

Abstract
We develop expressions for the power to detect associations between parental genotypes and offspring phenotypes for quantitative traits. Three different “indirect” experimental designs are considered: full-sib, half-sib, and full-sib–half-sib families. We compare the power of these designs to detect genotype–phenotype associations relative to the common, “direct,” approach of genotyping and phenotyping the same individuals. When heritability is low, the indirect designs can outperform the direct method. However, the extra power comes at a cost due to an increased phenotyping effort. By developing expressions for optimal experimental designs given the cost of phenotyping relative to genotyping, we show how the extra costs associated with phenotyping a large number of individuals will influence experimental design decisions. Our results suggest that indirect association studies can be a powerful means of detecting allelic associations in outbred populations of species for which genotyping and phenotyping the same individuals is impractical and for life history and behavioral traits that are heavily influenced by environmental variance and therefore best measured on groups of individuals. Indirect association studies are likely to be favored only on purely economical grounds, however, when phenotyping is substantially less expensive than genotyping. A web-based application implementing our expressions has been developed to aid in the design of indirect association studies.
PREDICTIONS of the evolutionary dynamics of quantitative traits are highly sensitive to the genetic architecture assumed to underlie them (Barton and Turelli 1989). Accordingly, in recent times, a large amount of empirical attention has been paid to the dissection of quantitative traits at the genomic level (Barton and Keightley 2002). Classical methods for trait dissection such as quantitative trait locus (QTL) analysis by linkage rely upon linkage disequilibrium between marker loci and functional polymorphisms within pedigrees and, although useful for determining whether a particular genomic region may affect a trait, suffer undesirable properties such as narrow sampling of naturally occurring allelic variation and a limited scope for distinguishing physical linkage from pleiotropy (Mackay 2001). As these weaknesses all concern critical aspects of genetic architecture (Hansen 2006), they must be overcome if we are to better understand the genetic architecture of quantitative traits. A common alternative to analysis via linkage is to test for direct association between single-nucleotide polymorphisms (SNPs) in candidate gene regions (or the entire genome) and quantitative traits. For example, when linkage disequilibrium is weak, an association study may be more useful for distinguishing linkage from pleiotropy (e.g., Carbone et al. 2006).
Typically, association studies are conducted on a sample of individuals that are both phenotyped and genotyped (Long and Langley 1999); we refer to this as the direct approach. Although requiring large sample sizes, direct approaches have identified candidate polymorphisms contributing to quantitative trait variation in a wide range of organisms including humans (Lettre et al. 2008; Weedon et al. 2008) and in model organisms such as Drosophila (Dworkin et al. 2003, 2005; Long et al. 1998, 2000; Robin et al. 2002), Arabidopsis (Aranzana et al. 2005), and mice (Liu et al. 2006). However, independent replication of significant results appears vital to eliminate false positives and population-specific effects (e.g., Macdonald and Long 2004; Gruber et al. 2007). The direct approach is suitable when a candidate gene's contribution to naturally occurring phenotypic variance is of interest (via the use of field-sampled individuals) and for traits that can be measured with minimal error, such as morphology. However, many fitness-related traits such as behavioral and life-history traits can be heavily influenced by environmental variance (Houle 1992), making them difficult to measure on single field-caught individuals. Such traits are often impossible to measure under field conditions and are more reliably measured on groups of individuals.
For some organisms, performing association studies in a panel of inbred lines (ILs) or doubled haploid lines (DHLs) can be one solution to the requirement of phenotyping multiple individuals and or traits per genotype (e.g., Dworkin et al. 2003). The IL approach has some disadvantages: the genetic structure of an IL panel does not accurately reflect that of an outbred population, the variance of quantitative traits is altered by inbreeding (Hill and Caballero 1992; Van Buskirk and Willi 2006), and for many species inbreeding is impractical. Clearly, flexible approaches are required that can be implemented in outbred natural populations.
One potential solution is an indirect approach whereby a set of parents are genotyped and multiple progeny are phenotyped. Associations are then tested between parental genotype and progeny mean phenotype. This approach was recently used to detect associations in natural populations of Drosophila (Weeks et al. 2002; Kennington et al. 2007; Rako et al. 2007). Indirect methods have also been suggested and applied before in the case of linkage analysis, i.e., the association between phenotypes and identity-by-descent (IBD) within pedigrees, in particular in livestock (Weller et al. 1990; Van Der Beek et al. 1995) and experimental populations (Hill 1998). The motivations for indirect approaches in linkage studies are similar to those for association studies. First, it may be impossible to measure the phenotype or genotype in certain individuals, for example, for sex-limited traits or when it is necessary to kill individuals to be genotyped before their phenotype can be measured. Second, due to the increased sample size per genotype, measuring phenotypes on relatives can increase the precision of estimating QTL effects, thereby increasing the power of detection. However, the extra precision can come at a cost due to incomplete linkage between marker and QTL (Van Der Beek et al. 1995). Moreover, linkage analyses require very large sample sizes, lack power to detect QTL that explain only a few percent of population variance, and have poor map resolution (Visscher and Goddard 2004).
While the practical utility of indirect approaches is obvious, within the context of association studies, what remains less clear is the cost one pays in statistical power by indirectly associating parental genotypes with offspring phenotypes. Past investigations of the power of indirect methods have focused on the case of linkage rather than association (Weller et al. 1990; Van Der Beek et al. 1995). Here, we derive analytical expressions for the power to detect associations between candidate polymorphisms and quantitative traits, in four breeding designs that involve genotyping different configurations of parents and phenotyping their offspring, comparing the performance of each with the direct approach of genotyping and phenotyping the same set of individuals. We then derive expressions for the optimal experimental designs given the relative costs of genotyping and phenotyping. Our results demonstrate that these designs can provide levels of power equal to, and in some cases better than, the direct approach, indicating that indirect association may be a potentially powerful tool for detecting quantitative trait nucleotides (QTNs) in outbred populations.
