

Abstract
Thoracic aortic aneurysms (TAAs) are potentially devastating, and due to their asymptomatic behavior, pose a serious health risk characterized by the lack of medical treatment options and high rates of surgical morbidity and mortality. Independent of the inciting stimuli (biochemical/mechanical), TAA development proceeds by a multifactorial process influenced by both cellular and extracellular mechanisms, resulting in alterations of the structure and composition of the vascular extracellular matrix (ECM). While the role of enhanced ECM proteolysis in TAA formation remains undisputed, little attention has been focused on the upstream signaling events that drive the remodeling process. Recent evidence highlighting the dysregulation of transforming growth factor-beta (TGF-β) signaling in ascending TAAs from Marfan syndrome patients has stimulated an interest in this intracellular signaling pathway. However, paradoxical discoveries have implicated both enhanced TGF-β signaling and loss of function TGF-β receptor mutations, in aneurysm formation; obfuscating a clear functional role for TGF-β in aneurysm development. In an effort to elucidate this subject, TGF-β signaling and its role in vascular remodeling and pathology will be reviewed, with the aim of identifying potential mechanisms of how TGF-β signaling may contribute to the formation and progression of TAA.
Keywords: TGF-β, aneurysm, signal transduction, extracellular matrix, remodeling
Introduction
Thoracic aortic aneurysms (TAAs) develop as a result of maladaptive remodeling of the vascular extracellular matrix (ECM). These malignant alterations cause weakening of the aortic ultrastructure and lead to an increased propensity for dilatation, dissection, and rupture.[1,2] ECM remodeling occurs by a highly regulated process involving both intracellular and extracellular mechanisms that function to balance matrix deposition and matrix degradation in order to maintain the structural integrity of the vascular wall.[3,4] In the aneurysmal aorta, this balance is disrupted in favor of enhanced proteolysis which results in the pathological remodeling of the vascular ECM.[2,5] While the majority of previous studies have focused on the dysregulation of extracellular protease systems in aneurysm formation, little attention has been focused on the small molecule mediators that drive multiple signaling pathways upstream and regulate the remodeling process. One upstream signaling protein known to alter the structure and composition of the ECM, and known to play an important role in vascular remodeling is transforming growth factor-beta (TGF-β).[6,7] TGF-β is a member of a superfamily of ligands and receptors that include the TGF-βs, bone morphogenetic proteins, and the activins/inhibins. These soluble peptide growth factors are produced by multiple cell types and participate in a wide array of cellular responses including: proliferation, angiogenesis, differentiation, apoptosis, inflammation, and wound healing.[8-10] While TGF-β is probably best known for its role in matrix deposition (e.g. collagen synthesis) related to fibrotic disease,[7] TGF-β has also been shown to regulate alternate pathways that can lead to matrix degradation[11-14]. Recent studies demonstrating altered TGF-β signaling in aneurysm formation have sparked an interest in how a well known pro-fibrotic growth factor can participate in a pathological process that is characterized by extensive matrix degradation. Thus, the goal of the present article is to review both the classical and alternative pathways of TGF-β signaling, its role in regulating ECM remodeling, and to propose mechanisms based on how this complex signaling pathway may be involved in thoracic aortic aneurysm development.
Thoracic Aortic Aneurysms
Each year 15,000 lives are claimed by aortic aneurysmal disease, making it the 13th leading cause of death in people 55 years of age or older in the United States (National Center for Health Statistics, 2000). A TAA is defined as a localized dilatation of the supra-diaphragmatic aorta exceeding 1.5 times its original diameter.[15] Anatomically, ascending TAAs are the most common, accounting for 40% of those diagnosed, while aneurysms of the descending thoracic aorta account for 35%, with the remaining composed of aneurysms of the aortic arch (15%) and the thoracoabdominal regions (10%).[16] TAAs occur most frequently in Caucasians, and they afflict men two-to-four times more frequently than women. The mean age at diagnosis is 60-70 years of age.[17] The risk factors for developing aneurysms are similar to those for heart disease (atherosclerosis, hypertension, smoking, advanced age, and family history), however the lack of aneurysm-specific symptoms often renders them unnoticed until the aorta ruptures, resulting in significant morbidity and mortality.[3,16,18-24] While the most common etiology of TAA development is related to idiopathic aortic degeneration in patients with tricuspid aortic valves, other types of aortic aneurysm syndromes are associated with specific genetic conditions that carry a predisposition for TAA formation; these include possessing a congenital bicuspid aortic valve, Marfan syndrome (MFS), Loeys-Dietz syndrome (LDS), Ehlers-Danlos syndrome, and familial thoracic aortic aneurysms and dissections (TAAD).[3,25] When aortic dilatation is discovered, independent of the aneurysm etiology, aortic diameter is serially monitored over time using noninvasive imaging techniques. A high risk surgical procedure is often the only treatment option. There are currently no noninvasive interventional treatments available for these patients. Surgery is considered when: 1) the patient begins to experience specific symptoms; 2) the rate of aortic dilatation is determined to be greater than average (1.0 cm/yr); or 3) the aortic diameter reaches a critical size (5.0-5.5 cm (ascending) or 6-6.5 cm (descending);[26] modified based on diagnosis, family history, or body surface area.[27] Thus, only when the risk of aortic rupture outweighs the risk of the surgical repair, is the patient treated. Although recent advancements, such as endovascular stent grafting and improvements in perioperative care have lessened the significant morbidity associated with open surgical repair, stenting may not permanently arrest aneurysm development, and continued disease progression may result in failure of the stent graft. Therefore, further diagnostic and therapeutic advancement is critical and especially relevant for those patients who have not yet reached surgical criteria.
Vascular Remodeling and Aneurysm Formation
Vascular remodeling collectively refers to the architectural alterations that occur in a vessel wall in response to hemodynamic changes or various forms of vascular injury. This adaptive process is induced to maintain the vessel lumen diameter and consistent blood flow under normal physiological conditions. Both clinical and basic research studies have characterized aneurysmal disease histologically based on the alterations that occur within the vascular extracellular matrix, primarily the pathological remodeling of collagen and elastin, the key structural proteins within the aortic vascular wall. This remodeling process is now understood to be driven by enhanced production of extracellular proteases and is accompanied by the loss of vascular smooth muscle cells (SMCs), together leading to pronounced medial atrophy.[2,5,28]
While the inciting pathological stimulus leading to aneurysm formation remains undefined, the development of experimental animal models that recapitulate many characteristics of human aneurysms, have allowed hypothesis-directed investigation of specific proteolytic mechanisms driving aberrant vascular remodeling and aneurysm development.[29-36] Initial studies focused on the role of the matrix metalloproteinases (MMPs) in mediating ECM degradation during aneurysm formation.[2,3,5,35,37-41] The MMPs are a family of 27 unique extracellular proteases that are capable of degrading all aortic ECM constituents. MMP abundance and activity within the vascular wall is regulated by several mechanisms including transcriptional regulation, post-translational activation and release, and the regulated production of endogenous tissue inhibitors (TIMPs). Within the normal aorta, a balance between these mechanisms is maintained in order to tightly control matrix degradation and matrix deposition. Within the aneurysmal aorta, however, this balance is disrupted by an over production of MMPs or an under production of TIMPs, favoring an enhanced proteolytic state and driving matrix degradation.[37,42,43] Thus, while the physiological remodeling process within the aortic vascular wall operates to maintain normal aortic function, pathological dysregulation can result in excessive degradation of critical ECM components leading to loss of mechanical strength and integrity. This results in aortic dilatation, dissection, or rupture.
While the role of enhanced ECM proteolysis in TAA formation remains undisputed, the fundamental mechanisms regulating the balance between matrix deposition and degradation remain largely undiscovered. With the majority of previous studies focusing on the role of specific end effectors of aneurysm development, such as the MMPs, little information exists regarding the upstream signaling pathways that manage the pathological remodeling process. Hence, interest is shifting toward a mechanistic understanding of aneurysm formation and progression with a focus on identifying critical upstream regulators.
TGF-β Signaling
Transforming growth factor-β is a soluble peptide growth factor that has been implicated in numerous divergent cellular processes including proliferation, angiogenesis, differentiation, apoptosis, and wound healing,[9,44-48] and is well described as a modifier of the structure and composition of the ECM. Best known for its role in stimulating collagen production and deposition, dysregulated TGF-β signaling has been implicated in pathological fibrosis of the heart, lung, and liver.[7,49-51]
Classical TGF-β Signaling Pathway
In the classical TGF-β signaling pathway, upon release from sites of extracellular sequestration, TGF-β (TGF-β1, TGF-β2, TGF-β3) dimerizes (forming predominantly homodimers) and binds to a heteromeric receptor complex consisting of two type I receptors and two type II receptors, both of which possess serine/threonine kinase activity (Figure 1).[52] Upon ligand binding, the type II receptor recruits a type I receptor and activates it through a transphosphorylation event.[53] The activated type I receptor then phosphorylates a receptor-Smad (R-Smad), a class of intracellular signaling intermediates named for their homologues in C. elegans (sma genes; SMAll, regulators of body size) and Drosophila (mad genes; mothers against decapentaplegic (dpp)). The R-Smad then interacts with a co-Smad (Smad4) forming a complex that is shuttled into the nucleus where, upon interaction with transcriptional coregulators (activators or repressors), it forms a competent transcription complex capable of inducing or repressing numerous genes.[45,48,54,55] Accordingly, the classical TGF-β signaling pathway involving the Smad-mediated signaling cascade has been characterized by signals that induce ECM deposition (e.g. collagen, elastin)[7,56,57] while also repressing ECM degradation (e.g. TIMP-1, TIMP-3).[58,59]
Figure 1. Classical TGF-β signaling pathway.

The classical profibrotic TGF-β signaling pathway is initiated upon binding of ligand to a homodimer of the type II TGF-β receptor (TGF-βRII) (1.). The type II receptor is autophosphorylated (2.), which then recruits and transphosphorylates a type I receptor (TGF-βRI) (3.). The activated type I receptor in turn phosphorylates and activates a receptor-Smad (R-Smad) (4.). The R-Smad then binds the common co-Smad (5.) and translocates to the nucleus (6.). Once in the nucleus, it binds transcriptional co-factors and forms an activated transcriptional complex capable of inducing transcription of profibrotic genes (7.).
