

Abstract
After migrant chromosomes enter a population, they are progressively sliced into smaller pieces by recombination. Therefore, the length distribution of “migrant tracts” (chromosome segments with recent migrant ancestry) contains information about historical patterns of migration. Here we introduce a theoretical framework describing the migrant tract length distribution and propose a likelihood inference method to test demographic hypotheses and estimate parameters related to a historical change in migration rate. Applying this method to data from the hybridizing subspecies Mus musculus domesticus and M. m. musculus, we find evidence for an increase in the rate of hybridization. Our findings could indicate an evolutionary trajectory toward fusion rather than speciation in these taxa.
AN accurate understanding of population history is essential for such diverse applications as the search for recent signatures of positive selection in population genetic data (e.g., Jensen et al. 2005), the study of admixed human populations to identify disease-associated genetic variants (e.g., Montana and Pritchard 2004; Patterson et al. 2004), and the definition of management units in conservation (Pearse and Crandall 2004). Patterns of genetic variation contain information about past changes in population size (e.g., Cornuet and Luikart 1996; Marth et al. 2004), the timing of population splitting events (e.g., Nielsen and Wakeley 2001), and levels of migration between populations (e.g., Beerli and Felsenstein 2001).
Since the advent of molecular markers, researchers have sought to gauge the genetic differentiation of populations and to draw conclusions about the level of migration between them. Wright's FST (Wright 1952) has served as the classic metric of population differentiation, and, under ideal conditions, the population migration rate can be estimated by 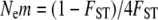 , where Ne is the effective population size, m is the per-generation probability of being a migrant, and Nem is thus equal to the number of migrants exchanged each generation. However, this relationship relies on several assumptions that may not be valid for most natural populations (reviewed in Whitlock and McCauley 1999), including that of a constant rate of migration. A given FST value between two populations could be produced by a constant level of migration over a long period of time, or by genetic drift following a relatively recent split between the two populations, or by recent admixture between historically isolated populations, or by any number of more complex scenarios. The isolation-migration (IM) inference framework (e.g., Nielsen and Wakeley 2001; Hey 2005) offers a way to differentiate ongoing migration between populations from lineage sorting in isolated populations, while estimating relevant demographic parameters.
, where Ne is the effective population size, m is the per-generation probability of being a migrant, and Nem is thus equal to the number of migrants exchanged each generation. However, this relationship relies on several assumptions that may not be valid for most natural populations (reviewed in Whitlock and McCauley 1999), including that of a constant rate of migration. A given FST value between two populations could be produced by a constant level of migration over a long period of time, or by genetic drift following a relatively recent split between the two populations, or by recent admixture between historically isolated populations, or by any number of more complex scenarios. The isolation-migration (IM) inference framework (e.g., Nielsen and Wakeley 2001; Hey 2005) offers a way to differentiate ongoing migration between populations from lineage sorting in isolated populations, while estimating relevant demographic parameters.
As in the case of IM, most population genetic methods that estimate demographic parameters assume that all sites or markers under study are either completely linked (no recombination) or completely unlinked (free recombination) (although see Becquet and Przeworski 2007). And correspondingly, most population genetic data have been collected with these criteria in mind. Assuming either full linkage among sites or else independence among loci can greatly simplify the task of modeling the histories of molecular markers. However, the bulk of the genome in most organisms consists of DNA that is subject to recombination, and, furthermore, the pattern of recombination events within a sample of chromosomes may hold valuable information concerning population history. For example, we know that haplotype statistics (Depaulis et al. 2003) and linkage disequilibrium (Wall et al. 2002) across short loci are quite sensitive to the effects of population bottlenecks. The recent availability of genome-scale polymorphism data should facilitate investigation of longer-range linkage patterns, which may shed new light on the recent histories of populations.
Patterns of diversity at partially linked markers may be especially informative concerning the historical pattern of migration between populations. Once a migrant chromosome enters a new population, recombination will break it down into progressively shorter segments. The lengths of these “migrant tracts”—or admixture “chunks” (Falush et al. 2003)—therefore contain information about how long ago migration occurred. This logic has been utilized to estimate the timing of recent admixture events (e.g., Patterson et al. 2004; Hoggart et al. 2004; Koopman et al. 2007), but its applicability should extend beyond such cases. We suggest that migrant tract lengths are expected to have a certain equilibrium distribution under a constant migration rate model. An excess of long migrant tracts would indicate a recent increase in migration rate, while the opposite pattern would suggest recently reduced gene flow. We use theoretical predictions and simulations to explore the migrant tract length distribution under a variety of demographic scenarios, and we assess the potential of this approach for inferring demographic parameters related to migration rate changes.