MATERIALS AND METHODS
We assume random mating, no segregation distortion, and a general model of gene action that may or may not include dominance. For the direct approach, we consider a number of unrelated individuals with a genotype and a phenotype. For the indirect approach, we consider a balanced design of k dams per sire and m progeny per dam. We assume a phenotypic standard deviation of unity and that a proportion, q2, of the phenotypic variance is due to additive genetic effects at the QTL. This quantity is sometimes called the (additive) QTL heritability (e.g., Almasy and Blangero 1998). For a biallelic QTL in Hardy–Weinberg equilibrium, with allele frequency of p and additive effect of a and dominance deviation of d, q2 = 2p(1 − p)[a + d(2p − 1)]2 (Falconer and Mackay 1996). In practice, the specific values of a, d, and p do not matter but their combination does, because power of detection using an additive model for analysis depends on q2 (Neimann-Sorensen and Robertson 1961; Lynch and Walsh 1998) . We note that although dominance can be present in this model (i.e., d ≠ 0), for reasons outlined below (see Modeling dominance effects), we model only the power to detect the additive genetic effect at the QTL. For the direct approach, the model for a phenotype is
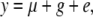 |
(1) |
with g the QTL and e the residual and var(y) = var(g) + var(e) = q2 + (1 − q2). For the indirect approach, the model is
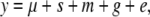 |
(2) |
with s and m the sire and dam effects and var(y) = s2 + m2 + q2 + (1 − s2 − m2 − q2) (Falconer and Mackay 1996). The (intraclass) correlation between full-sibs (FS) is 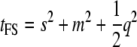 and that between half-sibs (HS) is
and that between half-sibs (HS) is  (via extension from Equations 18.33a,b in Lynch and Walsh 1998, p. 573). If all resemblance between relatives is due to additive genetic factors, then
(via extension from Equations 18.33a,b in Lynch and Walsh 1998, p. 573). If all resemblance between relatives is due to additive genetic factors, then 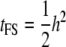 and
and 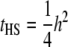 , where h2 is the narrow sense heritability that includes the effect of the QTL.
, where h2 is the narrow sense heritability that includes the effect of the QTL.
Power calculations:
We test associations by regressing the individual (direct approach) or family mean phenotype (indirect approach) on the individual or expected genotype, which is coded by an indicator variable (x) as the number of “A” alleles for a biallelic locus with alleles “A” and “B.” For individual genotypes (e.g., sires), x can have the value of 0, 1, or 2. The variance of x in the population is 2p(1 − p), with p the frequency of allele A. For the expected mean genotype in the progeny, x can have values 0, 0.5, 1, 1.5, and 2 for full-sib families, reflecting the different types of parental matings possible. For example, E(x) in the progeny is 2 if both parents have genotype AA.
We take an analytical approach to the analysis of power, a key feature of which is that our derivations focus on the noncentrality parameter (NCP) of the test of association rather than statistical power (1 − β) per se. The NCP of a particular design can be thought of as the amount of variation attributable to the model treatment effects (Lynch and Walsh 1998), which in our case is the additive effect of the polymorphism. There are several advantages to the NCP-based approach. First, unlike statistical power, the NCP does not depend upon any arbitrary choice of type 1 (α) error threshold, which is often contingent upon the type of study being conducted. Second, NCPs scale in a linear fashion with sample size, whereas power does not, making it far simpler to recalculate power for any variation in sample size without having to recalculate the NCP itself. The calculation of power itself remains straightforward under this approach. Once an NCP has been calculated, it can be used with the desired critical P-value to calculate statistical power.
The test statistic for association is the square of a simple t-test (i.e., an F-test). We assume that the sample size is large enough (N ≥ 60, Severo and Zelen 1960) so that the test statistic is approximately distributed as a central χ2 with 1 d.f. under the null hypothesis of no association. Under the alternative hypothesis, the test statistic is distributed as a noncentral χ2 (Searle 1971) with a NCP of λ. If α and β are the type-I and type-II error rates, then the power (= 1 − β) to detect an association is
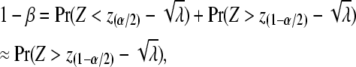 |
(3) |
with Z a standard normal variate and z(1−α/2) the threshold of a normal distribution corresponding to a type-I error rate of α [Pr(Z < z(α)) = α and Pr(Z > z(1−α)) = α] (e.g., Lynch and Walsh 1998, p. 870) The relationship between type-I error rate, power, and the NCP is
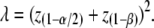 |
(4) |
General expression for the NCP:
When linear regression is used to estimate the effect of the marker on the quantitative trait, y = μ + bx + e, the general form of the NCP is 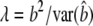 (Lynch and Walsh 1998, p. 881). From standard regression theory,
(Lynch and Walsh 1998, p. 881). From standard regression theory, 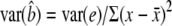 (Kendall and Stuart 1977). Treating the x as random,
(Kendall and Stuart 1977). Treating the x as random, 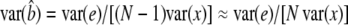 when N is large (Visscher and Hopper 2001; Visscher and Duffy 2006). Hence, λ = Nb2var(x)/var(e) = Nb2var(x)/[var(y) − b2var(x)], since b2var(x) is the variance removed by the regression. In our regression on the genotype indicator variable, this quantity is ∼cq2, with c the proportion of the QTL variance detected for a given design. A general expression of the NCP across our designs is then
when N is large (Visscher and Hopper 2001; Visscher and Duffy 2006). Hence, λ = Nb2var(x)/var(e) = Nb2var(x)/[var(y) − b2var(x)], since b2var(x) is the variance removed by the regression. In our regression on the genotype indicator variable, this quantity is ∼cq2, with c the proportion of the QTL variance detected for a given design. A general expression of the NCP across our designs is then
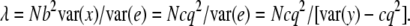 |
(5) |
For example, for the direct approach, var(y) = 1 and c = 1. For the indirect approach, both c and var(y) < 1 and their ratio determines the efficiency of the design. If cq2 is small (i.e., assuming that 0 < q2 < ∼0.05) relative to var(y), then λ ≈ Ncq2/var(y).
Direct method:
Individual genotypes and individual phenotypes:
The model for detecting an association is simply y = μ + x + e, with x the indicator variable for the genotype. If there are 2N individuals phenotyped and genotyped, then
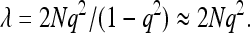 |
(6) |
This is a standard expression for the NCP of association between a SNP and a quantitative trait (Lynch and Walsh 1998). The NCP per genotyped individual is ∼q2. This is also the NCP per phenotyped individual.