Regulation of TGF-β Signaling
TGF-β was originally named for its ability to stimulate anchorage-independent growth in fibroblasts by inducing cellular transformation.[60,61] Not long afterwards, TGF-β was also shown to function as a potent growth suppressor.[62] Attempts to dissect the TGF-β pathway, separating its mitogenic responses from its growth suppression responses, initiated years of divergent results emphasizing the pleiotropic nature of TGF-β signaling. Since that time, the family of TGF-β signaling receptors and intermediates has dramatically expanded, and now defines TGF-β as one member of a superfamily of signaling components consisting of approximately 30 different ligands, 7 different type I receptors, 5 different type II receptors, and 8 different Smad proteins.[45,63]
TGF-β signaling is regulated at multiple levels including mechanisms such as, the extracellular regulation of ligand availability,[64,65] regulation at the transcriptional level by co-activators, co-repressors, and transcriptional terminators,[66] and by multiple feedback and cross-talk mechanisms that terminate or re-direct the intracellular signal.[10,46,67] For example, the extracellular TGF-β scavenging proteoglycan decorin binds TGF-β in the ECM and limits its ability to interact with TGF-β receptors. In a study by Coucke and coworkers, loss-of-function mutations were identified in the SLC2A10 gene that encodes the facilitative glucose transporter, GLUT10.[68] Patients deficient in this transporter develop arterial tortuosity syndrome (ATS), characterized by a twisting/contortion of the large and medium sized arteries (including the aorta), as well as aneurysms. Interestingly, the decorin gene is regulated in part by a glucose response element in its promoter. Vascular smooth muscle cells isolated from patients with ATS demonstrated severely reduced expression of decorin versus control cells.[68] In this example, the loss of decorin translated to an increase in the abundance of TGF-β available for signaling within the extracellular matrix, which may be an underlying cause for ATS development. In another example, TGF-β stimulation results in the induction of the inhibitory Smads (I-Smads; Smad6 and Smad7), which function to both mediate TGF-β signaling cross-talk with other signaling pathways and attenuate the TGF-β signaling response.[69] Smad6 has been described to bind directly to the type-I TGF-β receptor (TGF-βRI) preventing subsequent R-Smad phosphorylation.[70] This has been shown to interfere with the R-Smad:co-Smad complex formation,[71] attenuating nuclear translocation and the subsequent transcriptional activation mediated by the R-Smads. On the other hand, Smad7 interacts with the heteromeric TGF-β receptor complex, and recruits the E3 ubiquitin-ligases Smurf1 and Smurf2 (Smad ubiquitination regulatory factor -1 and -2), targeting the receptors for degradation, thus terminating the signaling response.[67,72,73] The R-Smads and the co-Smad have also been shown to be regulated by TGF-β induced Smurf activity.[48,74,75] Accordingly, these factors all work together to modify and direct the response to signals through this complex pathway (Table 1).
Table 1.
TGF-β Signaling Pathway Components and Mediators
| Abbreviation | Name | Description/Function |
|---|---|---|
| TGF-β Pathway Components: | ||
| TGF-β | Transforming growth factor beta | Multifunctional growth factor that regulates ECM structure and composition; TGF-β -1, -2, and -3 |
| LAP | Latency associated peptide | TGF-β is synthesized as a prepropolypeptide that is cleaved into mature growth factor and a latency-associated peptide |
| SLC | Small latent complex | Mature TGF-β and the LAP self-associated to form the small latent complex |
| LTBP | Latent TGF-β binding protein | Binding proteins that sequester the TGF-β SLC in the ECM |
| LLC | Large latent complex | LTBP binds the SLC to form the large latent complex |
| TGF-βRI | TGF-β receptor 1 | TGFBR1 gene; Type I TGF-β receptor with serine/threonine kinase activity; binds TGF-βRII & TGF-β |
| TGF-βRII | TGF-β receptor 2 | TGFBR2 gene; Type II TGF-β receptor with serine/threonine kinase activity; binds TGF-βRI & TGF-β |
| TGF-βRIII | TGF-β receptor 3 | Type III TGF-β receptor; accessory receptor that binds TGF-β and facilitates interaction with type I & II receptors |
| ALK-1 | Activin receptor-like kinase-1 | Type I TGF-β receptor that binds TGF-β and activin |
| Smad | Homologue to C. elegans SMA genes (SMAll, body size regulators) and drosophila MAD (mothers against decapentaplegic) genes | Family of TGF-β signaling intermediates that translate receptor signals into transcriptional responses |
| R-Smads | Receptor-Smads | Phosphorylated by direct interaction with TGF-β receptor complex (Smad1, 2, 3, 5, 8) |
| Phospho-Smad | Phosphorylated Smad | Phosphorylated R-Smad, phosphorylation regulates interactions with co-Smad and transcriptional co-regulators. |
| Co-Smad | Co-regulatory Smad | Smad4, binds phosphorylated R-Smad and translocates to the nucleus to regulate transcription |
| I-Smad | Inhibitory Smad | Smad6, Smad7; assist in the regulation/termination of Smad-mediated signaling |
| SARA | Smad anchor for receptor activation | Facilitates segregation of TGF-β receptor complexes into the clathrin-mediated endocytic pathway for recycling, and facilitates interactions with Smad2 |
| TGF-β Induced Signaling Mediators: | ||
| COL1A1 | Type 1 collagen alpha 1 chain | Type I and type III collagen; Major aortic fibrillar collagens induced by TGF-β |
| COL1A2 | Type 1 collagen alpha 2 chain | |
| COL3A1 | Type 3 collagen alpha 1 chain | |
| CTGF | Connective tissue growth factor | TGF-β-induced growth factor that induces fibroblast proliferation, migration, and ECM protein production |
| PAI-1 | Plasminogen activator inhibitor-1 | Inhibits the activation of uPA and tPA |
| uPA | Urokinase-type plasminogen activator | Plasminogen activators that are known to activate several matrix metalloproteinase species |
| tPA | Tissue-type plasminogen activator | |
| Smurf | Smad ubiquitination regulatory factor | E3 ubiquitin ligase induced by TGF-β; regulates TGF- β signaling by targeting TGF-β receptors, R-Smads, and co-Smads for proteasomal degradation |
| ERK1/2 | Extracellular regulated kinase 1/2 | Other signaling intermediates that are activated by TGF-β, primarily through alternate and non-Smad mediated signaling pathways. |
| p38MAPK | p38 mitogen activated protein kinase | |
| PI3K | Phosphatidylinositol-3-kinase | |
| PKC | Protein kinase C | |
Smad-Independent and Alternative TGF-β Signaling
A growing body of evidence now supports the hypothesis that TGF-β signaling can proceed by alternative mechanisms that bypass key mediators in the classical pathway (Figure 2).[47,76-78] For example, these responses include: 1) signals propagated directly by type II receptors without type I receptor involvement;[79,80] 2) type I receptor signals in the absence of Smad activity;[78,81-84] 3) R-Smad signaling to parallel pathways in the absence of co-Smad involvement;[85-87] and 4) activation of R-Smads by other signaling mediators in response to TGF-β, but not as a result of direct interaction with TGF-β receptors.[88,89] Thus in addition to the classical signaling pathway, many of the downstream effects of TGF-β may be mediated through alternative pathways that do not involve Smad-mediated transcriptional activity.
Figure 2. Alternative TGF-β signaling mechanisms.

Signaling directly mediated by the type II receptor without type I receptor function (1.), type I receptor signaling independent of Smad function (2.), R-Smad signaling independent of co-Smad interaction (3.), and R-Smad activation in response to TGF-β but in the absence of direct TGF-β receptor interaction (4.). Adapted from reviews by Derynck[76] and Moustakas.[47]
Thus, through the appreciation of these combinatorial receptor interactions, multiple regulatory mechanisms, and alternative signaling pathways, we are slowly gaining an understanding of the regulatory events that are responsible for the complexity and diversity of TGF-β signaling outcomes.
TGF-β Effects on the Extracellular Matrix
Just as TGF-β was identified as a bifunctional regulator of cell growth, TGF-β signaling has been attributed to many other opposing and disparate cellular functions. As such, TGF-β has been implicated not only in matrix deposition, but also in matrix degradation, positioning it as a critical mediator of the structure and composition of the ECM.
Early studies examining the biological effects of TGF-β on both primary and cultured cells demonstrated that TGF-β treatment could enhance the production of type I and type III collagen.[90-93] Subsequent studies revealed that collagen production was a result of enhanced collagen gene expression.[94-96] These studies defined a role for TGF-β in normal fibrogenesis. It was then postulated that TGF-β might provide a significant therapeutic target for the treatment of pathological fibrosis. Indeed, in a study by Smith et al., delivery of a recombinant soluble type II TGF-β receptor prevented adventitial fibrosis and collagen deposition in a rat carotid balloon-injury model.[97] In similar fashion, cells exposed to the proteoglycan decorin, which binds and sequesters TGF-β, dose-dependently inhibited collagen synthesis in isolated scar-derived fibroblasts.[98] As the TGF-β signaling pathway has become more defined, a clear role for TGF-β in collagen production and pathological fibrosis has now been well established.[99,100]
In addition to stimulating the production of matrix proteins, TGF-β has also been shown affect matrix deposition through other modalities. For example, other profibrotic genes are induced in response to TGF-β, including connective tissue growth factor (CTGF) which acts on fibroblasts to induce proliferation, migration, adhesion, and ECM protein production.[101] Furthermore, tropoelastin mRNA stability was increased in fibroblasts treated with TGF-β.[56] This response was shown to be dependent on Smad-mediated signaling, but also required PKC and p38MAPK activity.[57] As well, TGF-β has been shown to actively inhibit matrix degradation by at least two independent mechanisms. First, it was demonstrated that TGF-β could induce endogenous protease inhibitors. The expression of plasminogen activator inhibitor-1 (PAI-1), an endogenous inhibitor of the plasminogen activators (uPA and tPA) well known as upstream activators of the MMPs,[102] was found to be induced in response to TGF-β treatment.[12,103] Similarly, TGF-β was shown to induce several TIMP species which function as direct inhibitors of MMP activity. In fibrosarcoma cells, Kwak and coworkers demonstrated TGF-β-mediated induction of TIMP-1 through an ERK1/2-dependent pathway.[58] Likewise, work by Garciá-Alvarez et al. established that TGF-β1 could induce Timp-3 gene expression and protein production in primary lung fibroblasts.[59] Second, several of the MMP species (-1, -7, and -13) have promoter binding regions (termed TGF-β inhibitory elements) for transcription factors that mediate the direct repression of MMP gene expression.[104,105] Additionally, Kerr and coworkers also demonstrated TGF-β induced repression of MMP-3 mediated in part by c-fos-containing protein complexes.[106] Furthermore, Edwards et al. demonstrated that the co-treatment of fibroblasts with TGF-β1 could mediate the repression of growth factor induced-collagenase expression while synergistically enhancing TIMP expression.[107] Hence, TGF-β can drive matrix deposition by inhibiting matrix degrading enzymes (either directly or by blocking activation) as well as by repressing their transcription. In terms of the functional consequences of TGF-β-mediated inhibition of matrix degradation, it was suggested that overexpression of TGF-β may therefore be able to stabilize the degenerative ECM remodeling observed during aneurysm development. Indeed, in a study by Dai et al., virus-mediated overexpression of TGF-β1 in rat abdominal aortic aneurysms increased endogenous TGF-β1 levels, stabilized aortic dilatation, and attenuated vascular degeneration.[108]
Taken together, these studies suggest that the multiple outcomes of TGF-β signaling, specifically the production of matrix proteins along with the repression or inhibition of matrix degrading enzymes, work in concert to regulate matrix deposition (Table 2).
Table 2.
