

Abstract
TGF-β has pleiotropic effects on many cell types at different stages of their development, including mast cells. The present study examines the effects of TGF-β on human skin mast cells of the MCTC type. The expression of TGF-β receptors (TGF-R) was verified at the mRNA and protein levels for TGF-RI and TGF-RII, and at the mRNA level for accessory molecules β-glycan and endoglin. TGF-β did not affect mast cell viability after 1 wk at concentrations ≤10 ng/ml, but at 50 ng/ml caused significant cell death. TGF-β inhibited surface and total expression of Kit in a dose-dependent manner, whereas the surface expression of FcεRI, FcγRI, and FcγRII was not affected. TGF-β inhibited degranulation and cytokine production, but not PGD2 production. TGF-β diminished surface Kit expression through a TGF-RI kinase/Smad-dependent pathway by inhibiting new synthesis of Kit protein, which became evident following internalization and degradation of Kit after mast cells were exposed to the Kit ligand, stem cell factor. In contrast, addition of TGF-β had no discernible effect on surface Kit expression when administered 3 days after stem cell factor, by which time surface Kit levels had returned to baseline. Although both transcription and translation are important for de novo expression of Kit, Kit mRNA levels were not affected by TGF-β. Therefore, transcription of a gene other than Kit might be involved in Kit expression. Finally, activation of mast cells increased their susceptibility to TGF-β-mediated apoptosis, a process that might regulate the survival of activated mast cells in vivo.
Mast cells are key effector cells in IgE-dependent immediate hypersensitivity reactions. In response to FcεRI-mediated stimulation, mast cells secrete mediators such as histamine, PGD2, leukotriene C4, and cytokines that evoke clinical symptoms. Animal studies have revealed that mast cells also can act as APCs and express costimulatory molecules, defend against bacteria and viruses, participate in various types of hypersensitivity diseases (1, 2), and play a key role in allotypic tolerance mediated by regulatory T cells (3), indicating involvement in both innate and acquired forms of immunity. Also, mast cells may influence homeostatic processes such as wound healing, tissue fibrosis, and angiogenesis.
The growth, development, and mediator release of mast cells are regulated by stem cell factor (SCF)3 and other cytokines. Kit, the receptor for SCF, is expressed by progenitors of mast cells as well as by mature mast cells. Upon binding of SCF, Kit undergoes dimerization, followed by expression of tyrosine kinase activity. Downstream events include recruitment and tyrosine phosphorylation of Src homology 2-containing second messenger-generating enzymes (4) such as phospholipase C-γ and PI3K. The phosphorylated lipid products of these enzymes stimulate a variety of intracellular processes, including calcium mobilization and actin reorganization (5, 6). Mutations in Kit that lead to a gain of function, such as D816V, may increase the mast cell burden and cause mastocytosis, whereas loss of function mutations lead to mast cell deficiencies (7, 8). Various mechanisms are present to control the surface expression of Kit on mast cells as a means to avoid over-abundant or impaired signaling, which include regulation of transcription by various transcription factors (9, 10), regulation of translation of Kit mRNA by microRNAs (11), shedding of surface Kit (12, 13), internalization of surface Kit:SCF complexes, and ubiquitin-assisted degradation of internalized Kit (14, 15).
The TGF-β superfamily consists of structurally related polypeptide growth factors. These can be phylogenetically divided into three families, as follows: TGF-βs, activins, and bone morphogenetic proteins. They regulate development through their effects on cell proliferation, differentiation, and migration. TGF-β proteins also are involved in inflammation, immunity, fibrosis, and angiogenesis, as well as pathological process, including autoimmune/inflammatory diseases, atherosclerosis, fibrosis-associated disorders, and cancer in humans (16, 17). The effect of TGF-β on the immune system includes both inhibitory and stimulatory effects on T and B cells (17), as well as regulating Th and regulatory T cells (18). TGF-β is synthesized as an inactive precursor that dimerizes and is then processed to mature TGF-β dimers that remain non-covalently linked to their propeptides (latency-associated peptide). This complex then associates covalently with latent TGF-β-binding proteins (19) to form a larger latent TGF-β that is then secreted. The latent TGF-β-binding protein targets the inactive complex to specific receptors on cells, where the sequestered mature TGF-β can be released by the action of proteases, such as tissue plasminogen activator.
Free TGF-β binds to serine/threonine kinase receptors named type I and type II TGF-β receptor (TGF-RI and TGF-RII, respectively). Upon initially binding to TGF-RII, this receptor phosphorylates the juxtamembrane domain of TGF-RI (20). This in turn activates TGF-RI, which phosphorylates Smad2 and Smad3 (21), which then form a complex with Smad4. This heterotrimeric complex then translocates into the nucleus and transcriptionally regulates multiple effector genes (22). TGF-β also induces production of Smad6 and Smad7, which inhibit phosphorylation of Smad2 and Smad3 by competing for binding to TGF-RI. Smad7 also activates Smurf1 and Smurf2, members of the E3 ubiquitin ligase family that recognize TGF-β-activated receptors and target them for polyubiquitinylation and degradation (23, 24).
The complexity of TGF-β signaling is further complexified by cross-talk with other signaling pathways and by the capacity of each cell type to integrate these pathways into a unique transcriptional and biological response (25). For example, Smad-independent pathways are also activated by TGF-β (21, 26). Also, activated JNK can phosphorylate Smad3 and thereby enhance Smad-dependent activation by TGF-β (27). Protein kinase C (PKC) and TGF-β signaling pathways also can cross-modulate one another, although the details may vary considerably from one cell type to another (28–30).
TGF-β has both stimulatory and inhibitory effects on mast cells. TGF-β promotes the development of the murine mucosal type of mast cell (31, 32), as well as transcription of mouse mast cell proteases-6 and -7 (33, 34). TGF-β also promotes the survival and suppresses the proliferation of murine bone marrow-derived (grown in pokeweed mitogen-stimulated spleen cell conditioned medium) and of peritoneal-derived mast cells (35–37). It was previously reported that TGF-β inhibits a late stage of mast cell maturation, whereas later inducing apoptosis of murine bone marrow-derived and peritoneal-derived mast cells cultured with IL-3, potentially by inhibiting signaling through the IL-3R (38, 39). TGF-β also prevents rescue by SCF of murine bone marrow-derived mast cells that had been deprived of IL-3 (40). The positive vs negative impact of TGF-β on proliferation and survival of bone marrow-derived mast cells may depend in part on the precise culture conditions used. For example, in a mouse model of atopic dermatitis, Smad3−/− mice with attenuated TGF-β signaling compared with wild-type mice still exhibit increased mast cell numbers in the inflamed skin (41). FcεRI-dependent activation and mediator release by mouse bone marrow-derived and human cord blood-derived mast cells are attenuated by TGF-β, perhaps secondary to inhibiting FcεRI expression (42). With human mast cells, both HMC-1 and cord blood-derived mast cells migrate in response to TGF-β, whereas the proliferation of HMC-1 cells is reduced (43). Also, TGF-β diminishes surface Kit expression and up-regulates TIM-3 expression on cord blood-derived mast cells (44). However, human tissue-derived mast cells that have matured in vivo are distinguishable from HMC-1 and cord blood-derived mast cells. For example, HMC-1 cells do not express FcεRI (45, 46) and cord blood-derived mast cells express FcγRIIb, whereas skin-derived mast cells express FcγRIIa (47). Using skin mast cells, TGF-β together with SCF resulted in a marked decrease in surface Kit after 3 days along with an increase in cells having subdiploid amounts of DNA, suggesting increased apoptosis (39). Intestine-derived mast cells exposed to TGF-β exhibited attenuated degranulation and production of cysteinyl leukotrienes and augmented PGD2 production after FcεRI aggregation (48). In the present study using skin mast cells, TGF-β prevents de novo synthesis of Kit protein in a Smad-dependent manner, inhibits degranulation and cytokine production in response to aggregation of FcεRI, and promotes apoptosis, particularly following activation by FcεRI aggregation.
Materials and Methods
Reagents
The noncompeting IgG mAb anti-FcεRIα, 22E7 (49), was a gift from J. Kochan (Hoffmann-La Roche, Nutley, NJ). The following were obtained and used as described: IgG anti-CD117 mAb, TGF-β, and anti-TGF-RII mAb (extracellular epitope; BD Biosciences); anti-TGF-RI (intracellular epitope), anti-cyclooxygenase-1 (Cox-1) and -2 (Cox-2), anti-PGD synthase (Santa Cruz Biotechnology), anti-Smad3, and anti-P-Smad3/Smad1 (EMD Chemicals); anti-β-actin Ab (Cell Signaling Technology); anti-CD64 mAb, anti-CD16 mAb, soybean trypsin inhibitor, actinomycin D, and cycloheximide (Sigma-Aldrich); and anti-CD32a mAb (47). Pharmacologic inhibitors include SB431542 (TGF-RI kinase), SP600125 (JNK), bisindolylmaleimide (total PKC), and Gö6976 (classic PKC) (Sigma-Aldrich); and PD98059 (ERK), rottlerin (PCKδ), and SB202190 (p38 MAPK) (EMD Chemicals).
