

Abstract
The cardiac renin-angiotensin system (RAS) has been implicated in mediating myocyte hypertrophy, remodeling, and fibroblast proliferation in the hemodynamically overloaded heart. However, the intracellular signaling mechanisms responsible for regulation of angiotensinogen (Ao), a substrate of the RAS system, are largely unknown. Here we report the identification of JNK1/2 as a negative, and p38α as a major positive regulator of Ao gene expression. Isolated neonatal rat ventricular myocytes (NRVM) and fibroblasts (NRFB) plated on deformable membranes coated with collagen IV, were exposed to 20% equiaxial static-stretch (0-24 hr). Mechanical stretch initially depressed Ao gene expression (4 h), whereas after 8 h, Ao gene expression increased in a time-dependent manner. Blockade of JNK1/2 with SP600125 increased basal Ao gene expression in NRVM (10.52 ± 1.98 fold, P<0.001) and NRFB (13.32 ± 2.07 fold, P<0.001). Adenovirus-mediated expression of wild-type JNK1 significantly inhibited, whereas expression of dominant-negative JNK1 and JNK2 increased basal and stretch-mediated (24 h) Ao gene expression, showing that both JNK1 and JNK2 to be negative regulators of Ao gene expression in NRVM and NRFB. Blockade of p38α/β by SB202190 or p38α by SB203580 significantly inhibited stretch-induced (24 h) Ao gene expression, whereas expression of wild-type p38α increased stretch-induced Ao gene expression in both NRVM (8.41 ± 1.50 fold, P<0.001) and NRFB (3.39 ± 0.74 fold, P<0.001). Conversely, expression of dominant-negative p38α significantly inhibited stretch response. Moreover, expression of constitutively active MKK6b (E) significantly stimulated Ao gene expression in absence of stretch, indicating that p38 activation alone is sufficient to induce Ao gene expression. Taken together p38α was demonstrated to be a positive regulator, whereas JNK1/2 was found to be a negative regulator of Ao gene expression. Prolonged stretch diminished JNK1/2 activation, which was accompanied by a reciprocal increase in p38 activation and Ao gene expression. This suggests that a balance in JNK1/2 and p38α activation determines the level of Ao gene expression in myocardial cells.
Keywords: Angiotensinogen, Cardiac fibroblasts, Cardiac Myocytes, JNK, p38, MAP kinases, Mechanical stretch, Renin-angiotensin system
1. Introduction
Several lines of evidence from clinical and experimental studies indicate that the renin-angiotensin-system (RAS) has a key role in mediating ventricular hypertrophy in the pressure-and volume-overloaded heart. Angiotensinogen (Ao), a substrate of the RAS system, has been implicated in the pathogenesis of hypertension and congestive heart failure. Angiotensin II (Ang II), the most biologically active peptide of the RAS affects several aspects of cardiac function including contractility, cell metabolism, cellular growth, differentiation, apoptosis and gene expression [1]. Progression of heart failure is associated with steady increase of Ang II formation regardless of the underlying etiology [2-4]. It is generally accepted that activation of the RAS plays an important role in cardiac pathophysiology, since inhibition of Ang II production by angiotensin converting enzyme inhibitors or treatment with Ang II type-I receptor (AT1) blockers significantly improve cardiac function, reverse ventricular remodeling and reduce morbidity and mortality in patients with heart failure [5, 6]. We and others have shown that all components of RAS system (renin, Ao, ACE, Ang II, Ang II receptors) are present in the ventricular myocardium [7-11] and expressed by cardiac myocytes and fibroblasts. Elevated cardiac Ang II alone induced cardiac interstitial fibrosis under basal conditions and exacerbated cardiac remodeling and dysfunction and accelerated development of heart failure in mice with myocardial infarction, without affecting systemic hemodynamics [12]. Although the cardiac RAS is upregulated by increased mechanical load, the signaling pathways responsible remain to be determined.
MAP kinases p38 and JNK, sub-classified as stress-activated protein kinases (SAPKs), are specialized transducers of stress responses. Four genes encode p38 kinases (p38α, p38β, p38γ and p38δ), in which p38α is the major isoform expressed in the heart [13, 14]. The p38 cascade is initiated by MAP kinase kinase kinases (MAPKKK) at the level of the plasma membrane, which in turn promotes activation of the dual-specificity kinases, MKK3 and MKK6, which directly phosphorylate Thr and Tyr residues in the Thr-Gly-Tyr (TGY) motif of p38 kinases [14, 15]. Three JNK genes (JNK1, JNK2 and JNK3) have been identified, each of which gives rise to differentially-spliced isoforms [16]. Only JNK1 and JNK2 are present in the myocardium [15]. The JNK branch is initiated by MKKs at the plasma membrane, which promotes activation of dual-specificity kinases MKK4 and MKK7, which in turn directly phosphorylate Thr and Tyr residues in a Thr-Pro-Tyr (TPY) motif of JNK kinases [15]. While MKK7 is a specific activator of JNKs, MKK4 can also phosphorylate the TGY motif of p38 MAP kinases [17].
p38 and JNK cascades have been implicated in both cardiac protection and injury [14, 18-20]. Many pharmacological and molecular studies demonstrate that p38 activation enhances myocardial injury, whereas p38 inhibition is cardioprotective [13, 14, 21, 22]. Administration of p38 inhibitor has been shown to prevent left ventricular hypertrophy and dysfunction in hypertensive rats and improve cardiac function and attenuate left ventricular remodeling in rats with myocardial infarction [21]. Ventricular myocyte targeted over-expression of constitutively active MKK3 (MKK3bE) and MKK6 (MKK6bE) in transgenic mice produced a cardiomyopathic phenotype with extensive interstitial fibrosis [13]. However, many recent in-vivo studies suggest that JNK actually serves as negative regulator of mechanical load-induced cardiac growth-related responses [18, 19, 21, 23, 24]. In the present study, the objective was to determine the role of stress-activated kinases JNK and p38 in mediating mechanical load-induced Ao gene expression in neonatal rat ventricular myocytes (NRVM) and fibroblasts (NRFB).