MODELS AND METHODS
Constant migration rate:
A large set of different population genetic models converges to the same coalescence process as the population size becomes large (N → ∞; Kingman 1982a,b). In two-island models (Wright 1931), an ancestral process arises (e.g., Hudson 1983), which can be described by a Markov pure jump process {X(t), t ≥ 0} with state space on {0, … , n1} × {0, … , n2}\(0, 0), initial state (n1, n2), absorbing states (0, 1), (1, 0), and transition rates
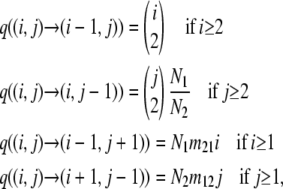 |
(1) |
where n1 and n2 are the sample sizes from populations 1 and 2, respectively, and N1 and N2 are the population sizes. Migration occurs from population 2 to 1, and from 1 to 2, at rates m21 and m12, respectively. Time is measured in units of N1 generations, and Njmij can be interpreted as the proportion of individuals in population j that are replaced with individuals from population i in each generation.
Consider the ancestry of a single lineage from population 1. The waiting time in number of generations until the last migration event for this lineage is exponentially distributed with mean 1/m (letting m = m21 here and in the following to simplify the notation). We now introduce recombination and measure distances in the genome as genetic distances. By using genetic distances, we may assume that recombination in each generation occurs according to a Poisson process with rate 1 along the chromosome. We assume that migrant tracts do not recombine together, we disallow back-migration events (i.e., assume m12 = 0), and we ignore the effect of the ends of the chromosome (but later we evaluate violations of these assumptions). Then, after t generations, the distribution of tracts lengths follows an exponential distribution with mean 1/t:
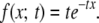 |
(2) |
Because we can reliably infer migrant tracts only over a certain length, we are interested in the distribution of tracts and the expected proportion of a chromosome in tracts larger than a certain threshold, C. The proportion of a migrant chromosome from time t that is in tracts on a size >C, pC, can be found from the convolution of two independent and identically distributed exponential random variables with parameter t:
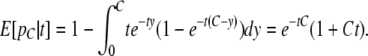 |
(3) |
These two variables represent, respectively, the distance to the left and right on the chromosome from the point of inspection to the nearest recombination event. Integrating over t, we find
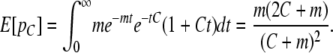 |
(4) |
The expected number of fragments in the population of a migrant chromosome of length L is
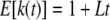 |
(5) |
after t generations; i.e., the contribution of migrant tracts from generation t to the population is proportional to me−mt(1 + Lt). Again ignoring recombination among migrant tracts, the density of tract lengths will be formed as a mixture distribution of tracts from different times,
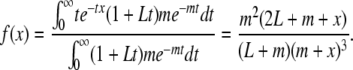 |
(6) |
The conditional tract length distribution of tracts of a length larger than C is then
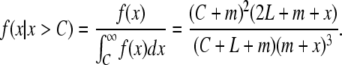 |
(7) |
These expressions do allow for genetic drift. However, they assume that recombination events between descendants of the same or different migration events contribute to the breakdown of chromosomes into smaller distinguishable tracts. In practice, we cannot distinguish between nonrecombinants and recombinants between copies of the same allele. The approximations we derive here are, therefore, expected to break down when t becomes so large compared to N1 that migrant alleles may have drifted to appreciably high allele frequencies, thereby allowing for recombination between migrant tracts. However, this is not a fundamental problem as we can infer only relatively large tracts that, with high probability, are descendants of recent migrants. If C is sufficiently large, it is highly probable that only fragments for which t is small have been sampled. The chance that a migrant allele of size >C has drifted to high frequencies is small if C ≫ 1/N (since recombination will break down tracts below this threshold before drift can substantially elevate them in frequency). Problems identifying recombinants between migrant alleles are, therefore, avoidable if C is sufficiently large. For the same reason, for large C, inferences based on Equation 7 should be relatively robust to violations of the assumption of no back migration; i.e., m12 = 0.