Outcomes of Classical TGF-β Signaling Affecting Matrix Deposition
| Mediator | Outcome (genes involved) | References |
|---|---|---|
| Effects on Matrix Synthesis: | ||
| Collagen | Enhanced collagen expression, synthesis, and deposition (COL1A1, COL1A2, COL3A1) | [90-93] |
| CTGF | Increased connective tissue growth factor expression (CTGF) | [101] |
| Elastin | Tropoelastin mRNA half-life is stabilized, increasing elastin expression (ELN) | [56,57] |
| Effects on Matrix Degradation: | ||
| PAI-1 | Increased PAI-1 expression; inhibits activation of uPA and tPA, activators of MMPs (PLAU and PLAT, respectively) | [12,103] |
| TIMP | Induction of TIMP-1, -3 expression, direct inhibitors of MMP activity (TIMP-1, TIMP-3) | [58,59] |
| MMPs | Direct repression of MMP expression mediated by TGF-β Inhibitory Elements (TIE) within the MMP promoter (MMP -1, -7, -13); MMP-3 repression by c-fos complexes | [104-106] |
In stark contrast to these results, TGF-β signaling has also been implicated in pathways directly leading to enhanced ECM degradation and the production of various MMP species. In human skin fibroblast cultures, TGF-β treatment induced the production of plasminogen activator.[12] In breast cancer cells, TGF-β treatment rapidly activated p38MAPK, leading to the production and release of MMP-2 and MMP-9.[11] In rat osteoblast cultures, TGF-β was shown to stimulate Mmp-13 expression, which was dependent on Smad2, p38MAPK, and ERK1/2 signaling.[14] Furthermore, Safina and coworkers, revealed that breast cancer cell invasion and tumor angiogenesis were dependent on signaling through the type-I TGF-β receptor, and resulted in MMP-9 production.[13] Interruption of the TGF-β signaling pathway by over-expressing a kinase-deficient type-I receptor attenuated both angiogenesis and MMP-9 expression. Thus, these data suggest that non-Smad TGF-β signaling pathways dependent on TGF-βRI, and often in combination with Smad-mediated signaling, can stimulate the proteolytic destruction of the vascular ECM (Figure 3).
Figure 3. Mechanisms for TGF-β induced matrix degradation.

TGF-β stimulates matrix degradation primarily through non-Smad-mediated pathways that may also involve Smad activation, and results in the increased expression of extracellular protease genes. (ERK1/2, extracellular regulated kinase 1 and 2; MMP, matrix metalloproteinase; PLAU/PLAT, urokinase-type plasminogen activator/tissue-type plasminogen activator).
TGF-β and Vascular Pathology
It is becoming apparent, as the above data illustrate, that TGF-β signaling can regulate the production of critical vascular matrix proteins as well as matrix degrading enzymes, and suggests that perturbations in the TGF-β signaling pathway may be detrimental to normal vascular function and architecture. Accordingly, alterations in normal TGF-β signaling have been implicated in the pathophysiology of several vascular disorders including atherosclerosis,[3,109-112] primary pulmonary hypertension,[113-116] and a host of aortic aneurysm syndromes which placed in context below.
Hereditary Hemorrhagic Telangiectasia
In addition to the type I and type II TGF-β receptors, the type III auxiliary receptors (endoglin, betaglycan; TGF-βRIII) bind TGF-β and function to sequester it in the ECM, thereby regulating ligand availability and its interaction with the type I and II receptors.[117] Recently, TGF-βRIII was shown to directly influence TGF-β-mediated growth inhibition by enhancing both Smad3- and p38MAPK- dependent signaling.[118] Interestingly, mutations in endoglin were directly linked to the development of hereditary hemorrhagic telangiectasia (HHT1, OMIM #187300), a disorder that results in vascular dysplasia and arteriovenous malformations. Similarly, mutations in ACVRL1 (activin A receptor, type II-like 1 or activin receptor-like kinase-1 (ALK-1)) gene, a type-I TGF-β receptor that binds TGF-β/activin and has enhanced expression in highly vascularized tissues and endothelial cells,[119,120] has been linked to the development of HHT2 (OMIM #600376).[119] Moreover, the targeted deletion of Acvrl1 (the Alk-1 gene in mice) has been associated with enhanced production of angiogenic factors and plasminogen activators; both of which serve to stimulate vascular remodeling through the induction and activation of extracellular proteases.[121] Interestingly, in the Acvrl1 deficient mice, dilatation of the yolk-sac vasculature was observed at day E9.5, prior to embryonic lethality at day E11.5.[121] Thus it was postulated that ALK-1 signaling negatively regulates angiogenesis and vascular remodeling. It is interesting to speculate that interruption of ALK-1 mediated signaling may induce angiogenic factors (i.e. specific MMPs) that contribute to the degradation of the vascular ECM and aortic dilatation. Indeed enhanced angiogenesis has been implicated in the development of abdominal aortic aneurysms.[122-124]
Marfan Syndrome
Marfan syndrome (MFS; OMIM #154700) is an inherited connective tissue disorder characterized by cardiovascular, skeletal, and ocular abnormalities, that display an autosomal dominant inheritance pattern with variable penetrance.[125] The primary gene defect lies on chromosome 15q21.1 within the coding sequence for the fibrillin-1 gene (FBN1); a principle component of the 10-12 nm microfibrils that form the scaffold for elastin assembly within the ECM.[125,126] The primary cause of morbidity and mortality in MFS patients relates to the common development of cardiovascular complications including annulo-aortic ectasia and dilatation of the aortic root leading to the formation of ascending aneurysms and dissections.[127,128] These lesions develop secondary to pathological remodeling events that occur within the medial and adventitial vascular ECM consisting of SMC loss, elastin breakdown, and accumulation of cyst-like structures containing mucopolysaccharide.[125]
Fibrillin-1 (and the closely related fibrillin-2) is a 350 kDa glycoprotein comprised of tandem repeats of an epidermal growth factor (EGF)-like motif; most of which contain a calcium binding sequence (cbEGF).[129] Fibrillin monomers self-assemble into macroaggregates that form the basic structure on which mature elastin fibers are synthesized from tropoelastin subunits. The cbEGF modules function to sequester extracellular calcium to protect against ECM proteolysis, mediate interactions between fibrillin monomers and other cellular components such as integrin αvβ3, and stabilize the structure of the microfibrils to favor lateral packing.[130,131] Thus, mutations in fibrillin-1 within the aorta result in weakened and disordered elastic fibers, as well as disruption of the microfibril network connecting the elastic lamellae to the adjacent interstitial cells.[132,133]
In addition to directing elastogenesis and providing structural integrity to the elastic lamellae, fibrillin-rich microfibrils have also been shown to sequester TGF-β within the ECM. TGF-β is synthesized as a prepropolypeptide that is cleaved in a post Golgi compartment to yield the mature growth factor and a latency-associated peptide (LAP). Homodimers of mature TGF-β and LAP form a tight biologically inactive complex termed the small latent complex (SLC).[134] The SLC is covalently bound to a latency-associated TGF-β binding protein (LTBP) through disulfide bonds that form between cysteine residues in the LAP and a cysteine rich motif in LTBP.[131,135,136] This large latent complex (LLC) is secreted from the cell and functions to target TGF-β to the ECM, and specifically to fibrillin microfibrils, which have been shown to directly bind LTBPs.[137,138] TGF-β can be made available for signaling upon release from the LAP through a number of activation mechanisms including: proteolysis of latent complexes, thrombospondin-1 competition with SLC for LTBP binding, integrin binding to embedded RGD peptide sequences within the LTBP (the central peptide sequence that binds αvβ3 integrin), or exposure to acidic/oxidative stress.[135,136,139] Thus, it was suggested that abnormalities in FBN-1 function, such as those observed in MFS, may lead to impaired sequestration of latent TGF-β complexes, thereby making more available for activation, and potentially leading to enhanced TGF-β signaling.[125] This hypothesis was confirmed by two key studies. First, Neptune et al. demonstrated that lung abnormalities, evident in the immediate postnatal period in mice deficient in fibrillin-1 (Fbn1mgΔ/mgΔ), were related to enhanced TGF-β activation and signaling, and that the perinatal administration of a TGF-β neutralizing antibody could rescue the defect in alveolar septation.[140] Second, in mice carrying a hypomorphic allele of the Fbn-1 gene (Fbn1C1039G/+), effectively recapitulating a mouse model of Marfan syndrome, Habashi and coworkers observed that treatment with a TGF-β neutralizing antibody or the angiotensin II type I receptor antagonist Losartan, was sufficient to attenuate spontaneous aortic root dilatation, elastic fiber fragmentation and Smad2 activation.[141] Together, these studies suggest that sequestration of TGF-β in the ECM is critical to its regulated activation, and mutations that functionally impair its sequestration likely contribute to the pathogenesis of MFS, and in particular the pathogenesis of ascending TAA.
Marfan Syndrome Type 2
The diagnosis of MFS has been based on a defined set of clinical criteria.[142-144] Interestingly, approximately 10% of patients classified as having MFS failed to show a defect in the FBN1 gene, suggesting that a second genetic locus is linked to MFS.[145-147] Originally reported in a French family by Bioleau and colleagues,[148] this syndrome was clinically very similar to classic MFS but with no ocular involvement. Subsequent linkage analysis by Collod and coworkers mapped the defect to a locus on chromosome 3p25-24.2 and designated the syndrome Marfan Syndrome Type 2 (MFS2; OMIM #154705).[145] When a Japanese individual with MFS was found to have a chromosome break at 3p24.1, within gene encoding the type II TGF-β receptor (TGFBR2), Mizuguchi et al. hypothesized that the TGFBR2 gene may be linked to the MFS2 locus.[149] Subsequent analysis revealed a mutation (1524G to A) which resulted in a synonymous amino acid substitution (Q508Q), but disrupted an RNA splice site, leading to early termination and truncation of the TGF-βRII protein.[149] Three other mutations in two unrelated families were also identified, all of which fell within the kinase domain of TGF-βRII and led to the loss of functional TGF-β signaling. Thus the MFS phenotype can be caused by mutations in both FBN1 and TGFBR2, and in both cases, the alterations in the TGF-β signaling may contribute to the underlying cause of aneurysm formation. While enhanced TGF-β signaling was associated with dilatation of the ascending aorta in a mouse model of MFS,[141] the loss of TGF-β signaling appeared to be linked to aneurysm formation in MFS2.[149]
Familial Thoracic Aortic Aneurysms and Dissections
In addition to the classified aneurysm syndromes that are directly associated with specific gene defects, such as Marfan syndrome as described above, an expanding collection of studies have identified thoracic aortic aneurysms and dissections that are not clearly associated with an identifiable syndrome. Of these individuals, approximately 20% display a genetic predisposition that was inherited in an autosomal dominant manner with decreased penetrance and variable expression.[150-152] These nonsyndromic cases have collectively been referred to as familial thoracic aortic aneurysms and dissections (TAAD). Currently six causal genetic loci have been identified and linked to TAAD: 11q23.3-q24 (AAT1; OMIM #607086), 5q13-q14 (AAT2; OMIM #607087), 3p24-25 (AAT3; OMIM #608967), 16p13.13-p13.12 (AAT4; OMIM #132900), 9q33-q34 (AAT5; OMIM #610380), and 10q22-q24 (ACTA2; OMIM #102620). Candidate genes mapping to these intervals have revealed several interesting mutations in genes encoding the smooth muscle myosin heavy chain (β) (Myh11; AAT4)[153], α-smooth muscle actin (α2; ACTA2)[154], and in both TGF-β receptors (TGFBR1, AAT5 and TGFBR2, AAT3).[155-157]
In a study by Pannu et al. four unrelated families were found to have mutations in the TGFBR2 gene, but tested negative for any signs of MFS.[155] In all four cases, structural analysis revealed mutations of the arginine residue at position 460; a key residue that was found within the highly conserved serine/threonine kinase domain and was predicted to disrupt receptor function.[158] In similar fashion, Matyas et al. screened 70 individuals that displayed phenotypes related to MFS, but tested negative for mutations in FBN-1. Of these individuals, 9 were identified to have TGFBR1 sequence variants that segregated within family groups and with TAAD disease, suggesting a causative link.[156] Thus taken together, the loss of signaling through TGF-β receptors Ι and ΙΙ may therefore be associated with the development of familial TAAD.
