

Abstract
Primary cilia are assembled and maintained by evolutionarily conserved intraflagellar transport (IFT) proteins that are involved in the coordinated movement of macromolecular cargo from the basal body to the cilium tip and back. The IFT machinery is organized in two structural complexes named complex A and complex B. Recently, inactivation in the mouse germline of Ift genes belonging to complex B revealed a requirement of ciliogenesis, or proteins involved in ciliogenesis, for Sonic Hedgehog (Shh) signaling in mammals. Here we report on a complex A mutant mouse, defective for the Ift122 gene. Ift122-null embryos show multiple developmental defects (exencephaly, situs viscerum inversus, delay in turning, hemorrhage and defects in limb development) that result in lethality. In the node, primary cilia were absent or malformed in homozygous mutant and heterozygous embryos, respectively. Impairment of the Shh pathway was apparent in both neural tube patterning (expansion of motoneurons and rostro-caudal level-dependent contraction or expansion of the dorso-lateral interneurons), and limb patterning (ectrosyndactyly). These phenotypes are distinct from both complex B IFT mutant embryos and embryos defective for the ciliary protein hennin/Arl13b, and suggest reduced levels of both Gli2/Gli3 activator and Gli3 repressor functions. We conclude that complex A and complex B factors play similar but distinct roles in ciliogenesis and Shh/Gli3 signaling.
Keywords: Intraflagellar transport, ciliogenesis, Sonic hedgehog, Gli3, neuronal patterning, limb patterning, situs viscerum inversus
Introduction
Increasing interest has been recently directed to primary cilia as specialized cell protrusions crucial in development and homeostasis (Michaud and Yoder, 2006; Pazour and Witman, 2003). Several developmental genetic diseases are associated with defects in cilia formation and function, including primary ciliary dyskinesia, polycystic kidney disease, pancreatic and liver cysts, hydrocephaly, skeletal abnormalities and Bardet-Biedl syndrome (Davenport and Yoder, 2005). Primary cilia represent sensory organelles where many signaling pathways are concentrated and may interact with each other to modulate different physiological processes (Michaud and Yoder, 2006; Pazour and Witman, 2003). In particular, the localization of somatostatin 2 receptor (Handel et al., 1999), integrin (Praetorius et al., 2004), taurine transport receptor (Christensen et al., 2005), angiopoietin receptor (Teilmann and Christensen, 2005) and platelet-derived growth-factor receptor (Schneider et al., 2005) to cilia membrane underlies the multifunction roles played by these organelles in cell physiology.
Biogenesis and maintenance of the primary cilium depends on intraflagellar transport (IFT), a process of bidirectional transport along the axoneme of biosynthetic and signaling proteins, as well as other cargos, to and from the cilium tip. IFT proteins are highly conserved and were initially identified in Chlamydomonas flagellar biosynthesis.
More recently, it has become apparent that, in mammalian cells, the presence of cilia, or minimally of proteins involved in cilia formation, such as IFT proteins, is required for signaling by Sonic Hedgehog (Shh), a master regulator involved in patterning during development (Huangfu and Anderson, 2005; Huangfu et al., 2003; Marszalek et al., 1999; Nonaka et al., 1998; Pazour et al., 2002). Specifically, inactivation of the Ift88/polaris, Ift172/wimple and Ift52/Ngd5 genes leads to embryonic lethality, with defects in Shh-dependent neural and limb patterning (Huangfu et al., 2003; Liu et al., 2005). This phenotype is also present in lethal embryos that are null for the IFT motors Kif3a, encoding a subunit of kinesin-2, involved in ciliary anterograde transport (Marszalek et al., 1999; Takeda et al., 1999), and Dnchc2, encoding a subunit of IFT dynein, involved in ciliary retrograde transport (Huangfu and Anderson, 2005; May et al., 2005). Genetic analyses indicate that these IFT proteins are involved in the Shh pathway at a step downstream of the Shh receptor Patched-1 (Ptc1) and the membrane protein Smoothened (Smo), and upstream of the transcription factors Gli2 and Gli3, that ultimately affect Shh signaling (Scholey and Anderson, 2006). Indeed, biochemical and morphological studies confirm that in IFT mutant embryos both the activator (primarily Gli2: Gli2A) and repressor (primarily Gli3: Gli3R) functions of these Shh effectors are compromised (Liu et al., 2005; Scholey and Anderson, 2006).
IFT proteins comprise a group of approximately 17 polypeptides organized in two large macromolecular complexes, A and B. It has been proposed that complexes A and B are dissociated at the base and tip of the cilium and re-associate during IFT (Pedersen et al., 2006). However, the two complexes are not functionally equivalent: complex A, including IFT122, mediates the direct association with the IFT motors kinesin-2 and dynein during anterograde and retrograde transport, respectively, while complex B, including IFT88, IFT172 and IFT52, is bound to complex A (Pedersen et al., 2006).
The mouse studies described above point to a critical requirement of IFT proteins for ciliogenesis and Shh signaling in mammals. However, with one recent exception – mice defective for THM1/IFT139 (Tran et al., 2008), all the IFT mouse mutants described to date are defective in complex B proteins. Here we describe the phenotype of mouse embryos null for the complex A protein Ift122. Characterization of Ift122-null embryos was the serendipitous result of studying the contrasting phenotypes of targeted alleles at the Med1/Mbd4 DNA repair gene previously generated by us and others (Cortellino et al., 2003; Millar et al., 2002; Wong et al., 2002). Unlike three other Med1/Mbd4 knock-out mice that are viable (Cortellino et al., 2003; Millar et al., 2002; Wong et al., 2002), embryos homozygous for the allele lacking exons 1-3 (Δ1-3) manifest mid-gestation lethality due to multiple developmental defects that resemble those seen in perturbations of the cilia and Shh pathways. Here we show that the Δ1-3 allele perturbs a neighboring gene, originally named Wdr10 (Gross et al., 2001), that shares a portion of its exon 1 with the Med1 exon 1 in the opposite direction. The fly Wdr10 ortholog, named Oseg1, was recently identified as a gene involved in ciliogenesis (Avidor-Reiss et al., 2004) and the Chlamydomonas homolog, named Ift122, is involved in IFT. Our results indicate that the murine Ift122/Wdr10 protein plays a role in cilia formation, and compromises Shh signaling in a similar, but distinct way from complex B mutants, confirming functional differences between the two IFT complexes.
Material and methods
Generation of Med1 mutant mice
Generation of Med1 Mutant Mice carrying the Δ1-3 and Δ2-5 alleles has been previously described (Cortellino et al., 2003). Briefly, the Med1 Δ1-3 targeting vector contained a 4.5 kb HindIII fragment immediately upstream of exon 1, and a 4.5 kb BglII fragment immediately downstream of exon 3, whereas the Med1 Δ2-5 targeting vector contained a 4.5 kb fragment harboring exon 1 and the first half of intron 1, and a 2.5 kb PCR fragment harboring intron 5 through part of intron 7. Targeting vectors were electroporated into W9.5 and R1 ES cells. Positive ES clones carrying the targeted Med1 locus were injected into C57/BL6 blastocysts to generate chimeric mice. Male chimeras were then mated to C57/BL6 females and the resulting Med1+/- F1 offspring backcrossed to C57/BL6. Initial experimental embryos, obtained from intercrosses of F2 Med1+/- mice, had a mixed genetic background that was approximately 75% C57/BL6 and 25% 129/Sv-J. Subsequent experiments were conducted after at least 8 backcrosses into C57/BL6, therefore the genetic background of the embryos was approximately 99% C57/BL6 and 1% 129/Sv-J.
Isolation and culture of mouse embryo fibroblasts (MEFs)
MEFs were prepared as previously described (Cortellino et al., 2003). Briefly, embryos were isolated from the uteri of day 12.5 p.c.-pregnant mice (E12.5). The embryo head was used for genotyping, while the body was minced and incubated in 2 ml of 0.25% trypsin at 37°C for 10 min. Dulbecco’s modified eagle medium (DMEM, 7ml) was added, and the cells were gently pelleted, resuspended in medium and plated for growth. MEFs were cultured in DMEM supplemented with 15% fetal calf serum, 1 mM sodium pyruvate, 2 mM glutamine, 10 units/ml penicillin and 10 μg/ml streptomycin. MEF cultures were kept on a 3T3 protocol, i.e., they were split 1/3 every three days.
RT-PCR
RNA was prepared from MEFs of different genotype in exponential growth from both the Δ1-3 and Δ2-5 colonies, using guanidinium/phenol extraction (Chomczynski and Sacchi, 1987). Total RNA was reverse-transcribed using an oligo-dT primer and Superscript (GIBCO-BRL). Med1 RT-PCR analysis was conducted with primers located in exon 1 (m1, 5′-GAG AGC CTA GTT CCA GAC CCG-3′) and exon 3 (m2, 5′-GAT GCT CCC TTT CGG CAG TAC-3′). Ift122 RT-PCR was performed with primers located in exon 1 (i1, 5′-GTG GAG AGA CAA AGC GGA-3′) and exon 3 (i2, 5′-CGC CAC ACA GTA CAC GGT-3′). PCR proceeded for 35 cycles for both Med1 and Ift122. Each cycle consisted of 94°C for 30 seconds, 52°C for 1 min and 72°C for 1 min.