Changes in the migration rate:
We now extend these results to the case where there has been a discrete change in the rate of migration. Again, we consider only migration into population 1, and assume that the current migration rate is m1, and that it before T generations ago was m2. We then have
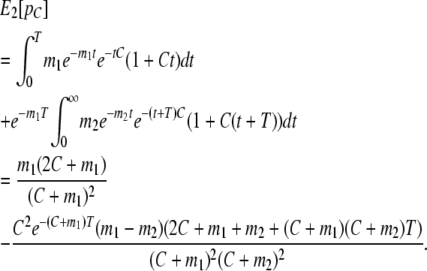 |
(8) |
Likewise, setting
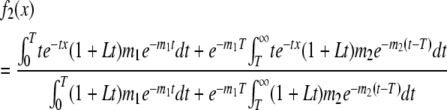 |
(9) |
and conditioning as in Equation 7, we find
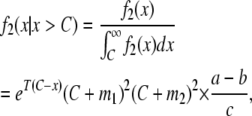 |
(10) |
where
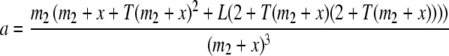 |
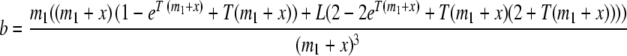 |
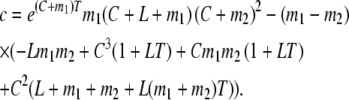 |
Inference:
We wish to estimate the parameters, m1, m2, and T from an observed tract length distribution. As only large tracts can be easily identified, we have to base inferences on Equations 8 and 10 and not on Equation 9. We define a composite-likelihood function by taking the product of Equation 10 among all tracts in the data above a prespecified threshold (C). The reason why we consider this a composite-likelihood function and not a true-likelihood function is that the same tract can be counted twice. However, for real data with C large, this will rarely happen and the estimation function is essentially a true-likelihood function.
Equation 10 contains only very little information about the overall amount of population subdivision, because we look only at the relative abundance of tracts with length greater than C. However, much of the information regarding the overall level of population subdivision is captured by our estimate of pC (Equation 8). We therefore do a constrained optimization of the likelihood function subject to the constraint
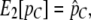 |
(11) |
where 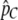 is the observed proportion of the genome in tracts larger than C. Specifically, we rearrange Equation 8 to express T as a function of C, m1, m2, and
is the observed proportion of the genome in tracts larger than C. Specifically, we rearrange Equation 8 to express T as a function of C, m1, m2, and 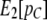 , and we then substitute
, and we then substitute 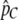 for
for 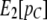 . We then perform a two-dimensional optimization for m1 and m2 while constraining T to take on the value given by the aforementioned equation. This approach reduces the number of parameters from Equation 10 to be estimated (from three to two) and adds information concerning the total proportion of migrant DNA observed (from
. We then perform a two-dimensional optimization for m1 and m2 while constraining T to take on the value given by the aforementioned equation. This approach reduces the number of parameters from Equation 10 to be estimated (from three to two) and adds information concerning the total proportion of migrant DNA observed (from 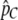 ). Constrained models with one of the two migration rates set to zero are evaluated similarly, via a one-dimensional optimization of the other migration rate. For the constant migration rate model, m can be estimated simply by setting m1 = m2 in Equation 8, and thus using
). Constrained models with one of the two migration rates set to zero are evaluated similarly, via a one-dimensional optimization of the other migration rate. For the constant migration rate model, m can be estimated simply by setting m1 = m2 in Equation 8, and thus using 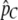 to solve for m.
to solve for m.
Comparison of likelihood scores from different models allows the testing of demographic hypotheses. Test 1 compares the maximum-log-likelihood score from the migration rate change model (with m1 and m2 allowed to vary) against the null hypothesis of a single, constant migration rate (with m inferred from 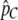 ). Test 2 compares the maximum-log-likelihood score of the migration rate change model against a model where either (A) m1 is constrained to be zero or (B) m2 is constrained to be zero. Generally, test 2A is performed when m1 < m2, and test 2B is performed if m1 > m2. Because the distributions of likelihood ratios are not well modeled by standard asymptotic theory for any of these tests, critical values are obtained using data simulated under the null hypothesis. For computational reasons, we obtain critical values using Nem = 0.1 for test 1, the true values of m2 and T for test 2A, and true m1 and T for test 2B (rather than using the estimated parameter values for each simulated replicate). In the analysis of empirical data, we use the estimated null model parameter values instead.
). Test 2 compares the maximum-log-likelihood score of the migration rate change model against a model where either (A) m1 is constrained to be zero or (B) m2 is constrained to be zero. Generally, test 2A is performed when m1 < m2, and test 2B is performed if m1 > m2. Because the distributions of likelihood ratios are not well modeled by standard asymptotic theory for any of these tests, critical values are obtained using data simulated under the null hypothesis. For computational reasons, we obtain critical values using Nem = 0.1 for test 1, the true values of m2 and T for test 2A, and true m1 and T for test 2B (rather than using the estimated parameter values for each simulated replicate). In the analysis of empirical data, we use the estimated null model parameter values instead.