Purification and culture of human skin-derived mast cells
Fresh samples of skin were obtained after breast reduction surgery or mastectomy for breast cancer or after abdominoplasty through the Cooperative Human Tissue Network of the National Cancer Institute or the National Disease Research Interchange, as approved by the Human Studies Institutional Review Board at Virginia Commonwealth University. Mast cells were obtained from human skin essentially as described (50). After removing s.c. fat by blunt dissection, residual tissue was cut into 1- to 2-mm fragments and digested with type 2 collagenase (1.5 mg/ml), hyaluronidase (0.7 mg/ml), and type 1 DNase (0.3 mg/ml) in HBSS for 2 h at 37°C. The dispersed cells were collected by filtering through a No. 80 mesh sieve and resuspended in HBSS containing 1% FCS and 10 mM HEPES. Cells were resuspended in HBSS and layered over 75% Percoll in an HBSS cushion and centrifuged at 700 × g at room temperature for 20 min. Nucleated cells were collected from the buffer/Percoll interface, whereas erythrocytes sediment to the bottom of the tube. Cells enriched by Percoll density-dependent sedimentation were resuspended at a concentration of 1 × 106 cells/ml in serum-free X-VIVO 15 medium (BioWhittaker) containing 100 ng/ml human rSCF (gift from Amgen, Thousand Oaks, CA). The culture medium was changed weekly, and cells were split when they reached a concentration of ~2 × 106 cells/ml. The percentages of mast cells were assessed cytochemically by metachromatic staining with toluidine blue, and by flow cytometry with anti-Kit (YB5.B8) and anti-FcεRI (22E7) mAbs. The protease phenotype of cultured cells was examined by immunocytochemistry with anti-tryptase and anti-chymase mAbs (G3 and B7 mAbs, respectively; Chemicon International) (51). Typically, mature mast cells approaching 100% purity were obtained by 6 wk of culture, and 8- to 16-wk-old mast cells were used in the experiments described below.
Cell viability assay
Human skin-derived mast cells were incubated in X-VIVO medium containing 100 ng/ml SCF with TGF-β at 0, 1, 10, and 50 ng/ml for 3, 5, and 7 days. Viable cells were identified at each time point by trypan blue exclusion and counted.
Flow cytometry analysis
For surface staining, human skin mast cells were first blocked in PBS containing 10% BSA for 30 min at 4°C. The cells were washed in PBS containing 1% BSA (FACS medium) and incubated with the primary Ab for 30 min at 4°C. After washing away unbound primary Ab, the cells were incubated with fluorescent dye-conjugated secondary Ab, washed again, and then analyzed by flow cytometry (FACScan; BD Biosciences). For intracellular staining, skin mast cells were first fixed in 4% paraformaldehyde at 4°C for 15 min. After washing in FACS medium, the cells were permeabilized in Perm/Wash solution (BD Biosciences) for 30 min at 4°C and then labeled with primary and secondary Abs, as above, before analysis by flow cytometry.
Western blotting
Mast cells were incubated with medium, TGF-β alone, SB431542 alone, or SB431542 for 2 h before the addition of TGF-β. Cells were then collected, washed in PBS, and lysed with 1% Triton X-100 in borate-buffered saline (lysis buffer) containing protease inhibitors (leupeptin, pepstatin A, aprotinin, PMSF, sodium pyrophosphate, sodium orthovanadate, sodium fluoride, octyl-β-glucoside, and soybean trypsin inhibitor). The lysates were centrifuged at 12,000 rpm × 10 min to remove large cell debris and were boiled in SDS sample buffer (Invitrogen). Samples were loaded onto poly-acrylamide gels, subjected to electrophoresis, and then transferred to a polyvinylidene difluoride membrane. After blocking in the manufacturer’s blocking buffer (Li-Cor Biosciences), the membrane was incubated with first Ab (rabbit anti-Smad, rabbit anti-P-Smad, rabbit anti-actin, mouse anti-Cox-1, mouse anti-Cox-2, and goat anti-PGD synthase Ab) overnight at 4°C and washed before incubation with IRDye-labeled donkey anti-rabbit, anti-mouse, or anti-goat Ab. The membrane was scanned in an Odyssey Infrared Imaging System (LI-COR Biosciences). Data were analyzed using Odyssey 2.1 software.
Mast cell activation
Human skin mast cells were washed, suspended at 106 cells/ml in X-VIVO medium, and activated in microfuge tubes by incubation with anti-FcεRI (22E7) mAb at 1 μg/ml in a total volume of 60 μl for 30 min at 37°C. Reactions were stopped by addition of 3 vol of cold PBS buffer (free of calcium and magnesium). Cells were then separated by centrifugation at 300 × g for 10 min at 4°C. Supernatants were removed, and cell pellets were resuspended in 240 μl of PBS. β-Hexosaminidase was assayed by measuring release of p-nitrophenol from the substrate p-nitrophenyl N-acetyl-β-D-glucosaminide, as described (52, 53). Absorbance values were read at 405 nm. Net percentage release values were calculated using the following formula: net percentage release = ((stimulated release − unstimulated release)/(stimulated (release + retained) − unstimulated release)) × 100.
Lipid and cytokine measurement
Skin mast cells (106 cells/ml) were incubated with TGF-β (10 ng/ml) for 2 days. The cells were then washed and activated with 22E7 mAb at 1 μg/ml for 45 min. Supernatants were removed by centrifugation. A PGD2 enzyme immunoassay kit (Cayman Chemical) was used to measure PGD2 levels, according to manufacturer’s instruction. The lower limit of detection was 10 pg/ml.
For cytokine measurements, skin mast cells (106 cells/ml) were incubated with TGF-β, as above, before the addition of 22E7 mAb and soybean trypsin inhibitor (100 μg/ml). Supernatants were removed after 24 h by centrifugation. Purified and biotinylated mouse or rat mAbs specific for each cytokine and standard recombinant cytokines were purchased from BD Biosciences. Assays were performed according to the manufacturer’s recommendations, but scaled down to 384-well flat-bottom microtiter plates, as described (54). The lower limit of detection for each of the cytokines under consideration was 16 pg/ml.
RNA preparation and measurement of specific mRNA
Total RNA was isolated from mast cells using the RNeasy Mini Kit (Qiagen). For standard RT-PCR, 1 μg of total RNA was converted to cDNA using the reverse-transcription system (Promega). cDNA was synthesized by incubating the RNA with Moloney murine leukemia virus reverse transcriptase and 20 pmol of oligo-dT primer for 1 h at 42°C. A portion of the cDNA (typically 1/20th vol) was used for standard PCR. Thirty-five cycles of PCR were performed with 2.5 U of TaqDNA polymerase and 20 pmol of gene-specific sense and antisense primers. A portion of each PCR product was separated on an agarose gel, stained with ethidium bromide (500 ng/ml), and photographed. Forward and reverse primers, respectively, were 5′-GCACCCTCTTCAAAAACTGG-3′ and 5′-TCGATGGTGAATGACA GTGC-3′ for TGF-RI; 5′-TGTGTGACTTTGGGCTTTCC-3′ and 5′-GA CATCGGTCTGCTTGAAGG-3′ for TGF-RII; 5′-TGTCTCACTTCATG CCTCCAGCT-3′ and 5′-AGGCTGTCCATGTTGAGGCAGT-3′ for endoglin; 5′-CCTGTCATTCCCAGCATACAACT-3′ and 5′-ATCACCT GACACCAGATCTTCATA-3′ for β-glycan; 5′-GTTGACCGCTCCTTG TATGG-3′ and 5′-CTGTGAATTCTTCCCCTTCC-3′ for Kit; and 5′-TC TCCTCTGACTTCAACAGC-3′ and 5′-CCTCTTCAAGGGGTCTACAT GG-3′ for G3PDH.
SYBR Green real-time PCR was performed using the SensiMix OneStep system (Quantace). Total RNA (200 ng) was first converted to cDNA, as above. The PCR step was then conducted in the presence of forward and reverse primers (200 nM each), 1× SYBR Green solution (Quantace), in a final volume of 50 μl. Kit and G3PDH primers were the same as those used above. Additional primers were as follows: 5′-TACCCCCAGGAGAAGA TTCC-3′ and 5′-TTTCAGCCATCTTTGGAAGG-3′ for IL-6; 5′-GGTC AACATCACCCAGAACC-3′ and 5′-TTTACAAACTGGGCCACCTC-3′ for IL-13; 5′-ATGTGAATGCCATCCAGGAG-3′ and 5′-GGGCAGTGC TGTTTGTAGTG-3′ for GM-CSF; 5′-TCAGCCTCTTCTCCTTCCTG-3′ and 5′-GGCTACAGGCTTGTCACTCG-3′ for TNF-α; 5′-CGGTGTCC AGTTCCAATACC-3′ and 5′-GGATGTGGTGGTCCATGTTC-3′ for Cox-1; 5′-TGAGCATCTACGGTTTGCTG-3′ and 5′-TGCTTGTCTGGA ACAACTGC-3′ for Cox-2; and 5′-CCCCATTTTGGAAGTTGATG-3′ and 5′-TGAGGCGCATTATACGTGAG-3′ for PGD synthase. Assays were performed in a MiniOpticon System (Bio-Rad) under the following conditions: initial denaturation for 10 min at 95°C; 35 cycles at 95°C for 1 min, 55°C for 1 min, and 72°C for 1 min. A melting curve analysis between 50 and 95°C was performed to confirm the specificity of each amplification product. Results were analyzed by the comparative cycle threshold (Ct) method, in which Ct is the number of cycles required to reach an arbitrary fluorescence threshold. The reproducibility of the Ct was analyzed by real-time RT-PCR of triplicate samples derived from various amounts of RNA. Further quantitation was calculated using 2−ΔΔCt method, in which ΔCt(experimental) = Ct target − Ct G3PDH for treated cells; ΔCt(control) = Ct target − Ct G3PDH for control cells; and ΔΔCt = ΔCt(experimental) − ΔCt(control).