Indirect designs:
Full-sib families (FS)–sire and dam genotyped and full-sib progeny phenotyped:
The model for the family mean (Y) is Y = μ + b E(xo) + en, with E(xo) the expected genotype indicator variable in the progeny. The variance of the family mean is var(Y) = [(1 − tFS)/m + tFS], with tFS the intraclass correlation of full-sibs (appendix a, Equation A2). E(xo) is simply the average of the parents,
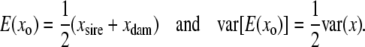 |
(7) |
The regression of Y on x0 is b = a, and therefore c, the proportion of QTL variance detected is  and var(y) is [(1 − tFS)/m + tFS]. The NCP for the test of association is, per full-sib family,
and var(y) is [(1 − tFS)/m + tFS]. The NCP for the test of association is, per full-sib family,
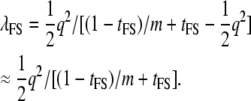 |
(8) |
The NCP per genotyped individual is 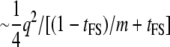 . The NCP per phenotyped individual is
. The NCP per phenotyped individual is  . For the limiting case of m = 1, the NCP per genotyped individual is one-quarter that of the direct approach and the NCP per phenotyped individual is one-half that of the direct approach because the ratio of genotypes to phenotypes is 2.
. For the limiting case of m = 1, the NCP per genotyped individual is one-quarter that of the direct approach and the NCP per phenotyped individual is one-half that of the direct approach because the ratio of genotypes to phenotypes is 2.
Full- and half-sib families (HS and FSHS1 designs)—sires genotyped, full- and half-sib offspring phenotyped:
If a sire is mated to k dams and each dam has m progeny (i.e., n = km), then the variance of the progeny average phenotype is (appendix a, Equation A1)
 |
(9) |
Only one-quarter of the QTL variance is detected by using the expected genotype in the progeny, so 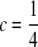 . The noncentrality parameter per sire family (and per genotyped individual) is
. The noncentrality parameter per sire family (and per genotyped individual) is
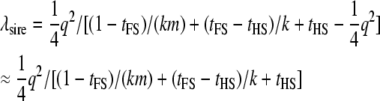 |
(10) |
and the NCP of an experiment with N sires is Nλsire. A special case of Equation 10 is the classical HS design in which sires are mated to k dams with a single progeny per dam (m = 1 and k = n). The NCP per genotyped sire for this design is
 |
(11) |
The expression for the NCP in the full-sib and the half-sib case is very similar, the difference being the value of c (=  for the full-sib design and
for the full-sib design and  for the half-sib design) and the variance of the family mean.
for the half-sib design) and the variance of the family mean.
Full- and half-sib families (FSHS2 design)—sires and dams genotyped, full- and half-sib offspring phenotyped:
When dams are nested within sires and all parents are genotyped, then the contribution of dams and sires to the NCP can be treated separately, taking account of the data structure. For a sire family with k dams, the contribution from each of the dams is (from Equation 8 but with 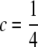 )
)
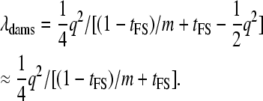 |
(12) |
The contribution from the half-sibs is (Equation 10)
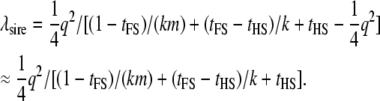 |
(13) |
And the total NCP per sire family is
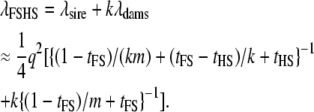 |
(14) |
When k = 1, the expression is equivalent to that for the full-sib design (Equation 8). When m = 1 (and therefore k = n), i.e., a half-sib design but all dams genotyped, the NCP is 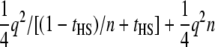 , the two terms corresponding to the contribution of the half-sib family (Equation 11) and n parent-offspring pairs, respectively.
, the two terms corresponding to the contribution of the half-sib family (Equation 11) and n parent-offspring pairs, respectively.
If all dams are genotyped, then there are k + 1 genotypes per sire family and km phenotypes per family. Hence, the NCPs per genotype and per phenotype are (λsire + kλdams)/(k + 1) and [λsire/(km) + λdams/m], respectively.
Validation of equations:
We performed a simulation study to verify our analytical results for the indirect design (Equations 8, 10, 11, and 14). Phenotypes for progeny were simulated as y = μ + s + m + g + e, for a given QTL heritability and a model in which all family resemblance was due to additive genetic factors. Input parameters were the numbers of sires, dams, and progeny, the proportion of variance explained by the QTL, the sire and dam intraclass correlations, the degree of dominance, and the allele frequency at the QTL. Sire and dam genotypes were simulated by sampling alleles from the population, using the binomial distribution (assuming Hardy–Weinberg equilibrium). Genotypes of progeny were simulated by sampling from the parental gametes, assuming Mendelian inheritance. Sire, dam, and residual random effects were simulated from a normal distribution with the appropriate standard deviation. Phenotypic observations on progeny were calculated by summation of the individual-specific terms (i.e., sire, dam, QTL, and residual). Data were analyzed using linear regression of the progeny means on SNP genotype. A total of 10,000 replicates were run for many combinations of parameters, and the average test statistic was recorded. The mean test statistic was found to be extremely close to our predictions (results not shown) and therefore simulations were not pursued further.
Modeling dominance effects:
Our coding for the indicator variable x reflects the expected value of the transmitted allele from the parent and therefore models the additive effects of alleles. An alternative parameterization is to code the expected dosage of the a and d effects in the progeny, given the observed genotype in the parents and the allele frequency in the population. For example, for parents with genotypes BB, AB, and AA, the expected mean values in the progeny due to the QTL are {−(1 − p)a + pd}, 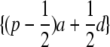 , and {pa + (1 − p)d}, respectively (Falconer and Mackay 1996). However, the coefficients for a and d are linear combinations of each other, so additive and dominance effects cannot be separated when genotypes are observed on a single parent and progeny are phenotyped, for example, in the case of half-sib designs. Therefore, only an allele substitution effect can be estimated, as coded, for example, by 0,
, and {pa + (1 − p)d}, respectively (Falconer and Mackay 1996). However, the coefficients for a and d are linear combinations of each other, so additive and dominance effects cannot be separated when genotypes are observed on a single parent and progeny are phenotyped, for example, in the case of half-sib designs. Therefore, only an allele substitution effect can be estimated, as coded, for example, by 0,  , and 1 for these genotypes. This makes sense because twice the expected value of the mean progeny phenotype is, when deviated from the population mean, the additive breeding value of the parent.