2. Materials and Methods
2.1. Isolation of cardiac cells and mechanical stretch
Primary cultures of NRVM and NRFB were prepared from 1 to 2-day-old Sprague Dawley rats as previously described [25]. Dispersed cardiac cells were separated using a discontinuous Percoll gradient, containing a density of 1.060 gm/L (nonmyocyte layer) and 1.086 gm/L (myocyte layer). The NRVM were plated on deformable membranes coated with collagen-IV (1 μg/cm2) on Bioflex plates (Flexcell International Corp, Hillsborough, NC), at a density of 0.75 × 106 cells/well in DMEM/M199 medium and maintained at 37°C in humid air with 5% CO2. Cytosine arabinoside (100 μM) was added to prevent cell division of nonmyocytes and culture media was changed to serum-free 12 h prior to initiation of experiments. The cultured myocytes were >95% pure, as revealed by microscopic observation of contractile characteristics and by flow cytometry, after staining with anti-desmin antibody (Sigma Chemical Co., St. Louis, MO). After Percoll separation, cells in the nonmyocyte band were transferred to culture flasks and incubated for 2 h. Unattached cells were removed and remaining nonmyocytes (primarily NRFB) were grown to confluence, trypsinized and plated on deformable membranes at a density of 0.50 × 106 cells/well (first passage) in DMEM/M199 medium and maintained at 37°C in humid air with 5% CO2. When cells were ~90% confluent, the culture medium was changed to serum-free DMEM and experiments were performed 12 h later. NRVM and NRFB were exposed to 20% equiaxial static-stretch for various times in a Flexercell FX-3000 strain unit equipped with loading posts. This study conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
2.2. Antibodies and reagents
Phospho-p38-T180/Y182 antibody (9211), p38 antibody (9217), phospho-SAPK/JNK-T183/Y185 antibody (9251), JNK antibody (9252) and horseradish peroxidase-conjugated secondary antibodies (7074) were obtained from Cell Signaling Technology, Inc. (Danvers, MA). Bovine serum albumin (BSA, diagnostic grade K) was obtained from Celluliance (Kanakee, IL). Enhanced chemiluminescence (ECL) reagent (Western Lightning™) was obtained from Perkin Elmer Life Science (Boston, MA).
2.3. Adenovirus expansion and infection of cells
Recombinant adenoviruses expressing GFP-p38-MAPK-alpha wild-type (Ad-p38α-WT), FLAG-tagged p38α dominant-negative (Ad-p38α-DN) and HA-tagged MKK6bE (Ad-MKK6bE) were gifts from Dr. Yibin Wang (Cardiovascular Research Laboratory, University of California, Los Angeles). Recombinant adenoviruses expressing JNK1-wild-type (Ad-JNK1-WT) and JNK1 dominant-negative (Ad-JNK1-DN) were purchased from Cell Biolabs Inc. (San Diego, CA). Dominant-negative JNK2 (Ad-JNK2-DN) was purchased from Seven Hills Bioreagents, (Cincinnati, OH). Amplification of adenovirus was performed in transformed 293 human embryonic kidney (HEK) cells (CRL-1573, ATCC, Manassas, VA), followed by purification on CsCl gradients. The viral MOI was determined by dilution assay in HEK-293 cells grown in 6-well clusters. Titration assays were used to determine the lowest MOI which would result in at least a two-fold increase in expressed protein and/or block endogenous target protein phosphorylation in NRVM and NRFB. Corresponding MOIs of adenoviruses expressing lacZ or GFP were used to control for viral effects. After 24 h of plating, NRVM were infected with following MOI of adenoviruses Ad-p38α-WT (50 MOI), Ad-p38α-DN (100 MOI), Ad-MKK6bE (100 MOI), Ad-JNK1-WT (50 MOI), Ad-JNK1-DN (50 MOI) and Ad-JNK2-DN (100 MOI), diluted in DMEM/medium 199. For NRFB transfections, adenoviruses were diluted in serum-free DMEM/medium 199 and the following MOI were used, Ad-p38α-WT (100 MOI), Ad-p38α-DN (100 MOI), Ad-MKK6bE (250 MOI), Ad-JNK1-WT (200 MOI), Ad-JNK1-DN (200 MOI) and Ad-JNK2-DN (100 MOI). At these concentrations, there were no obvious signs of toxicity (cell detachment, cellular vacuoles, cell rounding) of NRVM and NRFB. Levels of expressed proteins were determined using Western blot analysis (not shown). After 24 h of transfection, the medium was replaced with virus-free DMEM/medium 199, and cells were cultured for an additional 12 h prior to stretch experiments.
2.4. Preparation of cell lysates and western blotting
Cell lysates were obtained by scrapping NRVM and NRFB in assay lysis buffer (Cell Signaling) containing 10 μg/mL aprotinin, 10 μg/mL leupeptin, 1 mM 2-(2-aminoethyl)-benzenesulfonyl fluoride, hydrochloride and 1 mM sodium orthovanadate. Insoluble material was removed by centrifugation for 15 min at 14,000 g and samples were boiled with loading buffer and protein was determined using a kit (BioRad DC Protein Assay) according to the manufacturer’s recommendation. Western blot analysis was performed as previously described [25]. Briefly, equal amounts of protein (30 μg) from cell lysates were separated by SDS-PAGE and blotted onto PVDF transfer membranes. The membranes were blocked for 2 h using 5% BSA in TBST buffer (10 mM Tris, 100 mM NaCl, 0.1% Tween 20, pH 7.4). Blots were incubated with the primary antibodies in 5% BSA in TBST buffer overnight at 4°C with light agitation. Bound primary antibodies were visualized using horseradish peroxidase-labeled secondary antibodies and were detected using ECL. Densities of the protein bands were quantified using ImageQuant software. Signals from the phosphoproteins were normalized to total protein, obtained by stripping and reprobing blots with the corresponding total antibody. Blots were again stripped and probed with GAPDH antibody to confirm equal loading.
2.5. Quantitative measurement of Ao mRNA using real-time PCR
A commercial kit (Totally RNA™, Ambion) was used to isolate RNA from the NRVM and NRFB. First strand cDNA was reverse transcribed (RT) with random hexamer primer using the High Capacity cDNA Archive kit for RT-PCR (ABI Prism). Real-time RT-PCR was carried out in a MX3005P (Stratagene) thermocycler using Taqman Universal PCR Master Mix (ABI, USA). Absolute levels of Ao mRNA were quantified using 21-base sense (5’-AGCACGACTTCCTGACTTGGA-3’) and antisense primers (5’-TTGTAGGATCCCCGATTTCC-3’), which span the second intron of the genomic sequence and produce an 88 base-pair amplicon. Amplification of Ao DNA was performed using oligonucleotide (6FAM-5’-CCGTCTGACCCTGCCGCAGC- 3’-TAMRA), as the reporter probe. Parallel reactions, yeast RNA (carrier RNA) spiked with known amounts of synthetic Ao RNA, were used to convert sample determinations to absolute values. Ao primers and reporters were multiplexed with GAPDH, for a “houser-keeper” RNA (proprietary reagents supplied by Perkin-Elmer-Applied Biosystems). The multiplexing was used to control for RNA loading, degradation and PCR efficiency.
2.6. Statistics
Data are expressed as mean ±SEM. Differences among groups were tested by 1-way ANOVA and among groups before and after stretch by 2-way ANOVA, where appropriate. p<0.05 or less was considered to denote statistical significance (GraphPad prism Software Inc., San Diego, CA).