Northern blot analysis
RNA preparation and Northern blot analysis were performed as previously described (Delmas et al., 1999). Briefly, total RNA was prepared from mouse embryos at stage E4.5 to E18.5 (decidua is included for E4.5 to E6.5) using the Roti-Quick RNA purification kit (Roth), subjected to electrophoresis in 1% agarose formaldehyde gels under denaturing conditions, transferred to nylon membranes (Hybond-N+, Amersham) and probed with an Ift122 cDNA probe (obtained by RT-PCR using primers 5′-CTG TTC TAC CAA CAA CCC G-3′, located on exon 25, and 5′-GGC CAG TGT CGT CGA TAC-3′, located on exon 29) (NT_039353), the Med1 f5 probe (Bellacosa et al., 1998) and Gapdh (Cortellino et al., 2003). Probes were 32P radiolabeled by random priming (Neblot kit, Ozyme). Hybridization was in 2 × sodium citrate buffer (SSC), 1% (w/v) sodium dodecyl sulfate (SDS), 10% dextran sulfate, 0.5% (w/v) low fat milk at 65°C, overnight. Membranes were washed twice in 2 × SSC, 0.1% SDS at room temperature for 10-15 min, and twice in 0.2 × SSC, 0.1% SDS at 65°C for 40 min. Membranes were exposed to hyperfilm MP (Amersham) and revealed one week later.
Scanning electron microscopy (SEM)
For SEM imaging, E8 mouse embryos were fixed in 2% glutaraldehyde in 0.1M cacodylate buffer pH 7.2 at 4°C for 16 hours, washed in the buffer and post-fixed in 1% osmium tetroxide in the same buffer for 90 min. After two washes in the buffer and three more washes in double distilled water, the samples were stored in double distilled water for 16 hours, dehydrated in increasing concentration of ethanol replaced subsequently by increasing concentration of amyl acetate. After two incubations in 100% amyl acetate for 30 min and 1 hour respectively, the samples were stored in 100% amyl acetate for 48 hours. Critical point drying with liquid carbon dioxide (Anderson, 1951) was performed in Denton CPD-1 apparatus. The samples were then mounted on aluminum pins, sputter-coated with Pt-Pd and examined in an ETEC scanning electron microscope. Images were recorded and processed digitally.
Fixation and hematoxylin/eosin staining
Timed matings were set up between heterozygous mice and the embryos were dissected at different time points. Embryos were fixed in 4% paraformaldehyde overnight at 4°C, dehydrated in increasing ethanol series (70, 80, 90% and absolute) for 1 hr each step. Before embedding in paraffin, embryos were incubated with ethanol:xylene (50:50) and xylene for 1 hour each. Sections (10 μm) were prepared for hematoxylin and eosin staining. After deparaffinization and rehydration using xylene and decreasing ethanol series (absolute, 95, 80%), the sections were stained with hematoxylin for 3 minutes, rinsed in deionized water, destained in acid ethanol, and washed again with deionized water. Eosin staining was performed by incubating the sections in eosin for 30 seconds, ethanol 95% and absolute for 15 min each, and xylene for 45 min. Slides were mounted using Permount media.
Cryopreservation and immunohistochemistry
Embryos were fixed in 4% paraformaldehyde for 1 hr at 4°C, extensively washed with PBS for 2 hr at room temperature and incubated for 16 hours in 30% sucrose. Thereafter, embryos were equilibrated in OCT solution for 20 min and then snap-frozen in liquid nitrogen. Immunohistochemistry was performed on 10 μm cryosections using antibodies against Shh, Foxa2, Nkx2.2, MNR2, Isl1, Pax3 and Pax6, all from the Developmental Studies Hybridoma Bank (Iowa City, IA 52242, USA). Sections were blocked in 10% goat serum in PBS containing 0.1% triton-X 100 at room temperature for 1 hr to reduce nonspecific staining, followed by an overnight incubation at 4°C with the primary antibodies. Sections were washed with several rinses of PBS containing 0.1% Tween-20 (Sigma). Alexa 488 (green)-conjugated secondary antibodies (Molecular Probes) were applied at 1:600 dilutions for 1 hr at room temperature. The sections were washed again with 0.1% triton-X PBS and mounted with Vectashield medium (Vector Laboratories, Inc. Burlingame, CA 94010).
Immunofluorescence
MEFs were rinsed twice with PBS, and starved for 48 hours in medium without serum. The cells were washed with PBS, and fixed and permeabilized with cold methanol for 10 minutes at −20°C. After three washes with PBS, the cells were incubated with anti-acetylated tubulin, (1:500) and anti-γ-tubulin (1:200), both from Sigma. Secondary anti-mouse Alexa 488 (green)-or anti-rabbit Alexa 594 (red) secondary antibodies (Molecular Probes) were used at 1:1000 dilution. After rinsing in PBS, the slides were mounted with mounting media (Vector Laboratories, Inc. Burlingame, CA 94010) containing DAPI.
Whole-mount in situ hybridization
Embryonic gene expression was analyzed with nonradioactive single-stranded RNA probes. The Ift122 probe was the same used for Northern analysis, described above. 11-Digoxigenin-UTP (DIG-UTP) was incorporated during synthesis (Roche Diagnostics GmbH, Mannheim). The probes for Shh and Ptc1 were provided by Dr. A.P. McMahon (Harvard Univ.); the Col2A1 and Fgf4 probes were provided by Dr. M. Barna (UCSF); the Bmp2 and Bmp4 probes were provided by Dr. Ewa Wandzioch (Fox Chase Cancer Center).
Mouse embryos were fixed in 4% (v/v) paraformaldehyde at 4°C overnight, and, after several washes with 0.1% Tween-PBS (PBST), stored in methanol at −20°C. Embryos were rehydrated, bleached with 6% H2O2, and treated with proteinase K for 6 to 11 minutes, depending on embryonal days of age. Embryos were prehybridized in a hybridization buffer consisting of 50% (v/v) formamide, 5 × SSC pH 4.5, 1% (v/v) SDS, 50 μg/ml yeast tRNA and 50 μg/ml heparin, for 1 hour at 70°C. Hybridization was performed overnight at 70°C in the hybridization buffer with DIG-labeled antisense or sense RNA probes. After 1.5-hr washes with a solution of 50% (v/v) formamide, 2 × SSC pH 4.5 and 1%(v/v) SDS at 70°C, the embryos were incubated twice for 30 min in 10 mM Tris-HCl pH 7.5, 0.5 M NaCl, 0.1% Tween 20, 100 μg/ml RNAse at 37°C, washed three times for 20 min in 2 × SSC pH 4.5, 50% Formamide. 0.2% SDS at 70°C, and transferred to PBST at room temperature. The embryos were incubated for 3 hr in 2% (w/v) blocking reagent (100 mM maleic acid, 150 mM NaCl, 2 mM levamisole, 0.1% Tween20, 10% sheep serum, pH 7.5) and then with anti-DIG antibodies (1:8000, from Roche Diagnostics, Indianapolis, IN) in pre-blocking solution at 4°C overnight. After 3 washes in 100 mM maleic acid, 150 mM NaCl, 2 mM levamisole, 0.1% Tween20, pH 7.5 for 24 hours, changing the solution approximately every hour, the embryos were incubated in a staining buffer consisting of 100 mM Tris-HCl pH 9.5, 50 mM MgCl2, 100 mM NaCl and 0.1% (v/v) Tween 20, 2 mM levamisole, for varying time, and stained with NBT/BCIP (Roche Diagnostics, Indianapolis, IN). Staining reaction was stopped by washing the embryos in PBST pH 5,5.
Western blot
Detection of Gli3 processing was done by western blotting as previously described (Wang et al., 2000). Briefly, E10.5 whole mouse embryos were lysed with ice-cold RIPA buffer and 7% SDS-PAGE was employed to resolve Gli3-190kD from Gli3-83kD using an affinity-purified antibody directed against Gli3 (Wang et al., 2000). Densitometry was done with NIH Image.
Results
Lethality associated with homozygosity for the Med1 Δ1-3 allele but not Δ2-5 allele
We previously described the generation of engineered mouse strains bearing two different inactive alleles at the Med1/Mbd4 DNA repair locus, the Δ1-3 and Δ2-5 alleles, with targeted deletions of exons 1 through 3 and 2 through 5, respectively (Cortellino et al., 2003) (Fig. 1A). When F2 Med1+/- mice were interbred, the resulting offspring for the Δ2-5 allele were obtained according to the expected Mendelian ratio (Table 1); homozygous Δ2-5 mice are viable and fertile. To the contrary, no livebirth Δ1-3 homozygotes were derived, while wild type and Δ1-3 heterozygous mice were produced at a 1:2 ratio (Table 1). This indicates that embryonic lethality is associated with homozygosity for the Med1 Δ1-3 allele.
Figure 1. The targeted Med1 Δ1-3 allele impairs the expressions of both Ift122 and Med1.

(A) Schematic representation of wild type, Δ1-3 and Δ2-5 alleles and their corresponding targeting constructs. Arrow heads, labeled m1-m2 and i1-i2, correspond to oligonucleotides used for analysis of Med1 and Ift122 expression in lines of various genotypes. (B) Schematic of the organization of the overlapping 5′ ends of the murine Ift122 (in red) and Med1 (in blue) genes. This region is contained within the HindIII-BglII fragment that, in the Δ1-3 allele, is replaced by the selectable cassette upon homologous recombination in ES cells. Arrows indicate the direction of transcription. The position of the respective ATG codons is marked. The shading in Ift122 and Med1 exon 1 refers to the presence of multiple transcription start sites, whose position is schematically indicated by asterisks. (C) RT-PCR analysis of mRNA abundance of Ift122 and Med1 in MEFs with various genotypes. Size of the bands in base pairs is shown. Control lanes (c) are reactions in which cDNA was omitted. The asterisks mark primers.