, and 1 for these genotypes. This makes sense because twice the expected value of the mean progeny phenotype is, when deviated from the population mean, the additive breeding value of the parent.
There are therefore qualitative differences in the ability to model dominance effects between the different indirect designs. As it is possible to fit a model parameter for dominance only when all parents are genotyped, we confine our results to the 1-d.f. model that detects only additive effects. A possible consequence of this is that when designing a study, one may make design decisions on the assumption that all genetic effects are additive. Although the loss in additive genetic variance due to dominance is simple to estimate for a given genetic variance due to a QTL, it is important to verify that the indirect designs perform in line with these theoretical expectations when dominance is nonzero. We considered this issue using simulations and results are presented in appendix b.
Efficiency of designs:
The direct and indirect designs can differ greatly in the number of genotyped and phenotyped individuals. To compare the efficiency of the different designs when the cost of genotyping and phenotyping varies, we express the power (NCP) as a proportion of the cost of the experiment. If the cost per genotype is 1.0 unit and the relative cost of phenotyping to genotyping is Cp, then the total cost is
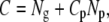 |
(15) |
with Ng and Np the number of individuals genotyped and phenotyped, respectively. For a given design we can express the cost-corrected noncentrality parameter (CNCP) as the NCP per “QTL heritability” and per dollar. That is,
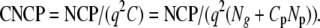 |
(16) |
Taking the approximate expressions for the NCP (i.e., assuming that 0 < q2 < ∼0.05), then the q2 drops out of the equation. The resulting CNCPs are then simple expressions of all these parameters.
For the direct approach,
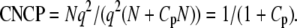 |
(17) |
For the classic HS design,
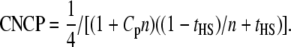 |
(18) |
For a given value of the heritability (and therefore tHS if all family resemblance is due to additive genetic factors), the optimum value of n is
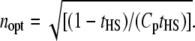 |
(19) |
Similarly for the FS design,
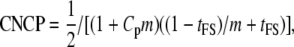 |
(20) |
and the optimum value of m is
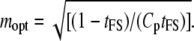 |
(21) |
For the nested design with only sires genotyped (FSHS1),
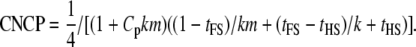 |
(22) |
If m is fixed, then the optimal value for k is 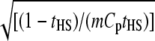 .
.
RESULTS
Statistical power:
Common genotyping effort:
It can be seen from the per genotype and per phenotype approximations (Table 1) that the powers of the indirect approaches relative to that of the direct approach are simple functions of two factors: first, the degree to which phenotypes within families are correlated (i.e., the intraclass correlations), which, in the absence of nongenetic and nonadditive genetic causes of family resemblance, is the narrow-sense heritability of the trait, and second, the numbers of progeny phenotyped per family when using an indirect design. There is no difference in the relative power of direct and indirect approaches as the QTN effect size varies. Thus, we explore performance of the indirect approaches due to variation in the narrow-sense heritability and progeny phenotyping effort, for an arbitrary QTN effect size while holding genotyping effort constant.
TABLE 1.
Approximate NCP/q2 per genotype and per phenotype for different indirect breeding designs
| Design | NCP per genotype | NCP per phenotype |
|---|---|---|
| Direct | 1 | 1 |
| Half-sib families (HS) | 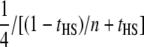 |
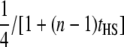 |
| Full-sib families (FS) |  |
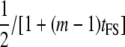 |
| Dams nested within sires; dams not genotyped (FSHS1) | 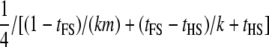 |
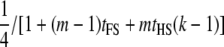 |
| Dams nested within sires; dams genotyped (FSHS2) | 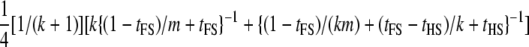 |
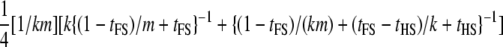 |
The power for three indirect designs (FS, HS, and FSHS1) and that for the corresponding direct design are plotted for different progeny numbers and narrow-sense heritabilities in Figure 1. For all three designs there are combinations of progeny number and heritability for which the power exceeds that of the corresponding direct design, with the indirect approach performing better as heritability declines (Figure 1, A–C). We investigated the FSHS2 design for a wide range of parameters but found that genotyping both sires and dams is not efficient because the additional information from the progeny of the dams is not compensated by the increase in the number of genotypes. Thus we do not consider this design further.
Figure 1.—

Effect of narrow-sense heritability and progeny number on the power (1 − β) of the full-sib (FS) (A), classic half-sib (HS) (B), and nested half-sib design in which only sires are genotyped (FSHS1) (C). In all cases, a QTN is assumed to explain 5% of the phenotypic variance (i.e., q2 = 0.05), the genotyping effort is set at 100 individuals, and α = 0.05. In all plots the solid horizontal line is the power for the direct design with identical genotyping effort and QTN effect size. For the full- and half-sib designs (A and B), m and n were fixed at the following values: 2, 4, 10, and 20. For the FSHS1 design different combinations of k and m were used (2, 2; 2, 5; 5, 2; and 4, 5).
Due to its effect on the phenotypic correlation among full- and half-sibs, heritability has a strong impact on the power of the indirect designs. While there is little difference in performance of the different indirect designs when heritability is low, all methods suffer reduced power when heritability is high. This is because the variance in progeny mean is relatively large, a factor that is particularly important for the performance of the full-sib design. Equating (6) and (8) for a particular value of tFS gives the number of progeny that need to be phenotyped to give equal power of the direct and indirect designs. That number is m = 4(1 − tFS)/(1 − 4tFS). Thus, for small values of tFS, four progeny per full-sib family need to be phenotyped to have equal power. Relative to the direct approach in which 2N individuals are phenotyped this is an increase of a factor of 2. For large values of tFS (tFS > 0.25, e.g., h2 >  ) there is no number of phenotyped progeny that can compensate for the loss of information. Thus, for highly heritable traits there will always be a higher genotyping effort for the full-sib design compared with the direct method. This limitation is not as severe for the half-sib case due to a lower expected correlation between half-sibs over a much wider range of heritabilities.
