

Abstract
Both mast cells and complement participate in innate and acquired immunity. The current study examines whether β-tryptase, the major protease of human mast cells, can directly generate bioactive complement anaphylatoxins. Important variables included pH, monomeric vs tetrameric forms of β-tryptase, and the β-tryptase-activating polyanion. The B12 mAb was used to stabilize β-tryptase in its monomeric form. C3a and C4a were best generated from C3 and C4, respectively, by monomeric β-tryptase in the presence of low molecular weight dextran sulfate or heparin at acidic pH. High molecular weight polyanions increased degradation of these anaphylatoxins. C5a was optimally generated from C5 at acidic pH by β-tryptase monomers in the presence of high molecular weight dextran sulfate and heparin polyanions, but also was produced by β-tryptase tetramers under these conditions. Mass spectrometry verified that the molecular mass of each anaphylatoxin was correct. Both β-tryptase-generated C5a and C3a (but not C4a) were potent activators of human skin mast cells. These complement anaphylatoxins also could be generated by β-tryptase in releasates of activated skin mast cells. Of further biologic interest, β-tryptase also generated C3a from C3 in human plasma at acidic pH. These results suggest β-tryptase might generate complement anaphylatoxins in vivo at sites of inflammation, such as the airway of active asthma patients where the pH is acidic and where elevated levels of β-tryptase and complement anaphylatoxins are detected.
Although levels of both C3a and C5a are increased in bronchoalveolar lavage fluid obtained from subjects with asthma (1–4), how they are produced in asthmatic airways is uncertain. Efficient generation of these anaphylatoxins along with C4a is well known to occur during the classical pathway of IgG/IgM Ag-dependent complement activation. However, because IgG/IgM immune complexes are not typically associated with asthma, other mechanisms of anaphylatoxin generation need to be considered. The lectin and alternative pathways could be involved, but several proteolytic enzymes outside of these complement pathways also can generate anaphylatoxin-like activity, including thrombin, elastase, monocyte protease, kallikrein, and house dust mite protease (5–10). Mast cells are known to be activated in asthma (11–14); mast cell hyperplasia in bronchial smooth muscle and mucus glands occurs in asthmatic lungs (15–17); and mast cells are a rich source of proteases (18, 19). Also, the acidic pH of the airways of asthma subjects (20) may promote β-tryptase proteolytic activity (21–23). We hypothesize that mast cell-released β-tryptase may be responsible for anaphylatoxin generation in the airways of asthma subjects.
Mast cell activation is triggered by several pathways. Classically, aggregation of FcεR1 by allergen and IgE triggers release of the preformed mediators by degranulation and of newly synthesized lipids, such as PGD2 and leukotriene C4, cytokines such as IL-5, IL-6, IL-8, IL-13, TNF-α, and GM-CSF and chemokines such as MIP-1α, MIP-1β, and MCP-1 (18). Stimulation through TLRs (24–28), the C5aR (CD88) and C3aR (29–32), and through FcγRIIa (33) also may activate human mast cells to release such mediators.
Human mast cells have been separated into two types based initially on the protease composition of their secretory granules; those from MCTC cells containing β-tryptase, chymase, carboxypeptidase A3, and cathepsin G (18), and MCT cells containing only β-tryptase and lacking chymase mRNA. These proteases are released along with histamine and proteoglycans during granule exocytosis. MCT cells are found in small intestinal mucosa and lung (particularly alveolar walls and near the surface of bronchi and bronchioles), whereas MCTC cells are the dominant type of mast cell in intestinal submucosa, skin and conjunctiva, and in bronchiolar smooth muscle and mucus glands of asthmatics. Functional surface expression of CD88, the C5a receptor, on MCTC but not MCT cells, is an additional distinguishing feature (34).
αβ-Tryptases in human mast cells are expressed by two genes on human chromosome 16, TPSAB1 and TPSB2 (35). TPSAB1 is allelic for α-tryptase and β-tryptase in an ~1:1 ratio, whereas TPSB2 encodes only β-tryptase. The α-tryptase gene is absent from the genome of ~25% of individuals, but when present yields a product that appears to be processed no further than to α-protryptase, which is spontaneously released and lacks significant proteolytic activity (36, 37). The β-tryptase gene is universally expressed by all human mast cells, and yields a product that is mostly processed to the fully mature, enzymatically active, heparin-stabilized tetramer, which then is stored in secretory granules until degranulation occurs. β-Tryptase cleaves at the carboxyl end of arginine and lysine residues in proteins, a specificity needed to generate complement anaphylatoxins that each has arginine in the C-terminal position.
The quaternary structure of the β-tryptase homotetramer (38) restricts access of both high m.w. (HMW)3 substrates and inhibitors to the active sites because these active sites face into the small central pore of the tetramer. Consequently, nearly all biological protease inhibitors fail to affect β-tryptase activity (39). The β-tryptase tetramer is proteolytically active in both acidic and neutral pH buffers, though greater fibrinogenolytic activity occurs at acidic than neutral pH. In contrast, a heparin-stabilized monomeric form of mature β-tryptase has been detected at low concentrations of β-tryptase, which is inactive at neutral pH but active at acidic (pH 6 – 6.5) conditions (22, 40). High concentrations of β-tryptase monomers can be formed with the anti-αβ-tryptase mAb, B12, as well as the B12 Fab fragment, which dissociates heparin-stabilized β-tryptase tetramers to monomers that, like free monomers, are inactive at neutral pH but active at acidic pH (23). Active β-tryptase monomers, compared with tetramers, exhibit an enhanced ability to cleave protein substrates and an enhanced susceptibility to inhibition by HMW serine protease inhibitors at acidic pH. A previous study revealed that complement C5 was not cleaved by β-tryptase at neutral pH, whereas C3 was slowly cleaved to yield the anaphylatoxin C3a in the absence of heparin (41). In the presence of heparin, cleavage of the C3α-chain subunit by β-tryptase was also evident, but enhanced degradation of C3a appeared to account for the absence of this anaphylatoxin in Western blots. The current study presents evidence that β-tryptase can generate anaphylatoxins C3a, C4a, and C5a in an acidic buffer in vitro, and speculates that this could be important in the pathogenesis of human asthma.
Materials and Methods
Reagents
MES, HEPES, BSA, HMW heparin from porcine intestinal mucosa, low m.w. (LMW) heparin from porcine intestinal mucosal (3 kDa), 500 kDa dextran sulfate (DS500K), 5 kDa dextran sulfate (DS5K), histone, protamine, poly-L-lysine, lactoferrin, chromogenic substrate TGPK (tosyl-Gly-Pro-Lys-p-nitroanilide), mouse IgG1 (MOPC 31C), 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium tablets, p-nitrophenyl phosphate tablets, Percoll (density = 1.13 g/ml; Sigma-Aldrich); avidin (EMD Biosciences); alkaline phosphatase-conjugated goat anti-mouse IgG (Fc specific; Jackson ImmunoResearch Laboratories); alkaline phosphatase-conjugated streptavidin (Roche Molecular Biochemicals); chromogenic substrate S-2288 IPR (H-D-Ile-Pro-Arg-p-nitroanilide; Chromogenix); protein A-agarose (Pierce); human complement C3, C4, and C5; anaphylatoxins C3a, C3adesArg, C4a, C4adesArg, and C5adesArg; rabbit anti-human C3a Ab, rabbit anti-human C4a Ab, rabbit anti-human C5a Ab, and human C3-depleted serum (42) (Complement Technology); anti-CD117 and rabbit anti-C5aR (CD88) (BD Biosciences) and recombinant human C5a (EMD Biosciences) were obtained as indicated. Mouse anti-C5aR mAb that inhibit C5a binding to C5aR (S5/1) was obtained from Serotec. Rabbit anti-human C3a Ab, rabbit anti-human C4a Ab, and rabbit anti-human C5a Ab are also provided as a gift from Dr. T. E. Hugli (Torrey Pines Institute for Molecular Studies, San Diego, CA). Mouse anti-human Factor I was obtained from GeneTex. Human stem cell factor is a gift from Amgen. Human β-tryptase was isolated from human lung using B2 mAb Affi-Gel and heparin Sepharose CL-6B (Amersham Biosciences) chromatography as described (22). Purified β-tryptase (100 –200 μg/ml) was stored in 10 mM MES buffer (pH 6.5), containing 0.8 M NaCl and 20% glycerol at −70°C. Mouse anti-human tryptase mAbs, B2, B12, G3, and G4 (all of these Abs are IgG1 isotype) were prepared as described (43). Fab fragments of B12 were prepared using immobilized papain digestion and were purified by protein A-agarose column (Pierce).
Measurements of β-tryptase activity and protein
Enzymatic activity of β-tryptase was measured by cleavage of TGPK as described using 0.1 mM TGPK in 0.05 M HEPES buffer (pH 7.4), containing 0.12 M NaCl (21). Released p-nitroanilide was monitored at 405 nm by a Cary 3E UV-Visible spectrophotometer (Varian). Activity measurements at pH 6.0 were performed using IPR substrate in PBS (pH 6.0), as described. β-Tryptase protein levels were determined by ELISA using B12 for capture, biotinylated G4 for detection, and alkaline phosphatase-conjugated streptavidin (1/2000 dilution) and p-nitrophenyl phosphate solution for color development (44).