Total protein synthesis
Skin mast cells (2 × 105) were incubated with TGF-β at 0, 1, 10, and 50 ng/ml for 3, 6, and 24 h. Cells were labeled with 5 μCi of [35S]methionine (PerkinElmer) for the last 3 h of incubation. Cells were then collected and washed. 35S-labeled protein was measured by liquid scintillation counting.
Apoptosis study
Apo Logix Carboxyfluorescein Caspase Detection Kit (Cell Signaling Technology) was used to detect apoptosis, according to manufacturer’s instructions. Skin mast cells (3 × 105) were incubated with carboxyfluorescein-Val-Ala-Asp-fluoromethyl ketone (FAM-VAD-FMK) for 1 h at 37°C, 6% CO2 in darkness. After washing twice, the cells were suspended in 400 μl of washing buffer. Fluorescent intensity on the FL-1 channel, which reflects caspase activity, was determined by flow cytometry.
Statistical analysis
Student’s t test was used to compare data between two treatment groups. One-way ANOVA was used to compare data among three or more different treatment groups. If significant differences were detected, post hoc testing was then used to compare all treatment groups vs the control group. Values of p ≤ 0.05 were considered to be significant.
Results
Human skin mast cells express TGF-β receptors
The expression of TGF-β receptors was examined at both mRNA and protein levels. Total RNA was isolated from skin mast cells, and RT-PCR was used to examine the specific transcripts of TGF-β receptors. As shown in Fig. 1A, transcripts for TGF-RI, TGF-RII, and the accessory TGF-R molecules endoglin and β-glycan were detected. The expression of TGF-RII and TGF-RI protein was demonstrated by flow cytometry in Fig. 1B.
Figure 1.

Expression of TGF-β receptor in human skin mast cells. A, RT-PCR. mRNAs for TGF-RI (213 bp), TGF-RII (158 bp), endoglin (378 bp), β-glycan (364 bp), and G3PDH (303 bp) were detected by RT-PCR in skin-derived mast cells. B, Expression of TGF-RI and TGF-RII. Skin mast cells were incubated with mouse anti-TGF-RII mAb (black) or isotype control IgG (gray), followed by FITC-conjugated rat anti-mouse IgG. For TGFRI, cells were first fixed and permeabilized, and then incubated with either rabbit IgG (gray) or rabbit anti-TGFRI Ab (black), which recognizes the intracellular portion of TGF-RI. Alexa Fluor 488-conjugated goat anti-rabbit IgG was used as the secondary Ab. One representative experiment of three with different samples is shown in A and B.
Effect of TGF-β on viability of and the surface expression of Kit and FcRs on human skin mast cells
Whether TGF-β affects cell viability of skin mast cells was then examined. Human skin mast cells were incubated with 0, 1, 10, and 50 ng/ml TGF-β for 3, 5, and 7 days. Viable cell numbers on days 3 and 5 showed no significant differences among the four groups, although cells exposed to 50 ng/ml TGF-β tended to decrease (27%). At day 7, cells exposed to 50 ng/ml TGF-β clearly demonstrated a significant decrease in viable cell numbers compared with the control group (1.0 ± 0.2 vs 2.1 ± 0.3 × 105 mast cells, p < 0.01) (Fig. 2A).
Figure 2.

Effect of TGF-β on the viability of and surface expression of Kit and FcεRI on skin mast cells. A, Cell viability. Human skin mast cells were incubated with the indicated concentrations of TGF-β for up to 7 days. Viable cells were counted on days 3, 5, and 7 using trypan blue exclusion. Data are presented as mean ± SE of three independent experiments, each performed in duplicate. *, p < 0.05 compared with control group. B, Flow cytometry. Mast cells were incubated with TGF-β at 0 (black), 0.01 (green), 0.1 (blue), 1 (purple), 10 (light blue), and 50 (yellow) ng/ml for 3 days along with fresh medium containing SCF (100 ng/ml). Cells were then harvested and incubated with PE-conjugated mouse anti-CD117 or isotype IgG mAbs. FcεRI was labeled with 22E7 and PE rat anti-mouse IgG. Isotype controls are in red. Data are representative of three separate experiments with similar results.
The effect of TGF-β on surface Kit and FcεRI expression was examined next. Skin mast cells were incubated with 0, 0.01, 0.1, 1, 10, and 50 ng/ml TGF-β along with SCF for 2 days, because cell viabilities were not significantly affected under these conditions, and then were labeled with anti-CD117 or 22E7 mAb. TGF-β diminished surface Kit expression in a dose-dependent pattern. The mean fluorescent intensities for Kit labeling were 90, 69, 11, 6.2, and 6.1% of controls (not exposed to TGF-β) with TGF-β concentrations of 0.01, 0.1, 1, 10, and 50 ng/ml, respectively (Fig. 2B). The highest dose of TGF-β (50 ng/ml) failed to show inhibition beyond that observed at 10 ng/ml TGF-β. In contrast, the surface expression of FcεRIα was not affected by TGF-β except at the highest dose used, which led to a modest reduction in expression of this receptor (Fig. 2B) (mean fluorescent intensities: 94, 105, 111, 108, and 62% of control, respectively). The expression of FcγRIIa (CD32a) was not affected by any of the TGF-β doses (data not shown). FcγRI (CD64) and FcγRIII were not detected in control cells, and treatment with TGF-β failed to induce their expression (data not shown).
Effect of TGF-β on degranulation, PGD2 release, and cytokine production by human skin mast cells
To determine the effect of TGF-β on human mast cell function, degranulation together with PGD2 and cytokine production were examined, as summarized in Fig. 3A. Mast cells were preincubated with TGF-β (10 ng/ml) for 2 days before activation with 22E7 mAb (1 μg/ml). Without exposure to TGF-β, net release of β-hexosaminidase (degranulation) values ranged from 42 to 64% after exposure to 1 μg/ml 22E7 for 30 min at 37°C. Spontaneous release values were <5% for isotype controls ± TGF-β. TGF-β-treated cells demonstrated a significant 40% reduction of β-hexosaminidase release. Interestingly, the release of PGD2 was not significantly affected by TGF-β, with control and TGF-β-treated mast cells producing 145 ± 30 ng and 130 ± 33 ng per 106 mast cells, respectively, 45 min after 22E7 stimulation at 37°C ( p = 0.66). In the absence of 22E7 stimulation, the PGD2 levels were below the lower limit of detection (10 pg/ml). The production of selected cytokines was assessed 24 h after stimulation in the cells pretreated with TGF-β or medium containing SCF alone. TGF-β caused significant reductions for each cytokine, as follows: 53% for TNF-α (control 394 ± 194 pg/106 cells), 78% for GM-CSF (control 1963 ± 399 pg/106 cells), 76% for IL-13 (control 1067 ± 398 pg/106 cells), and 43% for IL-6 (1007 ± 154 pg/106 cells). In the absence of 22E7, mast cells produced low levels of IL-6 (57 ± 6 pg/106 cells over 24 h). IL-13, GM-CSF, and TNF-α were not detected. Interestingly, the spontaneous levels of IL-6 were reduced in TGF-β-treated groups in a dose-dependent pattern (87, 65, and 18% of control with 0.1, 1, and 10 ng/ml TGF-β, respectively).
Figure 3.