) there is no number of phenotyped progeny that can compensate for the loss of information. Thus, for highly heritable traits there will always be a higher genotyping effort for the full-sib design compared with the direct method. This limitation is not as severe for the half-sib case due to a lower expected correlation between half-sibs over a much wider range of heritabilities.
The FSHS1 design requires a choice of both the number of dams, k, and the number of offspring per dam, m. For a given value of km it appears that there are marginal gains in power by increasing the number of dams rather than increasing the number of offspring per dam. For example, Figure 1C illustrates this point with km = 10. With sires mated to five dams and two full-sib progeny phenotyped per dam, power was always higher compared with the alternative situation in which a sire is mated to two dams and five progeny are phenotyped.
Common phenotyping effort:
The indirect designs can result in a large variation in the number of phenotyped progeny; thus we also analyzed the performance of the designs on a per phenotype basis. Under no circumstances is power better than that of the direct approach for a common phenotyping effort (Figure 2). The per phenotype NCP of each indirect design tends to plateau well below the corresponding NCP for the direct design. On a per phenotype basis, the full-sib design always performs better than the half-sib designs regardless of whether a nested design is used or not.
Figure 2.—

Effect of the number of full- and half-sib families on the power (1 − β) of the full-sib (FS) (A) and classic half-sib (HS) (B) designs for a common phenotyping effort of 1000 individuals where α = 0.05. As family number varies, the numbers of m and n have been scaled to reflect the common phenotyping effort; thus an analysis involving 100 families implies a value of m = 10 and so on. The QTN explains 5% of the phenotypic variance (i.e., q2 = 0.05). Corresponding power for the direct approach is 1.0 in both cases and profiles for three narrow-sense heritability values are shown.
Relative efficiency and economy:
Because cost is often the major limiting factor in an association study and the indirect designs can lead to a very large phenotyping effort compared with the direct method, we also modeled how the cost of phenotyping relative to genotyping affects the efficiency of different designs. Figure 3 illustrates the CNCPs for the two simplest cases, the full-sib and classic half-sib designs with each optimized for m and n, respectively, using (21) and (19). The full-sib design provides a much larger contribution to power per unit expenditure when Cp = 0.1, which represents a 10-fold lower cost of phenotyping relative to genotyping. For all other cases considered, equal costs and when genotyping is 10 times cheaper than phenotyping, the direct design is favored on economical grounds.
Figure 3.—

Efficiency, expressed as the cost-corrected noncentrality parameter (CNCP), of the direct (Equation 17), full-sib (FS, Equation 20), and classic half-sib designs (HS, Equation 18), when the cost of phenotyping varies relative to the cost of genotyping. For each indirect design, we first estimated the optimal values of m and n for the relative cost of phenotyping vs. genotyping (CP), using Equations 21 and 19, respectively. For this example, the narrow-sense heritability was set at 0.2 and the QTN explained 5% of the phenotypic variance.
DISCUSSION
We have provided analytical solutions for the power to detect genotype–quantitative trait associations using an indirect approach, in which parents are genotyped and different configurations of offspring are phenotyped in an outbred population. Inspired by the possibility that indirect approaches may represent a flexible and cost-effective strategy for traits that are difficult to measure on single individuals, our goal was to determine whether an indirect approach could provide power equal to or better than the direct approach of genotyping and phenotyping the same individuals. Upon finding that there are regions of parameter space for which the indirect approaches perform well, we explored how the cost of phenotyping relative to genotyping affects the efficiency of the designs.
Power considerations:
It is immediately apparent from the per genotype noncentrality parameter expressions and their approximations (Table 1) that the QTN heritability, q2, does not affect the power of indirect approaches relative to the direct method. Thus our discussion concerning the performance of different indirect designs has generality across the entire range of effect sizes. However, the power of indirect approaches is heavily influenced by the narrow-sense heritability of the trait, decreasing in all designs as heritability increases. When heritability is high, the contribution of environmental variance to trait values is low. Thus the benefit achieved via the indirect design, due to a decrease in the contribution of environmental variance to family means, is also diminished. By contrast, heritability has the opposite effect on the power of indirect linkage analysis, in full- and half-sib families (Van Der Beek et al. 1995). The likely source of this discrepancy is that, for linkage analysis, the power depends on the proportion of within-family variance explained by the QTL, so for a fixed QTL effect size, a larger heritability implies a larger proportion of within-family variance explained and therefore more power (Visscher and Hopper 2001).
Although any “best” design is likely to depend on the reproductive biology of the organism studied, heritability, and budget, four guiding principles emerge from our analyses. First, half-sib designs are more powerful on a per genotype basis than full-sib designs. Second, when using the FSHS1 design, it is always better to increase the number of dams rather than the number of offspring per dam. Because there are more independent half-sib families in this design, the power contribution to the test statistic comes from half- rather than full-sibs (see Equation 14). Third, on a per phenotype basis no indirect approach can match the power of the direct approach. Finally, there may be little to be gained from genotyping both dams and sires in a full-sib–half-sib design. The poor performance of the FSHS2 design is partially a consequence of the fact that, within a group of half-sibs, the genotyping effort allocated to the dams delivers little power gain compared with the alternative of genotyping more independent sires from the population.
One key difference between our approach and other considerations of the power of association studies is that we have assumed that the causal SNP is genotyped. For example, Long and Langley (1999) investigated the power of direct association studies to detect QTL in outbred populations as a function of sample size, the QTL heritability, and recombination rate between the causal and genotyped variants that are in linkage disequilibrium with the causal variant. Our power calculations can be easily adjusted for imperfect linkage disequilibrium by multiplying the noncentrality parameter of the test statistic by r2, the squared correlation between alleles at the causal and genotyped variant (Hill and Robertson 1968). In other words, if there is not perfect linkage disequilibrium between the causal and the genotyped variant, then the experimental sample size needs to increase by a factor of 1/r2 to achieve the same power.
Considerations due to dominance:
In our analyses we have fitted an additive model and therefore our power calculations are relevant for the proportion of additive genetic variance due to the QTN. The predictable degree to which parental genotype reflects progeny mean phenotype is altered by dominance. Dominance has been shown to influence the power of tests of association and linkage and in general terms can either increase or decrease power depending upon the magnitude of dominance variance relative to the cost of fitting an extra model parameter to account for it (Sham et al. 2000). Although it has been argued from theoretical grounds and empirical observations that dominance effects may be generally relatively weak (Hill et al. 2008), in the indirect case, as we have considered here, we model only the ability to detect additive effects, so it is likely that power will be compromised by dominance, an issue we consider below.
