, San Francisco.
- Stern, D. L., and V. Orgogozo, 2008. The loci of evolution: How predictable is genetic evolution? Evolution 62 2155–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao, H., D. Cox and K. Frazer, 2006. Allele-specific KRT1 expression is a complex trait. PLoS Genet. 2 e93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp, P. J., 2005. Genomic sources of regulatory variation in cis and in trans. Cell. Mol. Life Sci. 62 1779–1783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp, P., B. Haerum and A. Clark, 2004. Evolutionary changes in cis and trans gene regulation. Nature 430 85–88. [DOI] [PubMed] [Google Scholar]
- Wittkopp, P. J., B. K. Haerum and A. G. Clark, 2008. a Independent effects of cis- and trans-regulatory variation on gene expression in Drosophila melanogaster. Genetics 178 1831–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkopp, P. J., B. K. Haerum and A. G. Clark, 2008. b Regulatory changes underlying expression differences within and between Drosophila species. Nat. Genet. 40 346–350. [DOI] [PubMed] [Google Scholar]
- Wray, G. A., 2007. The evolutionary significance of cis-regulatory mutations. Nat. Rev. Genet. 8 206–216. [DOI] [PubMed] [Google Scholar]
- Yan, H., W. Yuan, V. E. Velculescu, B. Vogelstein and K. W. Kinzler, 2002. Allelic variation in human gene expression. Science 297 1143. [DOI] [PubMed] [Google Scholar]
- Zhuang, Y., and K. Adams, 2007. Extensive allelic variation in gene expression in populus F1 hybrids. Genetics 177 1987–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]