3. Results
3.1. Mechanical stretch upregulates Ao gene expression
We and others have demonstrated that cardiac myocytes and fibroblasts from neonate and adult rats express Ao mRNA and protein [8, 10, 11, 26]. Ao gene expression is increased in ventricular myocytes from acutely infarcted rat hearts [8] and hypertrophied and failing hearts of rats with spontaneous hypertension [7]. In the present study, NRVM and NRFB were used to determine the role of stress activated kinases JNK and p38 in mediating mechanical load-induced Ao gene expression. Interestingly, acute mechanical stretch (up to 4 h) decreased the Ao gene expression, at 4 h in NRVM (1.53 ± 0.10 fold, P<0.05) and NRFB (1.47 ± 0.13 fold, P<0.05). However, chronic mechanical stretch increased Ao gene expression in a time dependent manner in both NRVM and NRFB (Fig 1A & 1B), in which significant changes were first observed at 8 h for both NRVM (1.71 ± 0.17 fold, P<0.001) and NRFB (1.55 ± 0.23 fold, P<0.05).
Figure 1. Time-dependent Ao gene expression in stretched myocytes and cardiac fibroblasts.
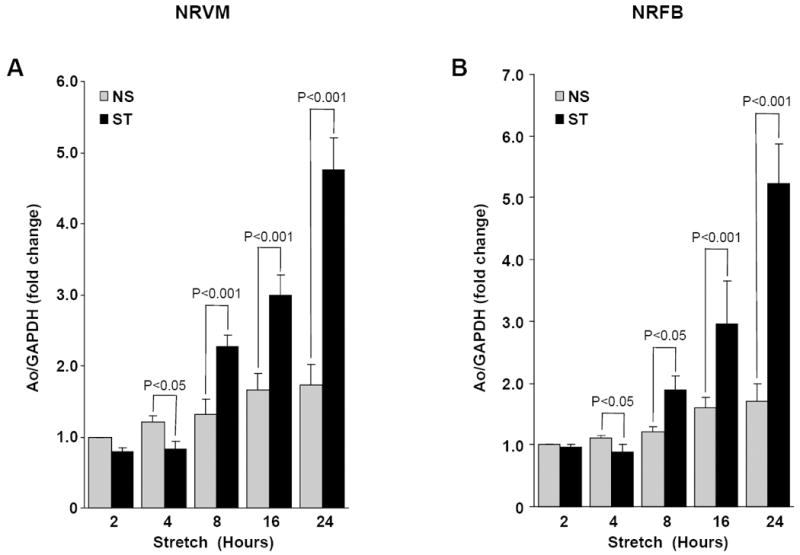
NRVM (A) and NRFB (B) were stretched (20%) for different time (2 – 24 h), as indicated. After stretch, cells were harvested and RNA was isolated, expression of Ao was determined by Taqman real time PCR in a multiplex system with GAPDH (for normalization). Results are expressed as means ± SEM. N=5 experiments. NS, No stretch; ST, Stretch; NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts.
3.2. JNK activation dominates in the acute and p38 in chronic phase of mechanical stretch
We have recently shown that mechanical stretch induces rapid activation of JNK and p38 in NRVM [25]. Because, stretch-induced Ao mRNA activation is delayed, a time-course of JNK and p38 activation was performed to test whether Ao gene expression may be related to ctivation of these two MAP kinases. In NRFB, stretch significantly increased JNK phosphorylation within 5 min (1.27±0.079 fold, P<0.05), which peaked at 15 min (2.01±0.092 fold, P <0.001) and returned to below basal levels after 30 min and remained depressed up to 24 h (Fig 2A & 2C). In NRVM, stretch induced maximal JNK phosphorylation at 5 min (2.68 ± 0.098 fold, P<0.001), which slowly declined during the remaining portion of the 24 h stretch period. Initial activation of p38 followed the same time-course as JNK in the respective cells types (Fig 2B & 2D). In contrast to JNK, which declined after peak activation, p38 phosphorylation was reactivated at 4 h and continued to increase with stretch (Fig 2B & 2D). Thus, in both cardiac cell types, JNK was initially more highly phosphorylated following stretch, whereas p38 activation continues to dominate afterwards.
Figure 2. Time-course of stretch-induced JNK and p38 activation in cardiac myocytes and fibroblasts.
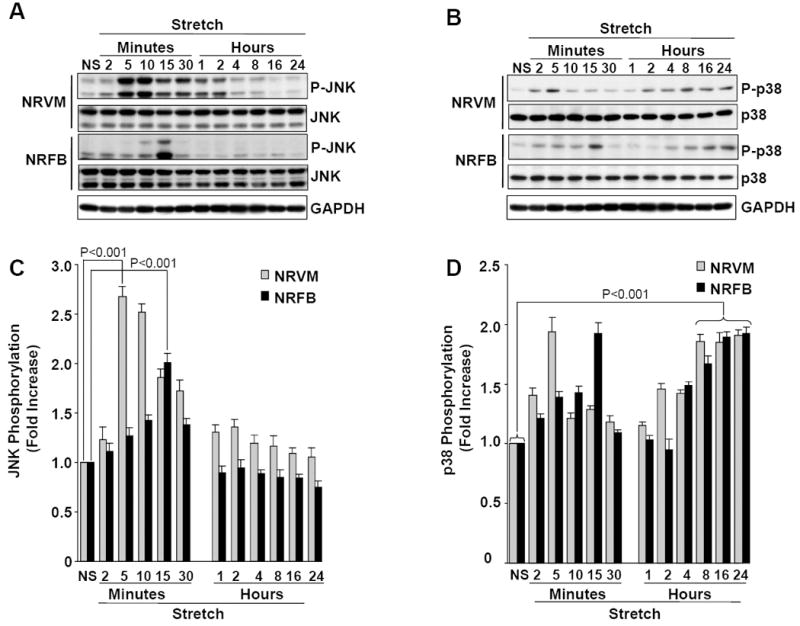
NRVM (A) and NRFB (B) were stretched (20%) for different time (2 min-24 h), as indicated. After stretch, cells were harvested and lysates were analyzed by immunoblotting with phospho-JNK and phospho-p38 antibodies, as indicated (A&B). C&D, Bar graphs show fold changes in JNK (C) and p38 (D) phosphorylation after stretch, when compared to no-stretch (NS). Results are expressed as means ± SEM. N=5 experiments. NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts.
3.3. JNK negatively regulates basal and stretch-induced Ao gene expression
To determine whether JNK may play a role in stretch-induced Ao gene expression, cells were pretreated (1 h) with a specific JNK1/2 inhibitor (SP600125). Incubation with SP600125 increased the basal expression of Ao several fold in NRVMs (10.52 ± 1.98 fold, P<0.001) and NRFBs (13.32 ± 2.07 fold, P<0.001) (Fig 3A & 3B), which was not further increased by mechanical stretch. These results suggested that JNK serves as a major negative regulator of Ao gene expression in NRVM and NRFB. Adenovirus overexpression experiments were performed to determine the role of JNK1 and/or JNK2 in this response. Expression of wild-type JNK1 significantly inhibited stretch-mediated Ao gene expression, whereas expression of dominant-negative JNK1 significantly increased NRVM basal (3.25 ± 0.32 fold, P<0.001) and stretch-mediated (4.32±0.44 fold, P<0.001) Ao gene expression and NRFB, basal (2.40 ± 0.26 fold, P<0.01) and stretch- mediated (3.98 ± 0.34 fold, P<0.001) Ao gene expression (Fig 3C & 3D). Expression of dominant-negative JNK2 significantly increased NRVM basal (2.58 ± 0.30 fold, P<0.01) and stretch-mediated (4.01 ± 0.25 fold, P<0.001) Ao gene expression and NRFB, basal (1.87 ± 0.19 fold, P<0.01) and stretch-mediated (3.77 ± 0.45 fold, P<0.001) Ao gene expression (Fig 3C & 3D). These results indicate that both JNK1 and JNK2 are major negative regulators of Ao gene expression.