Loeys-Dietz Syndrome
Work by Loeys and coworkers, as well as other laboratories, have recently described another condition that presented with symptoms similar to MFS, but included a greater cardiovascular risk.[159,160] This disorder was designated Loeys-Dietz syndrome (LDS, OMIM #609192), and was characterized by the enhanced development of aortic aneurysms and dissections that occur at a younger age and smaller aortic diameter.[159] Heterozygous mutations were identified in both TGFBR1 and TGFBR2 genes, thus it was initially suggested that the loss of TGF-β signaling may be the underlying cause of LDS. Interestingly, aortic tissues derived from affected patients showed elevated protein levels of collagen and CTGF; both of which are well-described indicators of active TGF-β signaling.[159] Even more compelling, the tissues displayed enhanced nuclear translocation of phospho-Smad2, suggesting that there was an enhancement, not a repression, of TGF-β signaling in these aortic specimens. Upon culturing fibroblasts from affected individuals it was hypothesized that, although the mutant TGF-β receptors were unable to transmit signals, the wild-type receptor (also expressed in a heterozygous individual) retained its acute responsiveness to TGF-β, and in fact displayed elevated activity.[159] Thus, consistent with the observations from the MFS patient studies and the MFS mouse model, the authors concluded that an enhancement of TGF-β signaling drove aortic dilatation and aneurysm formation in patients with LDS.
Ehlers-Danlos Syndrome Type IV
Ehlers-Danlos syndrome type IV (EDS, OMIM # 130050), also known as vascular type EDS, primarily affects the skin and large arteries and can lead to medial degenerative disease of the aorta resulting in acute dissection. The original cause was linked to a 3.3 kb DNA deletion in one allele of the type III procollagen gene (COL3A1), which results in a truncated procollagen monomer that has decreased thermal stability, cannot be proteolytically processed, and cannot be efficiently secreted.[161] The end effect on the large arteries (including the aorta) is a diminished collagen network, with a low intimal-medial thickness, increased wall stress, and a propensity for acute dissection and rupture.[162] Diagnosis of EDS type IV can be difficult since there is significant phenotypic overlap with patients presenting with LDS. Loeys et al., while characterizing 52 LDS affected families for mutations in TGFBR1 and TGFBR2 genes also assessed a cohort of EDS type IV patients that lacked the COL3A1 gene mutations and the craniofacial features of the typical LDS patient.[151] Interestingly, 12 EDS type IV probands were identified that possessed TGFBR1 or TGFBR2 mutations, suggesting a possible re-classification of this group as LDS type 2. Hence, in addition to COL3A1 mutations, this predisposition to acute aortic dissection may also be driven by mutations in TGF-β receptors. Like the LDS patients with TGF-β receptor mutations that display signs of elevated TGF-β signaling,[159] it is interesting to speculate whether EDS type IV patients with COL3A1 mutations, show enhanced TGF-β signaling in an effort to compensate for the loss of type III collagen within the aorta. In either case, within the aorta, the importance of proper regulation of the TGF-β signaling pathway and its downstream transcriptional targets is further emphasized.
Proposed Mechanisms of TGF-β-Mediated Aneurysm Formation
As put forward by the pathophysiological studies above, alterations in TGF-β signaling may therefore be an underlying factor contributing to the development of TAAs. While the majority of studies demonstrated that loss-of-function mutations within the kinase domain of the type I or type II TGF-β receptors were associated with TAA formation, the predominant emerging theory suggests that over stimulation of the TGF-β signaling is associated with enhanced proteolysis of the vascular ECM. Accordingly, based on additional in vitro and in vivo studies, some potential mechanisms have emerged that may help to explain 1) how the loss of TGF-βRII kinase activity can result in enhanced TGF-β signaling, and 2) how stimulation of the TGF-β signaling pathway, generally attributed to being profibrotic, can drive matrix degradation during aneurysm development.
Enhanced Signaling by Mutant Receptors
In the study by Loeys et al., LDS was associated with deficient TGF-βRII receptor kinase function, yet paradoxically displayed evidence of enhanced TGF-β signaling; including enhanced expression of PAI-1, collagen, and CTGF, as well as nuclear localization of phospho-Smad2.[159] Thus, other aneurysm syndromes associated with TGFBR2 mutations may also be a result of over stimulation of the TGF-β pathway. While difficult to reconcile how kinase-deficient TGF-β receptors could result in enhanced TGF-β signal transmission, there are several potential mechanisms that could account for these observations.
First, Denton and coworkers developed a transgenic mouse that expressed a fibroblast-restricted kinase-deficient TGF-βRII under control of the COL1A2 promoter.[163] Based on in vitro results characterizing the over expression of the mutant receptor, the authors had predicted that the transgene would have a dominant-negative effect in mice resulting in the fibroblast-specific suppression of TGF-β signaling. Surprisingly they observed the opposite; the mice developed dermal and pulmonary fibrosis, and isolated transgenic fibroblasts displayed hallmarks of enhanced TGF-β signaling. To explain these paradoxical results, the authors suggested that the dominant-negative TGF-β receptor may enhance TGF-β signaling in functional TGF-β receptor complexes by facilitating ligand interactions with the functional receptors, similar to type III TGF-β receptors endoglin and betaglycan.[164] They also suggested that the presence of the mutant TGF-βRII may modify the orientation of wild-type receptors in a manner that would more easily facilitate signaling. Thus, the enhanced TGF-β signaling observed in aneurysm syndromes associated with heterozygous TGF-β receptor mutations may be a result of mutant receptors functioning as accessory receptors and facilitating ligand binding.
Secondly, kinase-deficient receptors may enhance TGF-β signaling by altering the cellular dynamics of receptor stability. It is well known that endocytosis of cell surface receptors is an important regulatory step in the transduction of intracellular signals. De Guglielmo and colleagues described internalization of TGF-β receptors by both clathrin-mediated and caveolin-mediated endocytic pathways.[165] Interestingly, segregation into the clathrin-mediated pathway sustained receptor recycling and propagated receptor signaling, whereas segregation into the caveolin-mediated pathway led to receptor degradation and termination of TGF-β signaling. This process is regulated by receptor interactions with accessory proteins that direct the fate of TGF-β signaling. For example, Di Guglielmo et al. showed that the Smad anchor for receptor activation (SARA) interacted with TGF-βRII at the plasma membrane, facilitating segregation into the clathrin-dependent endocytic pathway, as well as facilitating the interaction of the receptor complex with Smad2.[165] This resulted in signal propagation and eventual receptor recycling to the plasma membrane. Alternatively, it was shown that Smad7 interacted with the heteromeric TGF-β receptor complex (requiring both receptors for efficient interaction), facilitating segregation into the caveolin-mediated endocytic pathway, and recruiting the E3 ubiquitin ligases Smurf1 and Smurf2.[165,166] This resulted in the ubiquitin-mediated proteasomal degradation of TGF-β receptors, and thus the attenuation of TGF-β signaling. Hence, it is possible that the TGFBR2 mutations observed in the above aneurysm syndromes, may facilitate interactions with SARA or diminish association with Smad7, preventing segregation into the caveolin-mediated pathway, subsequently resulting in the prolonged activation of TGF-β signal transmission (Figure 4).
Figure 4. Potential mechanism for enhancement TGF-β signaling in the presence of receptor mutations.
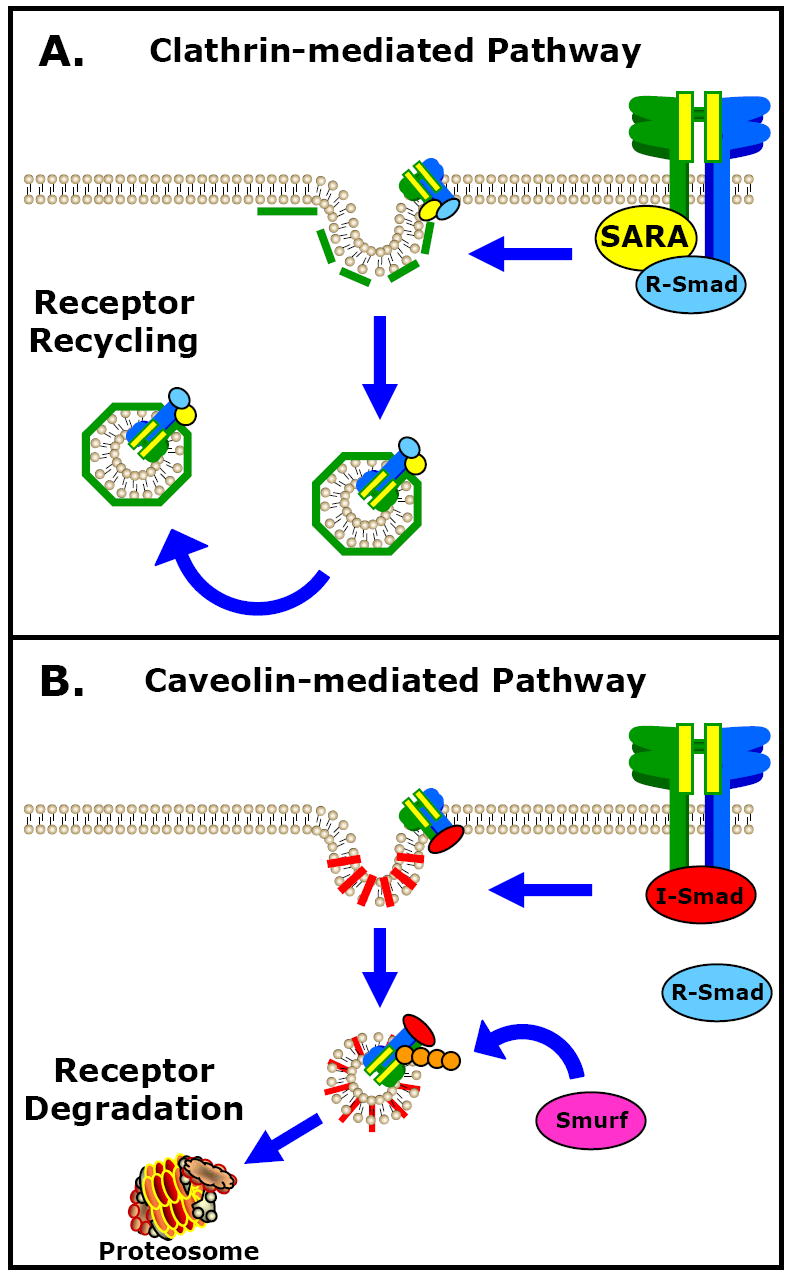
The endocytosis and subsequent recycling or degradation of TGF-β receptors is an important mechanism for regulating TGF-β signaling. There are two independent endocytic pathways, regulated by accessory protein interactions that exist to regulate TGF-β signaling: A.) the clathrin-mediated pathway leading to receptor recycling, and B.) the caveolin-mediated pathway leading to proteasomal degradation of TGF-β receptors. The TGF-βRII mutations identified in several aneurysm syndromes may contribute to the enhanced TGF-β signaling by facilitating interactions with SARA, or by diminishing association with Smad7, favoring receptor recycling and resulting in prolonged activation of TGF-β signals.