Table 1.
Genotype analysis of offspring from matings of Δ1-3 and Δ2-5 heterozygotes: the Δ1-3 allele is lethal in homozygosity
| +/Δ1-3 × +/Δ1-3 | |||
|---|---|---|---|
| Genotype | Livebirth mice | Percent of total observed | Percent of total expected |
| +/+ | 389 | 36.15 | 25 |
| +/Δ1-3 | 687 | 63.85 | 50 |
| Δ1-3/Δ1-3 | 0 | 0 | 25 |
| +/Δ2-5 × +/Δ2-5 | |||
| Genotype | Livebirth mice | Percent of total observed | Percent of total expected |
| +/+ | 37 | 26.2 | 25 |
| +/Δ2-5 | 64 | 45.4 | 50 |
| Δ2-5/Δ2-5 | 40 | 28.4 | 25 |
Structure of the Med1 locus: Med1 and the overlapping gene Wdr10/Oseg1/Ift122
Two other Med1/Mbd4 knock-out mice recently described, bearing an exon trapping cassette downstream of exon 1 (Millar et al., 2002; Wong et al., 2002) or an exon 3 deletion (Millar et al., 2002; Wong et al., 2002) are viable and fertile, much like mice homozygous for the Δ2-5 allele. One thing in common among these three viable alleles is that they do not compromise exon 1. This raises the possibility that, in addition to Med1 itself, the Δ1-3 allele perturbs a neighboring gene, closely linked to Med1, causing lethality.
Interrogation of the human and mouse genomic resources (NCBI Entrez Genome and Celera) revealed the existence of the poorly characterized Wdr10 gene closely linked to Med1 and oriented in the opposite direction. The human WDR10 gene was cloned in a search for novel steroid hormone receptor genes by degenerate PCR; however, it bears no resemblance to steroid hormone receptors. The human WDR10 and murine Wdr10 genes comprise 31 exons with alternatively spliced forms. The encoded protein isoforms have a predicted molecular weight of approximately 140 kDa and are characterized by seven WD-repeats at the N-terminus (Gross et al., 2001).
In the course of these studies, a comparative genomic analysis of ciliated vs. non-ciliated organisms led to the identification of approximately 200 genes highly conserved and specific for ciliated eukaryotes across several phyla, including the fly Wdr10 ortholog CG7161, that was renamed Oseg1, due to the localization of the encoded protein to the outer segment of D. melanogaster sensory cilia (Avidor-Reiss et al., 2004). It was later recognized that Wdr10/Oseg1 is the homolog of the Chlamydomonas Ift122 gene involved in flagella biogenesis (http://www.ncbi.nlm.nih.gov/sites/entrez?Db=gene&Cmd=ShowDetailView&TermToSearch=55764&ordinalpos=1&itool=EntrezSystem2.Pentrez.Gene.GeneResultsPanel.Gene_RVDocSum). An Ift122 ortholog exists also in C. elegans (the F23B2.4 gene), which, based on its mutants, is also known as DAF-10 for abnormal DAuer Formation, or OSM-4 for abnormal OSMotic avoidance.
Altered expression of Med1 and Ift122 mRNA in cells homozygous for the Δ1-3 allele
Careful sequence analysis of the NCBI Entrez Genome and Celera databases indicated that MED1 and IFT122 mRNAs are separated by a very small region of approximately 100-300 nucleotides in both humans and mice, and are oriented in opposite directions in a head-to-head arrangement (Fig. 1B). This region is likely to encompass a minimal, bidirectional promoter. Based on the sequence of expressed sequence tags (ESTs) present in the databases, it appears that there are multiple transcriptional initiation sites for both mRNAs in the human and mouse genome (Fig. 1B).
Remarkably, at least at the murine locus, an Ift122 transcript has been identified in which exon 1 sequences overlap in the opposite orientation with sequences from the Med1 exon 1 (Fig. 1B). Both the 100-300 nucleotides of the putative bidirectional promoter and the entire Med1 and Ift122 exon 1 are contained within the Δ1-3 deletion, specifically within the deleted HindIII-BglII fragment which is replaced by the neomycin cassette upon recombination in ES cells (Fig. 1B). With the exception of one isoform that originates from a promoter located in the intron 16 between exons 16 and 17, all the reported alternatively spliced Ift122 isoforms include exon 1.
To test the possibility that the Δ1-3 allele alters the expression of both Med1 and the Ift122 transcripts containing exon 1, we conducted reverse transcriptase (RT)-PCR experiments on mouse embryo fibroblasts (MEFs) of different Δ1-3 genotype using primers located on the exons 1 and 3 of each gene. We also analyzed the Med1 and Ift122 expression in MEFs of different Δ2-5 genotype, due to the possibility that altering the structure of Med1 downstream of its exon 1 may also have an impact on Ift122 transcription. The results indicated that indeed the Δ1-3 mutation abrogates the expression of both Med1 and Ift122, while the Δ2-5 allele inactivates Med1 alone (Fig. 1C).
Expression of Med1 and Ift122 in normal development
Human IFT122 mRNA is expressed in multiple tissues, with high levels of expression in testis and pituitary (Gross et al., 2001). In order to gain some insight in Ift122, we analyzed its expression during development. Northern blot analysis with probes specific for each gene was conducted on RNA extracted from mouse embryos ranging from E4.5 to E18.5 (E4.5 to E6.5 RNAs include decidua). With the latter caveat on the inclusion of decidua, results revealed expression of Ift122 mRNA in all the stages analyzed with lower levels at E7.5-E9.5 (Fig. 2A, top). At E15.5-18.5, detection of faster migrating band(s), in addition to the main band, is consistent with alternatively spliced Ift122 isoforms (Gross et al., 2001). Weaker expression of Med1 mRNA was observed, with very low/undetectable levels at E8.5-E9.5 (Fig. 2A, center). The similar pattern of expression of the two genes is consistent with their transcription from a single, bidirectional promoter. In situ hybridization using gene specific, non-overlapping probes for Ift122 and Med1 were performed on E11.5 embryos. The expression of these two genes appears to be ubiquitous at this stage (Fig. 2 B, C and data not shown).
Figure 2. Expression of Ift122 and Med1 during mouse development.

Northern blot analysis (A) with probes specific for Ift122 (top) and Med1 (center). Migration of the 28S and 18S rRNA is indicated, and their amount shown (bottom). Asterisks refer to the fact that embryo preparations from E4.5 to E6.5 include decidua. In situ hybridization reveals expression of Ift122 throughout an E11.5 embryo using an antisense probe (B); negative control hybridization with a sense probe is shown in (C). Similar labeling was observed on the right and left side of embryo.
Phenotype of embryos homozygous for the Δ1-3 allele
We next examined in detail the lethality of embryos homozygous for the Δ1-3 allele. Timed matings were set up between Δ1-3 heterozygotes, and pregnant mice were sacrificed at different gestation times, ranging from E8.5 to E14.5. Analysis of embryos at different gestation times (Table 2) revealed that while Δ1-3 homozygous E8.5 embryos are morphologically normal, morphological defects appear as early as E9.5, and are characterized by elongated neck region and delay in turning (Fig. 3). Lethality manifests in midgestation, between E11.5-E13.5 (Table 2). Embryos homozygous for the Δ1-3 allele ranging from E10.5 to E13.5 showed a complex phenotype characterized primarily by a defect in neural tube closure (Table 2) (Fig. 3). The rostral neural tube defect, or exencephaly (corresponding to human anencephaly), was particularly dramatic and was associated with altered eye development, defects of the ventral portion of the head and altered development of branchial arches. Additional macroscopic defects included delay in limb development, defect in abdominal wall closure, enlarged heart and presence of diffuse hemorrhagic lesions (Fig. 3).
Table 2.
Number, percent and phenotype of embryos obtained from matings between Δ1-3 heterozygotes: earlier manifestations of lethality occur at E11.5/E12.5.
| Genotype Stage |
+/+ | +/Δ1-3 | Δ1-3/Δ1-3 | Phenotype of Δ1-3 homozygous embryos |
|---|---|---|---|---|
| E8.5 | 6 | 15 | 4 | None |
| E9.5 | 8 | 17 | 6 | Elongated neck, turning defect |
| E10.5 | 73 (25.8%) |
143 (50.5%) |
67 (23.7%) |
Complex |
| E11.5 | 37 (29.4%) |
62 (49.2%) |
27 (21.4%) |
Complex |
| E12.5* | 73 (29.4%) |
130 (52.4%) |
45 (18.1%) |
Complex and resorption |
| E13.5* | 27 (36%) |
38 (50.7%) |
10 (13.3%) |
Complex and resorption |
| E14.5* | 4 | 6 | 0 | Resorption |
| E15.5* | 5 | 2 | 0 | Resorption |
Dissections of embryos at E12.5 and later reveal increasing evidence of resorption.
Figure 3. Macroscopic phenotype of Δ1-3 homozygous embryos.