Simulation:
A forward simulation program was written to allow the generation of migrant tract data. This program simulates each chromosome present in two populations and models the processes of genetic drift, migration, and recombination under a Wright–Fisher model (Fisher 1930; Wright 1931). It does not generate polymorphism data; instead it directly monitors migrant tract status along chromosomes. When an individual migrates, all previously nonmigrant chromosome sections become migrant tracts, and any previous migrant tracts become nonmigrant. Tracts are “forgotten” when recombination breaks them down to a size below the threshold length. The program initializes with no migrant tracts present, but goes through a “burn-in” period with migration at rate m2. For the analyses shown here, using a threshold tract length of C = 0.5 cM, the burn-in time was 2000 generations (results and theory indicated this was more than enough time to reach an equilibrium migrant tract length distribution) and Ne was 10,000. At the end of the burn-in, the migration rate switches to m1 and the program records all migrant tracts present in each population at a series of time points (T) after this change. An extension to this program allows migrant tracts to be sampled from a specific number of individuals. In testing the performance of the likelihood method, we simulated “genomes” containing 35 chromosomes, each 100 cM in length (3500 cM is close to the genetic map size of humans and many other mammals; Kong et al. 2002), and we sampled 100 haploid individuals from one population.
Application to empirical data:
The likelihood method was applied to genomewide single-nucleotide polymorphism (SNP) data from two hybridizing subspecies of the house mouse, Mus musculus domesticus and M. m. musculus. These data were produced by the Wellcome Trust Center for Human Genetics and are available at http://www.well.ox.ac.uk/mouse/INBREDS/. The strains examined here consist of seven from M. m. domesticus and eight from M. m. musculus, with varying geographic origins (see Harr 2006 for a summary). The data come from wild-derived, inbred mouse strains and are effectively haploid. The few apparently heterozygous sites were recoded as missing data, and invariant SNPs were removed. Since the X chromosome is expected to have a different history, all of the 9935 SNPs analyzed here were autosomal. The vast majority of these SNPs have inferred genetic map positions (Jensen-Seaman et al. 2004), and all analyses were done in terms of genetic distance, rather than physical position. These SNPs had been ascertained in laboratory lines of mixed origin and could be biased in terms of diversity levels and allele frequencies (Boursot and Belkhir 2006), but we do not expect a particular bias for the inference and analysis of migrant tracts.
In general, our likelihood inference method allows the user to decide how migrant tracts should be defined. The sample sizes of the mouse SNP data set seemed too small for published methods for identifying ancestry along recombining chromosomes (e.g., Falush et al. 2003). However, the task of tract identification is simplified by the high level of genetic differentiation between the two subspecies, which diverged perhaps 1 million generations ago and show very high levels of genetic differentiation (Baines and Harr 2007; Salcedo et al. 2007). We were therefore able to use a very simple set of criteria for defining migrant tracts in these data. Given the small sample sizes, an individual's SNP allele was deemed to provide evidence for a migrant tract only if it was otherwise absent from the individual's subspecies, but present in the in other subspecies (we call this a “positive SNP”). If an individual's SNP allele is otherwise present in both subspecies, this is a “neutral SNP” neither favoring nor opposing migrant tract status. And if an individual's SNP allele is not present in the other subspecies, it is taken as evidence against migrant tract status (a “negative SNP”). Migrant tracts consisted of two or more positive SNPs with no negative SNPs between them. The minimum tract length was considered to be the genetic distance spanning only the positive SNPs at each end of the tract. The maximum tract length included all sites up to the first negative SNPs flanking the tract.
Given the minimum and maximum length of a migrant tract, we were interested in estimating how far beyond the positive SNPs this tract is expected to extend. To do this we assume that the length of a tract is exponentially distributed with parameter λ. If marker Mi is in a tract, the probability that the next marker, Mi+1, is also in the same tract is 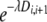 , where Di,i+1 is the genetic distance between markers Mi and Mi+1. A log-likelihood function for λ is then given by
, where Di,i+1 is the genetic distance between markers Mi and Mi+1. A log-likelihood function for λ is then given by
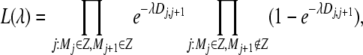 |
(12) |
where Z is the set of all markers in a migration tract. By entering the lengths of all SNP intervals (Di,i+1) where we remain in a migrant tract or leave one and then maximizing this function, we obtain a maximum-likelihood estimate of λ. Now the expected distance to add to a tract on the right side is
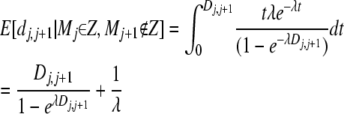 |
(13) |
and we similarly add
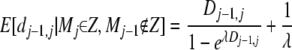 |
(14) |
to the left side.
Applying this method to the mouse SNP data, the resulting tract lengths were then used in the likelihood inference method described above. To ensure that undetected tracts did not lead to spurious rejection of the null model, migrant tracts from simulated data were subjected to the constraints of the mouse SNP data set. The probability that any given SNP allele is informative concerning migrant ancestry was estimated by replacing each SNP allele in one subspecies with each possible SNP allele from the other subspecies and monitoring the proportion of transplanted alleles that yielded positive evidence for migrant history under the criteria detailed above. Average SNP informativeness was estimated in this way for each subspecies separately. Tract lengths from constant migration rate simulations were randomly placed on the mouse SNP map, and each tract was detected only if two or more informative SNPs fell within it. This process was repeated until the number of tracts observed in the empirical data was matched.