Isolation of human skin mast cells
Human skin mast cells were isolated as described (45). Fresh skin was obtained from Cooperative Human Tissue Network of the National Cancer Institute or National Disease Research Interchange. Briefly, tissue was cut in fragments and incubated in a solution of HBSS with 1 mM CaCl2 containing type II collagenase (1.5 mg/ml), hyaluronidase (0.7 mg/ml), type I DNase (0.3 mg/ml), and 1% FCS for 2 h at 37°C. The dispersed cells were separated from residual tissues by filtration through 80-mesh sieve and suspended in HBSS containing 10 mM HEPES and 1% FCS. Then, cells were layered over Percoll and centrifuged at 700 × g at room temperature for 20 min. Nucleated cells were collected from the buffer/Percoll interface, and skin mast cells (MCTC cells) were resuspended in AIM-V medium or X-Vivo medium with 100 ng/ml recombinant human stem cell factor, and cultured up to 2–3 mo. The purity of mast cells was analyzed by toluidine blue staining, by flow cytometry using anti-CD88, anti-CD117, and anti-FcεR Ab, and by immunohistochemical staining with anti-tryptase G3 and anti-chymase mAb.
Degranulation assay of mast cells
Degranulation of human mast cells was determined by measuring β-hexosaminidase release (46, 47). Briefly, 10-μl samples are incubated with 50 μl of skin mast cells (1 × 106/ml) in Tyrodes buffer containing 10 mM HEPES (pH 7.35), 0.1% glucose, 0.05% gelatin, 1 mM MgCl2, and 2.5 mM CaCl2 at 37°C for 15–30 min. After adding 180 μl of cold Tyrodes buffer lacking CaCl2 and MgCl2, the mixture is centrifuged at 4000 rpm for 10 min at 4°C. Then, 5 μl of supernatant is transferred to a 96-well mi-croplate and 45 μl of p-nitrophenyl-N-acetyl β-D-glucosaminide solution in sodium citrate buffer (pH 4.5) are added. After 1.5 h at 37°C, 150 μl of 0.2 M glycine buffer (pH 10.7) is added and the OD405 is measured. The total amount of β-hexosaminidase activity retained in the mast cell pellet is measured in extracts obtained by sonication. To confirm the specificity for C5a, skin mast cells were treated with blocking anti-C5aR mAb, S5/1 (Serotec).
Anaphylatoxin generation by β-tryptase and analysis by Western blotting
An active monomer of tryptase is made by adding the Fab fragment of B12 to heparin-stabilized tryptase tetramer as described (23). LMW heparin, DS500K and DS5K were also used to stabilize tryptase. C3, C4, and C5 were treated with different forms of tryptase and with different stabilizers. Initially, C3 and tryptase stabilized with various polyanion was incubated in PBS (pH 6.0), with and without Fab fragment of B12 at 37°C. After using different incubation times, samples were mixed with SDS sample buffer containing 2% 2-ME, boiled for 3 min, and subjected to electrophoresis under denaturing conditions on a 16% polyacrylamide gel. Proteins were transferred onto a nitrocellulose membrane using a Novex system (Invitrogen) for 1 h at 50 V. After blocking with PBS (pH 7.4), containing 5% BSA and 0.05% Tween 20 for 1 h, C3a was labeled with rabbit anti-human C3a Ab (1/1000 dilution) for 1 h at room temperature followed incubations with alkaline phosphatase-conjugated goat anti-rabbit IgG Ab (1/2000 dilution; Jackson ImmunoResearch Laboratories) and developed with 5-bromo-4-chloro-3-indolyl phosphate/nitroblue tetrazolium solution (Sigma-Aldrich). Gels were also stained with Coomassie brilliant blue (Pierce) to confirm the cleavage of α-chain (115 kDa) of C3. C4a and C5a generation was examined as similar manner as C3a and detected using rabbit anti-C4a Ab and rabbit anti-C5a Ab, respectively.
ELISA for C3a
Mouse anti-C3a/C3adesArg Ab against a neo-epitope (1.25 μg/ml; Cell Sciences) was coated onto microplates for capture, and 50 μg/ml biotin-conjugated rabbit anti-C3a Ab was used for detection. Rabbit anti-C3a was biotinylated (Pierce). After incubation with avidin-peroxidase (1/1000; BD Biosciences), 3,3′,5,5′-tetramethylbenzidine solution (Sigma-Aldrich) was added and the reaction was stopped by adding 1 N H2SO4 and the OD measured at 450 nm using a SpectraMax 384 Plus UV-VIS plate reader (Molecular Devices).
Mass spectrometry analysis
Reaction mixtures of generated C3a- and C4a-like molecules were acidified by adding 2% acetic acid (final concentration) and concentrated by ZipTip C18, reversed-phase pipette tips (Millipore). After elution by 50% acetonitrile in 2% acetic acid, samples were examined by MALDI-TOF mass spectrometry at University of Virginia Biomolecular Research Facility (Charlottesville, VA) to determine the m.w. of anaphylatoxin-like molecules. Because the carbohydrate moiety on C5a interferes with the mass spectrometry analysis, C5a-like molecules were treated with N-glycosidase F (EMD Biosciences) in 50 mM phosphate buffer (pH 7.5) with or without 0.02% 2-ME and 200 μg/ml BSA for 17 h at 37°C. N-glycosidase F-treated C5a-like molecules were also concentrated using ZipTip C18. As for control, purified C3a, C4a, and C5adesArg were treated and analyzed as described.
Results
The strategy used to understand whether β-tryptase can generate complement anaphylatoxins can be divided into three portions. Initial experiments used purified components. In these experiments, LMW products of C3, C4, and C5 were referred to as anaphylatoxin-like fragments because Western blotting cannot reliably distinguish between intact anaphylatoxins and partially degraded, biologically inactive anaphylatoxin fragments. Other experiments evaluated whether these fragments represented intact anaphylatoxins by mass spectroscopy and biologic activity. The final experiments were designed to assess whether the process of anaphylatoxin generation by β-tryptase could occur in more complex biologic settings, albeit in vitro, by assessing whether a mast cell releasate or activated mast cells could generate anaphylatoxins, and whether β-tryptase might generate anaphylatoxins from human plasma.
Generation of C3a-like molecules by β-tryptase stabilized by different polyanions at acidic pH
Degradation of the C3a anaphylatoxin was previously noted to be enhanced by excess heparin (41). Whether this response was due to a direct affect on the conformation of C3a caused by heparin binding, or by an effect of heparin on tryptase beyond stabilizing its tetrameric conformation was uncertain. To begin to resolve this question, a limiting amount of polyanion was used to stabilize β-tryptase. β-Tryptase (3.5 μg/ml) was incubated with different amounts of different polyanions at pH 7.4 for 60 min at 37°C to determine the amounts needed to preserve 60% of the initial tryptase activity; residual peptidolytic activity was measured at pH 7.4 using TGPK. As a result, 0.8 μg/ml HMW heparin, 7 μg/ml LMW heparin, 0.7 μg/ml DS500K, and 10 μg/ml DS5K were used to stabilize 60% of the β-tryptase activity. Polyanion-stabilized β-tryptase was then incubated with C3 in the presence or absence of the B12 mAb Fab fragment (4 molar excess over β-tryptase) at pH 6.0 for 15 min at 37°C. B12 mAb Fab fragment converts β-tryptase tetramers to monomers that are proteolytically active at acidic but not neutral pH. As shown in Fig. 1A, LMW fragments of C3 approximating the size of C3a (9094 Da) were only detected in the presence of B12 Fab fragments (Fig. 1A, lanes 1, 3, 5, and 7). Both HMW heparin- and DS500K-stabilized β-tryptase produced smaller sized products or doublets, suggesting further metabolism (Fig. 1A, lanes 1 and 5). Extending these incubations from 15 min to 2 h did not appreciably change these patterns (data not shown). When the cleavage of C3 was examined by protein staining after SDS-PAGE, most of the C3α (120 kDa) had been converted to a smaller C3α′-like fragment (110 kDa), even in the absence of B12 Fab (Fig. 1B). DS500K-stabilized β-tryptase with B12 Fab seemed to result in the most complete conversion (Fig. 1B, lane 5). The presence of C3α′-like fragments without C3a-like molecules in the absence of B12 Fab suggested that C3a could have been generated and rapidly degraded by β-tryptase, even with limiting amounts of polyanions.
Figure 1.

C3 cleavage by β-tryptase stabilized with limiting amounts of different polyanions in the presence or absence of B12 Fab. A, C3a Western blotting. β-Tryptase (3.5 μg/ml) was mixed with equal volumes of HMW heparin (0.8 μg/ml), LMW heparin (7 μg/ml), DS500K (0.7 μg/ml), or DS5K (10 μg/ml) and incubated for 10 min at room temperature, then for another 15 min with a 4 molar excess of B12 Fab or MOPC Fab, as indicated, and finally for 15 min at 37°C with 33 μg/ml human C3 in PBS at pH 6.0. Samples were then subjected to SDS-PAGE in a 16% acrylamide gel followed by Western blotting using rabbit anti-C3a Ab as described. C3 (lane 9) and C3a (lane 10) controls are shown. B, Cleavage of C3α but not C3β by β-tryptase. Protein bands were detected by staining with Coomassie brilliant blue after SDS-PAGE in a 12% acrylamide gel. Lane contents were as described.