Figure 3. JNK negatively regulates Ao gene expression.
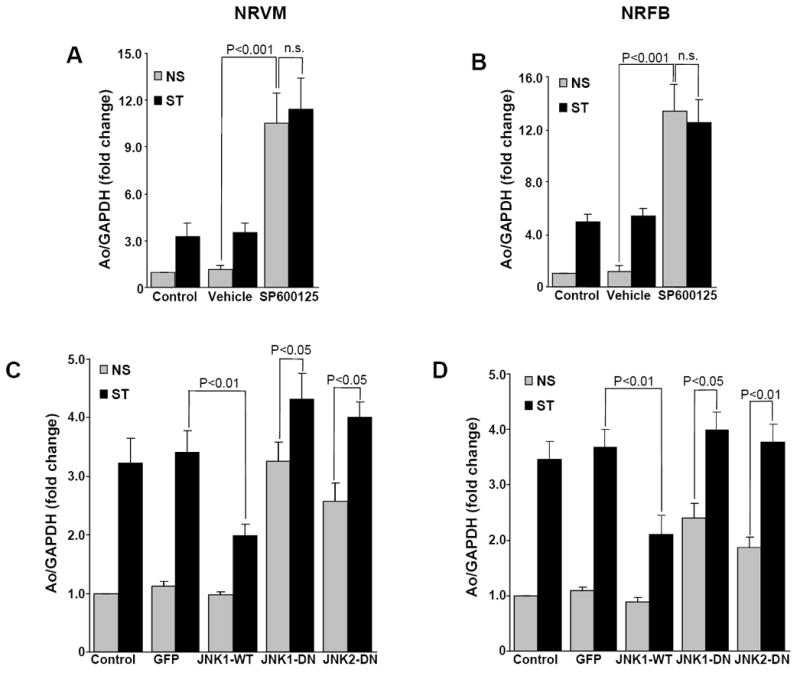
NRVM and NRFB were stretched in presence and absence of specific JNK1/2 inhibitors 20 μM, SP600125 for 24 h. After stretch, cells were harvested and RNA was isolated, and expression of Ao was determined. SP600125 treatment significantly increased basal Ao gene expression in myocytes (A) and NRFB (B). (C&D) Cells were transfected with adenoviruses expressing wild-type (JNK1-WT) or dominant-negative (JNK1-DN) forms of JNK1 or JNK2 (JNK2-DN). After 24 h of transfection, viral medium was replaced with serum free medium and allowed for overnight starvation before starting the stretch experiment. After stretching, cells were lysed, RNA was isolated and Ao expression was determined. Results are expressed as means ± SEM. N=5 experiments. NS, No-stretch; ST, Stretch; NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts; n.s., Not signficant.
3.4. p38α is required for stretch-induced Ao gene expression
In the above experiments, prolonged stretch diminished JNK activation, which was accompanied by a reciprocal increase in p38 activation. To test whether p38 may have a role in stretch-induced regulation of Ao gene expression, cells were pretreated (1 h) with 10 μM, SB202190, which inhibits both p38α (IC50 = 50 nM) and p38β (IC50 = 100 nM) isoforms expressed in cardiac tissue. The inhibitor SB202190 inhibited stretch-induced Ao gene expression in both NRVM (2.22 ±0.29 fold, P<0.001) and NRFB (2.54 ± 0.21 fold, P<0.001) (Fig 4A&B). Treatment with the p38α specific inhibitor SB203580 (IC50 = 50 nM for p38α and 500 nM for p38β nM) further suggested that p38α mediates stretch-induced Ao gene expression (Fig 4A & 4B). To further confirm this role of p38α, NRVM and NRFB were transfected with adenoviruses expressing wild-type or dominant-negative forms of p38α. Expression of wild-type p38α increased the stretch-induced Ao gene expression several fold in both NRVM (8.41 ± 1.50, P<0.001) and NRFB (3.39 ± 0.74 fold, P<0.001) (Fig 4C & 4D). However, expression of dominant-negative p38α significantly inhibited stretch-mediated Ao gene expression in NRVM (1.88 ± 0.22, P<0.01) and NRFB (2.44 ± 1.30, P<0.01) (Fig 4C & 4D), further indicating that p38α is a primary mediator of stretch-induced Ao gene expression in both myocardial cells.
Figure 4. p38 mediates stretch-induced Ao gene expression.
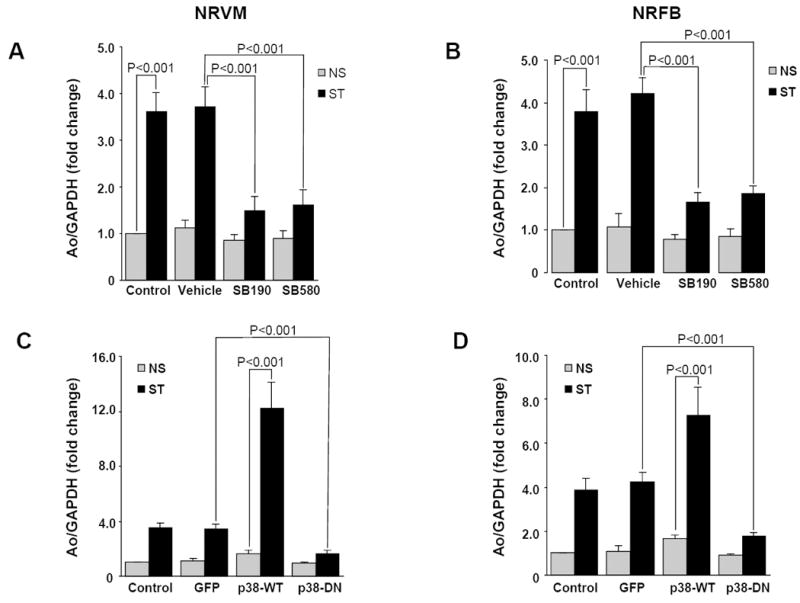
NRVM and NRFB were stretched in presence and absence of specific p38 inhibitors (10 μM, SB202190 and SB203580) for 24 h. Inhibition of p38 significantly inhibited stretch-mediated Ao gene expression both in myocytes (A) and NRFB (B). (C&D) Cells were transfected with adenoviruses expressing wild-type (p38-WT) and dominant-negative (p38-DN) forms of p38α. After 24 h of transfection, viral medium was replaced with serum free medium and allowed for overnight starvation before starting the stretch experiment. After stretch, cells were harvested and RNA was isolated, expression of Ao was determined (C&D) Results are expressed as means ± SEM. N=5 experiments. NS, No stretch; ST, Stretch; NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts; SB190, SB20190; SB580, SB203580.