Lastly, it is also possible that TGF-β receptor complexes that incorporate kinase-deficient receptors, while unable to signal directly, retain the ability to form functional signaling platforms and mediate interactions between other critical intracellular signaling components (Figure 5). For example, Runyan and coworkers showed that stimulation of human mesangial cells with TGF-β resulted in the induction collagen gene expression (COL1A2) that was dependent upon phosphoinositide 3-kinase (PI3K) activity.[88] Using a pharmacological inhibitor of PI3K (LY294002) in combination with phosphorylation site mutants of Smad3, they demonstrated that PI3K catalyzed collagen gene expression by directly phosphorylating Smad3 at residues other than those within the TGF-βRI target site. Thus, the binding of TGF-β induced a receptor signaling complex which included at least PI3K and Smad3, and led to Smad3-mediated collagen gene expression that was not dependent on phosphorylation by TGF-βRI. Therefore, TGF-βRII mutants that retain the ability to bind TGF-β may induce signaling through alternate pathways that are independent of receptor kinase activity.
Figure 5. Potential mechanism for enhancement of TGF-β signaling in the presence of non-functional receptors.

Ligand-induced receptor signaling complexes that are unable to transmit signals because of mutations within the receptor kinase domain (TGF-βRII/TGF-βRI/both), may still function by seeding active signaling platforms that localize other signaling components capable of inducing downstream signaling events that contribute to aneurysm development.
TGF-β-Induced Matrix Degradation
As detailed above, elevated TGF-β levels and/or evidence of functional TGF-β signaling have been associated thoracic aortic aneurysm development. Furthermore, it is becoming clear that TGF-β not only functions to induce matrix deposition, but also has been implicated in regulating matrix degradation, in part due to its ability to induce matrix degrading enzymes such as the MMPs (Figure 3). In fact, the majority of studies demonstrating TGF-β-dependent protease production implicate activated TGF-βRI and MAP kinases ERK1/2 and p38.[11-14] Hence, TGF-β-induced matrix degradation may be driven by non-Smad-mediated pathways, either alone or in combination with Smad-mediated signaling.[45,167,168] Whereas hypotheses explaining elevated TGF-β tissues levels have been proposed for some aneurysm syndromes,[125] the mechanisms determining the overall (or temporal) cellular response to TGF-β (matrix deposition vs. matrix degradation) remain unclear. Accordingly, while all cell types within aortic wall are known to respond to TGF-β, each cell type may respond differently. Thus the combined aortic response will ultimately be determined by the representative amounts of the constituitive cells present during aneurysm development.
There are three major endogenous cell types found within the aortic wall: 1) endothelial cells, found populating the intimal layer lining the lumen of the aorta; 2) SMCs, the predominant cell type found in the medial elastic layer; and 3) fibroblasts, found primarily in the adventitial layer. Importantly, it is well known that the cellular constituents within the aortic wall change during the course of aneurysm development. It has been well documented that aortic dilatation is accompanied by SMC apoptosis. [169-172] In fact, Fukui and colleagues demonstrated in human abdominal aortic aneurysm specimens that high levels of TGF-β expression colocalized with SMC markers that stained positive for fragmented DNA (TUNEL assay).[173] This suggested that SMC apoptosis during aneurysm formation might be mediated by TGF-β signaling pathways. Accordingly, as the aneurysm progresses the SMC content within the aortic wall decreases, leaving fibroblasts as the predominant cell type to manage the vascular remodeling process. Thus, the degradative changes occurring within the aortic wall may be a result of the differential responses to TGF-β mediated by the changing proportions of cellular constituents.
In addition to changes in the cellular make-up of the aortic wall during aneurysm formation, the adventitial fibroblasts themselves may also undergo phenotypic changes in response to vascular injury/stress.[174] This transition from fibroblast to myofibroblast confers SMC-like characteristics including increased expression of SMC markers (e.g. α-smooth muscle actin) and contractile properties. Interestingly, TGF-β may provide the signal to induce this fibroblast differentiation.[175] Shi and coworkers demonstrated a direct correlation between autocrine TGF-β production and the expression of α-smooth muscle actin in adventitial fibroblasts.[176] Likewise, Vaughan et al. demonstrated that TGF-β promoted both morphological and functional differentiation of myofibroblasts, and demonstrated that the differentiated phenotype was dependent on the presence of TGF-β; when TGF-β was removed, the morphological markers of myofibroblast differentiation resolved.[177] Mechanistically, studies in lung and coronary fibroblasts demonstrated that TGF-β induced myofibroblast differentiation was dependent on enhanced signaling through the classical TGF-β signaling pathway.[178,179] Pharmacologic inhibitors of ERK1/2, p38MAPK, and TGF-βRI were used to differentiate the Smad-dependent responses from the alternative signaling pathways. This was confirmed by a study in which the overexpression of a mutant TGF-βRI (lacking critical amino acid residues to interact with the Smads) in mammary epithelial cells, failed to induce myofibroblast differentiation in response to TGF-β, but maintained an ability to activate p38MAPK.[78] Interestingly, Shi and coworkers demonstrated that adventitial fibroblasts from coronary arteries displayed enhanced migratory properties characterized by increased MMP-2 and MMP-9 production.[180,181] Moreover, they demonstrated a differential response from medial SMCs, in which medial explants were characterized by diminished migration and enhanced TIMP production.
Taken together, it is interesting to speculate that Smad-mediated TGF-β signaling may drive SMC apoptosis and catalyze the differentiation of fibroblasts into myofibroblasts during aneurysm development. It is then conceivable that upon acquisition of the new functional properties, the myofibroblasts may respond differently to TGF-β signals, promoting non-Smad-mediated pathways that enhance matrix degrading enzyme production and cell migration (Figure 6). While myofibroblast transition has not been characterized in the aortic vascular wall during TAA formation, myofibroblasts have been identified in association with inflammatory aneurysms; a relatively rare subset of abdominal aortic aneurysms.[182] Accordingly, additional studies are needed to address the role of myofibroblast differentiation during TAA development, as well as the role of non-Smad mediated signaling in myofibroblast function.
Figure 6. Potential mechanism of TGF-β-induced changes in the cellular constituents of the vascular wall during aneurysm development.

A. Non-aneurysmal thoracic aorta showing normal homeostatic content of medial smooth muscle cells (SMCs) and adventitial fibroblasts. B. Thoracic aorta during aneurysm formation. TGF-β-mediated SMC apoptosis leaves the adventitial fibroblast as the predominate endogenous cell type in the vascular wall. TGF-β can induce adventitial fibroblasts to differentiate into myofibroblasts, conferring SMC-like characteristics including α-smooth muscle actin expression, enhanced contractile properties, and the ability to migrate. Migrating myofibroblasts respond to TGF-β stimulation by inducing MMP production and secretion. Thus, the resulting myofibroblast population may dominate the vascular remodeling process leading to enhanced matrix degradation.
Significance
Thoracic aortic aneurysm disease is a potentially devastating disease process which often causes death by rupture in the absence of symptoms. There are currently no effective non-surgical clinical treatment protocols available which will halt or reverse the aortic remodeling process once diagnosed. While it is clear that the pathological alterations in the structure and composition of the vascular ECM are associated with reduced aortic compliance and resiliency, and lead to aortic dysfunction, there remains a paucity of information regarding the regulation of specific upstream signaling intermediates and pathways involved in the remodeling process during aneurysm development. As highlighted here, mutations in key TGF-β signaling pathway components are invariably associated with vascular pathology, and have been directly implicated in the development of ascending TAAs. While the majority of aneurysm syndromes are associated with inactivating mutations in the kinase domain of TGF-β receptors (primarily TGF-βRII), direct evidence has implicated enhanced TGF-β signaling during TAA formation. As each cell-type within the aortic wall is capable of responding differently to TGF-β, it is conceivable that the homeostatic balance between matrix deposition and degradation are maintained through differential cellular responses. This may be driven by TGF-β-mediated effects on the cellular composition of the aortic wall. For example, TGF-β may induce apoptosis in the SMC population, while at the same time inducing adventitial fibroblasts to transform into myofibroblasts. Thus, newly acquired cellular functions (including the production of MMPs) may work in concert with alterations in the relative number of constituitive cells, to change the overall tissue response to TGF-β, shifting the balance toward matrix degradation and aneurysm development.
These studies demonstrating enhanced signaling in the presence of mutant receptors not only speak to the complexities of TGF-β signaling, but also underlie the importance of continued study of TGF-β in TAA development. Thus, it is likely that by enhancing our understanding of the complex events that mediate malignant vascular remodeling and aortic dilatation, we may also uncover interventional strategies to interrupt aneurysm formation.