RESULTS
Above, we described a theoretical framework for the distribution of migrant tract lengths and a forward whole-population simulation tool to generate migrant tract data. The simulation program enables several assumptions of the theory to be violated: by allowing back migration, recombinational joining of migrant tracts, and effects of the ends of chromosomes. In all cases examined, including those shown in Figure 1, simulated data closely matched theoretical predictions. Figure 1 depicts the migrant tract length distributions generated by a constant migration rate model and by admixture beginning 100, 200, or 300 generations ago. The contrasting migrant tract lengths generated by these histories suggested that such data could be informative for demographic inference. But Figure 1 is based on a large number of simulated replicates, and we were interested in testing whether individual data sets would contain enough information for demographic hypothesis testing and parameter inference.
Figure 1.—

The distribution of migrant tract lengths after the advent of admixture. Models where previously isolated populations begin exchanging migrants at rate Nem = 0.1 100, 200, or 300 generations ago are compared against the case in which populations exchange migrants at a constant rate Nem = 0.1 with no prior isolation (the single migration rate, “equilibrium” model). Depicted here is the relative abundance of migrant tracts for 0.01-cM histogram bins between 0.5 (the minimum/threshold tract length) and 5 cM. Also shown is the agreement between theoretical predictions (lines) and tracts from 1000 simulated replicates with Ne = 10,000 (shapes).
Large, genome-scale data sets were generated for population samples under various demographic histories, using the migrant tract simulation method described above. Genomes 3500 cM in size were generated for a sample size of 100, and a minimum tract length of 0.5 cM was used. Likelihood optimization was performed for each simulated data set under the migration rate change model, yielding estimates of m1, m2, and T. The highest log-likelihood value obtained for this model was compared against the log-likelihood score for the constant migration rate model, and the significance of likelihood ratios was assessed via comparison with data sets simulated under the constant rate model. Results are presented in Figure 2, A and B.
Figure 2.—

Power to test demographic hypotheses. Shown here first are tests comparing the migration rate change model to the null model of a constant migration rate, for histories involving decreasing (A) or increasing (B) migration rates. For histories involving decreasing migration rates, power to reject a model with m1 constrained to be zero is shown (C). For histories involving increasing migration rates, power to reject a model with m2 constrained to be zero is shown (D). Significance was gauged by comparing the difference in log-likelihood scores between models to data simulated under the null model. Each data set consisted of 100 simulated haploid genomes, and a threshold tract length of 0.5 cM was used.
The method was found to have high power to reject a constant rate model for a range of histories. The highest power often occurred within the first few hundred generations after a migration rate change—this is not surprising, as only tracts >0.5 cM are considered here, and recombination will typically break down migrant chromosomes to this size within ∼200 generations. In some cases, particularly for strong decreases in migration rate, the method's power lasted well beyond this expectation. Even for the most subtle migration rate changes considered (from Nem = 0.1 to Nem = 0.04 and vice versa), power was fairly high, particularly around the T = 200 to T = 500 time window.
For histories involving a migration rate decrease, a similar procedure was applied to test whether a model with no current migration (m1 = 0) could be rejected (test 2A). Here, power was often a bit lower than for test 1, but generally still quite high (Figure 2C). Conversely, for histories involving a migration rate increase, we tested whether a model with no migration before the rate change (m2 = 0) could be rejected (test 2B). Power for this test was high for very recent migration rate changes (i.e., 100–200 generations ago), but declined quickly from there (Figure 2D).
Accuracy of parameter estimation under the migration rate change model is shown in Figure 3. For a variety of demographic histories involving isolation, migration rate decreases, migration rate increases, and admixture, estimates of m1 and m2 were often quite precise. Although the method cannot always distinguish low migration rates from zero, higher migration rates of 1E−5 (Nem = 0.1) were estimated quite accurately, often with 95% confidence intervals extending only ∼30% above and below the true value. A similar degree of accuracy was observed for T, with confidence intervals spanning a factor of 2 or considerably less. Parameter estimates for migration rate changes beyond 500 generations ago typically became less precise (data not shown), which makes sense as these data sets become less informative, with few tracts >0.5 cM having arisen before the migration rate change.
Figure 3.—

Distribution of demographic parameter estimates. Results from the analysis of simulated migrant tract data are shown, including median estimates (diamonds) and 95% confidence intervals (the 2.5 and 97.5 percentiles of the distribution of estimates) for (A) m1, (B) m2, and (C) T. The order of parameter sets is the same in each panel (i.e., the far left estimates are for true values of m1 = 0, m2 = 1E−5, and T = 100).