TGF-β selectively regulates mediator release by FcεRI-stimulated skin mast cells. A, Production of cytokines and PGD2 by activated human skin mast cells. Mast cells were treated with TGF-β at 10 ng/ml for 2 days in the presence of 100 ng/ml SCF. The cells were washed in medium and activated with 1 μg/ml 22E7 mAb at 37°C. β-Hexosaminidase release and PGD2 and cytokine production were measured as described after 30 min, 45 min, and 24 h of stimulation, respectively. Data are displayed as mean ± SE values for cells treated with TGF-β as a percentage of control cells not exposed to TGF-β (n = 4 independent experiments for β-hexosaminidase and PGD2, n = 3 independent experiments for cytokine production). *, p < 0.05 compared with the control that was not exposed to TGF-β. B, Cellular mRNA levels of Cox-1, Cox-2, PGD synthase, and cytokines. Skin mast cells were treated with TGF-β for 2 days and then stimulated with 1 μg/ml 22E7. Enzyme mRNA levels were measured before stimulation, and cytokine levels were measured 3 h after stimulation by real-time RT-PCR. The 2−ΔΔCt values for TGF-β-treated cells were then calculated. Data are expressed as mean ± SE of three independent experiments, each performed in triplicate. *, p < 0.05 compared with the control that was not exposed to TGF-β. C, Cellular protein levels of Cox-1, Cox-2, and PGD synthase. These enzyme levels were measured by Western blotting 2 days after treatment of the cells with TGF-β and compared with medium. The blot shown is representative of three independent experiments.
Levels of cytokine mRNAs were determined by real-time RT-PCR. As shown in Fig. 3B, TGF-β treatment significantly reduced IL-6 and GM-CSF mRNA levels by 42 and 30%, respectively, indicating that TGF-β inhibits transcription or enhances turnover of these cytokines. However, mRNA levels of IL-13 and TNF-α were not significantly affected by TGF-β (117 and 113% of control, respectively), suggesting diminished translation or enhanced protein turnover might account for their diminished production. The mRNA levels of key enzymes involved in PGD2 synthesis were also determined. TGF-β did not affect Cox-1 and PGD synthase mRNA levels, but did up-regulate Cox-2 mRNA (195% of control, p < 0.05). Western blot was used to determine the protein levels of above molecules. As demonstrated in Fig. 3C, no significant changes in Cox-1, Cox-2, and PGD synthase protein levels were observed. After they were normalized to β-actin, the ratios of protein, after TGF-β to before TGF-β treatment, were 1.2 ± 0.1, 1.0 ± 0.1, and 1.1 ± 0.1, respectively.
TGF-β reduces Kit expression by inhibiting reappearance of Kit after SCF-mediated internalization and degradation
To further explore the mechanism of reduced Kit expression on TGF-β-treated mast cells, surface and intracellular Kit levels were assessed at different times after addition of SCF ± TGF-β, as shown in Fig. 4. Surface Kit levels were markedly and comparably reduced soon after SCF (100 ng/ml) ± TGF-β had been added to the cells. The nadir for surface Kit expression occurred by 6 h and persisted for at least 24 h after addition of SCF, regardless of whether TGF-β was also included (Fig. 4, top panel). By 72 h after addition of SCF alone, surface Kit expression had recovered to near the baseline value, whereas mast cells that had been exposed to TGF-β from 0 to 72 h and 24 –72 h failed to up-regulate Kit expression to baseline by 72 h.
Figure 4.

TGF-β inhibits re-expression of Kit after treatment with SCF. Skin mast cells were incubated with SCF (100 ng/ml) ± TGF-β (10 ng/ml) as indicated and harvested at the times shown. Cells were then fixed with paraformaldehyde and labeled with PE anti-CD117 mAb to detect surface Kit (top panel). For detection of intracellular plus surface Kit, fixed cells were permeabilized as described and then labeled with PE anti-CD117 mAb. Data presented as log mean fluorescent intensity (mean ± SE, n = 3 independent experiments). *, p < 0.05 TGF-β-treated cells compared with TGF-β-untreated cells at each time point.
To assess intracellular as well as surface expression of Kit, permeabilized mast cells were assessed for Kit expression (Fig. 4, lower panel). Although the time courses for the decline and re-expression of Kit ± TGF-β were similar to those observed for surface Kit alone, one difference was a higher total Kit expression detected at 0.5– 48 h, presumably reflecting internalization of surface Kit before its degradation, particularly at early time points, and possibly new synthesis of Kit at later time points that had not yet migrated to the surface. Importantly, the TGF-β-treated and untreated groups showed a similar nadir within 24 h, and the TGF-β group continued to show a marked suppression of total Kit expression through 72 h, excluding the possibility that TGF-β permitted the accumulation of newly synthesized Kit in an intracellular compartment while blocking its transport to the cell surface. Thus, TGF-β appears to affect primarily the de novo synthesis of Kit protein; it is uncertain as to whether this is a posttranscriptional and/or transcriptional effect.
TGF-β-mediated suppression of Kit re-expression is Smad dependent
To determine whether Smad-dependent or Smad-independent signaling pathways are required for TGF-β-mediated suppression of Kit re-expression, pharmacologic inhibitors were used. Mast cells were incubated with SB431542 (inhibitor of TGF-RI kinase) at 10 μM for 2 h at 37°C before the addition of TGF-β. Surface Kit expression was determined by flow cytometry at 48 h. As shown in Fig. 5A, SB431542 abolished almost completely the TGF-β-mediated suppression of Kit re-expression, indicating the involvement of a Smad-dependent pathway. SB431542 alone did not change the surface expression of Kit on skin mast cells (data not shown). To demonstrate that the Smad pathway is blocked, Western blotting was performed and demonstrated that neither Smad1 nor Smad3 was phosphorylated in SB431542-treated mast cells in response to TGF-β stimulation (Fig. 5B).
Figure 5.

A Smad-dependent pathway is required for TGF-β to suppress Kit re-expression. Skin mast cells were incubated with SCF (100 ng/ml) ± SB431542 (10 μM) for 2 h before the addition of TGF-β. A, Surface expression of Kit at 48 h by flow cytometry. SCF alone (green) and SCF with TGF-β (blue), SB431542 (purple, right), and SB431542 + TGF-β (purple, left). Unlabeled cells (red) and an isotype control (black) are also shown. One representative experiment of three is shown. SB431542 had no effect on Kit re-expression after SCF in the absence of TGF-β (data not shown). B, Smad3 and p-Smad1/3 protein. Smad3 and p-Smad1/3 were detected 30 min after TGF-β treatment by Western blotting. β-Actin was used as housekeeping control. One representative experiment of three independent experiments with similar results is shown.
To explore the potential role of a MAPK kinase pathway in TGF-β-mediated suppression of Kit re-expression, SP600125 (JNK inhibitor, 25 μM), PD98059 (ERK inhibitor, 20 μM), and SB202190 (p38 MAPK kinase inhibitor, 20 μM) were incubated with mast cells for 2 h before the addition of TGF-β. The expression of Kit was not affected by those inhibitors, implying that the MAPK kinase pathway is not important for this effect of TGF-β (data not shown).
TGF-β can affect cell growth and motility in some cells through Smad-independent PKC activation (29, 30). In contrast, the phosphorylation of Smad2 and Smad3 by PKC leads to the loss of their DNA-binding activity. To test whether PKC activation is required for TGF-β-mediated suppression of Kit re-expression, general and selective PKC inhibitors were added with SCF to mast cell cultures before the introduction of TGF-β. Bisindolylmaleimide, a general PKC inhibitor, permitted SCF-mediated internalization of Kit, but accelerated the subsequent re-expression of Kit, regardless as to whether TGF-β was present (data not shown). Levels of surface Kit returned to baseline in less than 1 day. This may indicate that a PKC isoenzyme down-regulates Kit expression, but is unlikely to account for TGF-β-mediated suppression of Kit re-expression. Gö6976, which inhibits conventional calcium-dependent PKC, and rottlerin, a PKCδ inhibitor, did not change the Kit re-expression in the presence or absence of TGF-β (data not shown). Therefore, other members of the novel PKC subfamily (ε, η,θ) or members of the atypical PKC subfamily (ζ,λ) can apparently suppress Kit re-expression, but none appear to prevent TGF-β-mediated suppression of Kit re-expression.
New Kit expression requires both transcription and translation
SCF (100 ng/ml) was added to human skin mast cells to induce Kit internalization, and after 24 h actinomycin D or cycloheximide was added to inhibit global transcription and translation, respectively. A separate group received TGF-β at 10 ng/ml concentration simultaneously with SCF, as in Fig. 4. Surface and total Kit expression was assessed in nonpermeabilized and permeabilized cells, as above. As before, a marked reduction in surface and total Kit expression was observed at 24 h (Fig. 6). Cells exposed only to SCF regained surface and total Kit levels by 72 h. Both actinomycin D (Fig. 6, left panels) and cycloheximide (Fig. 6, right panels) prevented re-expression of surface and total Kit in a dose-dependent manner, indicating both transcription and translation are required for Kit re-expression. The TGF-β-treated cells suppressed Kit re-expression, as in Fig. 4.
Figure 6.

Re-expression of Kit requires both transcription and translation. Skin mast cells were cultured with SCF (100 ng/ml) for 24 h before the addition of various concentrations (μM) of either actinomycin D or cycloheximide. A control culture had neither actinomycin D nor cycloheximide. In a parallel culture, skin mast cells were monitored after SCF (100 ng/ml) and TGF-β (10 ng/ml) were added together. Surface expression and surface plus intracellular expression of Kit were measured using PE anti-CD117 mAb. Data are expressed as mean ± SE, n = 3 experiments.