In each panel, the left side views of control littermate (left) and mutant embryo (right) are shown. (A) : E9.5, (B) : E10.5, (C) : E11.5 and (D) : E12.5. (E) Ventral and (F) dorsal views of E12.5 wild type and mutant embryos.
At later gestation times, Δ1-3 homozygous embryos are usually resorbed, because we observed only one Δ1-3 homozygous E14.5 embryo with exencephaly. Thus, a reduced (non-mendelian) number of Δ1-3/Δ1-3 homozygotes is observed from E14.5 to birth (Table 2). The causes of death are presumably related to the heart defect and diffuse hemorrhagic lesions.
Defects in left-right asymmetry in mutant embryos are associated with the lack of nodal cilia
We observed that approximately 40% of the mutant embryos showed left-ward looping of the heart and right-sided location of the stomach (Fig. 4). This defect, known as situs viscerum inversus, or localization of the internal organs in the opposite side with respect to the medial axis of the organism, underlies a failure in the left-right determination during gastrulation. In this phase, movement of nodal cilia is required to break symmetry and establish left-right asymmetry either by induction of fluid movement across the node sensed by mechanosensory cilia (the “two-cilia” hypothesis), or by unidirectional (leftward) transport of morphogen(s) (Tabin, 2006). In either case, immotile or defective cilia cannot break the symmetry, eliciting randomization of left-right determination, resulting in situs viscerum inversus (Tabin, 2006).
Figure 4. Randomization of left-right asymmetry and situs viscerum inversus in Δ1-3 homozygous embryos.
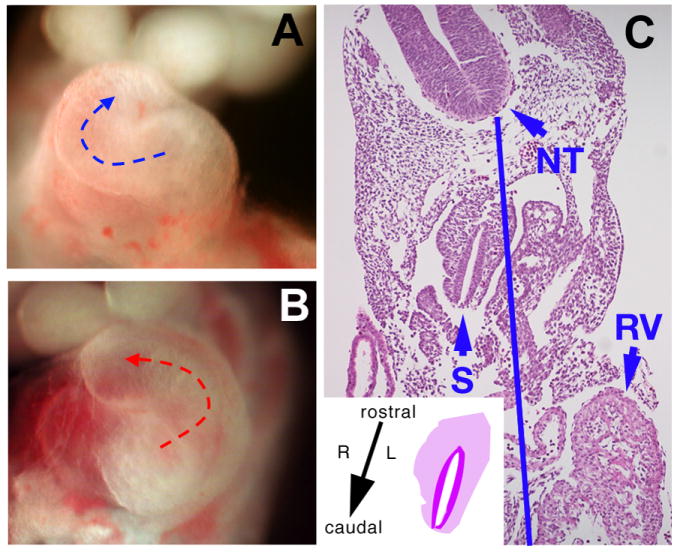
Close-up of the heart in dissected E10.5 wild type (A) and Δ1-3 homozygous embryos (B). Mutant embryos can show normal heart looping, as in wild type embryos – blue arrow - (A) or left-ward looping – red arrow - (B), resulting in dextrocardia. (C): histological section of an Ift122-mutant E10.5 embryo with situs viscerum inversus, showing the right-sided location of the stomach (S); the blue line is an ideal midline joining the center of the floor plate of the rostral (NT) and caudal (not shown) neural tube. In (C), the left-sided location of the right ventricle (RV) is also apparent. Inset in (C) shows a schematic of the transposed stomach with its rostro-caudal, left-right orientation, which is the mirror image of the normal rostro-caudal, right-left orientation.
The role of the homologous genes Oseg1 and Ift122 in ciliogenesis in Drosophila and Chlamydomonas, respectively, suggests that the asymmetry defect and situs viscerum inversus observed in the Δ1-3 homozygous embryos might be due to impairment of the Ift122 function in node ciliogenesis. Visualization of the node from E8.0 embryos performed by SEM revealed that the nodal surface of Δ1-3 homozygous embryos is devoid of cilia in contrast to the node of wild type embryos that is covered with normal cilia (Fig. 5). Nodal cilia appear to be shorter in Δ1-3 heterozygous embryos relative to wild type embryos (Fig. 5). Despite the shorter nodal cilia, no evidence of situs viscerum inversus was noted in midgestation Δ1-3/+ heterozygous embryos (or later, in Δ1-3/+ heterozygous adult mice).
Figure 5. Lack of nodal cilia in Δ1-3 homozygous embryos.

Scanning electron microscope images at different magnification of E8 wild type (a, d, g, j), Δ1-3 heterozygous (b, e, h, k) and Δ1-3 homozygous (c, f, i, l) embryos. Cilia are absent in Δ1-3 homozygous embryos, and are apparently shorter than wild type in Δ1-3 heterozygous embryos. On the nodal surface of Δ1-3 homozygous embryos, only small structures are present (spheres), that are either nodal vesicle particles (NVPs) or defective cilia. Scale bars: 50 μm (a, b, c), 20 μm (d, e, f), 10 μm (g, h, i), 2 μm (j, k, l)
In order to assess if the cilia defect was constrained to the node or affected other tissues, mouse embryo fibroblasts (MEFs) with different Δ1-3 genotype, prepared from E12.5 embryos, were studied. Serum starvation of the MEFs for 48 hours leads to cell cycle arrest and induces formation of cilia on the cell surface (Wheatley, 1995; Wheatley et al., 1996). In starved wild type MEFs, immunostaining with an antibody against acetylated tubulin revealed a single cilium adjacent to one of the basal bodies in the majority of cells (Fig. 6A-C); to the contrary, in Ift122-null, Δ1-3/Δ1-3 homozygous MEFs, the same antibody produced a network-like background staining but no evidence of cilia (Fig. 6E-F). Consistent with the lack of embryonic lethality associated with homozygosity for the Med1 Δ2-5 allele, cilia were present in Δ2-5/Δ2-5 homozygous MEFs (Fig. 6G-I).
Figure 6. Lack of primary cilia in Δ1-3 but not Δ2-5 homozygous mouse embryo fibroblasts.
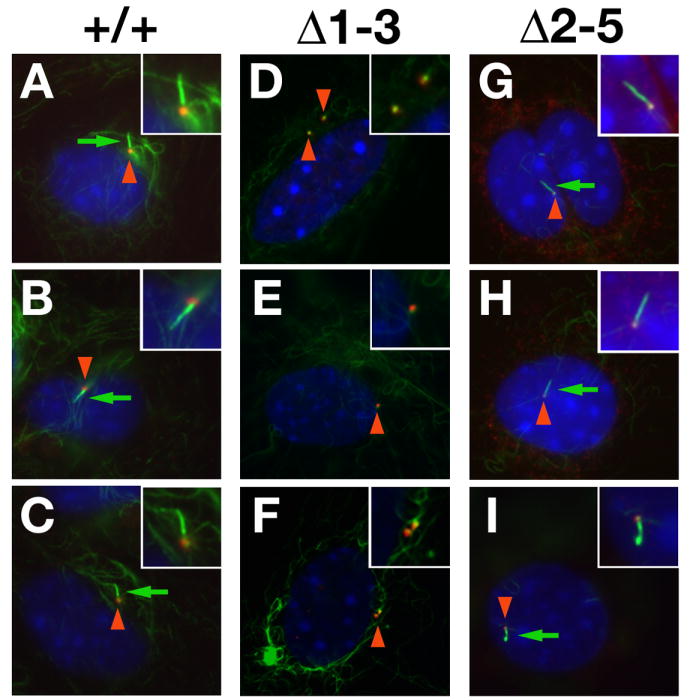
Wild type (A-C), Δ1-3 (D-F) and Δ2-5 homozygous (G-H) MEFs, serum starved for 48 hours, were immunostained with antibodies against acetylated tubulin (green) and γ-tubulin (red), whereas nuclei were counterstained with DAPI. Basal bodies (red arrowheads) were detected in both genotypes whereas primary cilia (green arrows) were only detected in wild type and Δ2-5 MEFs. Insets show enlargements of basal bodies and cilia.
Deletion of Ift122 impairs sonic hedgehog (Shh) signaling during neuronal patterning
Recently, it has been proposed that, in mammalian cells, ciliogenesis or, minimally, IFT proteins involved in cilia formation are required for signaling by Sonic Hedgehog (Shh) (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; Nonaka et al., 1998). Since the phenotypes observed in Δ1-3 homozygous embryos resemble those of complex B Ift mutant embryos, such as Ift88/polaris, Ift172/wimple, Ift52 (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; Nonaka et al., 1998), and the developmental processes aberrant in these embryos are mediated by the Shh pathway, we investigated whether the complex A protein Ift122 is required for Shh signaling.
We first examined neuronal patterning, because Shh plays a key role in the specification of neuronal cell fate during development. The release of Shh, initially from the notochord and later from floor plate cells, at the ventral midline of the neural tube, generates a gradient of this morphogen along the dorsal-ventral axis that leads to differentiation of progenitor neural cells in five classes of neurons: four ventral interneurons (V0-V3) and motor neurons (MN). Impairment of Shh signaling would cause dorsalization of neuronal patterning due to the lack of ventral neuron specification (Briscoe et al., 2000; Briscoe et al., 1999; Ericson et al., 1997a; Ericson et al., 1997b; Pierani et al., 1999).