Given the generally favorable performance of the likelihood inference method on simulated migrant tract data, we then sought to apply it to empirical data. Because a prerequisite for this method is a set of migrant tracts inferred with reasonable confidence, it is most applicable to populations or taxa that show a high degree of genetic differentiation. One such case is represented by the hybridizing house mouse subspecies M. m. domesticus and M. m. musculus in Europe. We used a simple set of criteria to define migrant (hybrid) tracts in genomewide SNP data from both subspecies and then applied the likelihood inference method. Due to the limited size of the data set, relatively small numbers of migrant tracts were found: 75 in M. m. domesticus and 60 in M. m. musculus. However, the length distributions seemed to contain an excess of long tracts relative to equilibrium expectations (Figure 4), and the inference method detected a signal for an increase in the rate of introgression for both subspecies (Table 1).
Figure 4.—

Migrant tract lengths found in M. m. domesticus and M. m. musculus, compared to constant migration rate expectations.
TABLE 1.
Parameter inference and hypothesis testing for house mouse data
| No. tracts | P | m1 | m2 | T | Test 1 | Test 2B | |
|---|---|---|---|---|---|---|---|
| Subspecies | |||||||
| domesticus | 75 | 0.01095 | 6.08E-05 | 0 | 202 | P = 0.08 | NA |
| musculus | 60 | 0.00684 | 3.29E-05 | 1.00E-06 | 234 | P = 0.03 | NS |
| Combined | 135 | 0.00876 | 4.71E-05 | 7.50E-07 | 206 | P = 0.02 | NS |
Listed for each subspecies (and for the combined analysis) is the number of tracts >0.5 cM; the proportion of the genome that included migrant tracts (P); parameter estimates for m1, m2, and T; and results of hypothesis tests. NA, test 2B is not applicable when the estimate of m2 is zero; NS, P-values not approaching significance.
In spite of having a larger likelihood-ratio statistic against the constant rate model than for M. m. musculus, test 1 was only marginally significant for M. m. domesticus, while being significant for M. m. musculus and for a combined analysis of tracts from both subspecies. The weaker result for M. m. domesticus is due to a lower level of SNP informativeness in this subspecies: only 18% of M. m. musculus alleles would be detected as migrant in M. m. domesticus, compared to 38% in the opposite direction. Therefore, smaller tracts more frequently went undetected in the simulations used to assess significance in M. m. domesticus (see models and methods for details), and likelihood ratios from these simulations were higher. We also confirmed that the combined tract length data set for both subspecies showed the same signal for increased hybridization when each tract was required to have a minimum of three SNPs favoring migrant ancestry, rather than two (P < 0.01; results not shown).
M. m. domesticus had an estimate of zero for m2, while the estimate for M. m. musculus was nonzero (Table 1), but in neither case were the data sufficient to differentiate between no hybridization vs. low hybridization prior to the inferred rate change. The estimated timing of the rate change was similar in both taxa (202 and 234 generations ago) and in the combined analysis (206 generations ago). The two subspecies' estimates of m1 differ by only about a factor of 2 (Table 1), and both suggest a high contemporary population migration rate between these subspecies.
DISCUSSION
The specific demographic parameter estimates obtained from the house mouse SNP data should be interpreted with caution in light of limitations in the data set. Sample sizes are small—seven and eight haploid genomes. Samples originate from various geographic locations (Harr 2006), and our quantitative estimates might depend on the proximity of samples to the hybrid zone. Thus, it would be worthwhile to confirm our conclusions and refine parameter estimates using full-genome sequence data from reasonably large population samples of both subspecies.
Still, it is interesting that both taxa yielded estimates of ∼200 generations for the time since an increase in hybridization rate. Particularly when using a minimum tract length as large as 0.5 cM (which is necessitated by the density of SNPs in this data set), the time scope of our inference method is limited to fairly recent events (Figure 2). Thus, the time of ∼200 generations may not represent the first contact between these subspecies in Europe, and, indeed, archaeological evidence suggests a more ancient date for this event (reviewed in Boursot et al. 1993). However, this timing may still represent an increase in the rate of hybridization. If these mice have ∼2 generations per year, the inference method suggests that hybridization increased ∼100 years ago, which seems generally coincident with an increased potential for human-mediated transport in Europe.