TGF-β does not inhibit Kit mRNA levels in skin mast cells
The effect of TGF-β on Kit mRNA expression was determined by RT-PCR and real-time RT-PCR, as shown in Fig. 7. RT-PCR shows Kit and GAPDH RT-PCR products of similar intensities at baseline and 1, 3, and 6 h after addition of SCF ± TGF-β (Fig. 7, top panel). This qualitative observation was confirmed by quantitative real-time RT-PCR, as shown in the lower panel of Fig. 7, whereby the medium control and TGF-β-treated groups had Ct values that were not significantly different from one another (9.2 ± 0.2 and 9.5 ± 0.4, respectively; p = 0.43).
Figure 7.
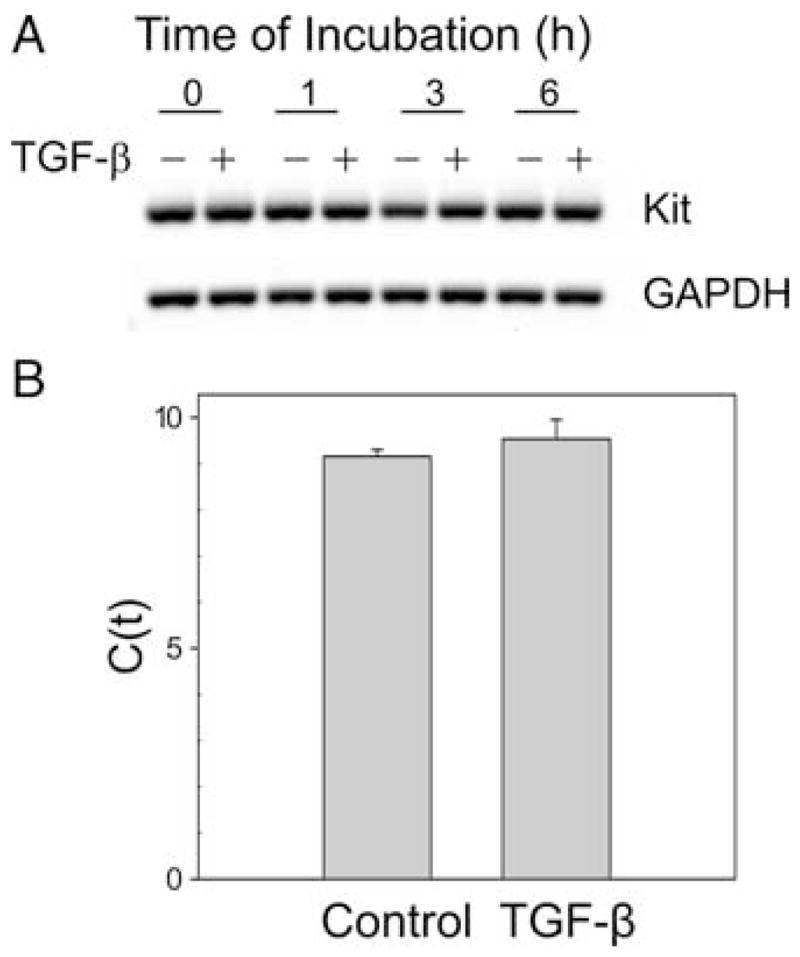
Kit messenger is unchanged after TGF-β treatment. A, RT-PCR. Skin-derived mast cells were treated with TGF-β for up to 6 h. Kit mRNA was assessed by RT-PCR. Representative data from one of three experiments are shown. B, Real-time RT-PCR. RNA from skin mast cells treated with TGF-β for 24 h was isolated and subjected to real-time RT-PCR using the SYBR Green method. Data are expressed as mean ± SE of three independent experiments with p = 0.43.
TGF-β inhibits mast cell protein synthesis
Because TGF-β inhibited Kit protein synthesis rather than mRNA expression, the effect on total protein synthesis was examined by [35S]methionine incorporation. As shown in Fig. 8, TGF-β concentrations of 1, 10, and 50 ng/ml caused a dose-dependent inhibition of [35S]methionine incorporation at three time intervals. The respective mean cpm for each TGF-β-treated group expressed as a percentage of the corresponding non-TGF-β-treated control are 77, 66, and 59% for labeling from 0 to 3 h; 74, 56, and 39% for labeling from 3 to 6 h; and 71, 61, and 50% for labeling from 21 to 24 h. These results indicate global protein synthesis was inhibited by TGF-β in a dose-dependent pattern.
Figure 8.
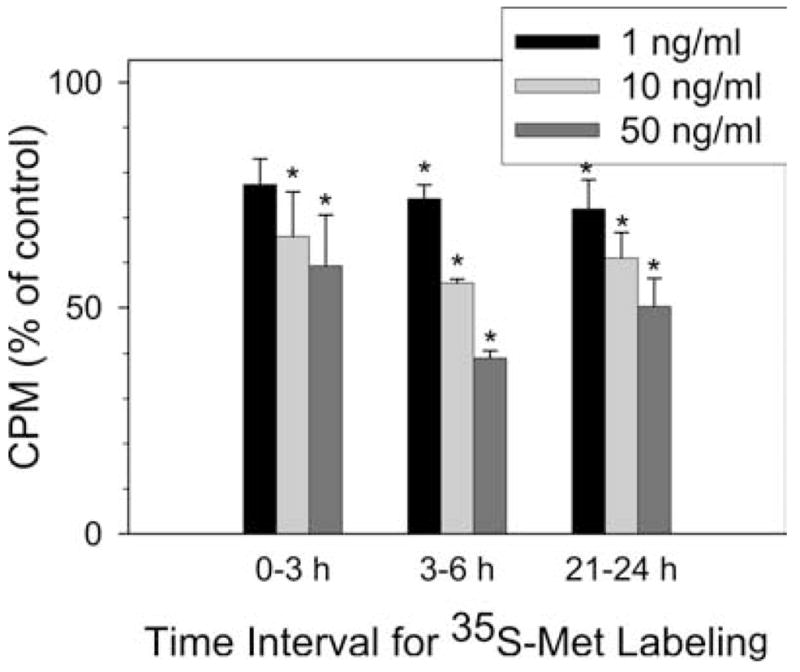
Mast cell protein synthesis is inhibited by TGF-β. Human skin mast cells were given fresh medium containing 100 ng/ml SCF alone or along with TGF-β at the concentration of 1, 10, and 50 ng/ml. [35S]Methionine (5 μCi) was added for 0 –3, 3– 6, or 21–24 h. Cells were then thoroughly washed and lysed. 35S cpm were measured as described. Data are expressed as mean ± SE of three independent experiments. *, p < 0.05 compared with control cells.
FcεRI-mediated activation of skin mast cells increases their susceptibility to TGF-β-induced apoptosis
Because Kit is needed for mast cell survival and TGF-β prevents expression of Kit after it is internalized with SCF, we postulated that activating mast cells by aggregating FcεRI in the presence of TGF-β might make these cells more susceptible to apoptosis. Cell viability was monitored for up to 7 days after activation ± TGF-β. As shown in Fig. 9A, viable numbers of activated cells were no different from those cultured in medium alone. However, activation in the presence of TGF-β significantly reduced viable cell numbers by ~50%. The percentage of mast cells with caspase activity was examined at day 7 by labeling cells with FAM-VAD-FMK. Labeled cells were clearly distinguished from unlabeled cells by flow cytometry (Fig. 9B). Fig. 9C summarizes the results of three independent experiments. TGF-β treatment (12.2 ± 1.6%) or FcεRI aggregation (12.7 ± 2.6%) alone caused a significant increase ( p = 0.02 and 0.03, respectively) in the percentage of apoptotic cells compared with a medium control (3.2 ± 1.3%). However, TGF-β treatment of FcεRI-activated cells markedly increased the percentage of apoptotic cells (34.3 ± 1.8%) compared with TGF-β-treated, but not activated cells ( p = 0.01); activated, but not TGF-β-treated ( p = 0.01); and control cells ( p = 0.01).
Figure 9.

TGF-β enhances apoptosis of activated skin mast cells. Human skin mast cells were given fresh medium containing SCF alone, or simultaneously stimulated with 22E7 (1 μg/ml) ± TGF-β (10 ng/ml). A, Viable cell numbers. Cells were assessed by trypan blue exclusion on days 1, 3, 5, and 7 (mean ± SE, n = 3). *, p < 0.05 compared with medium control cells and to those activated with 22E7, but not treated with TGF-β. B, Detection of apoptotic mast cells. Skin mast cells at day 7 after being treated as in A were labeled with FAM-VAD-FMK and assessed by flow cytometry. Graphs represent results from one of three independent experiments. C, Percentage of apoptotic mast cells. Percentages of apoptotic cells at day 7 of three independent experiments, each performed in duplicate, as described in A and B (mean ± SE, n = 3). *, p < 0.05 compared with control group. †, p < 0.05 compared with 22E7-alone group or TGF-β-alone group.