Effect of basic substances on the generation of intact size of C3a-like molecules by polyanion-stabilized β-tryptase
HMW polyanions may activate β-tryptase but enhance the degradation of anaphylatoxins if portions of these β-tryptase-bound polyanions are also available to bind anaphylatoxins and thereby make them more susceptible to proteolysis. To examine whether putative C3a could be protected from degradation by basic molecules that in theory would neutralize those negatively charged moieties on tryptase-stabilizing polyanions that were not already bound to positively charged amino acids in β-tryptase, several basic substances were added during the incubation of C3 with β-tryptase. Avidin, histone, protamine, lactoferrin, and poly-L-lysine were examined. To determine the proper amount of these substances, first their effects on the activity of HMW heparin-stabilized β-tryptase were examined. In dose-response experiments, high concentrations of each of these substance inhibited tryptase activity (pH 6.0, IPR substrate). The concentrations needed to inhibit 30 – 40% of the activity of 3.5 μg/ml β-tryptase were 12.5 μg/ml for avidin, 3.2 μg/ml for histone, 2 μg/ml for protamine, 100 μg/ml for lactoferrin and 16 ng/ml for poly-L-lysine. Fig. 2 shows the effect of 12.5 μg/ml of avidin on C3a-like molecule generation by β-tryptase in the presence of B12 Fab at pH 6.0. Avidin resulted in markedly less intact C3a-like material with HMW heparin (Fig. 2, lane 2 vs lane 1) and DS500K (Fig. 2, lane 6 vs lane 5), possibly by enhancing C3a-like molecule degradation, though there was no apparent effect on the sizes of C3a-like molecules detected. Putative C3a degradation fragments were observed with DS500K and heparin. Only with LMW heparin did avidin appear to shift the C3a-like product to the slower-migrating band (Fig. 2, lanes 4 and 3). This latter product exhibited a similar electrophoretic mobility to the C3a-like bands observed with DS5K both in the absence (Fig. 2, lane 7) and presence (Fig. 2, lane 8) of avidin. Histone and lactoferrin also showed no effect on the size of C3a-like molecules (data not shown). Protamine and poly-L-lysine showed marked inhibition of C3a-like molecule generation with all polyanions (data not shown). These results did not support the hypothesis that small basic substances might protect C3a from degradation by polyanion-stabilized β-tryptase, though conceivably other basic substances might do so. Furthermore, even if excess polyanions were used to stabilize β-tryptase, there was basically no difference in putative C3a degradation patterns, suggesting that such polyanions do not have a major effect on the susceptibility of C3a or C3a-like fragments to degradation (data not shown). Instead these data raise the possibility that polyanions of different charge density or size have subtle effects on β-tryptase conformation that in turn might affect β-tryptase-substrate interactions.
Figure 2.

Effect of avidin on generation of C3a by polyanion-stabilized β-tryptase in the presence of B12 Fab. β-Tryptase (3.5 μg/ml) stabilized with limiting amounts of polyanions was incubated with a 4 molar excess of B12 Fab and 33 μg/ml of C3 at 37°C for 20 min at pH 6.0 in PBS with and without 12.5 μg/ml of avidin. After SDS-PAGE in a 16% acryl-amide gel and Western blotting, C3a was probed with rabbit anti-C3a Ab. Contents were of HMW heparin (HMW-h), LMW heparin (LMW-h), commercial C3 control (C3), commercial C3a control (C3a), and degraded C3a (*).
Time course of C3a-like molecule generation by DS5K and HMW heparin-stabilized β-tryptase
The time course for generating C3a-like molecules from C3 was examined with β-tryptase stabilized with DS5K and HMW heparin at pH 6.0 in the presence of B12 Fab as shown in Fig. 3. C3a-like molecules were generated more rapidly by DS5K-stabilized β-tryptase (band apparent by 1 min) than by HMW heparin-stabilized β-tryptase (band apparent by 4 min). The band of C3a-like material initially generated by both conditions appeared to comigrate with the C3a control. By 60 min, the material generated by DS5K-stabilized β-tryptase exhibited a small amount of a faster migrating band, but even after 120 min of incubation, the major product comigrated with C3a. In contrast, the product generated by HMW heparin-stabilized β-tryptase showed comparable amounts of the C3a comigrating band and faster migrating bands at 60 and 120 min. Importantly, the maximal intensity of the C3a comigrating band was far higher for DS5K- than HMW heparin-stabilized β-tryptase. A reasonable interpretation of these results is that generation of C3a exceeds its degradation with DS5K-stabilized β-tryptase, whereas HMW heparin-stabilized β-tryptase enhances C3a degradations.
Figure 3.
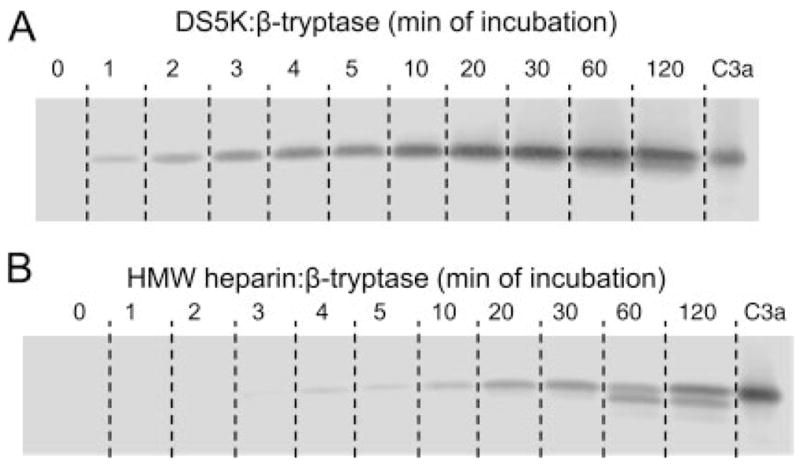
Time course of C3a-like molecule generation by β-tryptase stabilized with DS5K (A) and HMW heparin (B). β-Tryptase (3.5 μg/ml) was stabilized with 10 μg/ml DS5K or 0.8 μg/ml HMW heparin and incubated with a 4 molar excess of B12 Fab and then 33 μg/ml C3 in pH 6.0 PBS at 37°C at indicated time. After SDS-PAGE in 16% acrylamide gels, Western blotting was performed with rabbit anti-C3a Ab. Time points include 0, 1, 2, 3, 4, 5, 10, 20, 30, 60, and 120 min (lanes 1–11) with a commercial C3a control (C3a) (lane 12).
C4 metabolism by polyanion-stabilized β-tryptase
The ability of polyanion-stabilized β-tryptase to generate a C4a-like product was examined. Analogous to C3, generation of C4a-like molecules from C4 during a 60-min incubation was only observed in the presence of B12 Fab (Fig. 4A), indicating that this activity associates with β-tryptase monomers. Under these conditions DS5K- and HMW heparin-stabilized β-tryptase generated C4a-like molecules that exhibited similar electrophoretic mobility to that of native C4a, whereas those generated by DS500K-stabilized β-tryptase appeared to be smaller. An analysis of the time-dependent generation of C4a-like molecules (Fig. 4B) showed that the C4a-like products generated initially by DS500K-stabilized β-tryptase (5 min) and by HMW heparin-stabilized β-tryptase (30 min) had the same electrophoretic mobility as commercial C4a. However, DS500K-stabilized β-tryptase had completely degraded this product by 60 min, whereas degradation by HMW heparin-stabilized β-tryptase appeared to be minimal by 60 min. LMW heparin-stabilized and DS5K-stabilized β-tryptase appeared to have less C4a generating activity, producing C4a-like products only at the 60 min incubation time. When C4 degradation by polyanion-stabilized β-tryptase was examined, the C4α′ product was detected between the C4α and C4β bands both in the presence and absence of B12 Fab (Fig. 4C). However, a portion of the C4α remained intact, suggesting that generation of C4a-like molecules was submaximal under the experimental conditions used.
Figure 4.

C4 metabolism by β-tryptase stabilized with different polyanions in the presence or absence of B12 Fab. β-Tryptase (3.5 μg/ml) stabilized with HMW heparin (0.8 μg/ml), LMW heparin (7 μg/ml), DS500K (0.7 μg/ml), or DS5K (10 μg/ml) was incubated for 10 min at room temperature, then with or without a 4 molar excess of B12 Fab for 15 min and finally with 33 μg/ml human C4 in PBS at pH 6.0. After various incubation times at 37°C, samples were subjected to SDS-PAGE on either a 16% or 10% acrylamide gel. Western blotting was performed with rabbit anti-C4a Ab or goat anti-C4 Ab. A, C4a-like molecule generation. β-Tryptase was stabilized with HMW heparin, DS500K, and DS5K with or without B12 Fab, as indicated, and incubated with C4 for 60 min. A purified commercial human C4a control (C4a) is used. B, Time course of C4a-like molecule generation in the presence of B12 Fab. β-Tryptase was stabilized with HMW heparin, DS500K, LMW heparin, and DS5K for 5, 30, and 60 min as indicated. The C4 control (C4) is used. C, C4 cleavage. β-Tryptase was stabilized with HMW heparin, DS500K, DS5K, and heparin proteoglycan with or without B12 Fab, and incubated with C4 control (C4) as in A for 60 min.
C5 metabolism by polyanion-stabilized β-tryptase
β-Tryptase stabilized by different polyanions in the presence and absence of B12 Fab was incubated with C5 for 30 min at 37°C. As shown in Fig. 5A, the most intense bands of C5a-like product were generated by β-tryptase stabilized with HMW heparin and DS500K in the presence of B12 Fab. However, in contrast to C3 and C4 metabolism, C5a-like product also was detected with each of the polyanions in the absence of B12 Fab, indicating that β-tryptase tetramers as well as monomers could generate this product. All C5a-like products comigrated with plasma-derived C5adesArg. C5a is a glycosylated protein with an apparent molecular mass of ~11,000 Da, higher than the calculated molecular mass based only on its primary amino acid sequence of 8274 Da. Glycosylated C5a and C5adesArg, though, are essentially indistinguishable from one another by electrophoretic mobility in SDS-PAGE. Fig. 5B shows the time-dependent generation of a C5a-like molecule by DS500K-stabilized β-tryptase. This molecule was detected by 5 min in the presence of B12 Fab. However, in contrast to generation of C3a- and C4a-like molecules, no degradation of the C5a-like product was detected during the 120-min incubation time. Also, excess amounts of HMW heparin and DS500K did not affect the apparent size of the generated C5a-like molecule (data not shown), indicating enhanced stability from β-tryptase-catalyzed degradation relative to the other anaphylatoxins.