3.5. p38α activation alone is sufficient to induce Ao gene expression
Mechanical stretch is known to activate several signaling events, which could interact with p38 to activate Ao expression. To test whether the p38 cascade was sufficient to stimulate Ao gene expression, NRVM and NRFB were transfected with adenovirus expressing MKK6b (E), a constitutively active form of MKK6, an upstream activator of p38. The efficiency of MKK6b (E) expression on p38 phosphorylation was confirmed on whole cell lysate using antiphospho-p38 antibody (Fig 5A & B). Expression of MKK6b (E) significantly stimulated Ao gene expression in both NRVM (9.32 ± 2.01 fold, p<0.001), and NRFBs (3.36 ± 0.56 fold, p<0.001), indicating the direct role of p38 activation in Ao gene expression. MKK6b (E) expressing, NRVM and NRFBs were incubated with a p38 inhibitor (SB202190, 10 μM), which abolished the response and further confirmed the role of p38 in Ao gene expression.
Figure 5. Activation of p38 alone is sufficient to induce Ao gene expression.
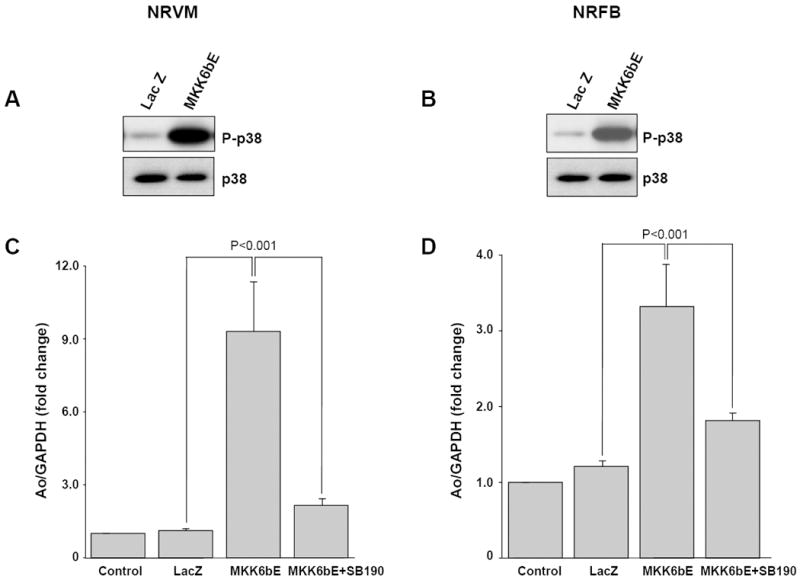
NRVM and NRFB were transfected with adenovirus expressing MKK6b (E), in the presence and absence of the specific p38 inhibitor SB202190 (10 μM). A&B, p38 activity was induced by MKK6b (E) expression in NRVM and NRFB as measured from immunoblot to detect levels of phospho-p38 in cell lysates. C&D, real-time PCR analysis of Ao gene expression reveals that MKK6b (E) expression alone significantly induced Ao gene expression and treatment with p38 inhibitor significantly inhibited this response in both cell types. Results are expressed as means ± SEM. N=5 experiments. NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts; SB190, SB202190.
3.6. JNK mediates the acute stretch-induced downregulation of Ao gene expression
The above results suggested that upregulation of JNK was responsible for negative regulation of Ao gene expression in the early phase of mechanical stretch. To test this hypothesis, cells were pretreated for 1 h with a specific JNK1/2 inhibitor (SP600125) prior to 20% mechanical stretch for 2 h and 4 h. Inhibition of JNK abolished mechanical stretch-induced down regulation of Ao gene expression at 2 h (data not shown). However, 4 h of mechanical stretch in the presence of JNK inhibitor significantly increased stretch-mediated NRVM (1.38±0.082 fold, P<0.05) and NRFB (1.36 ± 0.07 fold, P<0.05) Ao gene expression (Fig 6A & 6B). The reversal of the negative regulatory effects at 4 h of mechanical stretch, therefore, indicated that JNK is an important negative regulator of Ao gene expression during this time period.
Figure 6. Increased JNK activation is responsible for downregulation of Ao gene expression in response to acute mechanical stretch.
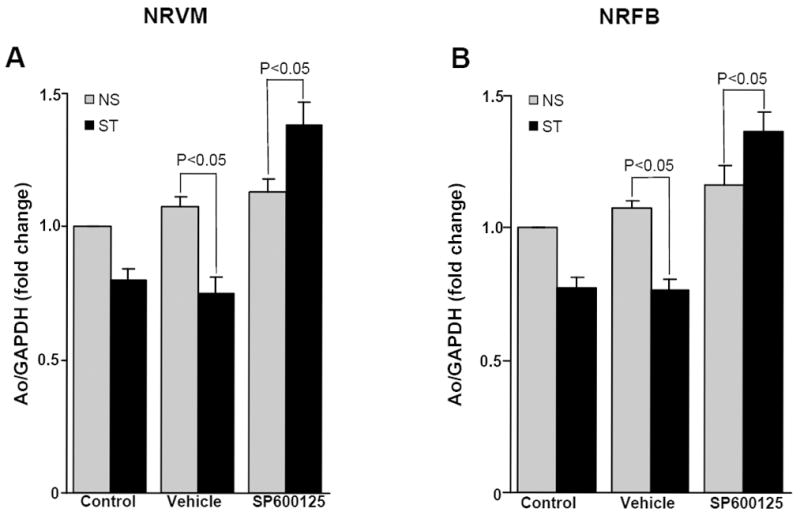
NRVM and NRFB were pretreated (1 h) with specific JNK1/2 inhibitor SP600125 (20 μM), then stretched for 4 h. After stretch, cells were harvested and RNA was isolated, and expression of Ao was determined. SP600125 treatment significantly increased stretch mediated Ao gene expression in NRVM (A) and NRFB (B). Results are expressed as means ± SEM. N=5 experiments. NS, No stretch; ST, Stretch; NRVM, Neonatal rat ventricular myocytes; NRFB, Neonatal rat ventricular fibroblasts.
4. Discussion
The importance of the RAS in the pathophysiology of heart failure has been highlighted by the vast number of clinical and experimental investigations [1, 2, 27-29]. We have previously reported that myocytes and fibroblasts isolated from neonatal and adult rat hearts express Ao, which is upregulated in cardiac myocytes in various forms of load-induced heart failure, including acute myocardial infarction, genetic hypertension and aortic constriction [8, 10, 30]. In the current study, we have focused on identifying the pivotal signaling mechanism responsible for mechanical stretch-induced upregulation of Ao gene expression in neonatal rat cardiac myocytes and fibroblasts. Results indicate that a balance in p38α and JNK1/2 activation primarily dictate Ao regulation in both cell types.