Discussion
In the present study, the effects of TGF-β on human mast cells that had matured in vivo in skin and were then grown in vitro in serum-free medium containing SCF were investigated. These mast cells are of the MCTC type that contain tryptase and chymase along with carboxypeptidase A3 and cathepsin G in their secretory granules and CD88 (C5aR) on their surface (50, 55, 56). Skin mast cell viability was not affected by the presence of TGF-β concentrations up to 10 ng/ml for 1 wk. Mast cells stimulated by FcεRI cross-linking exhibited less degranulation and cytokine production in the presence of TGF-β, whereas PGD2 production was unaltered. Furthermore, TGF-β markedly inhibited de novo synthesis of Kit, most notably after surface Kit binds SCF and is internalized and degraded. Perhaps related to Kit down-regulation was the observation that TGF-β potentiated apoptosis of mast cells activated by aggregating FcεRI.
The lack of inhibition of TGF-β on the proliferation of human skin mast cells compared with murine mast cells might be explained by the absence of a significant role for IL-3 in the human mast cell system (57), because IL-3 drives proliferation of murine mast cells. Indeed, although IL-3 can facilitate expansion of the numbers of human progenitor cells capable of becoming mast cells, it has no consistent or dramatic direct effects on human mast cell development, proliferation, and survival (58). Consequently, any inhibition of IL-3 signaling in humans should have no direct effect on mature mast cells.
At TGF-β concentrations ≤10 ng/ml, no significant effect on proliferation of human skin mast cells was detected, whereas a dramatic effect on their survival was only observed after activation of these cells by FcεRI aggregation. After exposure of activated skin mast cells to 10 ng/ml TGF-β in vitro, only ~50% of the cells survived 1 wk, and ~30% of these were undergoing apoptosis as reflected by caspase activation. Perhaps the increased energy requirements and stress of activation in the context of low surface levels of Kit make these cells more susceptible to apoptosis. Activated mast cells being more sensitive to TGF-β than those at rest might facilitate the resolution of inflammation caused by these cells. This is in contrast to murine models in which connective tissue-like mast cells derived from mouse bone marrow showed 20% higher viability upon IgE receptor cross-linking and withdrawal of growth factors (59). However, the observation on murine connective tissue mast cells occurred over a short time period (24 h) and was not reproduced in murine mucosal mast cells (59, 60). In terms of the mechanism of apoptosis induced by TGF-β, whether this involves direct effects of TGF-β, occurs indirectly due to diminished levels of Kit, or both is uncertain. TGF-β can directly induce apoptosis of other cell types through the Daxx adaptor pathway (61) and the Smad4-mediated pathway involving JNK (62). Although TGF-β levels can reach 50 ng/ml in serum, the actual concentrations in different organ or tissue sites, where mast cells reside, are unknown. Therefore, the actual concentrations of TGF-β to which mast cells are exposed in vivo may vary substantially. Perhaps TGF-β can regulate the survival of activated mast cells in vivo, thereby limiting the persistence of mast cell-mediated inflammation.
The increased apoptosis observed in this study with human skin mast cells can be further compared with murine systems. However, the effects of TGF-β on the survival and proliferation of murine mast cells vary considerably. In one case, SCF-dependent rescue of IL-3-deprived bone marrow-derived mast cells was blocked by TGF-β (39). In another case, both a higher dose of TGF-β (50 ng/ml) and a longer duration of exposure (3 wk) were used (38). The observed effects of TGF-β in murine systems using bone marrow-derived cells also may reflect effects on the maturation of mast cells. However, TGF-β had no inhibitory effect on the development of mast cells from bone marrow-derived progenitors grown with SCF, IL-9, and WEHI-conditioned medium with IL-3 after 7 days (32). Thus, the dose and duration of TGF-β treatment, lack of an IL-3-dependent pathway, and use of mast cells that have matured in vivo are features that distinguish the human skin mast cell from the murine systems.
The suppressive effect of TGF-β on degranulation and cytokine production by human skin mast cells in response to FcεRI aggregation cannot be explained by less FcεRI on the cell surface, because unlike murine mast cells (42), no down-regulation in FcεRI levels was observed by flow cytometry. However, other studies using bone marrow-derived murine mast cells did not find an effect of TGF-β on degranulation or cysteinyl leukotriene release (36, 37). In the current study, TGF-β-mediated inhibition of IL-6 and GM-CSF production by stimulated mast cells was associated with diminished cellular levels of the corresponding mRNAs, suggesting attenuated production or enhanced turnover of these mRNAs. Levels of TNF-α and IL-13 mRNAs remained unchanged, suggesting diminished translation or enhanced turnover of the protein to account for diminished production of these cytokines. Protein and mRNA levels of Cox-1 and PGD synthase and protein levels of Cox-2 were not significantly altered by TGF-β, whereas mRNA levels of Cox-2 were elevated. Overall, these data are consistent with the observed lack of a TGF-β effect on PGD2 production by activated skin mast cells. One study using murine bone marrow-derived mast cells found that TGF-β can up-regulate cytosolic phospholipase A2 and thereby up-regulate both PGD2 and leukotriene production in response to FcεRI aggregation (63). Using human intestinal mast cells, TGF-β was found to attenuate degranulation and production of cysteinyl leukotrienes in response to FcεRI aggregation, while up-regulating cellular levels of Cox-1 and Cox-2 and the consequent FcεRI-dependent production of PGD2 (48). Thus, TGF-β may have opposing functions on mast cell mediator release depending on the type of mast cell and its state of maturation and activation.
The present study clearly demonstrated a reduction in Kit expression on the cell surface of human skin mast cells with TGF-β treatment. Surface Kit is well known to be rapidly internalized after SCF treatment, and is then degraded (64). Consequently, it is replaced by de novo synthesis (65), which occurs over several days. The simultaneous surface and intracellular staining used in this study support this scenario, because Kit levels in permeabilized mast cells transiently increased early after the cells were exposed to SCF, when surface levels of Kit had already declined. Kit re-expression was then inhibited by TGF-β, which prolonged the deficiency of Kit after exposure of these mast cells to SCF. Both actinomycin D and cycloheximide also inhibited Kit re-expression. New synthesis of Kit protein is clearly needed. However, mRNA levels of Kit were not significantly affected by TGF-β, suggesting that another gene must be transcribed for Kit production to occur. These observations contrast with a study on human leukemic myeloblasts and hematopoietic progenitors in which TGF-β decreased Kit mRNA stability (66), but are somewhat analogous to a study with murine mast cells showing inhibition of FcεRIβ protein, but not mRNA synthesis by TGF-β (42).
TGF-β can affect cells through Smad-dependent and Smad-independent pathways. The incubation of mast cells with TGF receptor kinase inhibitor SB431542 prevented phosphorylation of Smad1/3 and normalized Kit re-expression after SCF-mediated internalization of Kit in those cells. In contrast, MAPK kinase pathways, such as ERK, JNK, and p38, are not required because specific inhibitors failed to alter this effect of TGF-β. The PKC family also could not account for this effect, because the inhibition of total PKC seemed to accelerate the re-expression of Kit on mast cells after SCF-mediated internalization and degradation. How inhibition of PKC accelerates Kit re-expression remains to be determined. Of possible interest is the ability of PKC to directly phosphorylate serine residues on Kit, thereby inhibiting SCF-mediated activation of Kit (5, 67). However, whether this affects Kit turnover in human mast cells is not precisely known. Thus, the Smad-dependent pathway is most likely required for TGF-β-mediated suppression of Kit re-expression.
In summary, the current study demonstrates that TGF-β affects human skin mast cells by attenuating degranulation and cytokine production in response to FcεRI aggregation without interfering with PGD2 production, represses de novo Kit synthesis after SCF-mediated internalization and degradation, and markedly increases apoptosis after activation of these cells by FcεRI aggregation. Thus, TGF-β may limit mast cell numbers and dampen mediator production at sites of allergic inflammation.
Acknowledgments
We thank Larry Okumoto for excellent technical assistance.
Footnotes
This work was supported in part by National Institutes of Health Grants R01-AI27517 (to L.B.S.) and K08-AI057357 (to W.Z.), and by a grant from Philip Morris USA and Philip Morris International (to L.B.S.).