Acknowledgments
This work was supported by NIH/NHLBI R01 HL075488-04
References
- 1.Liapis CD, Paraskevas KI. The pivotal role of matrix metalloproteinases in the development of human abdominal aortic aneurysms. Vasc Med. 2003;8:267–271. doi: 10.1191/1358863x03vm504ra. [DOI] [PubMed] [Google Scholar]
- 2.Thompson RW, Geraghty PJ, Lee JK. Abdominal aortic aneurysms: basic mechanisms and clinical implications. Curr Probl Surg. 2002;39:110–230. doi: 10.1067/msg.2002.121421. [DOI] [PubMed] [Google Scholar]
- 3.Isselbacher EM. Thoracic and abdominal aortic aneurysms. Circulation. 2005;111:816–828. doi: 10.1161/01.CIR.0000154569.08857.7A. [DOI] [PubMed] [Google Scholar]
- 4.Thompson RW. Reflections on the pathogenesis of abdominal aortic aneurysms. Cardiovasc Surg. 2002;10:389–394. doi: 10.1016/s0967-2109(02)00042-x. [DOI] [PubMed] [Google Scholar]
- 5.Tamarina NA, McMillan WD, Shively VP, Pearce WH. Expression of matrix metalloproteinases and their inhibitors in aneurysms and normal aorta. Surgery. 1997;122:264–271. doi: 10.1016/s0039-6060(97)90017-9. discussion 271-262. [DOI] [PubMed] [Google Scholar]
- 6.Bobik A. Transforming growth factor-betas and vascular disorders. Arterioscler Thromb Vasc Biol. 2006;26:1712–1720. doi: 10.1161/01.ATV.0000225287.20034.2c. [DOI] [PubMed] [Google Scholar]
- 7.Roberts AB, Sporn MB, Assoian RK, Smith JM, Roche NS, Wakefield LM, Heine UI, Liotta LA, Falanga V, Kehrl JH, et al. Transforming growth factor type beta: rapid induction of fibrosis and angiogenesis in vivo and stimulation of collagen formation in vitro. Proc Natl Acad Sci U S A. 1986;83:4167–4171. doi: 10.1073/pnas.83.12.4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dennler S, Goumans MJ, ten Dijke P. Transforming growth factor beta signal transduction. J Leukoc Biol. 2002;71:731–740. [PubMed] [Google Scholar]
- 9.Massague J. TGFbeta signaling: receptors, transducers, and Mad proteins. Cell. 1996;85:947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- 10.Massague J. How cells read TGF-beta signals. Nat Rev Mol Cell Biol. 2000;1:169–178. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 11.Kim ES, Kim MS, Moon A. TGF-beta-induced upregulation of MMP-2 and MMP-9 depends on p38 MAPK, but not ERK signaling in MCF10A human breast epithelial cells. Int J Oncol. 2004;25:1375–1382. [PubMed] [Google Scholar]
- 12.Laiho M, Saksela O, Keski-Oja J. Transforming growth factor beta alters plasminogen activator activity in human skin fibroblasts. Exp Cell Res. 1986;164:399–407. doi: 10.1016/0014-4827(86)90038-8. [DOI] [PubMed] [Google Scholar]
- 13.Safina A, Vandette E, Bakin AV. ALK5 promotes tumor angiogenesis by upregulating matrix metalloproteinase-9 in tumor cells. Oncogene. 2007;26:2407–2422. doi: 10.1038/sj.onc.1210046. [DOI] [PubMed] [Google Scholar]
- 14.Selvamurugan N, Kwok S, Alliston T, Reiss M, Partridge NC. Transforming growth factor-beta 1 regulation of collagenase-3 expression in osteoblastic cells by cross-talk between the Smad and MAPK signaling pathways and their components, Smad2 and Runx2. J Biol Chem. 2004;279:19327–19334. doi: 10.1074/jbc.M314048200. [DOI] [PubMed] [Google Scholar]
- 15.Santilli JD, Santilli SM. Diagnosis and treatment of abdominal aortic aneurysms. Am Fam Physician. 1997;56:1081–1090. [PubMed] [Google Scholar]
- 16.Bickerstaff LK, Pairolero PC, Hollier LH, Melton LJ, Van Peenen HJ, Cherry KJ, Joyce JW, Lie JT. Thoracic aortic aneurysms: a population-based study. Surgery. 1982;92:1103–1108. [PubMed] [Google Scholar]
- 17.Coady MA, Rizzo JA, Goldstein LJ, Elefteriades JA. Natural history, pathogenesis, and etiology of thoracic aortic aneurysms and dissections. Cardiol Clin. 1999;17:615–635. vii. doi: 10.1016/s0733-8651(05)70105-3. [DOI] [PubMed] [Google Scholar]
- 18.Elefteriades JA, Tranquilli M, Darr U, Cardon J, Zhu BQ, Barrett P. Symptoms plus family history trump size in thoracic aortic aneurysm. Ann Thorac Surg. 2005;80:1098–1100. doi: 10.1016/j.athoracsur.2004.02.130. [DOI] [PubMed] [Google Scholar]
- 19.Guo D, Hasham S, Kuang SQ, Vaughan CJ, Boerwinkle E, Chen H, Abuelo D, Dietz HC, Basson CT, Shete SS, Milewicz DM. Familial thoracic aortic aneurysms and dissections: genetic heterogeneity with a major locus mapping to 5q13-14. Circulation. 2001;103:2461–2468. doi: 10.1161/01.cir.103.20.2461. [DOI] [PubMed] [Google Scholar]
- 20.Davies RR, Goldstein LJ, Coady MA, Tittle SL, Rizzo JA, Kopf GS, Elefteriades JA. Yearly rupture or dissection rates for thoracic aortic aneurysms: simple prediction based on size. Ann Thorac Surg. 2002;73:17–27. doi: 10.1016/s0003-4975(01)03236-2. discussion 27-18. [DOI] [PubMed] [Google Scholar]
- 21.Chiesa R, Melissano G, Civilini E, de Moura ML, Carozzo A, Zangrillo A. Ten years experience of thoracic and thoracoabdominal aortic aneurysm surgical repair: lessons learned. Ann Vasc Surg. 2004;18:514–520. doi: 10.1007/s10016-004-0072-z. [DOI] [PubMed] [Google Scholar]
- 22.Fleck TM, Koinig H, Czerny M, Hutschala D, Wolner E, Ehrlich M, Grabenwoger M. Impact of surgical era on outcomes of patients undergoing elective atherosclerotic ascending aortic aneurysm operations. Eur J Cardiothorac Surg. 2004;26:342–347. doi: 10.1016/j.ejcts.2004.04.025. [DOI] [PubMed] [Google Scholar]
- 23.Ghansah JN, Murphy JT. Complications of major aortic and lower extremity vascular surgery. Semin Cardiothorac Vasc Anesth. 2004;8:335–361. doi: 10.1177/108925320400800406. [DOI] [PubMed] [Google Scholar]
- 24.Kawaharada N, Morishita K, Fukada J, Hachiro Y, Fujisawa Y, Saito T, Kurimoto Y, Abe T. Stroke in surgery of the arteriosclerotic descending thoracic aortic aneurysms: influence of cross-clamping technique of the aorta. Eur J Cardiothorac Surg. 2005;27:622–625. doi: 10.1016/j.ejcts.2004.11.028. [DOI] [PubMed] [Google Scholar]
- 25.Alexander JJ. The pathobiology of aortic aneurysms. J Surg Res. 2004;117:163–175. doi: 10.1016/j.jss.2003.11.011. [DOI] [PubMed] [Google Scholar]
- 26.Elefteriades JA. Natural history of thoracic aortic aneurysms: indications for surgery, and surgical versus nonsurgical risks. Ann Thorac Surg. 2002;74:S1877–1880. doi: 10.1016/s0003-4975(02)04147-4. discussion S1892-1878. [DOI] [PubMed] [Google Scholar]
- 27.Davies RR, Gallo A, Coady MA, Tellides G, Botta DM, Burke B, Coe MP, Kopf GS, Elefteriades JA. Novel measurement of relative aortic size predicts rupture of thoracic aortic aneurysms. Ann Thorac Surg. 2006;81:169–177. doi: 10.1016/j.athoracsur.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 28.Dobrin PB, Mrkvicka R. Failure of elastin or collagen as possible critical connective tissue alterations underlying aneurysmal dilatation. Cardiovasc Surg. 1994;2:484–488. [PubMed] [Google Scholar]
- 29.Allaire E, Hasenstab D, Kenagy RD, Starcher B, Clowes MM, Clowes AW. Prevention of aneurysm development and rupture by local overexpression of plasminogen activator inhibitor-1. Circulation. 1998;98:249–255. doi: 10.1161/01.cir.98.3.249. [DOI] [PubMed] [Google Scholar]
- 30.Boyle JR, McDermott E, Crowther M, Wills AD, Bell PR, Thompson MM. Doxycycline inhibits elastin degradation and reduces metalloproteinase activity in a model of aneurysmal disease. J Vasc Surg. 1998;27:354–361. doi: 10.1016/s0741-5214(98)70367-2. [DOI] [PubMed] [Google Scholar]
- 31.Carrell TW, Smith A, Burnand KG. Experimental techniques and models in the study of the development and treatment of abdominal aortic aneurysm. Br J Surg. 1999;86:305–312. doi: 10.1046/j.1365-2168.1999.01092.x. [DOI] [PubMed] [Google Scholar]
- 32.Chiou AC, Chiu B, Pearce WH. Murine aortic aneurysm produced by periarterial application of calcium chloride. J Surg Res. 2001;99:371–376. doi: 10.1006/jsre.2001.6207. [DOI] [PubMed] [Google Scholar]
- 33.Freestone T, Turner RJ, Higman DJ, Lever MJ, Powell JT. Influence of hypercholesterolemia and adventitial inflammation on the development of aortic aneurysm in rabbits. Arterioscler Thromb Vasc Biol. 1997;17:10–17. doi: 10.1161/01.atv.17.1.10. [DOI] [PubMed] [Google Scholar]
- 34.Gertz SD, Kurgan A, Eisenberg D. Aneurysm of the rabbit common carotid artery induced by periarterial application of calcium chloride in vivo. J Clin Invest. 1988;81:649–656. doi: 10.1172/JCI113368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Longo GM, Xiong W, Greiner TC, Zhao Y, Fiotti N, Baxter BT. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J Clin Invest. 2002;110:625–632. doi: 10.1172/JCI15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Pyo R, Lee JK, Shipley JM, Curci JA, Mao D, Ziporin SJ, Ennis TL, Shapiro SD, Senior RM, Thompson RW. Targeted gene disruption of matrix metalloproteinase-9 (gelatinase B) suppresses development of experimental abdominal aortic aneurysms. J Clin Invest. 2000;105:1641–1649. doi: 10.1172/JCI8931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barbour JR, Spinale FG, Ikonomidis JS. Proteinase systems and thoracic aortic aneurysm progression. J Surg Res. 2007;139:292–307. doi: 10.1016/j.jss.2006.09.020. [DOI] [PubMed] [Google Scholar]
- 38.Ikonomidis JS, Barbour JR, Amani Z, Stroud RE, Herron AR, McClister DM, Jr, Camens SE, Lindsey ML, Mukherjee R, Spinale FG. Effects of deletion of the matrix metalloproteinase 9 gene on development of murine thoracic aortic aneurysms. Circulation. 2005;112:I242–248. doi: 10.1161/CIRCULATIONAHA.104.526152. [DOI] [PubMed] [Google Scholar]