Analysis of neuronal patterning was performed on E10.5 embryos, using immunohistochemical detection of the markers Shh, Foxa2, the three homeodomain proteins Nkx2.2, Pax3 and Pax6, and the two motoneuron markers MNR2 and Isl1. Normally, expression of Nkx2.2 is triggered by high concentrations of Shh and gives rise to V3 interneurons, whereas Pax6 is expressed dorsally and up to the V0-V2 interneurons (Briscoe and Ericson, 2001; Ericson et al., 1997b; Jessell, 2000; Shirasaki and Pfaff, 2002; Tanabe et al., 1998). In the rostral neural tube (at forelimb level) of Ift122-null Δ1-3 homozygous embryos, Shh was detected only at the level of the notochord and was absent in the floor plate (Fig. 7D). Consistent with the reduced Shh expression, there were no Foxa2- and Nkx2.2-expressing V3 interneural cells (Fig. 7E-F), with ventral expansion of the dorso-lateral neurons expressing Pax6 and Pax3 (Fig. 7M-N). Ventral expansion of the Pax6-expressing neurons did not occur in caudal neural tube (at hind limb level) (Fig. 7R-T), where Nkx2.2 expression was maintained, albeit at reduced levels compared to wild type (Fig. 7R).
Figure 7. Impairment of Sonic hedgehog-mediated neuronal patterning in Ift122-null embryos.

Immunohistochemical detection of the indicated markers in transversal sections of neural tube from E10.5 wild type (+/+) and Ift122 mutant (-/-) embryos at rostral (forelimb level) and caudal (hind limb level) sites. In panels A-N, the boundaries of the neural tube are indicated by the dotted line; in panels A-B, the outline of the notochord is also marked. In mutant embryos, at rostral levels of the neural tube, Shh expression is absent in the floor plate (fp) and accompanied by undetectable Foxa2 and Nkx-2.2 expression, leading to ventral expansion of lateral neurons expressing Pax6 and Pax3, while the MNR2- and Isl1-expressing motoneurons expand both ventrally and laterally. At caudal levels of the neural tube, Nkx2.2-expressing neurons are present in mutant embryos, preventing ventral expansion of the Pax6-expressing neurons, whereas ventral and lateral expansion of the MNR2- and Isl1-expressing motoneurons does take place.
This phenotype is similar to the clear dorsalization of the neural tube observed in Ift complex B mutant embryos, which is accompanied by the disappearance of ventral motoneurons (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; May et al., 2005; Nonaka et al., 1998). However, staining with the motoneuron markers MNR2 and Isl1 revealed unexpectedly that motoneurons expand ventrally and laterally in neural tubes from Ift122-null embryos (Fig. 7K-L, S-T). This ventrolateral expansion of motoneurons has been previously reported for hennin embryos, defective for the ciliary protein Arl13b (Caspary et al., 2007). However, unlike hennin embryos, the motoneuron expansion was apparent both in the rostral (Fig. 7K-L) and caudal (Fig. 7S-T) regions of the neural tube.
These observations demonstrate a complex Shh neuronal patterning defect in Ift122-null embryos; in particular, ventral expansion of dorso-lateral neurons with absence of Nkx2.2-expressing cells and ventrolateral expansion of motoneurons are consistent with reduced production of both Gli2A/Gli3A and Gli3R in neural tubes from Ift122-null embryos.
Inhibition of Shh signaling underlies alterations of limb patterning in Ift122-null embryos
The Shh signaling pathway plays a key role in anterior-posterior limb patterning and digit formation. Specifically, the posteriorly located zone of polarizing activity (ZPA), generates a spatial-temporal gradient of Shh that affects the expression of transcription factors along the antero-posterior axis. In particular, high concentration of Shh inhibits the proteolytic processing of full length Gli3 (Gli3A, a transcriptional activator) into Gli3 repressor (Gli3R), which is a major player of Shh signaling in the determination of anterior posterior patterning (McGlinn and Tabin, 2006).
Whereas lack of Shh during limb bud morphogenesis in mouse results in overt delay of limb development and complete loss of the ulna and digits 2 to 5 (Chiang et al., 2001; Kraus et al., 2001), in IFT mutant embryos, such as embryos defective for Ift88/polaris, Ift172/wimple, Ift52, as well as dynein heavy chain Dnchc2, or the Kif3A and Kif3B subunits of kinesin II, the proteolytic processing of Gli3 into Gli3R is compromised, and the reduced levels of Gli3R lead to extra digit formation (polydactyly) (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; May et al., 2005; Nonaka et al., 1998). Indeed, the limb phenotype observed in IFT mutant embryos resembles that of Gli3-null embryos (Hui and Joyner, 1993).
We evaluated the limb phenotype in Ift122-null embryos. In E10.5 Δ1-3 homozygous embryos, Shh was expressed in its normal posterior domain, which appeared to be shifted distally, due to the developmental delay in limb outgrowth (Fig. 8B, B’), as Ift122 mutant limbs are much smaller than wild type limbs. At E11.5, in addition to its normal posterior expression, ectopic (anterior) Shh expression was detected (Fig. 8D, D’); ectopic expression of Shh was also noticed in the hind limbs of E12.5 embryos mutant, whereas at this stage Shh expression is shut down in wild type fore- and hind limbs (Fig. 8E-F). However, both at the ZPA and the ectopic domain, Shh signaling was impaired, as shown by reduced expression of the Shh target gene Ptc1 in mutant limbs (Fig. 8H, H’), consistent with reduced Gli2A function, similar to IFT88 hypomorphic mice (Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; May et al., 2005; Nonaka et al., 1998). Because of the patterning defect, we evaluated the expression of the homeotic genes Hoxd11 and Hoxd13. In Ift122-mutant limbs, expression of these patterning genes was shifted anteriorly from the normal expression in the posterior mesenchyme (Fig. 8I, J and data not shown)). Similarly, when we analyzed the apical ectodermal ridge (AER), the other major signaling center together with ZPA, Fgf4 expression was expanded to the entire AER, instead of being limited to its posterior portion (Fig. 8K, L).
Figure 8. Inhibition of Sonic hedgehog signaling and alterations of limb patterning in Ift122-null embryos.

Detection by in situ hybridization of Shh (A-F’),Ptc1 (G-H’), Hoxd13 (I-J), Fgf4 (K-L), Col2a1 (M-N’), Fgf8 (O-P), Bmp4 (Q-R) and Bmp2 (S-T), in limbs of wild type and Δ1-3 homozygous embryos at the indicated developmental stages. Forelimbs are shown in A-F and G-H’, whereas hind limbs are shown in A’-F’ and I-T. In all cases, right limbs are shown with the exception of G, H, M and N, in which the left limb is shown. In panels N and N’, marking of the cartilage with a Col2a1 probe in Ift122-null embryos reveals polydactylous syndactyly (indicated by arrowheads) and features of ectrodactyly (marked by an asterisk). In panels S and T, the boundaries of Bmp2 expression in the AER are indicated by arrowheads.
In order to assess the number and morphology of the digits, we conducted an in situ hybridization with a Col2a1 (collagen II a1) probe, an early cartilage marker (Ng et al., 1997). While no Col2a1 was detected in mutant forelimbs, the pattern of expression in the hind limbs revealed features of fusion of digits that were supernumerary (polydactylous syndactyly) (Fig. 8N, N’, arrowheads). Malformed, claw-like digits were also present (ectrodactyly) (Fig. 8N’, asterisk).
Since the outgrowth of the Ift122-null limbs is delayed, we analyzed the integrity of the AER, whose main function is to regulate development along the proximal-distal axis of the limb. Of the four mouse FGF genes (Fgf4, -8, -9, and -17) known to display AER-specific expression domains within the limb bud (reviewed by Martin, 1998), only Fgf8 is expressed throughout the AER and its inactivation affects skeletal patterning, with a substantial reduction in limb-bud size, suggesting a unique function early in limb development (Lewandoski et al., 1997). Ift122-mutant embryos show a reduced expression of Fgf8 in the AER (Fig. 8P), suggesting that the reduced limb outgrowth is due to low Fgf8 expression. It remains to be determined whether the reduced Fgf8 expression is a reflection of the general developmental delay of the embryos.
In the developing limb, a positive feedback loop between Shh and Fgf family members maintains the expression of the former in the ZPA and of the latter in the AER, contributing to AER integrity (McGlinn and Tabin, 2006). Bone morphogenetic proteins (BMPs) play a different and antagonist roles at different stage of limb development, however at AER level inhibit Fgf signaling and interrupt the positive loop with Shh (McGlinn and Tabin, 2006). We then analyzed the expression of Bmp2 and Bmp4, which are normally expressed in the AER and in the posterior part of the limb mesenchyme at 11.5E (Fig. 8Q, S). In Ift122-mutant limbs, Bmp4 and Bmp2 expression was nearly normal or reduced in the AER, respectively, but completely absent in the posterior mesenchyme (Fig. 8R, T). These findings suggest that the Shh/Fgf loop is likely perturbed in Ift122-mutant limbs.
Since the levels of Gli3R are critical in anterior-posterior limb patterning, we investigated the processing of Gli3 in wild type and Δ1-3 homozygous embryos by Western blotting. Lack of Ift122 impairs dramatically the formation of Gli3R in comparison to wild-type embryos (Fig. 9A). Furthermore, quantification of the unprocessed and repressor forms of Gli3 revealed that this ratio in Ift122-null embryos (Fig. 9B) is comparable to that observed in embryos mutant for other IFT proteins (Huangfu and Anderson, 2005; Liu et al., 2005; Wang et al., 2000).