Figure 5.
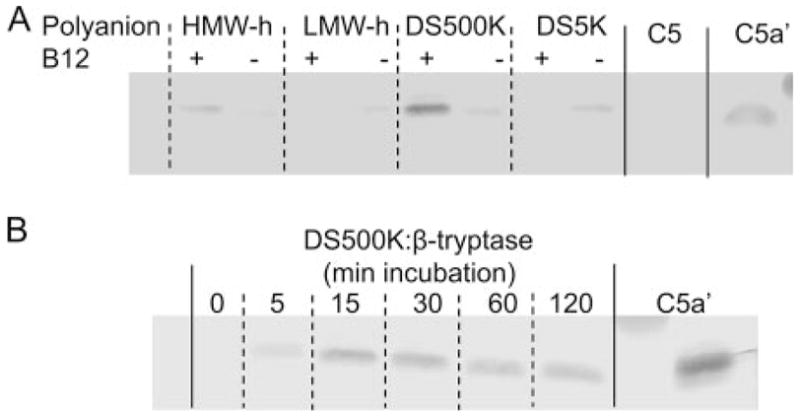
C5a-like molecule generation by polyanion-stabilized β-tryptase. A, Dependence of C5a-like molecule generation by β-tryptase on the polyanion and presence of B12 Fab. β-Tryptase (3.5 μg/ml), stabilized with HMW heparin (0.8 μg/ml), LMW heparin (7 μg/ml), DS500K (0.7 μg/ml), or DS5K (10 μg/ml), was incubated with 4 molar excess B12 Fab and then with C5 (33 μg/ml) in PBS (pH 6.0) at 37°C for 30 min. Incubation mixtures were subjected to SDS-PAGE in 16% acrylamide gels and Western blotting with rabbit anti-C5a Ab. A, β-Tryptase was stabilized with HMW heparin, LMW heparin, DS500K, and DS5K with (+) and without (−) B12 Fab (lanes 1–8). C5 (lane 9) and C5adesArg (C5a′) (lane 10) are shown. Commercial C5adesArg had been purified from human plasma, and thus was glycosylated. B, Time course of C5a-like molecule generation by DS500K-stabilized β-tryptase with B12 Fab. Lanes correspond to incubation times of 0, 5, 15, 30, 60, and 120 min, including C5adesArg (C5a′) (last lane).
Mass spectrometry analysis of anaphylatoxin-like molecules
A C3a-like molecule was generated as shown in Fig. 3A by DS5K-stabilized β-tryptase with B12 Fab incubated with C3 for 20 min. Approximately 0.5 μg of this product was concentrated with a ZipTip C18 cartridge and eluted with 50% acetonitrile in 2% acetic acid. After evaporation of acetonitrile, the sample was analyzed by mass spectrometry using a MALDI system. Purified commercial C3a, treated similarly, served as a control. The expected size of C3a, a nonglycosylated peptide, is 9094 Da. A C4a-like molecule was generated as shown in Fig. 4B by LMW heparin-stabilized β-tryptase with B12 Fab incubated with C4 for 60 min and concentrated for mass spectroscopy as for C3a. A C5a-like molecule was generated as shown above in Fig. 5B by DS500K-stabilized β-tryptase with B12 Fab incubated with C5 for 40 min. After adding 2-ME (0.02%), the C5a-like sample was treated with N-gly-cosidase F for 18 h at 37°C to remove N-linked carbohydrate that could interfere with the mass spectroscopic analysis, and then concentrated as described. Fig. 6 shows the mass spectroscopic data. The major peak associated with the C5a-like molecule occurred at a mass-to-charge ratio 8273 m/z, nearly identical with the calculated molecular mass of 8274 Da for C5a. Other less prominent peaks occurred at ~78 Da intervals due to different amounts of 2-ME (used during deglycosylation) attached to the seven cysteine residues. The C4a-like molecule spectrum is composed of two prominent peaks at 8048 and 8759 m/z. The peak of 8759 m/z corresponds to the expected molecular mass of 8764 Da for C4a, whereas the 8048 m/z peak appears to reflect the removal of seven amino acids from the C terminus of C4a. The one major peak associated with the C3a-like molecule appears at 9092 m/z, which corresponds to the calculated molecular mass of 9094 Da for C3a. In addition, a less intense peak at 8286 m/z might correspond to the thrombin-mediated cleavage fragment next to Arg69, 8 aa from the C-terminal of intact C3a (48).
Figure 6.
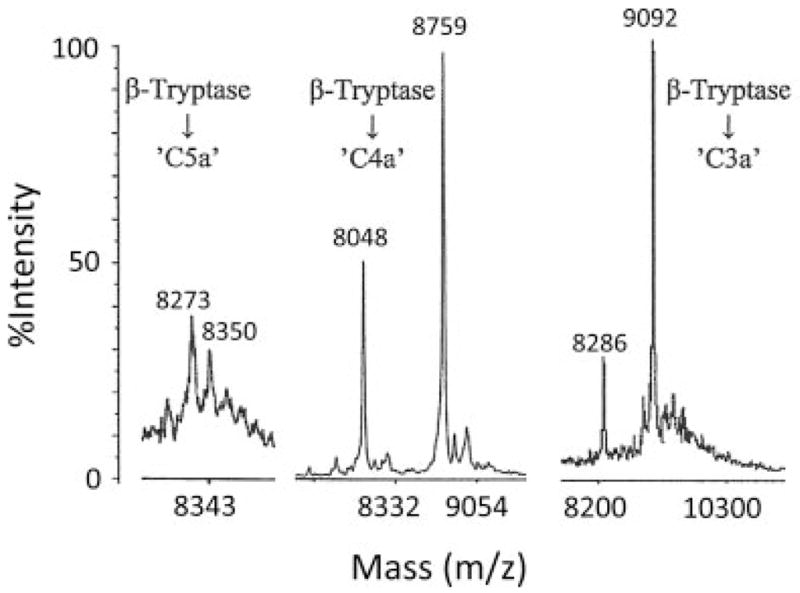
Mass spectrometry analysis of anaphylatoxin-like molecules. C5a-like, C4a-like, and C3a-like molecules, respectively, were generated by β-tryptase with B12 Fab in PBS (pH 6.0) that had been stabilized with DS500K, LMW heparin, and DS5K and then incubated with parent complement proteins for 40, 60, and 20 min. C5a-like molecules were further treated with N-glycosidase F for 18 h. These anaphylatoxin-like molecules were concentrated by ZipTip C18 reversed-phase chromatography and subjected to mass spectrometry using a MALDI system.
Biological activity of C5a-like and C3a-like molecules generated by polyanion-stabilized β-tryptase
Although mass spectral analysis confirmed the C5a-like product generated by β-tryptase to be C5a based on molecular mass, determination of biological activity is another important criterion for authentic C5a. C5a is well known to activate human skin mast cells to degranulate and release mediators such as β-hexosaminidase. The C5a degradation product, C5adesArg, is at least 1000-fold less potent than C5a at stimulating human skin mast cells to degranu-late (31). The β-tryptase-generated C5a product was tested accordingly as shown in Fig. 7A. C5 was treated with DS500K-stabilized β-tryptase with B12 Fab for 40 min at 37°C as mentioned. The concentration of C5a was estimated by comparing Western blots of the generated C5a to commercial C5a. As a positive control, 22E7 (mouse anti-FcεRI mAb) stimulated ~50% release. As another positive control, recombinant human C5a (0.1–10 μg/ml) stimulated skin mast cells in a dose-dependent manner to release from 13% to 42% of their β-hexosaminidase. β-Tryptase-generated C5a (0.15 and 0.3 μg/ml) also stimulated release of 19% and 32%, respectively, comparable to the magnitude of degranulation caused by comparable doses of recombinant human C5a. To validate the specificity of C5a stimulation, skin mast cells were preincubated with the neutralizing anti-C5aR Ab (5S/1) before adding recombinant human C5a or β-tryptase-generated C5a. In both cases, degranulation was effectively inhibited. Fig. 7B shows the ability of C3a-like molecules generated by DS5K-stabilized β-tryptase incubated with B12 Fab and C3 for 30 min at 37°C to release β-hexosaminidase from different preparations of skin mast cells, which exhibited ~65% degranulation to the 22E7 mAb. The concentrations of tryptase-generated C3a were determined by ELISA. The β-tryptase-generated C3a (0.02, 0.04, and 0.08 μg/ml) and commercial C3a (0.005–1 μg/ml) had similar dose-response relationships with respect to degranulation. Because C3adesArg is much less potent than C3a at stimulating degranulation of human skin mast cells (31), like C5adesArg/C5adesArg, these results indicate that functionally intact C3a is generated by β-tryptase. In contrast, the same concentration range from C4a to C4adesArg did not degranulate skin mast cells (data not shown).
Figure 7.

Degranulation of human skin mast cells in response to β-tryptase-generated C5a-like molecules and C3a-like molecules. Skin mast cells (1 × 106/ml) were incubated with different stimulants at 37°C for 20 min and the net release of β-hexosaminidase activity determined. C5a-like molecules were generated by incubating DS500K-stabilized β-tryptase with B12 Fab and C5 for 40 min as in Fig. 5B. C3a-like molecules were generated by incubating DS5K-stabilized β-tryptase with B12 Fab and C3. A, 22E7 mAb (1 μg/ml); 5S/1 Ab (160 μg/ml); recombinant human C5a (rC5a); β-tryptase (1.7 μg/ml) (β-Try); C5 (33 μg/ml); and β-tryptase-generated C5a (TC5a) were included as indicated. Spontaneous release was 1.8%. B, Purified commercial C3a, C3 (33 μg/ml), and β-tryptase-generated C3a (TC3a) are shown. Spontaneous release was 7.1%.