- 39.Ikonomidis JS, Gibson WC, Butler JE, McClister DM, Sweterlitsch SE, Thompson RP, Mukherjee R, Spinale FG. Effects of deletion of the tissue inhibitor of matrix metalloproteinases-1 gene on the progression of murine thoracic aortic aneurysms. Circulation. 2004;110:II268–273. doi: 10.1161/01.CIR.0000138384.68947.20. [DOI] [PubMed] [Google Scholar]
- 40.Jones JA, Barbour JR, Lowry AS, Bouges S, Beck C, McClister DM, Jr, Mukherjee R, Ikonomidis JS. Spatiotemporal expression and localization of matrix metalloproteinase-9 in a murine model of thoracic aortic aneurysm. J Vasc Surg. 2006;44:1314–1321. doi: 10.1016/j.jvs.2006.07.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Longo GM, Buda SJ, Fiotta N, Xiong W, Griener T, Shapiro S, Baxter BT. MMP-12 has a role in abdominal aortic aneurysms in mice. Surgery. 2005;137:457–462. doi: 10.1016/j.surg.2004.12.004. [DOI] [PubMed] [Google Scholar]
- 42.Brophy CM, Marks WH, Reilly JM, Tilson MD. Decreased tissue inhibitor of metalloproteinases (TIMP) in abdominal aortic aneurysm tissue: a preliminary report. J Surg Res. 1991;50:653–657. doi: 10.1016/0022-4804(91)90058-t. [DOI] [PubMed] [Google Scholar]
- 43.Koullias GJ, Korkolis DP, Ravichandran P, Psyrri A, Hatzaras I, Elefteriades JA. Tissue microarray detection of matrix metalloproteinases, in diseased tricuspid and bicuspid aortic valves with or without pathology of the ascending aorta. Eur J Cardiothorac Surg. 2004;26:1098–1103. doi: 10.1016/j.ejcts.2004.07.050. [DOI] [PubMed] [Google Scholar]
- 44.Bertolino P, Deckers M, Lebrin F, ten Dijke P. Transforming growth factor-beta signal transduction in angiogenesis and vascular disorders. Chest. 2005;128:585S–590S. doi: 10.1378/chest.128.6_suppl.585S. [DOI] [PubMed] [Google Scholar]
- 45.Feng XH, Derynck R. Specificity and Versatility in TGF-beta signaling through Smads. Annu Rev Cell Dev Biol. 2005;21:659–693. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 46.Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–644. [PubMed] [Google Scholar]
- 47.Moustakas A, Heldin CH. Non-Smad TGF-beta signals. J Cell Sci. 2005;118:3573–3584. doi: 10.1242/jcs.02554. [DOI] [PubMed] [Google Scholar]
- 48.Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. J Cell Sci. 2001;114:4359–4369. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- 49.Bartram U, Speer CP. The role of transforming growth factor beta in lung development and disease. Chest. 2004;125:754–765. doi: 10.1378/chest.125.2.754. [DOI] [PubMed] [Google Scholar]
- 50.Bataller R, Brenner DA. Liver fibrosis. J Clin Invest. 2005;115:209–218. doi: 10.1172/JCI24282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor-beta(1) Mol Genet Metab. 2000;71:418–435. doi: 10.1006/mgme.2000.3032. [DOI] [PubMed] [Google Scholar]
- 52.Wrana JL, Attisano L, Carcamo J, Zentella A, Doody J, Laiho M, Wang XF, Massague J. TGF beta signals through a heteromeric protein kinase receptor complex. Cell. 1992;71:1003–1014. doi: 10.1016/0092-8674(92)90395-s. [DOI] [PubMed] [Google Scholar]
- 53.Wrana JL, Attisano L, Wieser R, Ventura F, Massague J. Mechanism of activation of the TGF-beta receptor. Nature. 1994;370:341–347. doi: 10.1038/370341a0. [DOI] [PubMed] [Google Scholar]
- 54.Leivonen SK, Chantry A, Hakkinen L, Han J, Kahari VM. Smad3 mediates transforming growth factor-beta-induced collagenase-3 (matrix metalloproteinase-13) expression in human gingival fibroblasts. Evidence for cross-talk between Smad3 and p38 signaling pathways. J Biol Chem. 2002;277:46338–46346. doi: 10.1074/jbc.M206535200. [DOI] [PubMed] [Google Scholar]
- 55.Ottaviano AJ, Sun L, Ananthanarayanan V, Munshi HG. Extracellular matrix-mediated membrane-type 1 matrix metalloproteinase expression in pancreatic ductal cells is regulated by transforming growth factor-beta1. Cancer Res. 2006;66:7032–7040. doi: 10.1158/0008-5472.CAN-05-4421. [DOI] [PubMed] [Google Scholar]
- 56.Kahari VM, Olsen DR, Rhudy RW, Carrillo P, Chen YQ, Uitto J. Transforming growth factor-beta up-regulates elastin gene expression in human skin fibroblasts. Evidence for post-transcriptional modulation. Lab Invest. 1992;66:580–588. [PubMed] [Google Scholar]
- 57.Kucich U, Rosenbloom JC, Abrams WR, Rosenbloom J. Transforming growth factor-beta stabilizes elastin mRNA by a pathway requiring active Smads, protein kinase C-delta, and p38. Am J Respir Cell Mol Biol. 2002;26:183–188. doi: 10.1165/ajrcmb.26.2.4666. [DOI] [PubMed] [Google Scholar]
- 58.Kwak HJ, Park MJ, Cho H, Park CM, Moon SI, Lee HC, Park IC, Kim MS, Rhee CH, Hong SI. Transforming growth factor-beta1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol Cancer Res. 2006;4:209–220. doi: 10.1158/1541-7786.MCR-05-0140. [DOI] [PubMed] [Google Scholar]
- 59.Garcia-Alvarez J, Ramirez R, Checa M, Nuttall RK, Sampieri CL, Edwards DR, Selman M, Pardo A. Tissue inhibitor of metalloproteinase-3 is up-regulated by transforming growth factor-beta1 in vitro and expressed in fibroblastic foci in vivo in idiopathic pulmonary fibrosis. Exp Lung Res. 2006;32:201–214. doi: 10.1080/01902140600817481. [DOI] [PubMed] [Google Scholar]
- 60.de Larco JE, Todaro GJ. Growth factors from murine sarcoma virus-transformed cells. Proc Natl Acad Sci U S A. 1978;75:4001–4005. doi: 10.1073/pnas.75.8.4001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roberts AB, Frolik CA, Anzano MA, Sporn MB. Transforming growth factors from neoplastic and nonneoplastic tissues. Fed Proc. 1983;42:2621–2626. [PubMed] [Google Scholar]
- 62.Roberts AB, Anzano MA, Wakefield LM, Roche NS, Stern DF, Sporn MB. Type beta transforming growth factor: a bifunctional regulator of cellular growth. Proc Natl Acad Sci U S A. 1985;82:119–123. doi: 10.1073/pnas.82.1.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Suszko MI, Woodruff TK. Cell-specificity of transforming growth factor-beta response is dictated by receptor bioavailability. J Mol Endocrinol. 2006;36:591–600. doi: 10.1677/jme.1.01936. [DOI] [PubMed] [Google Scholar]
- 64.Lopez-Casillas F, Payne HM, Andres JL, Massague J. Betaglycan can act as a dual modulator of TGF-beta access to signaling receptors: mapping of ligand binding and GAG attachment sites. J Cell Biol. 1994;124:557–568. doi: 10.1083/jcb.124.4.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang XF, Lin HY, Ng-Eaton E, Downward J, Lodish HF, Weinberg RA. Expression cloning and characterization of the TGF-beta type III receptor. Cell. 1991;67:797–805. doi: 10.1016/0092-8674(91)90074-9. [DOI] [PubMed] [Google Scholar]
- 66.Xu L. Regulation of Smad activities. Biochim Biophys Acta. 2006;1759:503–513. doi: 10.1016/j.bbaexp.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wicks SJ, Grocott T, Haros K, Maillard M, ten Dijke P, Chantry A. Reversible ubiquitination regulates the Smad/TGF-beta signalling pathway. Biochem Soc Trans. 2006;34:761–763. doi: 10.1042/BST0340761. [DOI] [PubMed] [Google Scholar]
- 68.Coucke PJ, Willaert A, Wessels MW, Callewaert B, Zoppi N, De Backer J, Fox JE, Mancini GM, Kambouris M, Gardella R, Facchetti F, Willems PJ, Forsyth R, Dietz HC, Barlati S, Colombi M, Loeys B, De Paepe A. Mutations in the facilitative glucose transporter GLUT10 alter angiogenesis and cause arterial tortuosity syndrome. Nat Genet. 2006;38:452–457. doi: 10.1038/ng1764. [DOI] [PubMed] [Google Scholar]
- 69.Park SH. Fine tuning and cross-talking of TGF-beta signal by inhibitory Smads. J Biochem Mol Biol. 2005;38:9–16. doi: 10.5483/bmbrep.2005.38.1.009. [DOI] [PubMed] [Google Scholar]
- 70.Imamura T, Takase M, Nishihara A, Oeda E, Hanai J, Kawabata M, Miyazono K. Smad6 inhibits signalling by the TGF-beta superfamily. Nature. 1997;389:622–626. doi: 10.1038/39355. [DOI] [PubMed] [Google Scholar]
- 71.Hata A, Lagna G, Massague J, Hemmati-Brivanlou A. Smad6 inhibits BMP/Smad1 signaling by specifically competing with the Smad4 tumor suppressor. Genes Dev. 1998;12:186–197. doi: 10.1101/gad.12.2.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477–12480. doi: 10.1074/jbc.C100008200. [DOI] [PubMed] [Google Scholar]
- 73.Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365–1375. doi: 10.1016/s1097-2765(00)00134-9. [DOI] [PubMed] [Google Scholar]
- 74.Moren A, Hellman U, Inada Y, Imamura T, Heldin CH, Moustakas A. Differential ubiquitination defines the functional status of the tumor suppressor Smad4. J Biol Chem. 2003;278:33571–33582. doi: 10.1074/jbc.M300159200. [DOI] [PubMed] [Google Scholar]
- 75.Moren A, Imamura T, Miyazono K, Heldin CH, Moustakas A. Degradation of the tumor suppressor Smad4 by WW and HECT domain ubiquitin ligases. J Biol Chem. 2005;280:22115–22123. doi: 10.1074/jbc.M414027200. [DOI] [PubMed] [Google Scholar]
- 76.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–584. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 77.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 78.Yu L, Hebert MC, Zhang YE. TGF-beta receptor-activated p38 MAP kinase mediates Smad-independent TGF-beta responses. Embo J. 2002;21:3749–3759. doi: 10.1093/emboj/cdf366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Choy L, Derynck R. The type II transforming growth factor (TGF)-beta receptor-interacting protein TRIP-1 acts as a modulator of the TGF-beta response. J Biol Chem. 1998;273:31455–31462. doi: 10.1074/jbc.273.47.31455. [DOI] [PubMed] [Google Scholar]
- 80.Ozdamar B, Bose R, Barrios-Rodiles M, Wang HR, Zhang Y, Wrana JL. Regulation of the polarity protein Par6 by TGFbeta receptors controls epithelial cell plasticity. Science. 2005;307:1603–1609. doi: 10.1126/science.1105718. [DOI] [PubMed] [Google Scholar]