Mechanical stretch initially stimulated JNK activation, which was accompanied by a decrease in Ao gene expression in both NRVM and NRFBs. However, with prolonged stretch there was diminished JNK activation, which was accompanied by reciprocal increases in p38 activation and Ao gene expression. Thus, in both cell types, JNK was initially more highly phosphorylated following stretch, whereas p38 activation continues to dominate afterwards. Cardiac ventricular volume-overload studies remain to be performed to determine whether these mechanisms are operational in vivo. However, it is interesting to note that our observations are in agreement with those obtained from mouse and rat models of ventricular pressure overload [31, 32]. Exposito et al. [31] have shown that thoracic aortic constriction (TAC)-induced cardiac hypertrophy in the mouse, JNKs are activated as early as 7 h post-TAC, whereas p38 phosphorylation is primarily elevated after 3 days. Recently, it has been shown that in Wistar rats with acute left ventricular pressure overload, transient JNK phosphorylation occurred within the first 24 h and declined [32]. Taken together, these studies indicate that JNK plays an important role in the early phase of mechanical stretch.
Our results demonstrate that JNK1/2 negatively regulates Ao gene expression. SP600125 inhibits all three isoforms of JNK (JNK1/2/3) and upregulates Ao gene expression several fold. Similar results were obtained in a recent study using transgenic mice, in which cardiac selective deletion of JNK genes revealed that JNK1 preserved cardiac function in the early phase of acute hemodynamic stress [23]. Pharmacological inhibition of JNK pathway by SP600125 or adenovirus-mediated expression of dominant-negative JNK1/2 increased basal Ao gene expression several fold and conversely expression of wild-type JNK1 significantly inhibited stretch-mediated Ao gene expression, suggesting that JNK1/2 has at least two roles in the regulation of Ao expression in cardiac cells. One role is to preserve basal low-level expression of Ao, whereas the second is to counterbalance the stimulatory effects on Ao expression with increased mechanical load. The data implicating a causative role for JNK signaling in mediating the cardiac growth response has been controversial [15, 19, 21, 23, 24]. These results are intriguing in light of some acute in vitro studies, which argue in favor of a pro-hypertrophic role for JNK in cardiac myocytes [18, 19, 33, 34]. However, a number of recent studies have not only challenged the proposed role of JNK as a pro-hypertrophic signaling effector in cardiac myocytes, but also suggest that JNK serves as a negative regulator of this response [15, 21, 24, 35]. At present, this discrepancy is poorly understood. While compelling evidence indicates that JNK mediates cardiac protection, the present study is the first to demonstrate the molecular mechanism of action and suggests that JNK activation exerts its protective effect by preserving the low level of local RAS in acute phase of mechanical load. This is further supported by the fact that a local increase in Ao gene expression alone has a significant impact on cardiac pathophysiology [36, 37]. Over-expression of Ao in the myocardium mediates phenotypic cardiac myocyte remodeling, which leads to age-dependent cardiac dysfunction and failure [36]. Consistent with over-expression, adenovirus-mediated delivery of Ao antisense attenuates hypertension and cardiac hypertrophy [37]. Although cardiac myocytes represent the bulk of the myocardial volume, cardiac fibroblasts are the most numerous cell types in the heart and play an essential role in myocardial function. Previously we have demonstrated the expression of Ao in cardiac fibroblasts and its upregulation in different pathological conditions [38-40]. Although there is differential expression of mechanoreceptors in cardiac myocytes and fibroblasts [41], it was interesting to note that similar molecular mechanisms of Ao gene regulation occur at the MAP kinase level in both types of cardiac cells in response to mechanical stretch.
Although the underlying cellular and molecular mechanisms remain to be defined, several studies using pharmacological inhibitors and genetic manipulations suggest a detrimental role for p38α in mediating events associated with cardiac hypertrophy, remodeling and contractile dysfunction[13, 14, 21, 22]. Results from the present study implicate the role of RAS system upregulation in p38α-mediated myocardial effects. Expression of MKK6b (E) significantly stimulated Ao gene expression, which indicates that activation of p38 by itself is sufficient to stimulate Ao gene expression. Taken together, these data suggest that p38 is required for stimulation of Ao gene expression by mechanical stretch, and activation of p38 alone is sufficient for upregulation of Ao gene expression. Given the detrimental effect of Ao gene expression in the myocardium [36, 37], we speculate that p38 exerts its detrimental effects at least in part via upregulation of local Ao gene expression. This hypothesis is further supported by several studies which convincingly demonstrate that angiotensin II-induced hypertension, negative inotropy, reactive oxygen species production and organ damage are mediated by p38 [22, 42]. The downstream effectors responsible for mediating p38α effects on Ao gene expression remain to be determined. In isolated rat kidney cells, cyclic AMP response element binding protein (CREB) has been shown to be an important regulator of Ao gene expression [43]. Phosphorylated CREB has been shown to enhance Ao gene expression by binding to CRE in the 5’-flanking region of the rat Ao gene [44]. In the myocardium, CREB has been shown to be activated by mitogen and stress-activated kinase 1 (MSK-1), a downstream target of p38 [20]. Studies are ongoing in our laboratory to identify the upstream activator/s and the downstream transcription factor/s as molecular targets of p38 that mediate the observed upregulation of Ao gene expression in cardiac myocytes and fibroblasts. Inhibition of JNK1/2 by SP600125 resulted in stretch-induced upregulation of Ao gene expression at 4 h, suggesting that JNK activation is responsible for the initial suppression of Ao gene expression in mechanically stretched NRVM and NRFB.
Although, Results clearly indicate that JNK1/2 suppresses and p38α mediates stretch-induced Ao gene expression in isolated cardiac cells, it remains to be determined whether this mechanism is operational in the hemodynamically overloaded myocardium. Isolated cardiac cells may not completely mimic in vivo conditions since cells were grown as monolayer on a single extracellular matrix. Future in vivo studies employing pharmacological inhibitors and transgenic animal models of p38α and JNK1/2 are therefore warranted.
In summary, we have identified two important regulators of mechanical stretch-induced Ao gene expression in cardiac cells. Our results demonstrate that stress-activated kinases JNK1/2 and p38α have opposing roles in mechanical stretch-induced Ao gene expression; p38 mediates, and JNK suppresses, Ao gene expression. This study also reveals that prolonged stretch causes an imbalance in activation of JNK1/2 and p38α, resulting in upregulation of Ao gene expression in cardiac cells. To our knowledge, this is the first study to demonstrate that a balance between JNK1/2 and p38α is an important regulatory determinant of Ao, an essential precursor of the cardiac RAS. Given the importance of the RAS in mediating cardiac hypertrophy and remodeling, this suggests that both JNK1/2 and p38α may be viable therapeutic targets for the treatment of heart disease.