Anaphylatoxin generation by the supernatant from 22E7-stimulated skin mast cells
In addition to testing the ability of β-tryptase that had been stabilized with various commercial polyanions in vitro, β-tryptase-proteoglycan complexes formed in situ by cultured human skin mast cells were also examined. Such cells were stimulated with 22E7 (1 μg/ml) at 37°C for 30 min and supernatants were collected. β-Hexosaminidase release was 20%. Based on a β-tryptase content of 21 × 106 ng/cells, the β-tryptase concentration in the releasate was estimated to be 4 μg/ml, presumably in a complex with heparin proteoglycan. C3, C4, and C5 were each incubated with a portion this supernatant at acidic pH. As shown in Fig. 8, Western blotting indicated that C3a-, C4a-, and C5a-like molecules were detected in the presence of B12 Fab, whereas C4a- and C5a-like molecules were detected in the absence of B12 Fab. Most of the C3a-like fragments detected in the presence of B12 Fab and the C4a-like molecules detected without B12 Fab were of smaller apparent size than the standard C3a and C4a molecules, presumably because degradation follows generation of authentic C3a and C4a. Importantly, the size of the C5a-like product comigrated with commercial C5adesArg both in the presence and absence of B12 Fab.
Figure 8.

Generation of anaphylatoxin-like molecules by the releasate of 22E7-stimulated skin mast cells. Supernatants were collected from two different batches of skin mast cells that had been activated by 22E7 (1 μg/ml) for 30 min at 37°C and concentrated 2-fold with a Microcon YM-100 microconcentrator. Releasates were adjusted to pH 6.0 by adding 1/10 volume of 0.5 M MES buffer (pH 6.0). Approximately 8 μl (60 ng of β-tryptase) of this releasate was incubated with 1 μg of C3, C4, or C5, with or without B12 Fab (4 molar excess to β-tryptase) for 1 h at 37°C and then subjected to SDS-PAGE in 16% acrylamide gels. Western blotting was performed with rabbit anti-C3a, anti-C4a, or anti-C5a Ab, respectively. C5a′ is commercial C5adesArg.
Ex vivo generation of C5a-like molecules by incubation of stimulated skin mast cells and C5
To better gauge the ability of human skin mast cells, when activated, to generate C5a, such cells were stimulated with anti-FcεRI mAb in the presence of C5, and the time course for generating C5a-like molecules was monitored. C5a-like fragments were detected in the presence (Fig. 9A) and absence (Fig. 9B) of B12 Fab. However, the C5a generated appeared to be more stable in the presence of B12 Fab than its absence. In the presence of B12 Fab, a diminished band intensity was not evident until the 8-h time point, whereas in the absence of B12 Fab, the newly generated C5a fragment was clearly diminished by 1 h and nearly absent by 2 h. To directly study the effect of B12 Fab on C5a stability, purified C5adesArg was incubated with stimulated skin mast cells for up to 5 h in the presence and absence of B12 Fab (Fig. 9C). In the presence of B12 Fab, the intensity of the C5adesArg band appears to be stable for 5 h. In contrast, in the absence of B12 Fab, the C5adesArg band intensity is clearly diminished by 2 h and nearly gone by 5 h. This indicates that β-tryptase tetramer has a greater ability to degrade C5a than the B12 Fab to β-tryptase monomer.
Figure 9.

Generation of C5a by activated human skin mast cells. A, Generation of C5a-like molecules by human skin mast cells activated with anti-FcεRI mAb. Human skin mast cells (2 × 105) were incubated with 22E7 mAb (0.3 μg), C5 (3 μg), and B12 Fab (14 μg) in 35 μl of MES-buffered AIM-V medium (pH 6.2). After the indicated incubation times, supernatants were subjected to SDS-PAGE on 16% acrylamide gels, Western blotted and probed by rabbit anti-C5a Ab. B, Generation of C5a-like molecules by 22E7-stimulated skin mast cells in the absence of B12 Fab. Skin mast cells, 22E7 and C5 were incubated as in A, but without B12 Fab. C, Stability of purified C5adesArg with 22E7-stimulated skin mast cells. Skin mast cells were stimulated as above with 22E7 in the presence (+) B12 Fab (left) or absence (−) of B12 Fab (right) and in the presence of 500 ng of C5adesArg for the times indicated.
Plasma C3 conversion and C3a generation by polyanion-stabilized tryptase
To examine β-tryptase-catalyzed conversion of C3 to C3a under a somewhat more physiologic condition, human plasma was used as a source of C3. When different amounts of DS500K-stabilized β-tryptase were incubated with plasma in the presence of B12 Fab at pH 6.0, the intensity of the C3α band decreased in a dose-dependent manner (Fig. 10A). Interestingly, a band corresponding to the C3α′ was not detected, suggesting it had been degraded. As shown in Fig. 10B, C3a-like molecules are generated in a time-dependent manner.
Figure 10.

C3 conversion and generation of C3a in plasma by β-tryptase. A, β-Tryptase-catalyzed conversion of C3 in plasma to C3a in the presence of B12 Fab at pH 6.0 in PBS. Samples were subjected to SDS-PAGE in 8% acrylamide gels, and Western blotted using goat anti-C3 Ab. Human plasma (1 μl) was incubated with 320, 150, and 80 ng of DS500K-stabilized β-tryptase for 30 min. B, Time-dependent generation of C3a from plasma C3 by DS500K-stabilized β-tryptase (320 ng) detected by SDS-PAGE in 16% acrylamide gels followed by Western blotting using rabbit anti-C3a Ab. C3a is commercial C3a control.
To investigate further the possibility that C3α′ formed by β-tryptase in plasma was then degraded by endogenous proteases, plasma was pretreated at 56°C for 30 min. Now the β-tryptase-generated C3α′ was detected and stable for up to 30 min (Fig. 11A), indicating a heat-sensitive factor is necessary for this degradation of C3α′. To examine the possible involvement of thrombin, kallikrein, and plasmin, plasma was treated with inhibitors of these proteases, namely hirudin, budellin, and aprotinin, respectively. As seen in Fig. 11B, none of these inhibitors prevented the disappearance of β-tryptase-generated C3α′. Other serine protease inhibitors were then examined for their abilities to inhibit C3α′ degradation. As shown in Fig. 11C, the general protease inhibitor suramin effectively inhibited C3α′ degradation. Because suramin has been reported to inhibit the proteolytic activity of factor I (49, 50), we next examined the effect of depletion of factor I from plasma using anti-Factor I Ab. C3-depleted serum was treated with anti-Factor I Ab and then Abs were removed with protein G-agarose. When compared with the control protein G-agarose treatment, Factor I Ab-treated C3-depleted serum partially prevented C3α′ degradation, implicating Factor I, at least in part, in this process (Fig. 11D).
Figure 11.

C3 conversion in plasma by DS500K-stabilized tryptase. C3 conversion was detected by SDS-PAGE in 10% acrylamide gels followed by Western blotting with goat anti-C3 Ab. A, 320 ng of DS500K-stabilized β-tryptase was incubated with 1 μl of human plasma in the presence of B12 Fab at pH 6.0 PBS. Human plasma was heat-treated before use (56°C, 30 min). Incubations were for 0, 1, 15, and 30 min. B, Effect of protease inhibitors on the β-tryptase-generated C3b degradation. Human plasma was pretreated with inhibitors of thrombin (hirudin (H, 50 U/ml), kallikrein (budellin (B), 50 μg/ml), and plasmin (aprotinin (A), 1 μg/ml)), for 1 h at 4°C and incubated with DS500K-stabilized tryptase with B12 Fab at 37°C for 30 min. Heat-treated plasma (Plasmaht) is shown. C, Effect of serine protease inhibitors on the β-tryptase-generated C3b degradation. Human C3-depleted serum (C3DS) was pretreated with leupeptin (L, 50 μm), antipain (AP, 100 μM), suramin (S, 1 mM), Pefabloc SC (P, 250 μM), and soybean trypsin inhibitor (SBTI, 50 μM) for 1 h at 4°C and incubated with DS500K-stabilized tryptase and C3 (1 μg) with B12 Fab at 37°C for 30 min. D, Effect of Factor I depletion on the tryptase-generated C3b degradation. C3-depleted serum was treated sequentially with either MOPC or anti-Factor I Ab, followed by protein G-agarose to remove Ab, and then was incubated with C3 and β-tryptase-B12 Fab. Anti-FI, anti-Factor I Ab. Note that C3β but not C3α is detected in C3-depleted serum alone (C and D); the result of C3α being degraded after inactivation of the thioester bond by methylamine (42), the process used to deplete functional C3 from serum.
Discussion
The current study finds that β-tryptase generates C3a from C3, C4a from C4, and C5a from C5 at acidic pH. With β-tryptase monomers formed with B12 anti-tryptase Fab (23), C3 and C4 are efficiently cleaved to generate the corresponding anaphylatoxins, which are then slowly degraded. With β-tryptase tetramers the α-chains of C3 and C4 are cleaved, but any anaphylatoxins produced appear to be degraded before they are detected. The authenticity of the C3a and C4a generated by β-tryptase monomers was confirmed by mass spectroscopy and, in the case of C3a, by activation of human skin MCTC cells to degranulate.
In the current study, C3a and C4a were both generated and then eventually degraded by β-tryptase monomers. Such might not be the case in vivo, where these anaphylatoxins would be free to diffuse away from their sites of generation, thereby escaping from degradation by the β-tryptase that generated them. Also, inhibitors of monomeric β-tryptase might preferentially suppress the degradation of these anaphylatoxins because their degradation appears to occur more slowly than their generation. This could result in the activation of nearby mast cells or other cell types that express the corresponding anaphylatoxin receptors, thus amplifying the direct effects of the mediators released by activated mast cells at specific tissue sites.