We first considered the question of whether the decrease in power as a consequence of dominance is any worse than when using the equivalent direct method (i.e., a 1-d.f. model). From theory, power loss due to dominance can be substantial for both designs when dominance is strong (a = d) and the common allele is dominant (appendix b). Simulations suggest, however, that the direct and indirect approaches do not differ systematically in their sensitivity to this effect (appendix b). However, one has the option of fitting a model with a specific coding variable for d (i.e., a 2-d.f. model) when using the direct approach, an option available only for indirect designs in which both parents are genotyped. For the indirect FS design, dominance can be separated from additive effects because the progeny genotype can be predicted, in some cases without error. For example, AA × AA matings always give AA progeny and AA × BB matings always give AB progeny. Progeny from AB × AB matings have the same expectation for a as progeny from AA × BB matings but differ in their expectation for d ( and 1, respectively). In practice, however, the power to detect dominance will be lower than that of the direct approach for two reasons. First, the variance of the indicator variable for d is lower for the indirect case in a manner that depends upon allele frequency. For the direct approach this is Var(xddirect) = H(1 − H) whereas for the indirect full-sib case
and 1, respectively). In practice, however, the power to detect dominance will be lower than that of the direct approach for two reasons. First, the variance of the indicator variable for d is lower for the indirect case in a manner that depends upon allele frequency. For the direct approach this is Var(xddirect) = H(1 − H) whereas for the indirect full-sib case 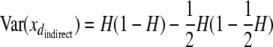 , where H is the heterozygosity (= 2p(1 − p)). Second, when allele frequencies are nonsymmetrical, the coding variables for a and d become correlated with each other. This has the effect of significantly reducing the amount of dominance that is recoverable after fitting an additive term. Thus, although it is theoretically possible to recover some information on dominance using the full-sib design, it is likely to be small unless moderate-frequency SNPs are being tested. In summary, it appears that the main impact on power between the direct and indirect methods due to dominance will be a function of the limited ability to fit a 2 d.f.-model in some cases rather than any systematic difference between the approaches in their ability to detect additive effects.
, where H is the heterozygosity (= 2p(1 − p)). Second, when allele frequencies are nonsymmetrical, the coding variables for a and d become correlated with each other. This has the effect of significantly reducing the amount of dominance that is recoverable after fitting an additive term. Thus, although it is theoretically possible to recover some information on dominance using the full-sib design, it is likely to be small unless moderate-frequency SNPs are being tested. In summary, it appears that the main impact on power between the direct and indirect methods due to dominance will be a function of the limited ability to fit a 2 d.f.-model in some cases rather than any systematic difference between the approaches in their ability to detect additive effects.
Relative efficiency and economy:
As power relative to the direct approach is a function of the phenotyping effort, the indirect designs can require considerable phenotyping effort to gain equal or better power than the direct approach. Thus, by developing expressions for the noncentrality parameter per dollar (CNCP) and optimal sizes for the three simplest indirect designs, we were able to investigate how variability in the relative costs of genotyping and phenotyping may influence design choice. Only when phenotyping is cheaper than genotyping (i.e., Cp < 1) is it possible for the indirect approach to be more economical. In fact, often phenotyping may have to be considerably cheaper than genotyping. For the limit of Cp → 0, i.e., very cheap phenotyping relative to genotyping, the comparison between designs is for the same number of genotypes (see Table 1). For the case considered in Figure 3, where heritability is 0.2, CNCP(HS) → 1/h2 = 5 and CNCP(FS) → 1/2h2 = 2.5, both for a large (strictly infinite) number of progeny.
Conclusion:
We have developed exact expressions and approximations for the statistical power of tests of association, in which parental genotypes are associated with progeny mean phenotypes, and compared their performance with a direct approach in which associations are tested between genotypes and phenotypes from the same individuals. For situations in which both direct and indirect approaches are feasible, our results suggest that the indirect approaches are more powerful when traits have low heritability but are more economical to implement only when genotyping costs far outweigh phenotyping costs. For studies in which the specific trait or study organism precludes a direct association study, the indirect approach nonetheless remains a viable option. We have implemented power calculations for both the direct method and the indirect designs considered here on a web-based application, power calculator for indirect association studies (PIAS), which can be accessed by the wider community (http://www.chenowethlab.org/pias/index.html).
Acknowledgments
We thank Bill Hill, Ary Hoffmann, and Emma Hine for comments on the manuscript. S.F.C. is supported by the Australian Research Council, and P.M.V. is supported by the Australian National Health and Medical Research Council.
APPENDIX A: DERIVATION OF VARIANCE AMONG FAMILY MEANS
From Falconer and Mackay (1996, p. 167), the variance of sire means is given by
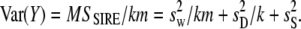 |
But assuming VP = 1,
 |
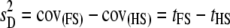 |
 |
Hence,
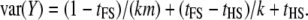 |
(A1) |
For the full-sib case, k = 1 and there is no contribution from half-sibs; thus, tHS = 0. Hence,
 |
(A2) |
APPENDIX B: ASSOCIATION WITH DOMINANCE
For a standard quantitative genetic model, with mean values of −a, d, and a for genotypes AA, AB, and BB, using k = d/a as the degree of dominance, p as the allele frequency of allele B, and H as the heterozygosity (2p(1 − p)),
 |
(B1) |
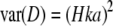 |
(B2) |
(Falconer and Mackay 1996; Lynch and Walsh 1998). Let the phenotypic variance be 1 and the total proportion of variance due to the QTL be Q2 = var(A) + var(D). As before, the proportion of phenotypic variance due to additive variance at the QTL is q2 = var(A). For a direct design, the NCP for an additive (1-d.f.) model is ∼Nq2/(1 − q2) and the NCP for a 2-d.f. model is ∼NQ2/(1 − Q2) (e.g., Sham et al. 2000). Some examples, keeping the total proportion of variance due to the QTL constant (Q2 = 0.05), are given in Table A1.
TABLE A1.