Figure 9. Altered Gli3 processing in Δ1-3 homozygous embryos.

(A) Detection by western blotting with an antibody directed against Gli3 of the full-length unprocessed (190 kD) and repressor (83 kD) forms of Gli3. Western blotting with an antibody directed against α-tubulin provides the loading control. (B) Densitometric quantitation of the western blotting data in (A) of the ratio Gli3-190kD/Gli3-83kD in wild type (0.29) and Ift122-null embryos (1.39). In comparison, the ratio Gli3-190kD/Gli3-83kD in wild type and Ift88-null embryos is 0.39 and 1.13, respectively (Huangfu and Anderson, 2005; Liu et al., 2005; Wang et al., 2000).
We conclude that the reduction in Gli2A and Gli3R levels in Ift122-null embryos, and of Fgf8 and Bmp2/4 may contribute to the severe phenotype observed in forelimbs, distinct from the simple polydactyly observed in other IFT mutant embryos.
Discussion
Until few years ago the primary cilia were considered only vestigial structures in higher organisms without any specific functions. Recently these organelles were reconsidered and revalued for their role in physiology and development (Davenport and Yoder, 2005; Rosenbaum and Witman, 2002). Our work adds to the functions of cilia formation in development and signaling pathways by characterizing the role of the complex A intraflagellar transport protein IFT122 in the mouse and demonstrating its requirement for both cilia formation and Shh signaling.
Disruption of Ift122 in mouse embryos prevents the formation of nodal cilia causing randomization of the left-right asymmetry and situs viscerum inversus in approximately 40% of the mutant embryos. Randomization of the left-right asymmetry could be due to the absence of the cilia-generated leftward fluid movement across the node or result from leftward transport of morphogen(s) (Tabin, 2006). Either process appears to be effected normally by the short cilia detected in the node of heterozygous embryos, because no evidence of situs viscerum inversus was found in heterozygous embryos or adult mice. Thus, the analysis of Ift122-null and heterozygous embryos may help disclose the precise role of ciliary proteins in establishing left-right asymmetry.
Neuronal patterning was impaired in Ift122-null embryos with a neural tube level-dependent phenotype, suggesting a complex impairment of Shh activity. In caudal neural tube of Ift122-null embryos, the phenotype resembled that of the caudal neural tube of hennin (hnn) mutant embryos (Tabin, 2006)): i) the Nkx2.2-expressing V3 interneurons were specified but reduced in number; ii) the domain of Pax6-expressing lateral interneurons was confined to the dorsal one third of the neural tube; iii) the motoneurons expanded ventrolaterally to occupy the ventral two thirds of the neural tube. The interpretation of this phenotype, based on a Ptc1-lacZ reporter, has been that in the caudal neural tube of hnn mice, Shh activity has a shallow ventro-dorsal gradient on an expanded domain of expression (Caspary et al., 2007). This may result from an altered GliA/GliR ratio, and in particular from reduced production of Gli2A and Gli3A in hnn (Arl13b-defective) cilia, whereas processing of Gli3R is normal (Caspary et al., 2007). It is possible that a similar explanation holds true also for Ift122-mutant mice, at least at the level of the caudal neural tube. It should be emphasized that some of the phenotypic differences between the Ift122-mutant and hnn embryos might be due to the genetic background, being C57/BL6 for the former and C3H for the latter (Caspary et al., 2007).
However, in the rostral neural tube of Ift122-null embryos, a unique phenotype is detected, characterized by a double expansion: while motoneurons expand ventrolaterally as in the caudal neural tube of Ift122-mutant and hnn embryos, Pax6- and Pax3-expressing lateral neurons also expand ventrally (and Nkx2.2 V3 interneurons are not specified), as in IFT complex B mutant embryos (Corbit et al., 2005; Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; Nonaka et al., 1998; Tanaka et al., 2005). This unique Ift122-mutant phenotype may also derive from altered GliA/GliR ratio, and specifically from reduced production of both Gli2A/Gli3A, as well as Gli3R in Ift122-null embryos (Fig. 9), whereas in IFT complex B mutant embryos the reduced production of GliR may be accompanied by no production of GliA (Corbit et al., 2005; Huangfu and Anderson, 2005; Huangfu et al., 2003; Liu et al., 2005; Marszalek et al., 1999; Nonaka et al., 1998; Tanaka et al., 2005).
It is important to emphasize that the unique phenotype of Ift122-null embryos is also distinct from that of the THM1/Ift139 complex A mutant embryos that exhibit hyperactivation of Shh and ventralization of the neural tube (Tran et al., 2008). In the latter mutant, ciliary anterograde transport is functional whereas retrograde transport is defective, leading to the hypothesis that anterograde transport is necessary for GliA function and retrograde transport is required for modulation of the activation step (Tran et al., 2008). Based on these considerations and the likely reduced production of GliA in Ift122-mutant embryos, it is possible that, as a complex A component, IFT122 is involved in retrograde transport similar to THM1/IFT139, but, unlike THM1/IFT139 is also involved in anterograde transport.
Whereas the aberrant anterior-posterior patterning leads to polydactyly in other IFT mutant embryos, a severe delay in forelimb development and ectrosyndactyly occur specifically in Ift122-null embryos, suggesting a possible functional hierarchy among IFTs. Since Gli3 processing, as revealed by Western Blot analysis, appears to be altered in Ift122-null embryos at approximately the same extent (Fig. 9) compared to other IFT mutant embryos, such as Ift88 mutant embryos (Huangfu and Anderson, 2005; Liu et al., 2005), it is likely that the low amount of Gli3R in Δ1-3 homozygous embryos is the main determinant of the severely impaired limb development. Indeed, the human GLI3 gene is mutated in Greig cephalopolysyndactyly syndrome (Vortkamp et al., 1991) and polydactylous syndactyly has been described for mice bearing Brachyphalangy (Johnson, 1969), an allele of Gli3 (Hui and Joyner, 1993). A possible explanation of the differences with the IFT complex B mutants is that some aspects of the limb phenotype in Ift122-null embryos, including the syndactyly, may be caused by the low levels of Bmp2 and Bmp4 in the limb mesenchyme (Guha et al., 2002). In addition, it is possible that the Ift122 deficiency interferes with the Shh-Fgf positive loop, leading to reduced Fgf8 expression and limb outgrowth.
The role of IFT in Shh signaling can be explained by a model, proposed by Yoder and colleagues (Haycraft et al., 2005), in which IFT particles have the main function to localize components of Shh pathway, along with the proteolytic machinery involved in Gli processing, to the tips of cilia. In this model, IFT particles affect indirectly the Shh pathway, as the absence of cilia would lead to a failure in the processing of Gli proteins that will be dispersed in the cytoplasm in their full-length form. This model fails to explain why different IFT mutants, including Ift122 and the motor proteins Kif3 and Dnchc2, affect the processing of Gli3 with considerable variation, unless we hypothesize a different impact of the various mutants on anterograde and retrograde transport, or on the type of cargo transported.
In an alternative model, considering that in Chlamydomonas IFT particles take place in signaling organization in the flagella (Wang et al., 2006), we cannot exclude that IFT particles are involved not only in the assembling of cilia but also in the organization of signaling pathways in vertebrate cells. Given that the phenotype of Ift122-null embryos is different from other IFT mutants, one possible explanation could be that each IFT protein carries out specific role(s) in different pathways in addition to their role in Shh signaling. Some possible pathways specifically regulated by Ift122 may include Indian hedgehog and Desert hedgehog (Scholey and Anderson, 2006). Also, given the interplay of ciliogenesis and planar cell polarity/Wnt pathway (Scholey and Anderson, 2006; Wallingford, 2006), it is possible that the latter is compromised in Ift122 mutant embryos. Finally, the syndactyly and ectrodactyly may suggest a possible involvement of the Notch (Sidow et al., 1997) and p63 (Rinne et al., 2007) pathways, respectively.
Another hypothesis to take into account is that IFT particles may be involved in other processes, as recently demonstrated for IFT20 which is also involved in transport of ciliary membrane proteins from the Golgi complex to the cilium (Follit et al., 2006). It is possible that the unique defects observed in Ift122-null embryos might be due the lack of specific cilia components processed and produced in other cellular organelles.
Finally, another possibility is that the phenotypic differences might be due to the type of gene inactivation: whereas the Ift122 mutant embryos described here appear to be null (Fig. 1), the limb defects have been previously studied in embryos bearing hypomorphic alleles of Ift88 and Ift52; the limb phenotype of the Ift172-null embryos has not been described. However, simple polydactyly without features of ectrosyndactyly is present in embryos null for Kif3A, Kif 3B, THM1/IFT139 and Dnchc2.