Metabolism of C5 by β-tryptase differs from that of C3 and C4 in that C5a was generated by both tetrameric and monomeric (B12 Fab-induced) forms of this protease at acidic pH. Furthermore, little, if any, degradation of C5a was detected under the experimental conditions used. Perhaps the carbohydrate on C5a protects potential cleavage sites from β-tryptase; alternatively, β-tryptase may not have access to such sites due to the conformation of C5a. The authenticity of C5a generated by β-tryptase was demonstrated by mass spectroscopy after deglycosylation and by its ability to degranulate skin MCTC cells. Importantly, the releasate from skin MCTC cells stimulated by aggregation of FcεRI also exhibited the ability to generate C5a from C5, both in the presence and absence of B12 Fab. C3a from C3 and C4a from C4 were generated only by β-tryptase monomers, the formation of which in these releasates was facilitated by addition of B12 Fab. Also, these β-tryptase-generated anaphylatoxins were not overtly susceptible to degradation by the other proteases released from skin mast cells, including carboxypeptidase A3, cathepsin G and chymase.
HMW and LMW forms of heparin glycosaminoglycans and Dextran sulfate were used to activate the monomeric and tetrameric forms of β-tryptase. Dextran sulfate has a higher negative charge density than heparin, but is not physiologic. The LMW heparin used in this study is not as effective as HMW heparin at filling the two polycationic grooves of tetrameric β-tryptase, each spanning two of the subunits. Nevertheless, LMW heparin is adequate to activate β-tryptase monomers (22, 23). Why HMW forms of heparin and dextran sulfate were preferred for generation of C4a and C5a, whereas LMW forms of these polyanions were preferred for generation of C3a is not understood. Perhaps there are subtle conformational differences in active tryptase according to the size of the polyanion that affect macromolecular substrate recognition. Alternatively, HMW polyanions might facilitate attraction of C4 and C5 to β-tryptase monomers. In vivo, heparin proteoglycan (51–53) or possibly chondroitin sulfate E proteoglycan (54) might be the stabilizing polyanion, in which case several glycosaminoglycans will be attached to the same core peptide. Such large β-tryptase-proteoglycan complexes might retard the diffusion of β-tryptase from its tissue site of release. Furthermore, the relative amount of β-tryptase monomers and tetramers at such sites is unknown, though both forms of the enzyme could coexist in equilibrium. As β-tryptase diffuses into airway fluid of asthmatic subjects, the low concentrations expected, based on in vitro measurements, might push this equilibrium to favor monomeric over tetrameric β-tryptase (22).
In vivo, β-tryptase, released from mast cells in tissues, would likely encounter C3, C4, or C5 in the context of a plasma transudate that enters these tissues in response to the corelease of vasoactive factors such as histamine, PGD2 and leukotriene C4. To see whether monomeric β-tryptase could generate C3a in plasma at acidic pH in vitro, human C3 and β-tryptase were added to C3-deficient plasma. C3a was rapidly generated, and at least over the 30-min time course of the experiment, no degradation was observed, suggesting that inhibitors present in plasma might inactivate monomeric β-tryptase before such degradation occurs. Also of interest was the finding that the C3α′ generated by monomeric β-tryptase was degraded in plasma. This had not occurred with either purified monomeric β-tryptase or mast cell releasate in buffer. This degradation of C3α′ in plasma apparently was Factor I-dependent because degradation was prevented by heat-inactivating the plasma, treating the plasma with suramin (49, 50) or removing Factor I from the plasma with anti-Factor I Ab.
These observations raise the possibilities that mast cell activation in vivo at sites of acidic pH could lead to the generation of C5a by tetrameric β-tryptase, and that if monomeric β-tryptase formed at such sites, generation of C3a and C4a might occur along with enhanced generation of C5a. However, diffusion of either monomeric or tetrameric β-tryptase from such sites into one at neutral pH would result in a loss of anaphylatoxin-generating activity. Acidic pH occurs at sites of inflammation such as the asthmatic airway (20) and also at areas of wound healing and solid tumor growth (55), all places where mast cell hyperplasia (56–59) has been noted. Indeed, mast cells may support wound healing through several pathways (60–63), and neo-vascularization associated with tumor growth (64–70). Generation of anaphylatoxins in the asthmatic airway is of great interest because levels of C5a and/or C3a are elevated in asthmatic bronchoalveolar lavage fluids (1–4) and appear to be involved in mouse models of allergic pulmonary disease (1, 71–73). C5a levels also are reportedly elevated in induced asthmatic sputum (74). Furthermore, receptors for these anaphylatoxins are elevated on the airway epithelium and bronchial smooth muscle of living asthmatics (72) on submucosal vessels, airway epithelium and smooth muscle cells in fatal asthma (75).
These results may explain at least in part how complement convertase-independent generation of complement anaphylatoxins may occur in a mast cell-dependent IgG immune complex-independent disease such as asthma. Increased numbers of mast cells in the epithelium, mucosal glands and smooth muscle may facilitate generation of complement anaphylatoxins at these sites by β-tryptase. In conclusion, both tetrameric and monomeric forms of β-tryptase can generate C5a from C5 at a mildly acidic pH, whereas both C3a from C3 and C4a from C4 are generated only by the monomeric form of β-tryptase under such conditions, and these activities may be important in diseases involving mast cells such as asthma.
Footnotes
This work was supported in part by Grant RO1-AI27517 (to L.B.S.) from the National Institutes of Health and by the American Lung Association (to Y.F.).
Abbreviations used in this paper: HMW, high molecular weight; LMW, low molecular weight; DS5K, dextran sulfate of 5000 Da; DS500k, dextran sulfate of 500,000 Da.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Humbles AA, Lu B, Nilsson CA, Lilly C, Israel E, Fujiwara Y, Gerard NP, Gerard C. A role for the C3a anaphylatoxin receptor in the effector phase of asthma. Nature. 2000;406:998–1001. doi: 10.1038/35023175. [DOI] [PubMed] [Google Scholar]
- 2.Krug N, Tschernig T, Erpenbeck VJ, Hohlfeld JM, Kohl J. Complement factors C3a and C5a are increased in bronchoalveolar lavage fluid after segmental allergen provocation in subjects with asthma. Am J Respir Crit Care Med. 2001;164:1841–1843. doi: 10.1164/ajrccm.164.10.2010096. [DOI] [PubMed] [Google Scholar]
- 3.Teran LM, Campos MG, Begishvilli BT, Schroder JM, Djukanovic R, Shute JK, Church MK, Holgate ST, Davies DE. Identification of neutrophil chemotactic factors in bronchoalveolar lavage fluid of asthmatic patients. Clin Exp Allergy. 1997;27:396–405. [PubMed] [Google Scholar]
- 4.van de Graaf EA, Jansen HM, Bakker MM, Alberts C, Schattenkerk JK Eeftinck, Out TA. ELISA of complement C3a in bronchoalveolar lavage fluid. J Immunol Methods. 1992;147:241–250. doi: 10.1016/s0022-1759(12)80014-7. [DOI] [PubMed] [Google Scholar]
- 5.Huber-Lang M, Younkin EM, Sarma JV, Riedemann N, McGuire SR, Lu KT, Kunkel R, Younger JG, Zetoune FS, Ward PA. Generation of C5a by phagocytic cells. Am J Pathol. 2002;161:1849–1859. doi: 10.1016/S0002-9440(10)64461-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Huber-Lang M, Sarma JV, Zetoune FS, Rittirsch D, Neff TA, McGuire SR, Lambris JD, Warner RL, Flierl MA, Hoesel LM, et al. Generation of C5a in the absence of C3: a new complement activation pathway. Nat Med. 2006;12:682–687. doi: 10.1038/nm1419. [DOI] [PubMed] [Google Scholar]
- 7.Wetsel RA, Kolb WP. Expression of C5a-like biological activities by the fifth component of human complement (C5) upon limited digestion with noncomplement enzymes without release of polypeptide fragments. J Exp Med. 1983;157:2029–2048. doi: 10.1084/jem.157.6.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward PA, Hill JH. C5 chemotactic fragments produced by an enzyme in lysosomal granules of neutrophils. J Immunol. 1970;104:535–543. [PubMed] [Google Scholar]