Examples of power loss due to dominance when fitting an additive (1-d.f.) model using the direct approach
| NCP (N = 100)
|
||||
|---|---|---|---|---|
| p | k | q2 | 1-d.f. fit (a) | 2-d.f. fit (a + d) |
| 0.1 | 0.0 | 0.050 | 5.26 | 5.26 |
| 0.5 | 0.049 | 5.13 | 5.26 | |
| 1.0 | 0.047 | 4.97 | 5.26 | |
| 0.5 | 0.0 | 0.050 | 5.26 | 5.26 |
| 0.5 | 0.044 | 4.65 | 5.26 | |
| 1.0 | 0.033 | 3.45 | 5.26 | |
| 0.7 | 0.0 | 0.050 | 5.26 | 5.26 |
| 0.5 | 0.042 | 4.39 | 5.26 | |
| 1.0 | 0.023 | 2.36 | 5.26 | |
Note that the power of the 2-d.f. model is unaffected by dominance when using this approach.
Only if there is strong dominance and the common allele is dominant (e.g., p = 0.1, k = −1) is there substantial loss in power by fitting an additive model. If Q2 is small, then the ratio of the NCP for fitting a or fitting a + d is ∼q2/Q2. This ratio can be expressed as
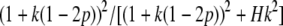 |
(B3) |
and depends on k and p.
In the indirect model we detect  or
or 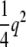 when we fit an additive model; thus apart from this reduction in the amount of q2 detected, there is no theoretical reason for expecting the performance of a 1-d.f. test to be different from that of the direct approach. We tested this prediction using simulations.
when we fit an additive model; thus apart from this reduction in the amount of q2 detected, there is no theoretical reason for expecting the performance of a 1-d.f. test to be different from that of the direct approach. We tested this prediction using simulations.
We simulated a quantitative trait according to a model in which the trait was influenced by both additive and dominance effects. Phenotypes for progeny were simulated as y = μ + s + m + g + e, for a given QTL heritability and a model in which all family resemblance was due to both additive and dominance effects (given by a value of k). Data were analyzed using linear regression of the progeny means on SNP genotype. A total of 10,000 replicates were run for each combination of parameters, and the average test statistic was recorded. We considered both the FS and the classic HS designs with parameter values similar to those in Figure 1, A and B: that is, a sample of 100 genotypes, total QTL variance of 0.05, and trait heritability of 0.4. We considered three allele frequencies (p = 0.1, 0.5, and 0.7) for the cases of no (k = 0), partial (k = 0.5), and complete (k = 1) dominance. We then compared predicted (theory) test statistics with simulated values for each set of parameter values. For both designs, simulated results were very similar to theory and in no case did theoretical test statistics significantly exceed simulated ones (Tables A2 and A3).
TABLE A2.
Theory and simulation results for power loss due to dominance in an indirect FS design
| p | k | a | D | Predicted mean test statistic: F-ratio | Simulation mean test statistic: F-ratio |
|---|---|---|---|---|---|
| 0.1 | 0.0 | 0.527 | 0.000 | 5.90 | 5.98 |
| 0.5 | 0.372 | 0.186 | 5.79 | 5.93 | |
| 1.0 | 0.285 | 0.285 | 5.63 | 5.85 | |
| 0.5 | 0.0 | 0.316 | 0.000 | 5.90 | 6.01 |
| 0.5 | 0.298 | 0.149 | 5.33 | 5.50 | |
| 1.0 | 0.258 | 0.258 | 4.21 | 4.34 | |
| 0.7 | 0.0 | 0.345 | 0.000 | 5.90 | 6.09 |
| 0.5 | 0.400 | 0.200 | 5.18 | 5.15 | |
| 1.0 | 0.391 | 0.391 | 3.20 | 3.20 |
A 1-d.f. model is fitted for all tests of association. Simulation details are given in the text. SEs from runs of 10,000 replicates ranged from 0.4 to 0.5.
TABLE A3.
Theory and simulation results for power loss due to dominance in an indirect HS design
| p | k | a | D | Predicted mean test statistic: F-ratio | Simulation mean test statistic: F-ratio |
|---|---|---|---|---|---|
| 0.1 | 0.0 | 0.527 | 0.000 | 8.04 | 8.18 |
| 0.5 | 0.372 | 0.186 | 7.87 | 8.05 | |
| 1.0 | 0.285 | 0.285 | 7.65 | 7.78 | |
| 0.5 | 0.0 | 0.316 | 0.000 | 8.04 | 8.13 |
| 0.5 | 0.298 | 0.149 | 7.21 | 7.22 | |
| 1.0 | 0.258 | 0.258 | 5.59 | 5.62 | |
| 0.7 | 0.0 | 0.345 | 0.000 | 8.04 | 8.10 |
| 0.5 | 0.400 | 0.200 | 6.99 | 7.09 | |
| 1.0 | 0.391 | 0.391 | 4.13 | 4.16 |
A 1-d.f. model is fitted for all tests of association. Simulation details given in the text. SEs from runs of 10,000 replicates ranged from 0.4 to 0.6.
References
- Almasy, L., and J. Blangero, 1998. Multipoint quantitative-trait linkage analysis in general pedigrees. Am. J. Hum. Genet. 62 1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aranzana, M. J., S. Kim, K. Y. Zhao, E. Bakker, M. Horton et al., 2005. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PloS Genet. 1 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton, N. H., and P. D. Keightley, 2002. Understanding quantitative genetic variation. Nat. Rev. Genet. 3 11–21. [DOI] [PubMed] [Google Scholar]
- Barton, N. H., and M. Turelli, 1989. Evolutionary quantitative genetics: how little do we know. Annu. Rev. Genet. 23 337–370. [DOI] [PubMed] [Google Scholar]
- Carbone, M. A., K. W. Jordan, R. F. Lyman, S. T. Harbison, J. Leips et al., 2006. Phenotypic variation and natural selection at Catsup, a pleiotropic quantitative trait gene in Drosphila. Curr. Biol. 16 912–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dworkin, I., A. Palsson, K. Birdsall and G. Gibson, 2003. Evidence that Egfr contributes to cryptic genetic variation for photoreceptor determination in natural populations of Drosophila melanogaster. Curr. Biol. 13 1888–1893. [DOI] [PubMed] [Google Scholar]
- Dworkin, I., A. Palsson and G. Gibson, 2005. Replication of an Egfr-wing shape association in a wild-caught cohort of Drosophila melanogaster. Genetics 169 2115–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconer, D. S., and T. F. C. Mackay, 1996. Introduction to Quantitative Genetics. Longman, Essex, UK.