Acknowledgments
This work was supported by a grant from the National Institutes of Health (R01-HL-68838) and Scott and White Hospital.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Xu J, Carretero OA, Lin CX, Cavasin MA, Shesely EG, Yang JJ, et al. Role of cardiac overexpression of ANG II in the regulation of cardiac function and remodeling postmyocardial infarction. American journal of physiology. 2007 Sep;293(3):H1900–7. doi: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kumar R, Singh VP, Baker KM. The intracellular renin-angiotensin system: implications in cardiovascular remodeling. Curr Opin Nephrol Hypertens. 2008 Mar;17(2):168–73. doi: 10.1097/MNH.0b013e3282f521a8. [DOI] [PubMed] [Google Scholar]
- 3.Serneri GG, Boddi M, Cecioni I, Vanni S, Coppo M, Papa ML, et al. Cardiac angiotensin II formation in the clinical course of heart failure and its relationship with left ventricular function. Circ Res. 2001 May 11;88(9):961–8. doi: 10.1161/hh0901.089882. [DOI] [PubMed] [Google Scholar]
- 4.Wollert KC, Drexler H. The renin-angiotensin system and experimental heart failure. Cardiovasc Res. 1999 Sep;43(4):838–49. doi: 10.1016/s0008-6363(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 5.Jorde UP. Suppression of the renin-angiotensin-aldosterone system in chronic heart failure: choice of agents and clinical impact. Cardiol Rev. 2006 Mar-Apr;14(2):81–7. doi: 10.1097/01.crd.0000201550.94389.50. [DOI] [PubMed] [Google Scholar]
- 6.Granger CB, McMurray JJ, Yusuf S, Held P, Michelson EL, Olofsson B, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003 Sep 6;362(9386):772–6. doi: 10.1016/S0140-6736(03)14284-5. [DOI] [PubMed] [Google Scholar]
- 7.Dostal DE, Baker KM. The cardiac renin-angiotensin system: conceptual, or a regulator of cardiac function? Circ Res. 1999 Oct 1;85(7):643–50. doi: 10.1161/01.res.85.7.643. [DOI] [PubMed] [Google Scholar]
- 8.Zhang X, Dostal DE, Reiss K, Cheng W, Kajstura J, Li P, et al. Identification and activation of autocrine renin-angiotensin system in adult ventricular myocytes. Am J Physiol. 1995 Nov;269(5 Pt 2):H1791–802. doi: 10.1152/ajpheart.1995.269.5.H1791. [DOI] [PubMed] [Google Scholar]
- 9.Baker KM, Booz GW, Dostal DE. Cardiac actions of angiotensin II: Role of an intracardiac renin-angiotensin system. Annu Rev Physiol. 1992;54:227–41. doi: 10.1146/annurev.ph.54.030192.001303. [DOI] [PubMed] [Google Scholar]
- 10.Dostal DE, Rothblum KN, Chernin MI, Cooper GR, Baker KM. Intracardiac detection of angiotensinogen and renin: a localized renin-angiotensin system in neonatal rat heart. Am J Physiol. 1992 Oct;263(4 Pt 1):C838–50. doi: 10.1152/ajpcell.1992.263.4.C838. [DOI] [PubMed] [Google Scholar]
- 11.Shivakumar K, Dostal DE, Boheler K, Baker KM, Lakatta EG. Differential response of cardiac fibroblasts from young adult and senescent rats to ANG II. American journal of physiology. 2003 Apr;284(4):H1454–9. doi: 10.1152/ajpheart.00766.2002. [DOI] [PubMed] [Google Scholar]
- 12.Xu J, Carretero OA, Lin CX, Cavasin MA, Shesely EG, Yang JJ, et al. Role of Cardiac Overexpression of Angiotensin II in the Regulation of Cardiac Function and Remodeling Post-Myocardial Infarction. American journal of physiology. 2007 Jun 22; doi: 10.1152/ajpheart.00379.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liao P, Georgakopoulos D, Kovacs A, Zheng M, Lerner D, Pu H, et al. The in vivo role of p38 MAP kinases in cardiac remodeling and restrictive cardiomyopathy. Proc Natl Acad Sci U S A. 2001 Oct 9;98(21):12283–8. doi: 10.1073/pnas.211086598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bassi R, Heads R, Marber MS, Clark JE. Targeting p38-MAPK in the ischaemic heart: kill or cure? Curr Opin Pharmacol. 2008 Apr;8(2):141–6. doi: 10.1016/j.coph.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 15.Liang Q, Bueno OF, Wilkins BJ, Kuan CY, Xia Y, Molkentin JD. c-Jun N-terminal kinases (JNK) antagonize cardiac growth through cross-talk with calcineurin-NFAT signaling. Embo J. 2003 Oct 1;22(19):5079–89. doi: 10.1093/emboj/cdg474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Xu B, et al. MAP kinases. Chem Rev. 2001 Aug;101(8):2449–76. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 17.Davis RJ. Signal transduction by the JNK group of MAP kinases. Cell. 2000 Oct 13;103(2):239–52. doi: 10.1016/s0092-8674(00)00116-1. [DOI] [PubMed] [Google Scholar]
- 18.Petrich BG, Wang Y. Stress-activated MAP kinases in cardiac remodeling and heart failure; new insights from transgenic studies. Trends Cardiovasc Med. 2004 Feb;14(2):50–5. doi: 10.1016/j.tcm.2003.11.002. [DOI] [PubMed] [Google Scholar]
- 19.Liang Q, Molkentin JD. Redefining the roles of p38 and JNK signaling in cardiac hypertrophy: dichotomy between cultured myocytes and animal models. J Mol Cell Cardiol. 2003 Dec;35(12):1385–94. doi: 10.1016/j.yjmcc.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 20.Nagy N, Shiroto K, Malik G, Huang CK, Gaestel M, Abdellatif M, et al. Ischemic preconditioning involves dual cardio-protective axes with p38MAPK as upstream target. J Mol Cell Cardiol. 2007 May;42(5):981–90. doi: 10.1016/j.yjmcc.2007.02.010. [DOI] [PubMed] [Google Scholar]
- 21.Wang Y. Mitogen-activated protein kinases in heart development and diseases. Circulation. 2007 Sep 18;116(12):1413–23. doi: 10.1161/CIRCULATIONAHA.106.679589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Park JK, Fischer R, Dechend R, Shagdarsuren E, Gapeljuk A, Wellner M, et al. p38 mitogen-activated protein kinase inhibition ameliorates angiotensin II-induced target organ damage. Hypertension. 2007 Mar;49(3):481–9. doi: 10.1161/01.HYP.0000256831.33459.ea. [DOI] [PubMed] [Google Scholar]
- 23.Tachibana H, Perrino C, Takaoka H, Davis RJ, Naga Prasad SV, Rockman HA. JNK1 is required to preserve cardiac function in the early response to pressure overload. Biochem Biophys Res Commun. 2006 May 19;343(4):1060–6. doi: 10.1016/j.bbrc.2006.03.065. [DOI] [PubMed] [Google Scholar]