- 9.Wiggins RC, Giclas PC, Henson PM. Chemotactic activity generated from the fifth component of complement by plasma kallikrein of the rabbit. J Exp Med. 1981;153:1391–1404. doi: 10.1084/jem.153.6.1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maruo K, Akaike T, Ono T, Okamoto T, Maeda H. Generation of anaphylatoxins through proteolytic processing of C3 and C5 by house dust mite protease. J Allergy Clin Immunol. 1997;100:253–260. doi: 10.1016/s0091-6749(97)70233-1. [DOI] [PubMed] [Google Scholar]
- 11.Wenzel SE, Fowler AA, III, Schwartz LB. Activation of pulmonary mast cells by bronchoalveolar allergen challenge: in vivo release of histamine and tryptase in atopic subjects with and without asthma. Am Rev Respir Dis. 1988;137:1002–1008. doi: 10.1164/ajrccm/137.5.1002. [DOI] [PubMed] [Google Scholar]
- 12.Liu MC, Bleecker ER, Lichtenstein LM, Kagey-Sobotka A, Niv Y, McLemore TL, Permutt S, Proud D, Hubbard WC. Evidence for elevated levels of histamine, prostaglandin D2, and other bronchoconstricting prostaglandins in the airways of subjects with mild asthma. Am Rev Respir Dis. 1990;142:126–132. doi: 10.1164/ajrccm/142.1.126. [DOI] [PubMed] [Google Scholar]
- 13.Polosa R, Ng WH, Crimi N, Vancheri C, Holgate ST, Church MK, Mistretta A. Release of mast-cell-derived mediators after endobronchial adenosine challenge in asthma. Am J Respir Crit Care Med. 1995;151:624–629. doi: 10.1164/ajrccm/151.3_Pt_1.624. [DOI] [PubMed] [Google Scholar]
- 14.Bradding P, Walls AF, Holgate ST. The role of the mast cell in the pathophysiology of asthma. J Allergy Clin Immunol. 2006;117:1277–1284. doi: 10.1016/j.jaci.2006.02.039. [DOI] [PubMed] [Google Scholar]
- 15.Brightling CE, Bradding P, Symon FA, Holgate ST, Wardlaw AJ, Pavord ID. Mast-cell infiltration of airway smooth muscle in asthma. N Engl J Med. 2002;346:1699–1705. doi: 10.1056/NEJMoa012705. [DOI] [PubMed] [Google Scholar]
- 16.Carroll NG, Mutavdzic S, James AL. Increased mast cells and neutrophils in submucosal mucous glands and mucus plugging in patients with asthma. Thorax. 2002;57:677–682. doi: 10.1136/thorax.57.8.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen FH, Samson KT, Miura K, Ueno K, Odajima Y, Shougo T, Yoshitsugu Y, Shioda S. Airway remodeling: a comparison between fatal and nonfatal asthma. J Asthma. 2004;41:631–638. doi: 10.1081/jas-200026405. [DOI] [PubMed] [Google Scholar]
- 18.Schwartz LB. Mast cells and basophils. In: Zweiman B, Schwartz LB, editors. Inflammatory Mechanisms in Allergic Diseases. Marcel Dekker, Inc; New York: 2002. p. 3. [Google Scholar]
- 19.Caughey GH. Mast cell tryptases and chymases in inflammation and host defense. Immunol Rev. 2007;217:141–154. doi: 10.1111/j.1600-065X.2007.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hunt JF, Fang K, Malik R, Snyder A, Malhotra N, Platts-Mills TA, Gaston B. Endogenous airway acidification: implications for asthma pathophysiology (see comments) Am J Respir Crit Care Med. 2000;161:694–699. doi: 10.1164/ajrccm.161.3.9911005. [DOI] [PubMed] [Google Scholar]
- 21.Ren S, Lawson AE, Carr M, Baumgarten CM, Schwartz LB. Human tryptase fibrinogenolysis is optimal at acidic pH and generates anticoagulant fragments in the presence of the anti-tryptase monoclonal antibody B12. J Immunol. 1997;159:3540–3548. [PubMed] [Google Scholar]
- 22.Fukuoka Y, Schwartz LB. Human β-tryptase: detection and characterization of the active monomer and prevention of tetramer reconstitution by protease inhibitors. Biochemistry. 2004;43:10757–10764. doi: 10.1021/bi049486c. [DOI] [PubMed] [Google Scholar]
- 23.Fukuoka Y, Schwartz LB. The B12 anti-tryptase monoclonal antibody disrupts the tetrameric structure of heparin-stabilized β-tryptase to form monomers that are inactive at neutral pH and active at acidic pH. J Immunol. 2006;176:3165–3172. doi: 10.4049/jimmunol.176.5.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kulka M, Alexopoulou L, Flavell RA, Metcalfe DD. Activation of mast cells by double-stranded RNA: evidence for activation through Toll-like receptor 3. J Allergy Clin Immunol. 2004;114:174–182. doi: 10.1016/j.jaci.2004.03.049. [DOI] [PubMed] [Google Scholar]
- 25.McCurdy JD, Olynych TJ, Maher LH, Marshall JS. Cutting edge: distinct Toll-like receptor 2 activators selectively induce different classes of mediator production from human mast cells. J Immunol. 2003;170:1625–1629. doi: 10.4049/jimmunol.170.4.1625. [DOI] [PubMed] [Google Scholar]
- 26.Varadaradjalou S, Feger F, Thieblemont N, Hamouda NB, Pleau JM, Dy M, Arock M. Toll-like receptor 2 (TLR2) and TLR4 differentially activate human mast cells. Eur J Immunol. 2003;33:899–906. doi: 10.1002/eji.200323830. [DOI] [PubMed] [Google Scholar]
- 27.Qiao H, Andrade MV, Lisboa FA, Morgan K, Beaven MA. FcεR1 and toll-like receptors mediate synergistic signals to markedly augment production of inflammatory cytokines in murine mast cells. Blood. 2006;107:610–618. doi: 10.1182/blood-2005-06-2271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Okumura S, Kashiwakura JI, Tomita H, Matsumoto K, Nakajima T, Saito H, Okayama Y. Identification of specific gene expression profile in human mast cells via Toll-like receptor 4 and FcεRI. Blood. 2003;102:2547–2554. doi: 10.1182/blood-2002-12-3929. [DOI] [PubMed] [Google Scholar]
- 29.Ember JA, Hugli TE. Complement factors and their receptors. Immunopharmacology. 1997;38:3–15. doi: 10.1016/s0162-3109(97)00088-x. [DOI] [PubMed] [Google Scholar]
- 30.Gerard C, Gerard NP. C5A anaphylatoxin and its seven transmembrane-segment receptor. Annu Rev Immunol. 1994;12:775–808. doi: 10.1146/annurev.iy.12.040194.004015. [DOI] [PubMed] [Google Scholar]
- 31.El-Lati SG, Dahinden CA, Church MK. Complement peptides C3a- and C5a-induced mediator release from dissociated human skin mast cells. J Invest Dermatol. 1994;102:803–806. doi: 10.1111/1523-1747.ep12378589. [DOI] [PubMed] [Google Scholar]
- 32.Huey R, Fukuoka Y, Hoeprich PD, Jr, Hugli TE. Cellular receptors to the anaphylatoxins C3a and C5a. Biochem Soc Symp. 1986;51:69–81. [PubMed] [Google Scholar]
- 33.Zhao W, Kepley CL, Morel PA, Okumoto LM, Fukuoka Y, Schwartz LB. Fcγ;RIIa, not FcγRIIb, is constitutively and functionally expressed on skin-derived human mast cells. J Immunol. 2006;177:694–701. doi: 10.4049/jimmunol.177.1.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oskeritzian CA, Zhao W, Min HK, Xia HZ, Pozez A, Kiev J, Schwartz LB. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol. 2005;115:1162–1168. doi: 10.1016/j.jaci.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caughey GH. Tryptase genetics and anaphylaxis. J Allergy Clin Immunol. 2006;117:1411–1414. doi: 10.1016/j.jaci.2006.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schwartz LB, Min HK, Ren S, Xia HZ, Hu J, Zhao W, Moxley G, Fukuoka Y. Tryptase precursors are preferentially and spontaneously released, whereas mature tryptase is retained by HMC-1 cells, mono-mac-6 cells, and human skin-derived mast cells. J Immunol. 2003;170:5667–5673. doi: 10.4049/jimmunol.170.11.5667. [DOI] [PubMed] [Google Scholar]
- 37.Sakai K, Ren S, Schwartz LB. A novel heparin-dependent processing pathway for human tryptase: autocatalysis followed by activation with dipeptidyl peptidase I. J Clin Invest. 1996;97:988–995. doi: 10.1172/JCI118523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pereira PJ, Bergner A, Macedo-Ribeiro S, Huber R, Matschiner G, Fritz H, Sommerhoff CP, Bode W. Human β-tryptase is a ring-like tetramer with active sites facing a central pore. Nature. 1998;392:306–311. doi: 10.1038/32703. [DOI] [PubMed] [Google Scholar]
- 39.Alter SC, Kramps JA, Janoff A, Schwartz LB. Interactions of human mast cell tryptase with biological protease inhibitors. Arch Biochem Biophys. 1990;276:26–31. doi: 10.1016/0003-9861(90)90005-j. [DOI] [PubMed] [Google Scholar]
- 40.Fajardo I, Pejler G. Formation of active monomers from tetrameric human β-tryptase. Biochem J. 2003;369:603–610. doi: 10.1042/BJ20021418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Schwartz LB, Kawahara MS, Hugli TE, Vik D, Fearon DT, Austen KF. Generation of C3a anaphylatoxin from human C3 by human mast cell tryptase. J Immunol. 1983;130:1891–1895. [PubMed] [Google Scholar]
- 42.Isenman DE, Kells DI, Cooper NR, Muller-Eberhard HJ, Pangburn MK. Nucleophilic modification of human complement protein C3: correlation of conformational changes with acquisition of C3b-like functional properties. Biochemistry. 1981;20:4458–4467. doi: 10.1021/bi00518a034. [DOI] [PubMed] [Google Scholar]