- Gruber, J. D., A. Genissel, S. J. Macdonald and A. D. Long, 2007. How repeatable are associations between polymorphisms in achaete-scute and bristle number variation in Drosophila? Genetics 175 1987–1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen, T. F., 2006. The evolution of genetic architecture. Annu. Rev. Ecol. Evol. Syst. 37 123–157. [Google Scholar]
- Hill, W. G., 1998. Selection with recurrent backcrossing to develop congenic lines for quantitative trait loci analysis. Genetics 148 1341–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and A. Caballero, 1992. Artificial selection experiments. Annu. Rev. Ecol. Syst. 23 287–310. [Google Scholar]
- Hill, W. G., and A. Robertson, 1968. Linkage disequilbrium in finite populations. Theor. Appl. Genet. 38 183–201. [DOI] [PubMed] [Google Scholar]
- Hill, W. G., M. E. Goddard and P. M. Visscher, 2008. Data and theory point to mainly additive genetic variance for complex traits. PloS Genet. 4 e1000008. [DOI] [PMC free article] [PubMed]
- Houle, D., 1992. Comparing evolvability and variability of quantitative traits. Genetics 130 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kendall, M. G., and A. Stuart, 1977. The Advanced Theory of Statistics. Macmillan, New York.
- Kennington, W. J., A. A. Hoffmann and L. Partridge, 2007. Mapping regions within cosmopolitan inversion In(3R)Payne associated with natural variation in body size in Drosophila melanogaster. Genetics 177 549–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lettre, G., A. U. Jackson, C. Gieger, F. R. Schumacher, S. I. Berndt et al., 2008. Identification of ten loci associated with height highlights new biological pathways in human growth. Nat. Genet. 40 584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, P. Y., Y. Wang, H. Vikis, A. Maciag, D. L. Wang et al., 2006. Candidate lung tumor susceptibility genes identified through whole-genome association analyses in inbred mice. Nat. Genet. 38 888–895. [DOI] [PubMed] [Google Scholar]
- Long, A. D., and C. H. Langley, 1999. The power of association studies to detect the contribution of candidate genetic loci to variation in complex traits. Genome Res. 9 720–731. [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., R. F. Lyman, C. H. Langley and T. F. C. Mackay, 1998. Two sites in the Delta gene region contribute to naturally occurring variation in bristle number in Drosophila melanogaster. Genetics 149 999–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long, A. D., R. F. Lyman, A. H. Morgan, C. H. Langley and T. F. C. Mackay, 2000. Both naturally occurring insertions of transposable elements and intermediate frequency polymorphisms at the achaete-scute complex are associated with variation in bristle number in Drosophila melanogaster. Genetics 154 1255–1269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M., and B. Walsh, 1998. Genetics and Analysis of Quantitative Traits. Sinauer, Sunderland, MA.
- Macdonald, S. J., and A. D. Long, 2004. A potential regulatory polymorphism upstream of hairy is not associated with bristle number variation in wild-caught Drosophila. Genetics 167 2127–2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay, T. F. C., 2001. The genetic architecture of quantitative traits. Annu. Rev. Genet. 35 303–339. [DOI] [PubMed] [Google Scholar]
- Neimann-Sorensen, A., and A. Robertson, 1961. The association between blood groups and several production characters in three Danish cattle breeds. Acta Agric. Scand. 11 163–196. [Google Scholar]
- Rako, L., M. J. Blacket, S. W. McKechnie and A. A. Hoffmann, 2007. Candidate genes and thermal phenotypes: identifying ecologically important genetic variation for thermotolerance in the Australian Drosophila melanogaster cline. Mol. Ecol. 16 2948–2957. [DOI] [PubMed] [Google Scholar]
- Robin, C., R. F. Lyman, A. D. Long, C. H. Langley and T. F. C. Mackay, 2002. Hairy: a quantitative trait locus for Drosophila sensory bristle number. Genetics 162 155–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Searle, S. R., 1971. Linear Models. John Wiley & Sons, New York.
- Severo, N. C., and M. Zelen, 1960. Normal approximation to the chi-square and non-central F-probability functions. Biometrika 47 411–416. [Google Scholar]
- Sham, P. C., S. S. Cherny, S. Purcell and J. K. Hewitt, 2000. Power of linkage versus association analysis of quantitative traits, by use of variance-components models, for sibship data. Am. J. Hum. Genet. 66 1616–1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Buskirk, J., and Y. Willi, 2006. The change in quantitative genetic variation with inbreeding. Evolution 60 2428–2434. [PubMed] [Google Scholar]
- Van Der Beek, S., J. A. M. Van Arendonk and A. F. Groen, 1995. Power of 2-generation and 3-generation QTL mapping experiments in an outbred population containing full-sib or half-sib families. Theor. Appl. Genet. 91 1115–1124. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., and D. L. Duffy, 2006. The value of relatives with phenotypes but missing genotypes in association studies for quantitative traits. Genet. Epidemiol. 30 30–36. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., and M. E. Goddard, 2004. Prediction of the confidence interval of quantitative trait loci location. Behav. Genet. 34 477–482. [DOI] [PubMed] [Google Scholar]
- Visscher, P. M., and J. L. Hopper, 2001. Power of regression and maximum likelihood methods to map QTL from sib-pair and DZ twin data. Ann. Hum. Genet. 65 583–601. [DOI] [PubMed] [Google Scholar]
- Weedon, M. N., H. Lango, C. M. Lindgren, C. Wallace, D. M. Evans et al., 2008. Genome-wide association analysis identifies 20 loci that influence adult height. Nat. Genet. 40 575–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks, A. R., S. W. McKechnie and A. A. Hoffmann, 2002. Dissecting adaptive clinal variation: markers, inversions and size/stress associations in Drosophila melanogaster from a central field population. Ecol. Lett. 5 756–763. [Google Scholar]
- Weller, J. I., Y. Kashi and M. Soller, 1990. Power of daughter and granddaughter designs for determining linkage between marker loci and quantitative trait loci in dairy-cattle. J. Dairy Sci. 73 2525–2537. [DOI] [PubMed] [Google Scholar]