- 24.Sadoshima J, Montagne O, Wang Q, Yang G, Warden J, Liu J, et al. The MEKK1-JNK pathway plays a protective role in pressure overload but does not mediate cardiac hypertrophy. J Clin Invest. 2002 Jul;110(2):271–9. doi: 10.1172/JCI14938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lal H, Verma SK, Smith M, Guleria RS, Lu G, Foster DM, et al. Stretch-induced MAP kinase activation in cardiac myocytes: differential regulation through beta1-integrin and focal adhesion kinase. J Mol Cell Cardiol. 2007 Aug;43(2):137–47. doi: 10.1016/j.yjmcc.2007.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fukuzawa J, Booz GW, Hunt RA, Shimizu N, Karoor V, Baker KM, et al. Cardiotrophin-1 increases angiotensinogen mRNA in rat cardiac myocytes through STAT3 : an autocrine loop for hypertrophy. Hypertension. 2000 Jun;35(6):1191–6. doi: 10.1161/01.hyp.35.6.1191. [DOI] [PubMed] [Google Scholar]
- 27.Bader M. Role of the local renin-angiotensin system in cardiac damage: a minireview focussing on transgenic animal models. J Mol Cell Cardiol. 2002 Nov;34(11):1455–62. doi: 10.1006/jmcc.2002.2077. [DOI] [PubMed] [Google Scholar]
- 28.Palomeque J, Sapia L, Hajjar RJ, Mattiazzi A, Vila Petroff M. Angiotensin II-induced negative inotropy in rat ventricular myocytes: role of reactive oxygen species and p38 MAPK. American journal of physiology. 2006 Jan;290(1):H96–106. doi: 10.1152/ajpheart.00324.2005. [DOI] [PubMed] [Google Scholar]
- 29.Mel’nikova NP, Timoshin SS, Jivotova EY, Pelliniemi LJ, Jokinen E, Abdelwahid E. Angiotensin-II activates apoptosis, proliferation and protein synthesis in the left heart ventricle of newborn albino rats. Int J Cardiol. 2006 Sep 20;112(2):219–22. doi: 10.1016/j.ijcard.2005.09.003. [DOI] [PubMed] [Google Scholar]
- 30.Dostal DE, Rothblum KC, Lin CC, Baker KM. Regulation of renin and angiotensinogen in cultured neonatal rat cardiac fibroblasts by adrenergic, glucocorticoids, and angiotensin II stimulation. Hypertension. 1995;26:A562. [Google Scholar]
- 31.Esposito G, Prasad SV, Rapacciuolo A, Mao L, Koch WJ, Rockman HA. Cardiac overexpression of a G(q) inhibitor blocks induction of extracellular signal-regulated kinase and c-Jun NH(2)-terminal kinase activity in in vivo pressure overload. Circulation. 2001 Mar 13;103(10):1453–8. doi: 10.1161/01.cir.103.10.1453. [DOI] [PubMed] [Google Scholar]
- 32.Roussel E, Gaudreau M, Plante E, Drolet MC, Breault C, Couet J, et al. Early responses of the left ventricle to pressure overload in Wistar rats. Life sciences. 2008 Jan 30;82(56):265–72. doi: 10.1016/j.lfs.2007.11.008. [DOI] [PubMed] [Google Scholar]
- 33.Wang Y, Su B, Sah VP, Brown JH, Han J, Chien KR. Cardiac hypertrophy induced by mitogen-activated protein kinase kinase 7, a specific activator for c-Jun NH2-terminal kinase in ventricular muscle cells. J Biol Chem. 1998 Mar 6;273(10):5423–6. doi: 10.1074/jbc.273.10.5423. [DOI] [PubMed] [Google Scholar]
- 34.Choukroun G, Hajjar R, Fry S, del Monte F, Haq S, Guerrero JL, et al. Regulation of cardiac hypertrophy in vivo by the stress-activated protein kinases/c-Jun NH(2)-terminal kinases. J Clin Invest. 1999 Aug;104(4):391–8. doi: 10.1172/JCI6350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kyoi S, Otani H, Matsuhisa S, Akita Y, Tatsumi K, Enoki C, et al. Opposing effect of p38 MAP kinase and JNK inhibitors on the development of heart failure in the cardiomyopathic hamster. Cardiovasc Res. 2006 Mar 1;69(4):888–98. doi: 10.1016/j.cardiores.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 36.Domenighetti AA, Wang Q, Egger M, Richards SM, Pedrazzini T, Delbridge LM. Angiotensin II-mediated phenotypic cardiomyocyte remodeling leads to age-dependent cardiac dysfunction and failure. Hypertension. 2005 Aug;46(2):426–32. doi: 10.1161/01.HYP.0000173069.53699.d9. [DOI] [PubMed] [Google Scholar]
- 37.Kimura B, Mohuczy D, Tang X, Phillips MI. Attenuation of hypertension and heart hypertrophy by adeno-associated virus delivering angiotensinogen antisense. Hypertension. 2001 Feb;37(2 Part 2):376–80. doi: 10.1161/01.hyp.37.2.376. [DOI] [PubMed] [Google Scholar]
- 38.Sanghi S, Kumar R, Smith M, Baker KM, Dostal DE. Activation of protein kinase A by atrial natriuretic peptide in neonatal rat cardiac fibroblasts: role in regulation of the local renin-angiotensin system. Regul Pept. 2005 Dec 15;132(13):1–8. doi: 10.1016/j.regpep.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 39.Dostal DE, Booz GW, Baker KM. Regulation of angiotensinogen gene expression and protein in neonatal rat cardiac fibroblasts by glucocorticoid and beta-adrenergic stimulation. Basic Res Cardiol. 2000 Dec;95(6):485–90. doi: 10.1007/s003950070025. [DOI] [PubMed] [Google Scholar]
- 40.Booz GW, Dostal DE, Baker KM. Paracrine actions of cardiac fibroblasts on cardiomyocytes: implications for the cardiac renin-angiotensin system. Am J Cardiol. 1999 Jun 17;83(12A):44H–7H. doi: 10.1016/s0002-9149(99)00257-x. [DOI] [PubMed] [Google Scholar]
- 41.Lal H, Guleria RS, Foster DM, Lu G, Watson LE, Sanghi S, et al. Integrins: novel therapeutic targets for cardiovascular diseases. Cardiovasc Hematol Agents Med Chem. 2007 Apr;5(2):109–32. doi: 10.2174/187152507780363223. [DOI] [PubMed] [Google Scholar]
- 42.Sabri A, Lucchesi PA. ANG II and cardiac myocyte contractility: p38 is not stressed out! American journal of physiology. 2006 Jan;290(1):H72–3. doi: 10.1152/ajpheart.00873.2005. [DOI] [PubMed] [Google Scholar]
- 43.Hsieh TJ, Fustier P, Zhang SL, Filep JG, Tang SS, Ingelfinger JR, et al. High glucose stimulates angiotensinogen gene expression and cell hypertrophy via activation of the hexosamine biosynthesis pathway in rat kidney proximal tubular cells. Endocrinology. 2003 Oct;144(10):4338–49. doi: 10.1210/en.2003-0220. [DOI] [PubMed] [Google Scholar]
- 44.Narayanan CS, Cui Y, Kumar S, Kumar A. cAMP increases the expression of human angiotensinogen gene through a combination of cyclic AMP responsive element binding protein and a liver specific transcription factor. Mol Cell Biochem. 2000 Sep;212(12):81–90. [PubMed] [Google Scholar]