Abbreviations used in this paper: SCF, stem cell factor; Cox, cyclooxygenase; Ct, cycle threshold; FAM-VAD-FMK, carboxyfluorescein-Val-Ala-Asp-fluoromethyl ketone; PKC, protein kinase C.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Bischoff SC. Role of mast cells in allergic and non-allergic immune responses: comparison of human and murine data. Nat Rev Immunol. 2007;7:93–104. doi: 10.1038/nri2018. [DOI] [PubMed] [Google Scholar]
- 2.Metz M, Maurer M. Mast cells: key effector cells in immune responses. Trends Immunol. 2007;28:234–241. doi: 10.1016/j.it.2007.03.003. [DOI] [PubMed] [Google Scholar]
- 3.Lu LF, Lind EF, Gondek DC, Bennett KA, Gleeson MW, Pino-Lagos K, Scott ZA, Coyle AJ, Reed JL, Van Snick J, et al. Mast cells are essential intermediaries in regulatory T-cell tolerance. Nature. 2006;442:997–1002. doi: 10.1038/nature05010. [DOI] [PubMed] [Google Scholar]
- 4.Ullrich A, Schlessinger J. Signal transduction by receptors with tyrosine kinase activity. Cell. 1990;61:203–212. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 5.Blume-Jensen P, Siegbahn A, Stabel S, Heldin CH, Ronnstrand L. Increased Kit/SCF receptor induced mitogenicity but abolished cell motility after inhibition of protein kinase C. EMBO J. 1993;12:4199–4209. doi: 10.1002/j.1460-2075.1993.tb06104.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Blume-Jensen P, Claesson-Welsh L, Siegbahn A, Zsebo KM, Westermark B, Heldin CH. Activation of the human c-kit product by ligand-induced dimerization mediates circular actin reorganization and chemotaxis. EMBO J. 1991;10:4121–4128. doi: 10.1002/j.1460-2075.1991.tb04989.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Valent P, Sperr WR, Schwartz LB, Horny HP. Diagnosis and classification of mast cell proliferative disorders: delineation from immunologic diseases and non-mast cell hematopoietic neoplasms. J Allergy Clin Immunol. 2004;114:3–11. doi: 10.1016/j.jaci.2004.02.045. [DOI] [PubMed] [Google Scholar]
- 8.Kitamura Y, Hirota S, Nishida T. A loss-of-function mutation of c-kit results in depletion of mast cells and interstitial cells of Cajal, while its gain-of-function mutation results in their oncogenesis. Mutat Res. 2001;477:165–171. doi: 10.1016/s0027-5107(01)00117-8. [DOI] [PubMed] [Google Scholar]
- 9.Tsujimura T, Morii E, Nozaki M, Hashimoto K, Moriyama Y, Takebayashi K, Kondo T, Kanakura Y, Kitamura Y. Involvement of transcription factor encoded by the mi locus in the expression of c-kit receptor tyrosine kinase in cultured mast cells of mice. Blood. 1996;88:1225–1233. [PubMed] [Google Scholar]
- 10.Vandenbark GR, Chen Y, Friday E, Pavlik K, Anthony B, deCastro C, Kaufman RE. Complex regulation of human c-kit transcription by promoter repressors, activators, and specific myb elements. Cell Growth Differ. 1996;7:1383–1392. [PubMed] [Google Scholar]
- 11.Felli N, Fontana L, Pelosi E, Botta R, Bonci D, Facchiano F, Liuzzi F, Lulli V, Morsilli O, Santoro S, et al. MicroRNAs 221 and 222 inhibit normal erythropoiesis and erythroleukemic cell growth via kit receptor down-modulation. Proc Natl Acad Sci USA. 2005;102:18081–18086. doi: 10.1073/pnas.0506216102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yee NS, Langen H, Besmer P. Mechanism of kit ligand, phorbol ester, and calcium-induced down-regulation of c-kit receptors in mast cells. J Biol Chem. 1993;268:14189–14201. [PubMed] [Google Scholar]
- 13.Cruz AC, Frank BT, Edwards ST, Dazin PF, Peschon JJ, Fang KC. Tumor necrosis factor-α-converting enzyme controls surface expression of c-kit and survival of embryonic stem cell-derived mast cells. J Biol Chem. 2004;279:5612–5620. doi: 10.1074/jbc.M312323200. [DOI] [PubMed] [Google Scholar]
- 14.Yee NS, Hsiau CW, Serve H, Vosseller K, Besmer P. Mechanism of down-regulation of c-kit receptor: roles of receptor tyrosine kinase, phosphatidylinositol 3′-kinase, and protein kinase C. J Biol Chem. 1994;269:31991–31998. [PubMed] [Google Scholar]
- 15.Shimizu Y, Ashman LK, Du Z, Schwartz LB. Internalization of kit together with stem cell factor on human fetal liver-derived mast cells: new protein and RNA synthesis are required for reappearance of kit. J Immunol. 1996;156:3443–3449. [PubMed] [Google Scholar]
- 16.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor β in human disease. N Engl J Med. 2000;342:1350–1358. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 17.Letterio JJ, Roberts AB. Regulation of immune responses by TGF-β. Annu Rev Immunol. 1998;16:137–161. doi: 10.1146/annurev.immunol.16.1.137. [DOI] [PubMed] [Google Scholar]
- 18.Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM. Conversion of peripheral CD4+CD25− naive T cells to CD4+CD25+ regulatory T cells by TGF-β induction of transcription factor Foxp3. J Exp Med. 2003;198:1875–1886. doi: 10.1084/jem.20030152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chin D, Boyle GM, Parsons PG, Coman WB. What is transforming growth factor-β (TGF-β)? Br J Plast Surg. 2004;57:215–221. doi: 10.1016/j.bjps.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 20.Derynck R, Akhurst RJ, Balmain A. TGF-β signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 21.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 22.Xu L, Kang Y, Col S, Massague J. Smad2 nucleocytoplasmic shuttling by nucleoporins CAN/Nup214 and Nup153 feeds TGF-β signaling complexes in the cytoplasm and nucleus. Mol Cell. 2002;10:271–282. doi: 10.1016/s1097-2765(02)00586-5. [DOI] [PubMed] [Google Scholar]
- 23.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF β receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 24.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf1 interacts with transforming growth factor-β type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 25.Massague J. How cells read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 26.Deaton RA, Su C, Valencia TG, Grant SR. Transforming growth factor-β1-induced expression of smooth muscle marker genes involves activation of PKN and p38 MAPK. J Biol Chem. 2005;280:31172–31181. doi: 10.1074/jbc.M504774200. [DOI] [PubMed] [Google Scholar]
- 27.Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-β-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 28.Yakymovych I, ten Dijke P, Heldin CH, Souchelnytskyi S. Regulation of Smad signaling by protein kinase C. FASEB J. 2001;15:553–555. doi: 10.1096/fj.00-0474fje. [DOI] [PubMed] [Google Scholar]
- 29.Kim YK. TGF-β1 induction of p21WAF1/cip1 requires smad-independent protein kinase C signaling pathway. Arch Pharm Res. 2007;30:739–742. doi: 10.1007/BF02977636. [DOI] [PubMed] [Google Scholar]
- 30.Chow JY, Dong H, Quach KT, Van Nguyen PN, Chen K, Carethers JM. TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am J Physiol. 2008;294:G899–G905. doi: 10.1152/ajpgi.00411.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller HR, Wright SH, Knight PA, Thornton EM. A novel function for transforming growth factor-β1: up-regulation of the expression and the IgE-independent extracellular release of a mucosal mast cell granule-specific β-chymase, mouse mast cell protease-1. Blood. 1999;93:3473–3486. [PubMed] [Google Scholar]
- 32.Wright SH, Brown J, Knight PA, Thornton EM, Kilshaw PJ, Miller HR. Transforming growth factor-β1 mediates coexpression of the integrin subunit αE and the chymase mouse mast cell protease-1 during the early differentiation of bone marrow-derived mucosal mast cell homologues. Clin Exp Allergy. 2002;32:315–324. doi: 10.1046/j.1365-2222.2002.01233.x. [DOI] [PubMed] [Google Scholar]
- 33.Funaba M, Ikeda T, Murakami M, Ogawa K, Nishino Y, Tsuchida K, Sugino H, Abe M. Transcriptional regulation of mouse mast cell protease-7 by TGF-β. Biochim Biophys Acta. 2006;1759:166–170. doi: 10.1016/j.bbaexp.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 34.Funaba M, Ikeda T, Murakami M, Ogawa K, Abe M. Up-regulation of mouse mast cell protease-6 gene by transforming growth factor-β and activin in mast cell progenitors. Cell Signal. 2005;17:121–128. doi: 10.1016/j.cellsig.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 35.Funaba M, Nakaya K, Ikeda T, Murakami M, Tsuchida K, Sugino H. Requirement of Smad3 for mast cell growth. Cell Immunol. 2006;240:47–52. doi: 10.1016/j.cellimm.2006.06.002. [DOI] [PubMed] [Google Scholar]