A final consideration concerns the bi-directional organization of the genomic locus containing Ift122 and Med1/Mbd4. It is certainly intriguing that MED1MBD4 and IFT122 share the same promoter region. The close linkage of these two genes is evolutionarily conserved from mouse to rat to humans and seems to have arisen in the lineage leading to mammals. In fact, biased organization was found only in mammals and not in other eukaryotes (Adachi and Lieber, 2002). The function and physiological consequences of this organization is currently unclear. Given the role of MED1 in DNA repair and its nuclear localization, it would seem unlikely that the two proteins belong to a common signaling pathway and have a functional relationship. On the other hand, it is intriguing that left-right dynein was shown recently to be involved in non-random segregation of chromatids in murine ES cells (Armakolas and Klar, 2007). Thus, although highly speculative, MED1 and IFT122 might functionally interact during some DNA transactions. Be as it may, we cannot formally rule out the possibility that the phenotype of Δ1-3 homozygous embryos is caused by inactivation of both Ift122 and Med1, although this possibility seems remote. Future studies will be based on the generation and characterization of embryos/mice bearing Ift122 inactive alleles that do not compromise Med1 expression.
Acknowledgments
We thank M. Fanciulli, E. Golemis, W. Kruger and C. Passananti, for critical reading of the manuscript; S. Litwin for statistical analyses; A. McMahon and M. Barna for ISH probes; and R. Sonlin for secretarial assistance. We thank the following core facilities and personnel at the Fox Chase Cancer Center: Cell Culture (S. Howard), Transgenic and Knock-out (S. Hua), Laboratory Animal (K. Baxter-Jones, T. Lerro, K. Lauff, A.M. Pimble, T. Poole, M. Schoell and J. Stephan), Electron Microscopy (R. Chen) and the Fannie E. Rippel Biotechnology Facility (P. Do, G. Miller and A. Yeung). This work was supported by NIH grants CA78412 and CA06927 and an appropriation from the Commonwealth of Pennsylvania to the Fox Chase Cancer Center; Dr. S. Cortellino is supported in part by an American-Italian Cancer Foundation Fellowship.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adachi N, Lieber MR. Bidirectional gene organization: a common architectural feature of the human genome. Cell. 2002;109:807–809. doi: 10.1016/s0092-8674(02)00758-4. [DOI] [PubMed] [Google Scholar]
- Anderson TF. Technique for the preservation of three-dimensional structure in preparing specimens for the electron microscope. Trans NY Acad Sci Ser 2. 1951;13:130–134. [Google Scholar]
- Armakolas A, Klar AJ. Left-right dynein motor implicated in selective chromatid segregation in mouse cells. Science. 2007;315:100–101. doi: 10.1126/science.1129429. [DOI] [PubMed] [Google Scholar]
- Avidor-Reiss T, Maer AM, Koundakjian E, Polyanovsky A, Keil T, Subramaniam S, Zuker CS. Decoding cilia function: defining specialized genes required for compartmentalized cilia biogenesis. Cell. 2004;117:527–539. doi: 10.1016/s0092-8674(04)00412-x. [DOI] [PubMed] [Google Scholar]
- Bellacosa A, Chan TO, Ahmed NN, Datta K, Malstrom S, Stokoe D, McCormick F, Feng J, Tsichlis P. Akt activation by growth factors is a multiple-step process: the role of the PH domain. Oncogene. 1998;17:313–325. doi: 10.1038/sj.onc.1201947. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Ericson J. Specification of neuronal fates in the ventral neural tube. Curr Opin Neurobiol. 2001;11:43–49. doi: 10.1016/s0959-4388(00)00172-0. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Pierani A, Jessell TM, Ericson J. A homeodomain protein code specifies progenitor cell identity and neuronal fate in the ventral neural tube. Cell. 2000;101:435–445. doi: 10.1016/s0092-8674(00)80853-3. [DOI] [PubMed] [Google Scholar]
- Briscoe J, Sussel L, Serup P, Hartigan-O’Connor D, Jessell TM, Rubenstein JL, Ericson J. Homeobox gene Nkx2.2 and specification of neuronal identity by graded Sonic hedgehog signalling. Nature. 1999;398:622–627. doi: 10.1038/19315. [DOI] [PubMed] [Google Scholar]
- Caspary T, Larkins CE, Anderson KV. The graded response to Sonic Hedgehog depends on cilia architecture. Dev Cell. 2007;12:767–778. doi: 10.1016/j.devcel.2007.03.004. [DOI] [PubMed] [Google Scholar]
- Chiang C, Litingtung Y, Harris MP, Simandl BK, Li Y, Beachy PA, Fallon JF. Manifestation of the limb prepattern: limb development in the absence of sonic hedgehog function. Dev Biol. 2001;236:421–435. doi: 10.1006/dbio.2001.0346. [DOI] [PubMed] [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Christensen ST, Voss JW, Teilmann SC, Lambert IH. High expression of the taurine transporter TauT in primary cilia of NIH3T3 fibroblasts. Cell Biol Int. 2005;29:347–351. doi: 10.1016/j.cellbi.2005.02.003. [DOI] [PubMed] [Google Scholar]
- Corbit KC, Aanstad P, Singla V, Norman AR, Stainier DY, Reiter JF. Vertebrate Smoothened functions at the primary cilium. Nature. 2005;437:1018–1021. doi: 10.1038/nature04117. [DOI] [PubMed] [Google Scholar]
- Cortellino S, Turner D, Masciullo V, Schepis F, Albino D, Daniel R, Skalka AM, Meropol NJ, Alberti C, Larue L, Bellacosa A. The base excision repair enzyme MED1 mediates DNA damage response to antitumor drugs and is associated with mismatch repair system integrity. Proc Natl Acad Sci U S A. 2003;100:15071–15076. doi: 10.1073/pnas.2334585100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davenport JR, Yoder BK. An incredible decade for the primary cilium: a look at a once-forgotten organelle. Am J Physiol Renal Physiol. 2005;289:F1159–1169. doi: 10.1152/ajprenal.00118.2005. [DOI] [PubMed] [Google Scholar]
- Delmas V, Pla P, Feracci H, Thiery JP, Kemler R, Larue L. Expression of the cytoplasmic domain of E-cadherin induces precocious mammary epithelial alveolar formation and affects cell polarity and cell-matrix integrity. Dev Biol. 1999;216:491–506. doi: 10.1006/dbio.1999.9517. [DOI] [PubMed] [Google Scholar]
- Ericson J, Briscoe J, Rashbass P, van Heyningen V, Jessell TM. Graded sonic hedgehog signaling and the specification of cell fate in the ventral neural tube. Cold Spring Harb Symp Quant Biol. 1997a;62:451–466. [PubMed] [Google Scholar]
- Ericson J, Rashbass P, Schedl A, Brenner-Morton S, Kawakami A, van Heyningen V, Jessell TM, Briscoe J. Pax6 controls progenitor cell identity and neuronal fate in response to graded Shh signaling. Cell. 1997b;90:169–180. doi: 10.1016/s0092-8674(00)80323-2. [DOI] [PubMed] [Google Scholar]
- Follit JA, Tuft RA, Fogarty KE, Pazour GJ. The intraflagellar transport protein IFT20 is associated with the Golgi complex and is required for cilia assembly. Mol Biol Cell. 2006;17:3781–3792. doi: 10.1091/mbc.E06-02-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross C, De Baere E, Lo A, Chang W, Messiaen L. Cloning and characterization of human WDR10, a novel gene located at 3q21 encoding a WD-repeat protein that is highly expressed in pituitary and testis. DNA Cell Biol. 2001;20:41–52. doi: 10.1089/10445490150504684. [DOI] [PubMed] [Google Scholar]
- Guha U, Gomes WA, Kobayashi T, Pestell RG, Kessler JA. In vivo evidence that BMP signaling is necessary for apoptosis in the mouse limb. Dev Biol. 2002;249:108–120. doi: 10.1006/dbio.2002.0752. [DOI] [PubMed] [Google Scholar]
- Handel M, Schulz S, Stanarius A, Schreff M, Erdtmann-Vourliotis M, Schmidt H, Wolf G, Hollt V. Selective targeting of somatostatin receptor 3 to neuronal cilia. Neuroscience. 1999;89:909–926. doi: 10.1016/s0306-4522(98)00354-6. [DOI] [PubMed] [Google Scholar]
- Haycraft CJ, Banizs B, Aydin-Son Y, Zhang Q, Michaud EJ, Yoder BK. Gli2 and Gli3 localize to cilia and require the intraflagellar transport protein polaris for processing and function. PLoS Genet. 2005;1:e53. doi: 10.1371/journal.pgen.0010053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Anderson KV. Cilia and Hedgehog responsiveness in the mouse. Proc Natl Acad Sci USA. 2005;102:11325–11330. doi: 10.1073/pnas.0505328102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huangfu D, Liu A, Rakeman AS, Murcia NS, Niswander L, Anderson KV. Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature. 2003;426:83–87. doi: 10.1038/nature02061. [DOI] [PubMed] [Google Scholar]