- 43.Schwartz LB, Bradford TR, Lee DC, Chlebowski JF. Immunologic and physicochemical evidence for conformational changes occurring on conversion of human mast cell tryptase from active tetramer to inactive monomer: Production of monoclonal antibodies recognizing active tryptase. J Immunol. 1990;144:2304–2311. [PubMed] [Google Scholar]
- 44.Schwartz LB, Bradford TR, Rouse C, Irani AM, Rasp G, Van der Zwan JK, Van Der Linden PWG. Development of a new, more sensitive immunoassay for human tryptase: use in systemic anaphylaxis. J Clin Immunol. 1994;14:190–204. doi: 10.1007/BF01533368. [DOI] [PubMed] [Google Scholar]
- 45.Kambe N, Kambe M, Kochan JP, Schwartz LB. Human skin-derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood. 2001;97:2045–2052. doi: 10.1182/blood.v97.7.2045. [DOI] [PubMed] [Google Scholar]
- 46.Schwartz LB, Austen KF, Wasserman SI. Immunologic release of β-hexosaminidase and β-glucuronidase from purified rat serosal mast cells. J Immunol. 1979;123:1445–1450. [PubMed] [Google Scholar]
- 47.Schwartz LB, Lewis RA, Seldin D, Austen KF. Acid hydrolases and tryptase from secretory granules of dispersed human lung mast cells. J Immunol. 1981;126:1290–1294. [PubMed] [Google Scholar]
- 48.Hugli TE. Complement factors and inflammation: Effects of α-thrombin on components C3 and C5. In: Lunblad RL, Fenton JW, Mann KG, editors. Chemistry and Biology of Thrombin. Ann Arbor Science Publishers, Inc; Ann Arbor: 1977. p. 346. [Google Scholar]
- 49.Tsiftsoglou SA, Sim RB. Human complement factor I does not require cofactors for cleavage of synthetic substrates. J Immunol. 2004;173:367–375. doi: 10.4049/jimmunol.173.1.367. [DOI] [PubMed] [Google Scholar]
- 50.Tsiftsoglou SA, Willis AC, Li P, Chen X, Mitchell DA, Rao Z, Sim RB. The catalytically active serine protease domain of human complement factor I. Biochemistry. 2005;44:6239–6249. doi: 10.1021/bi047680t. [DOI] [PubMed] [Google Scholar]
- 51.Craig SS, Irani AMA, Metcalfe DD, Schwartz LB. Ultra-structural localization of heparin to human mast cells of the MCTC and MCT types by labeling with antithrombin III-gold. Lab Invest. 1993;69:552–561. [PubMed] [Google Scholar]
- 52.Metcalfe DD, Soter NA, Wasserman SI, Austen KF. Identification of sulfated mucopolysaccharides including heparin in the lesional skin of a patient with mastocytosis. J Invest Dermatol. 1980;74:210–215. doi: 10.1111/1523-1747.ep12541737. [DOI] [PubMed] [Google Scholar]
- 53.Metcalfe DD, Lewis RA, Silbert JE, Rosenberg RD, Wasserman SI, Austen KF. Isolation and characterization of heparin from human lung. J Clin Invest. 1979;64:1537–1543. doi: 10.1172/JCI109613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Thompson HL, Schulman ES, Metcalfe DD. Identification of chondroitin sulfate E in human lung mast cells. J Immunol. 1988;140:2708–2713. [PubMed] [Google Scholar]
- 55.Gillies RJ, Raghunand N, Karczmar GS, Bhujwalla ZM. MRI of the tumor microenvironment. J Magn Reson Imaging. 2002;16:430–450. doi: 10.1002/jmri.10181. [DOI] [PubMed] [Google Scholar]
- 56.Artuc M, Hermes B, Steckelings UM, Grutzkau A, Henz BM. Mast cells and their mediators in cutaneous wound healing–active participants or innocent bystanders? Exp Dermatol. 1999;8:1–16. doi: 10.1111/j.1600-0625.1999.tb00342.x. [DOI] [PubMed] [Google Scholar]
- 57.Hermes B, Feldmann-Boddeker I, Welker P, Algermissen B, Steckelings MU, Grabbe J, Henz BM. Altered expression of mast cell chymase and tryptase and of c-Kit in human cutaneous scar tissue. J Invest Dermatol. 2000;114:51–55. doi: 10.1046/j.1523-1747.2000.00837.x. [DOI] [PubMed] [Google Scholar]
- 58.Humphreys TR, Monteiro MR, Murphy GF. Mast cells and dendritic cells in basal cell carcinoma stroma. Dermatologic Surgery. 2000;26:200–203. doi: 10.1046/j.1524-4725.2000.09207.x. [DOI] [PubMed] [Google Scholar]
- 59.Huttunen M, Aalto ML, Harvima RJ, Horsmanheimo M, Harvima IT. Alterations in mast cells showing tryptase and chymase activity in epithelializating and chronic wounds. Exp Dermatol. 2000;9:258–265. doi: 10.1034/j.1600-0625.2000.009004258.x. [DOI] [PubMed] [Google Scholar]
- 60.Levi-Schaffer F, Kupietzky A. Mast cells enhance migration and proliferation of fibroblasts into an in vitro wound. Exp Cell Res. 1990;188:42–49. doi: 10.1016/0014-4827(90)90275-f. [DOI] [PubMed] [Google Scholar]
- 61.Numata Y, Terui T, Okuyama R, Hirasawa N, Sugiura Y, Miyoshi I, Watanabe T, Kuramasu A, Tagami H, Ohtsu H. The accelerating effect of histamine on the cutaneous wound-healing process through the action of basic fibroblast growth factor. J Invest Dermatol. 2006;126:1403–1409. doi: 10.1038/sj.jid.5700253. [DOI] [PubMed] [Google Scholar]
- 62.Reed JA, Albino AP, McNutt NS. Human cutaneous mast cells express basic fibroblast growth factor. Lab Invest. 1995;72:215–222. [PubMed] [Google Scholar]
- 63.Trautmann A, Toksoy A, Engelhard E, Brocker EB, Gillitzer R. Mast cell involvement in normal human skin wound healing: expression of monocyte chemoattractant protein-I is correlated with recruitment of mast cells which synthesize interleukin-4 in vivo. J Pathol. 2000;190:100–106. doi: 10.1002/(SICI)1096-9896(200001)190:1<100::AID-PATH496>3.0.CO;2-Q. [DOI] [PubMed] [Google Scholar]
- 64.Blair RJ, Meng H, Marchese MJ, Ren SL, Schwartz LB, Tonnesen MG, Gruber BL. Human mast cells stimulate vascular tube formation: tryptase is a novel, potent angiogenic factor. J Clin Invest. 1997;99:2691–2700. doi: 10.1172/JCI119458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Coussens LM, Raymond WW, Bergers G, Laig-Webster M, Behrendtsen O, Werb Z, Caughey GH, Hanahan D. Inflammatory mast cells up-regulate angiogenesis during squamous epithelial carcinogenesis. Genes Dev. 1999;13:1382–1397. doi: 10.1101/gad.13.11.1382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dethlefsen SM, Matsuura N, Zetter BR. Mast cell accumulation at sites of murine tumor implantation: implications for angiogenesis and tumor metastasis. Invasion Metastasis. 1994;14:395–408. [PubMed] [Google Scholar]
- 67.Imada A, Shijubo N, Kojima H, Abe S. Mast cells correlate with angiogenesis and poor outcome in stage I lung adenocarcinoma. Eur Respir J. 2000;15:1087–1093. doi: 10.1034/j.1399-3003.2000.01517.x. [DOI] [PubMed] [Google Scholar]
- 68.Ribatti D, Vacca A, Marzullo A, Nico B, Ria R, Roncali L, Dammacco F. Angiogenesis and mast cell density with tryptase activity increase simultaneously with pathological progression in B-cell non-Hodgkin’s lymphomas. Int J Cancer. 2000;85:171–175. [PubMed] [Google Scholar]
- 69.Ribatti D, Molica S, Vacca A, Nico B, Crivellato E, Roccaro AM, Dammacco F. Tryptase-positive mast cells correlate positively with bone marrow angiogenesis in B-cell chronic lymphocytic leukemia. Leukemia. 2003;17:1428–1430. doi: 10.1038/sj.leu.2402970. [DOI] [PubMed] [Google Scholar]
- 70.Takanami I, Takeuchi K, Naruke M. Mast cell density is associated with angiogenesis and poor prognosis in pulmonary adenocarcinoma. Cancer. 2000;88:2686–2692. [PubMed] [Google Scholar]
- 71.Karp CL, Grupe A, Schadt E, Ewart SL, Keane-Moore M, Cuomo PJ, Kohl J, Wahl L, Kuperman D, Germer S, et al. Identification of complement factor 5 as a susceptibility locus for experimental allergic asthma. Nat Immunol. 2000;1:221–226. doi: 10.1038/79759. [DOI] [PubMed] [Google Scholar]
- 72.Drouin SM, Kildsgaard J, Haviland J, Zabner J, Jia HP, McCray PB, Jr, Tack BF, Wetsel RA. Expression of the complement anaphylatoxin C3a and C5a receptors on bronchial epithelial and smooth muscle cells in models of sepsis and asthma. J Immunol. 2001;166:2025–2032. doi: 10.4049/jimmunol.166.3.2025. [DOI] [PubMed] [Google Scholar]
- 73.Taube C, Rha YH, Takeda K, Park JW, Joetham A, Balhorn A, Dakhama A, Giclas PC, Holers VM, Gelfand EW. Inhibition of complement activation decreases airway inflammation and hyperresponsiveness. Am J Respir Crit Care Med. 2003;168:1333–1341. doi: 10.1164/rccm.200306-739OC. [DOI] [PubMed] [Google Scholar]
- 74.Marc MM, Korosec P, Kosnik M, Kern I, Flezar M, Suskovic S, Sorli J. Complement factors C3a, C4a, and C5a in chronic obstructive pulmonary disease and asthma. Am J Respir Cell Mol Biol. 2004;31:216–219. doi: 10.1165/rcmb.2003-0394OC. [DOI] [PubMed] [Google Scholar]
- 75.Fregonese L, Swan FJSA, van Dolhnikoff M, Santos MA, Daha MR, Stolk J, Tschernig T, Sterk PJ, Hiemstra PS, Rabe KF, Mauad T. Expression of the anaphylatoxin receptors C3aR and C5aR is increased in fatal asthma. J Allergy Clin Immunol. 2005;115:1148–1154. doi: 10.1016/j.jaci.2005.01.068. [DOI] [PubMed] [Google Scholar]