The evolutionary trajectories of these hybridizing house mouse subspecies will depend on a variety of factors, but one potential predictor is the current rate of hybridization in terms of Nem1. The true values of current Ne for European populations of M. m. domesticus and M. m. musculus are unknown, but long-term effective sizes on the order of 1 million have been inferred for ancestral range populations of both subspecies (Baines and Harr 2007). Given the successful relationship of these mice with humans, it seems very plausible that the current Ne is at least this large. If we therefore take 1 million as an estimate for Ne in both taxa, the m1 estimates obtained here imply that M. m. domesticus is currently receiving ∼61 immigrants from M. m. musculus each generation, while M. m. musculus is receiving ∼33 immigrants per generation from M. m. domesticus (on the basis of the estimate of Nem1 for each subspecies). Since both of these estimates give 4Nem1 ≫ 1, these results could indicate that M. m. domesticus and M. m. musculus are currently on a path toward fusion rather than speciation. However, the presence of partial incompatibilities between these taxa, particularly on the X chromosome (e.g., Good et al. 2008), suggests that certain portions of the genome may resist homogenization.
Our analysis of simulated data showed that, given the lengths of migrant tracts from a population sample of genomes, the likelihood inference method presented here has high power to detect historical changes in migration (Figure 2), even for rather subtle shifts in migration rate (i.e., 2.5-fold changes), and should be useful in testing hypotheses and estimating parameters related to migration rate changes. This approach is conceptually related to methods that estimate the timing of recent admixture events (Hoggart et al. 2004; Patterson et al. 2004), but it allows for a greater variety of historical scenarios. In terms of its temporal scale, our method falls in between methods that identify very recent migration events (e.g., Rannala and Mountain 1997) and those that estimate long-term migration rates (e.g., Beerli and Felsenstein 2001). Although the results presented here suggest that our method is most relevant for detecting migration rate changes within the past 1000 generations, in many cases it may be possible to use a lower threshold tract length (C) than the 0.5 cM used in this study, and the temporal scope should expand with the inverse of C. The main assumption of the method is that recombination will break down tracts to below the threshold length before genetic drift can lift them to high frequency. Values of C that are <1/Ne are therefore recommended, but the choice of C will also depend on the level of diversity, the degree of population differentiation, and the density of markers (all of which constrain the inference of short migrant tracts). For M. musculus, a smaller threshold tract length would be a viable option with a denser SNP data set.
Our method does not address the inference of population ancestry along a recombining chromosome and requires that migrant tracts be identified beforehand. Published methods exist for this purpose (e.g., Falush et al. 2003) and the optimal method may depend on the data set being analyzed. Tract length data obtained from such methods can be used as the input for our analysis, and for methods that allow sampling from a posterior distribution of tract lengths, uncertainty in the tract length inference can be directly incorporated in the likelihood method. Without the use of such methods, the need for confident identification of migrant tracts would make this approach difficult to apply to weakly differentiated populations, but for more strongly differentiated populations or hybridizing subspecies, this method should be very useful in its current form.
To derive the tract length distributions, a number of assumptions were needed. The most troublesome of these, the lack of recombination among migrant tracts, would be very difficult to relax in the current framework. A full treatment of the problem would require analysis of an ancestral recombination graph in a subdivided population for whole-genome data. Another simplifying assumption is made by ignoring the ends of the chromosome. This assumption is much easier to relax and can be done by considering the conditional distribution in Equation 2. However, as this leads to a considerably less tractable algebraic representation, and since the current approximation performs very well for realistic chromosome lengths, we have chosen not to pursue this further.
The inference method described here may be applicable in a number of biological contexts. As demonstrated by our analysis of the M. musculus SNP data, the migrant tract approach may be especially relevant in testing hypotheses about historical trends of gene flow across hybrid zones, perhaps shedding light on the evolutionary trajectories of hybridizing taxa. The inferences enabled by this method may also find particular relevance in conservation: to test the effect of a new barrier (such as a highway) on the dispersal of an organism with a short generation time or to infer the rate of migration over relatively recent timescales (rather than over the past 4Ne generations) to guide management strategies for species with fragmented habitats. In this context it is important to note that inferences are done at a timescale more relevant to conservation genetics and that estimates of time in number of generations are obtained directly and do not rely on inferences of effective population sizes.
For optimal power, this method requires reasonably dense, genomewide polymorphism data from moderate to large sample sizes. It also requires information about the genetic map position of each marker, which can be estimated by genotyping related individuals such as parent–offspring trios. In light of rapidly improving DNA sequencing technology, we are optimistic that the inferences described here will be possible for both model and nonmodel organisms in the near future.
Acknowledgments
This research was supported by a National Institutes of Health (NIH) Kirschstein–National Research Service Award Postdoctoral Fellowship (F32 HG004182) to J.E.P. and a NIH research grant (UO1HL084706) to R.N.