- Hui CC, Joyner AL. A mouse model of greig cephalopolysyndactyly syndrome: the extra-toesJ mutation contains an intragenic deletion of the Gli3 gene. Nat Genet. 1993;3:241–246. doi: 10.1038/ng0393-241. [DOI] [PubMed] [Google Scholar]
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1:20–29. doi: 10.1038/35049541. [DOI] [PubMed] [Google Scholar]
- Johnson DR. Brachyphalangy, an allele of extra-toes in the mouse. Genet Res. 1969;13:275–280. doi: 10.1017/s0016672300002962. [DOI] [PubMed] [Google Scholar]
- Kraus P, Fraidenraich D, Loomis CA. Some distal limb structures develop in mice lacking Sonic hedgehog signaling. Mech Dev. 2001;100:45–58. doi: 10.1016/s0925-4773(00)00492-5. [DOI] [PubMed] [Google Scholar]
- Lewandoski M, Wassarman KM, Martin GR. Zp3-cre, a transgenic mouse line for the activation or inactivation of loxP-flanked target genes specifically in the female germ line. Curr Biol. 1997;7:148–151. doi: 10.1016/s0960-9822(06)00059-5. [DOI] [PubMed] [Google Scholar]
- Liu A, Wang B, Niswander LA. Mouse intraflagellar transport proteins regulate both the activator and repressor functions of Gli transcription factors. Development. 2005;132:3103–3111. doi: 10.1242/dev.01894. [DOI] [PubMed] [Google Scholar]
- Marszalek JR, Ruiz-Lozano P, Roberts E, Chien KR, Goldstein LS. Situs inversus and embryonic ciliary morphogenesis defects in mouse mutants lacking the KIF3A subunit of kinesin-II. Proc Natl Acad Sci USA. 1999;96:5043–5048. doi: 10.1073/pnas.96.9.5043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin GR. The roles of FGFs in the early development of vertebrate limbs. Genes Dev. 1998;12:1571–1586. doi: 10.1101/gad.12.11.1571. [DOI] [PubMed] [Google Scholar]
- May SR, Ashique AM, Karlen M, Wang B, Shen Y, Zarbalis K, Reiter J, Ericson J, Peterson AS. Loss of the retrograde motor for IFT disrupts localization of Smo to cilia and prevents the expression of both activator and repressor functions of Gli. Dev Biol. 2005;287:378–389. doi: 10.1016/j.ydbio.2005.08.050. [DOI] [PubMed] [Google Scholar]
- McGlinn E, Tabin CJ. Mechanistic insight into how Shh patterns the vertebrate limb. Curr Opin Genet Dev. 2006;16:426–432. doi: 10.1016/j.gde.2006.06.013. [DOI] [PubMed] [Google Scholar]
- Michaud EJ, Yoder BK. The primary cilium in cell signaling and cancer. Cancer Res. 2006;66:6463–6467. doi: 10.1158/0008-5472.CAN-06-0462. [DOI] [PubMed] [Google Scholar]
- Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B, Keightley PD, Bishop SM, Clarke AR, Bird A. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403–405. doi: 10.1126/science.1073354. [DOI] [PubMed] [Google Scholar]
- Ng LJ, Wheatley S, Muscat GE, Conway-Campbell J, Bowles J, Wright E, Bell DM, Tam PP, Cheah KS, Koopman P. SOX9 binds DNA, activates transcription, and coexpresses with type II collagen during chondrogenesis in the mouse. Dev Biol. 1997;183:108–121. doi: 10.1006/dbio.1996.8487. [DOI] [PubMed] [Google Scholar]
- Nonaka S, Tanaka Y, Okada Y, Takeda S, Harada A, Kanai Y, Kido M, Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- Pazour GJ, Baker SA, Deane JA, Cole DG, Dickert BL, Rosenbaum JL, Witman GB, Besharse JC. The intraflagellar transport protein, IFT88, is essential for vertebrate photoreceptor assembly and maintenance. J Cell Biol. 2002;157:103–113. doi: 10.1083/jcb.200107108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazour GJ, Witman GB. The vertebrate primary cilium is a sensory organelle. Curr Opin Cell Biol. 2003;15:105–110. doi: 10.1016/s0955-0674(02)00012-1. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Geimer S, Rosenbaum JL. Dissecting the molecular mechanisms of intraflagellar transport in chlamydomonas. Curr Biol. 2006;16:450–459. doi: 10.1016/j.cub.2006.02.020. [DOI] [PubMed] [Google Scholar]
- Pierani A, Brenner-Morton S, Chiang C, Jessell TM. A sonic hedgehog-independent, retinoid-activated pathway of neurogenesis in the ventral spinal cord. Cell. 1999;97:903–915. doi: 10.1016/s0092-8674(00)80802-8. [DOI] [PubMed] [Google Scholar]
- Praetorius HA, Praetorius J, Nielsen S, Frokiaer J, Spring KR. Beta1-integrins in the primary cilium of MDCK cells potentiate fibronectin-induced Ca2+ signaling. Am J Physiol Renal Physiol. 2004;287:F969–978. doi: 10.1152/ajprenal.00096.2004. [DOI] [PubMed] [Google Scholar]
- Rinne T, Brunner HG, van Bokhoven H. p63-associated disorders. Cell Cycle. 2007;6:262–268. doi: 10.4161/cc.6.3.3796. [DOI] [PubMed] [Google Scholar]
- Rosenbaum JL, Witman GB. Intraflagellar transport. Nat Rev Mol Cell Biol. 2002;3:813–825. doi: 10.1038/nrm952. [DOI] [PubMed] [Google Scholar]
- Schneider L, Clement CA, Teilmann SC, Pazour GJ, Hoffmann EK, Satir P, Christensen ST. PDGFRalphaalpha signaling is regulated through the primary cilium in fibroblasts. Curr Biol. 2005;15:1861–1866. doi: 10.1016/j.cub.2005.09.012. [DOI] [PubMed] [Google Scholar]
- Scholey JM, Anderson KV. Intraflagellar transport and cilium-based signaling. Cell. 2006;125:439–442. doi: 10.1016/j.cell.2006.04.013. [DOI] [PubMed] [Google Scholar]
- Shirasaki R, Pfaff SL. Transcriptional codes and the control of neuronal identity. Annu Rev Neurosci. 2002;25:251–281. doi: 10.1146/annurev.neuro.25.112701.142916. [DOI] [PubMed] [Google Scholar]
- Sidow A, Bulotsky MS, Kerrebrock AW, Bronson RT, Daly MJ, Reeve MP, Hawkins TL, Birren BW, Jaenisch R, Lander ES. Serrate2 is disrupted in the mouse limb-development mutant syndactylism. Nature. 1997;389:722–725. doi: 10.1038/39587. [DOI] [PubMed] [Google Scholar]
- Tabin CJ. The key to left-right asymmetry. Cell. 2006;127:27–32. doi: 10.1016/j.cell.2006.09.018. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yonekawa Y, Tanaka Y, Okada Y, Nonaka S, Hirokawa N. Left-right asymmetry and kinesin superfamily protein KIF3A: new insights in determination of laterality and mesoderm induction by kif3A-/- mice analysis. J Cell Biol. 1999;145:825–836. doi: 10.1083/jcb.145.4.825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe Y, William C, Jessell TM. Specification of motor neuron identity by the MNR2 homeodomain protein. Cell. 1998;95:67–80. doi: 10.1016/s0092-8674(00)81783-3. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Okada Y, Hirokawa N. FGF-induced vesicular release of Sonic hedgehog and retinoic acid in leftward nodal flow is critical for left-right determination. Nature. 2005;435:172–177. doi: 10.1038/nature03494. [DOI] [PubMed] [Google Scholar]
- Teilmann SC, Christensen ST. Localization of the angiopoietin receptors Tie-1 and Tie-2 on the primary cilia in the female reproductive organs. Cell Biol Int. 2005;29:340–346. doi: 10.1016/j.cellbi.2005.03.006. [DOI] [PubMed] [Google Scholar]
- Tran PV, Haycraft CJ, Besschetnova TY, Turbe-Doan A, Stottmann RW, Herron BJ, Chesebro AL, Qiu H, Scherz PJ, Shah JV, Yoder BK, Beier DR. THM1 negatively modulates mouse sonic hedgehog signal transduction and affects retrograde intraflagellar transport in cilia. Nat Genet. 2008;40:403–410. doi: 10.1038/ng.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vortkamp A, Gessler M, Grzeschik KH. GLI3 zinc-finger gene interrupted by translocations in Greig syndrome families. Nature. 1991;352:539–540. doi: 10.1038/352539a0. [DOI] [PubMed] [Google Scholar]
- Wallingford JB. Planar cell polarity, ciliogenesis and neural tube defects. Hum Mol Genet. 2006;15(Spec No2):R227–234. doi: 10.1093/hmg/ddl216. [DOI] [PubMed] [Google Scholar]
- Wang B, Fallon JF, Beachy PA. Hedgehog-regulated processing of Gli3 produces an anterior/posterior repressor gradient in the developing vertebrate limb. Cell. 2000;100:423–434. doi: 10.1016/s0092-8674(00)80678-9. [DOI] [PubMed] [Google Scholar]
- Wang Q, Pan J, Snell WJ. Intraflagellar transport particles participate directly in cilium-generated signaling in Chlamydomonas. Cell. 2006;125:549–562. doi: 10.1016/j.cell.2006.02.044. [DOI] [PubMed] [Google Scholar]
- Wheatley DN. Primary cilia in normal and pathological tissues. Pathobiology. 1995;63:222–238. doi: 10.1159/000163955. [DOI] [PubMed] [Google Scholar]
- Wheatley DN, Wang AM, Strugnell GE. Expression of primary cilia in mammalian cells. Cell Biol Int. 1996;20:73–81. doi: 10.1006/cbir.1996.0011. [DOI] [PubMed] [Google Scholar]
- Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K, Fazzari M, Jin B, Brown AM, Lipkin M, Edelmann W. Mbd4 inactivation increases C→transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci USA. 2002;99:14937–14942. doi: 10.1073/pnas.232579299. [DOI] [PMC free article] [PubMed] [Google Scholar]