- 36.Broide DH, Wasserman SI, Alvaro-Gracia J, Zvaifler NJ, Firestein GS. Transforming growth factor-β1 selectively inhibits IL-3-dependent mast cell proliferation without affecting mast cell function or differentiation. J Immunol. 1989;143:1591–1597. [PubMed] [Google Scholar]
- 37.Toyota N, Hashimoto Y, Matsuo S, Iizuka H. Transforming growth factor β1 inhibits IL-3- and IL-4-dependent mouse connective tissue-type mast cell proliferation. Arch Dermatol Res. 1995;287:198–201. doi: 10.1007/BF01262332. [DOI] [PubMed] [Google Scholar]
- 38.Kashyap M, Bailey DP, Gomez G, Rivera J, Huff TF, Ryan JJ. TGF-β1 inhibits late-stage mast cell maturation. Exp Hematol. 2005;33:1281–1291. doi: 10.1016/j.exphem.2005.07.001. [DOI] [PubMed] [Google Scholar]
- 39.Norozian F, Kashyap M, Ramirez CD, Patel N, Kepley CL, Barnstein BO, Ryan JJ. TGFβ1 induces mast cell apoptosis. Exp Hematol. 2006;34:579–587. doi: 10.1016/j.exphem.2006.02.003. [DOI] [PubMed] [Google Scholar]
- 40.Mekori YA, Metcalfe DD. Transforming growth factor-β prevents stem cell factor-mediated rescue of mast cells from apoptosis after IL-3 deprivation. J Immunol. 1994;153:2194–2203. [PubMed] [Google Scholar]
- 41.Anthoni M, Wang G, Deng C, Wolff HJ, Lauerma AI, Alenius HT. Smad3 signal transducer regulates skin inflammation and specific IgE response in murine model of atopic dermatitis. J Invest Dermatol. 2007;127:1923–1929. doi: 10.1038/sj.jid.5700809. [DOI] [PubMed] [Google Scholar]
- 42.Gomez G, Ramirez CD, Rivera J, Patel M, Norozian F, Wright HV, Kashyap MV, Barnstein BO, Fischer-Stenger K, Schwartz LB, et al. TGF-β1 inhibits mast cell FcεRI expression. J Immunol. 2005;174:5987–5993. doi: 10.4049/jimmunol.174.10.5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Olsson N, Piek E, ten Dijke P, Nilsson G. Human mast cell migration in response to members of the transforming growth factor-β family. J Leukocyte Biol. 2000;67:350–356. doi: 10.1002/jlb.67.3.350. [DOI] [PubMed] [Google Scholar]
- 44.Wiener Z, Kohalmi B, Pocza P, Jeager J, Tolgyesi G, Toth S, Gorbe E, Papp Z, Falus A. TIM-3 is expressed in melanoma cells and is up-regulated in TGF-β stimulated mast cells. J Invest Dermatol. 2007;127:906–914. doi: 10.1038/sj.jid.5700616. [DOI] [PubMed] [Google Scholar]
- 45.Nilsson G, Blom T, Kusche-Gullberg M, Kjellen L, Butterfield JH, Sundstrom C, Nilsson K, Hellman L. Phenotypic characterization of the human mast-cell line HMC-1. Scand J Immunol. 1994;39:489–498. doi: 10.1111/j.1365-3083.1994.tb03404.x. [DOI] [PubMed] [Google Scholar]
- 46.Butterfield JH, Weiler D, Dewald G, Gleich GJ. Establishment of an immature mast cell line from a patient with mast cell leukemia. Leukemia Res. 1988;12:345–355. doi: 10.1016/0145-2126(88)90050-1. [DOI] [PubMed] [Google Scholar]
- 47.Zhao W, Kepley CL, Morel PA, Okumoto LM, Fukuoka Y, Schwartz LB. FcγRIIa, not FcγRIIb, is constitutively and functionally expressed on skin-derived human mast cells. J Immunol. 2006;177:694–701. doi: 10.4049/jimmunol.177.1.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gebhardt T, Lorentz A, Detmer F, Trautwein C, Bektas H, Manns MP, Bischoff SC. Growth, phenotype, and function of human intestinal mast cells are tightly regulated by transforming growth factor β1. Gut. 2005;54:928–934. doi: 10.1136/gut.2004.054650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Riske F, Hakimi J, Mallamaci M, Griffin M, Pilson B, Tobkes N, Lin P, Danho W, Kochan J, Chizzonite R. High affinity human IgE receptor (FcεRI): analysis of functional domains of the α-subunit with monoclonal antibodies. J Biol Chem. 1991;266:11245–11251. [PubMed] [Google Scholar]
- 50.Kambe N, Kambe M, Kochan JP, Schwartz LB. Human skin-derived mast cells can proliferate while retaining their characteristic functional and protease phenotypes. Blood. 2001;97:2045–2052. doi: 10.1182/blood.v97.7.2045. [DOI] [PubMed] [Google Scholar]
- 51.Irani AM, Bradford TR, Kepley CL, Schechter NM, Schwartz LB. Detection of MCT and MCTC types of human mast cells by immunohistochemistry using new monoclonal anti-tryptase and anti-chymase antibodies. J Histochem Cytochem. 1989;37:1509–1515. doi: 10.1177/37.10.2674273. [DOI] [PubMed] [Google Scholar]
- 52.Schwartz LB, Lewis RA, Seldin D, Austen KF. Acid hydrolases and tryptase from secretory granules of dispersed human lung mast cells. J Immunol. 1981;126:1290–1294. [PubMed] [Google Scholar]
- 53.Schwartz LB, Austen KF, Wasserman SI. Immunologic release of β-hexosaminidase and β-glucuronidase from purified rat serosal mast cells. J Immunol. 1979;123:1445–1450. [PubMed] [Google Scholar]
- 54.Zhao W, Oskeritzian CA, Pozez AL, Schwartz LB. Cytokine production by skin-derived mast cells: endogenous proteases are responsible for degradation of cytokines. J Immunol. 2005;175:2635–2642. doi: 10.4049/jimmunol.175.4.2635. [DOI] [PubMed] [Google Scholar]
- 55.Irani AA, Schechter NM, Craig SS, DeBlois G, Schwartz LB. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci USA. 1986;83:4464–4468. doi: 10.1073/pnas.83.12.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oskeritzian CA, Zhao W, Min HK, Xia HZ, Pozez A, Kiev J, Schwartz LB. Surface CD88 functionally distinguishes the MCTC from the MCT type of human lung mast cell. J Allergy Clin Immunol. 2005;115:1162–1168. doi: 10.1016/j.jaci.2005.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Valent P, Besemer J, Sillaber C, Butterfield JH, Eher R, Majdic O, Kishi K, Klepetko W, Eckersberger F, Lechner K, Bettelheim P. Failure to detect IL-3-binding sites on human mast cells. J Immunol. 1990;145:3432–3437. [PubMed] [Google Scholar]
- 58.Kirshenbaum AS, Goff JP, Kessler SW, Mican JM, Zsebo KM, Metcalfe DD. Effect of IL-3 and stem cell factor on the appearance of human basophils and mast cells from CD34+ pluripotent progenitor cells. J Immunol. 1992;148:772–777. [PubMed] [Google Scholar]
- 59.Ekoff M, Strasser A, Nilsson G. FcεRI aggregation promotes survival of connective tissue-like mast cells but not mucosal-like mast cells. J Immunol. 2007;178:4177–4183. doi: 10.4049/jimmunol.178.7.4177. [DOI] [PubMed] [Google Scholar]
- 60.Xiang Z, Moller C, Nilsson G. IgE-receptor activation induces survival and Bfl-1 expression in human mast cells but not basophils. Allergy. 2006;61:1040–1046. doi: 10.1111/j.1398-9995.2006.01024.x. [DOI] [PubMed] [Google Scholar]
- 61.Perlman R, Schiemann WP, Brooks MW, Lodish HF, Weinberg RA. TGF-β-induced apoptosis is mediated by the adapter protein Daxx that facilitates JNK activation. Nat Cell Biol. 2001;3:708–714. doi: 10.1038/35087019. [DOI] [PubMed] [Google Scholar]
- 62.Dai JL, Bansal RK, Kern SE. G1 cell cycle arrest and apoptosis induction by nuclear Smad4/Dpc4: phenotypes reversed by a tumorigenic mutation. Proc Natl Acad Sci USA. 1999;96:1427–1432. doi: 10.1073/pnas.96.4.1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sanchez Mejia RO, Lam BK, Arm JP. Matrix-associated transforming growth factor-β1 primes mouse bone marrow-derived mast cells for increased high-affinity Fc receptor for immunoglobulin E-dependent eicosanoid biosynthesis. Am J Respir Cell Mol Biol. 2000;22:557–565. doi: 10.1165/ajrcmb.22.5.3902. [DOI] [PubMed] [Google Scholar]
- 64.Miyazawa K, Toyama K, Gotoh A, Hendrie PC, Mantel C, Broxmeyer HE. Ligand-dependent polyubiquitination of c-kit gene product: a possible mechanism of receptor down-modulation in M07e cells. Blood. 1994;83:137–145. [PubMed] [Google Scholar]
- 65.Broudy VC, Lin NL, Buhring HJ, Komatsu N, Kavanagh TJ. Analysis of c-kit receptor dimerization by fluorescence resonance energy transfer. Blood. 1998;91:898–906. [PubMed] [Google Scholar]
- 66.Heinrich MC, Dooley DC, Keeble WW. Transforming growth factor β1 inhibits expression of the gene products for steel factor and its receptor (c-kit) Blood. 1995;85:1769–1780. [PubMed] [Google Scholar]
- 67.Blume-Jensen P, Wernstedt C, Heldin CH, Ronnstrand L. Identification of the major phosphorylation sites for protein kinase C in kit/stem cell factor receptor in vitro and in intact cells. J Biol Chem. 1995;270:14192–14200. doi: 10.1074/jbc.270.23.14192. [DOI] [PubMed] [Google Scholar]