References
- Baines, J. F., and B. Harr, 2007. Reduced X-linked diversity in derived populations of house mice. Genetics 175 1911–1921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becquet, C., and M. Przeworski, 2007. A new approach to estimate parameters of speciation models with application to apes. Genome Res. 17 1505–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, P., and J. Felsenstein, 2001. Maximum likelihood estimation of migration matrix and effective population sizes in n subpopulations by using a coalescent approach. Proc. Natl. Acad. Sci. USA 98 4563–4568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boursot, P., and K. Belkhir, 2006. Mouse SNPs for evolutionary biology: beware of ascertainment biases. Genome Res. 16 1191–1192. [DOI] [PubMed] [Google Scholar]
- Boursot, P., J.-C. Auffray, J. Britton-Davidian and F. Bonhomme, 1993. The evolution of house mice. Annu. Rev. Ecol. Syst. 24 119–152. [Google Scholar]
- Cornuet, J. M., and G. Luikart, 1996. Description and power analysis of two tests for detecting population bottlenecks from allele frequency data. Genetics 144 2001–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Depaulis, F., S. Mousset and M. Veuille, 2003. Power of neutrality tests to detect bottlenecks and hitchhiking. J. Mol. Evol. 57 S190–S200. [DOI] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher, R. A., 1930. The Genetical Theory of Natural Selection. Clarendon Press, Oxford.
- Good, J. M., M. D. Dean and M. W. Nachman, 2008. A complex genetic basis to X-linked hybrid male sterility between two species of house mice. Genetics 179 2213–2228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harr, B., 2006. Genomic islands of differentiation between house mouse subspecies. Genome Res. 16 730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hey, J., 2005. On the number of New World founders: a population genetic portrait of the peopling of the Americas. PLoS Biol. 3 e193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoggart, C. J., M. D. Shriver, R. A. Kittles, D. G. Clayton and P. M. McKeigue, 2004. Design and analysis of admixture mapping studies. Am. J. Hum. Genet. 74 965–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., 1983. Properties of a neutral allele model with intragenic recombination. Theor. Popul. Biol. 23 183–201. [DOI] [PubMed] [Google Scholar]
- Jensen, J. D., Y. Kim, V. Bauer DuMont, C. F. Aquadro and C. D. Bustamante, 2005. Distinguishing between selective sweeps and demography using DNA polymorphism data. Genetics 170 1401–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen-Seaman, M. I., T. S. Furey, B. A. Payseur, Y. Lu, K. M. Roskin et al., 2004. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 14 528–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingman, J. F. C., 1982. a The coalescent. Stoch. Proc. Appl. 13 235–248. [Google Scholar]
- Kingman, J. F. C., 1982. b On the genealogy of large populations. J. Appl. Probab. 19A 27–43. [Google Scholar]
- Kong, A., D. F. Gudbjartsson, J. Sainz, G. M. Jonsdottir, S. A. Gudjonsson et al., 2002. A high-resolution map of the human genome. Nature 31 241–247. [DOI] [PubMed] [Google Scholar]
- Koopman, W. J., Y. Li, E. Coart, W. E. Van De Weg, B. Vosman et al., 2007. Linked vs. unlinked markers: multilocus microsatellite haplotype-sharing as a tool to estimate gene flow and introgression. Mol. Ecol. 16 243–256. [DOI] [PubMed] [Google Scholar]
- Marth, G. T., E. Czabarka, J. Murvai and S. T. Sherry, 2004. The allele frequency spectrum in genomewide human variation data reveals signals of differential demographic history in three large world populations. Genetics 166 351–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montana, G., and J. K. Pritchard, 2004. Statistical tests for admixture mapping with case-control and cases-only data. Am. J. Hum. Genet. 75 771–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen, R., and J. Wakeley, 2001. Distinguishing migration from isolation: a Markov chain Monte Carlo approach. Genetics 158 885–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson, N., N. Hattangadi, B. Lane, K. E. Lohmueller, D. A. Hafler et al., 2004. Methods for high-density admixture mapping of disease genes. Am. J. Hum. Genet. 74 979–1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearse, D. E., and K. A. Crandall, 2004. Beyond FST: analysis of population genetic data for conservation. Conserv. Genet. 5 585–602. [Google Scholar]
- Rannala, B., and J. L. Mountain, 1997. Detecting immigration by using multilocus genotypes. Proc. Natl. Acad. Sci. USA 94 9197–9201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salcedo, T., A. Geraldes and M. W. Nachman, 2007. Nucleotide variation in wild and inbred mice. Genetics 177 2277–2291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wall, J. D., P. Andolfatto and M. Przeworski, 2002. Testing models of selection and demography in Drosophila simulans. Genetics 162 203–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlock, M. C., and D. E. McCauley, 1999. Indirect measures of gene flow and migration: FST ≠ 1/(4Nm + 1). Heredity 82 117–125. [DOI] [PubMed] [Google Scholar]
- Wright, S., 1931. Evolution in Mendelian populations. Genetics 16 97–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright, S., 1952. The theoretical variance within and among subdivisions of a population that is in a steady state. Genetics 37 312–323. [DOI] [PMC free article] [PubMed] [Google Scholar]