- 81.Dumont N, Bakin AV, Arteaga CL. Autocrine transforming growth factor-beta signaling mediates Smad-independent motility in human cancer cells. J Biol Chem. 2003;278:3275–3285. doi: 10.1074/jbc.M204623200. [DOI] [PubMed] [Google Scholar]
- 82.Griswold-Prenner I, Kamibayashi C, Maruoka EM, Mumby MC, Derynck R. Physical and functional interactions between type I transforming growth factor beta receptors and Balpha, a WD-40 repeat subunit of phosphatase 2A. Mol Cell Biol. 1998;518:6595–6604. doi: 10.1128/mcb.18.11.6595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Itoh S, Thorikay M, Kowanetz M, Moustakas A, Itoh F, Heldin CH, ten Dijke P. Elucidation of Smad requirement in transforming growth factor-beta type I receptor-induced responses. J Biol Chem. 2003;278:3751–3761. doi: 10.1074/jbc.M208258200. [DOI] [PubMed] [Google Scholar]
- 84.Wilkes MC, Murphy SJ, Garamszegi N, Leof EB. Cell-type-specific activation of PAK2 by transforming growth factor beta independent of Smad2 and Smad3. Mol Cell Biol. 2003;23:8878–8889. doi: 10.1128/MCB.23.23.8878-8889.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. Embo J. 1999;18:1345–1356. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Imamichi Y, Waidmann O, Hein R, Eleftheriou P, Giehl K, Menke A. TGF beta-induced focal complex formation in epithelial cells is mediated by activated ERK and JNK MAP kinases and is independent of Smad4. Biol Chem. 2005;386:225–236. doi: 10.1515/BC.2005.028. [DOI] [PubMed] [Google Scholar]
- 87.Remy I, Montmarquette A, Michnick SW. PKB/Akt modulates TGF-beta signalling through a direct interaction with Smad3. Nat Cell Biol. 2004;6:358–365. doi: 10.1038/ncb1113. [DOI] [PubMed] [Google Scholar]
- 88.Runyan CE, Schnaper HW, Poncelet AC. The phosphatidylinositol 3-kinase/Akt pathway enhances Smad3-stimulated mesangial cell collagen I expression in response to transforming growth factor-beta1. J Biol Chem. 2004;279:2632–2639. doi: 10.1074/jbc.M310412200. [DOI] [PubMed] [Google Scholar]
- 89.Yakymovych I, Ten Dijke P, Heldin CH, Souchelnytskyi S. Regulation of Smad signaling by protein kinase C. Faseb J. 2001;15:553–555. doi: 10.1096/fj.00-0474fje. [DOI] [PubMed] [Google Scholar]
- 90.Ignotz RA, Massague J. Transforming growth factor-beta stimulates the expression of fibronectin and collagen and their incorporation into the extracellular matrix. J Biol Chem. 1986;261:4337–4345. [PubMed] [Google Scholar]
- 91.Varga J, Jimenez SA. Stimulation of normal human fibroblast collagen production and processing by transforming growth factor-beta. Biochem Biophys Res Commun. 1986;138:974–980. doi: 10.1016/s0006-291x(86)80591-5. [DOI] [PubMed] [Google Scholar]
- 92.Varga J, Rosenbloom J, Jimenez SA. Transforming growth factor beta (TGF beta) causes a persistent increase in steady-state amounts of type I and type III collagen and fibronectin mRNAs in normal human dermal fibroblasts. Biochem J. 1987;247:597–604. doi: 10.1042/bj2470597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wrana JL, Maeno M, Hawrylyshyn B, Yao KL, Domenicucci C, Sodek J. Differential effects of transforming growth factor-beta on the synthesis of extracellular matrix proteins by normal fetal rat calvarial bone cell populations. J Cell Biol. 1988;106:915–924. doi: 10.1083/jcb.106.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Eghbali M, Tomek R, Sukhatme VP, Woods C, Bhambi B. Differential effects of transforming growth factor-beta 1 and phorbol myristate acetate on cardiac fibroblasts. Regulation of fibrillar collagen mRNAs and expression of early transcription factors. Circ Res. 1991;69:483–490. doi: 10.1161/01.res.69.2.483. [DOI] [PubMed] [Google Scholar]
- 95.Ghosh AK. Factors involved in the regulation of type I collagen gene expression: implication in fibrosis. Exp Biol Med (Maywood) 2002;227:301–314. doi: 10.1177/153537020222700502. [DOI] [PubMed] [Google Scholar]
- 96.Verrecchia F, Mauviel A. TGF-beta and TNF-alpha: antagonistic cytokines controlling type I collagen gene expression. Cell Signal. 2004;16:873–880. doi: 10.1016/j.cellsig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 97.Smith JD, Bryant SR, Couper LL, Vary CP, Gotwals PJ, Koteliansky VE, Lindner V. Soluble transforming growth factor-beta type II receptor inhibits negative remodeling, fibroblast transdifferentiation, and intimal lesion formation but not endothelial growth. Circ Res. 1999;84:1212–1222. doi: 10.1161/01.res.84.10.1212. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Z, Li XJ, Liu Y, Zhang X, Li YY, Xu WS. Recombinant human decorin inhibits cell proliferation and downregulates TGF-beta1 production in hypertrophic scar fibroblasts. Burns. 2007;33:634–641. doi: 10.1016/j.burns.2006.08.018. [DOI] [PubMed] [Google Scholar]
- 99.Ihn H. Pathogenesis of fibrosis: role of TGF-beta and CTGF. Curr Opin Rheumatol. 2002;14:681–685. doi: 10.1097/00002281-200211000-00009. [DOI] [PubMed] [Google Scholar]
- 100.Verrecchia F, Mauviel A. Transforming growth factor-beta and fibrosis. World J Gastroenterol. 2007;13:3056–3062. doi: 10.3748/wjg.v13.i22.3056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Moussad EE, Brigstock DR. Connective tissue growth factor: what’s in a name? Mol Genet Metab. 2000;71:276–292. doi: 10.1006/mgme.2000.3059. [DOI] [PubMed] [Google Scholar]
- 102.Lijnen HR. Plasmin and matrix metalloproteinases in vascular remodeling. Thromb Haemost. 2001;86:324–333. [PubMed] [Google Scholar]
- 103.Keski-Oja J, Raghow R, Sawdey M, Loskutoff DJ, Postlethwaite AE, Kang AH, Moses HL. Regulation of mRNAs for type-1 plasminogen activator inhibitor, fibronectin, and type I procollagen by transforming growth factor-beta. Divergent responses in lung fibroblasts and carcinoma cells. J Biol Chem. 1988;263:3111–3115. [PubMed] [Google Scholar]
- 104.Chakraborti S, Mandal M, Das S, Mandal A, Chakraborti T. Regulation of matrix metalloproteinases: an overview. Mol Cell Biochem. 2003;253:269–285. doi: 10.1023/a:1026028303196. [DOI] [PubMed] [Google Scholar]
- 105.Yan C, Boyd DD. Regulation of matrix metalloproteinase gene expression. J Cell Physiol. 2007;211:19–26. doi: 10.1002/jcp.20948. [DOI] [PubMed] [Google Scholar]
- 106.Kerr LD, Miller DB, Matrisian LM. TGF-beta 1 inhibition of transin/stromelysin gene expression is mediated through a Fos binding sequence. Cell. 1990;61:267–278. doi: 10.1016/0092-8674(90)90807-q. [DOI] [PubMed] [Google Scholar]
- 107.Edwards DR, Murphy G, Reynolds JJ, Whitham SE, Docherty AJ, Angel P, Heath JK. Transforming growth factor beta modulates the expression of collagenase and metalloproteinase inhibitor. Embo J. 1987;6:1899–1904. doi: 10.1002/j.1460-2075.1987.tb02449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dai J, Losy F, Guinault AM, Pages C, Anegon I, Desgranges P, Becquemin JP, Allaire E. Overexpression of transforming growth factor-beta1 stabilizes already-formed aortic aneurysms: a first approach to induction of functional healing by endovascular gene therapy. Circulation. 2005;112:1008–1015. doi: 10.1161/CIRCULATIONAHA.104.523357. [DOI] [PubMed] [Google Scholar]
- 109.Bobik A, Agrotis A, Kanellakis P, Dilley R, Krushinsky A, Smirnov V, Tararak E, Condron M, Kostolias G. Distinct patterns of transforming growth factor-beta isoform and receptor expression in human atherosclerotic lesions. Colocalization implicates TGF-beta in fibrofatty lesion development. Circulation. 1999;99:2883–2891. doi: 10.1161/01.cir.99.22.2883. [DOI] [PubMed] [Google Scholar]
- 110.Cipollone F, Fazia M, Mincione G, Iezzi A, Pini B, Cuccurullo C, Ucchino S, Spigonardo F, Di Nisio M, Cuccurullo F, Mezzetti A, Porreca E. Increased expression of transforming growth factor-beta1 as a stabilizing factor in human atherosclerotic plaques. Stroke. 2004;35:2253–2257. doi: 10.1161/01.STR.0000140739.45472.9c. [DOI] [PubMed] [Google Scholar]
- 111.Lutgens E, Gijbels M, Smook M, Heeringa P, Gotwals P, Koteliansky VE, Daemen MJ. Transforming growth factor-beta mediates balance between inflammation and fibrosis during plaque progression. Arterioscler Thromb Vasc Biol. 2002;22:975–982. doi: 10.1161/01.atv.0000019729.39500.2f. [DOI] [PubMed] [Google Scholar]
- 112.Syed M, Lesch M. Coronary artery aneurysm: a review. Prog Cardiovasc Dis. 1997;40:77–84. doi: 10.1016/s0033-0620(97)80024-2. [DOI] [PubMed] [Google Scholar]
- 113.Gaine SP, Rubin LJ. Primary pulmonary hypertension. Lancet. 1998;352:719–725. doi: 10.1016/S0140-6736(98)02111-4. [DOI] [PubMed] [Google Scholar]
- 114.Loscalzo J. Genetic clues to the cause of primary pulmonary hypertension. N Engl J Med. 2001;345:367–371. doi: 10.1056/NEJM200108023450511. [DOI] [PubMed] [Google Scholar]
- 115.Newman JH, Trembath RC, Morse JA, Grunig E, Loyd JE, Adnot S, Coccolo F, Ventura C, Phillips JA, 3rd, Knowles JA, Janssen B, Eickelberg O, Eddahibi S, Herve P, Nichols WC, Elliott G. Genetic basis of pulmonary arterial hypertension: current understanding and future directions. J Am Coll Cardiol. 2004;43:33S–39S. doi: 10.1016/j.jacc.2004.02.028. [DOI] [PubMed] [Google Scholar]
- 116.Rudarakanchana N, Flanagan JA, Chen H, Upton PD, Machado R, Patel D, Trembath RC, Morrell NW. Functional analysis of bone morphogenetic protein type II receptor mutations underlying primary pulmonary hypertension. Hum Mol Genet. 2002;11:1517–1525. doi: 10.1093/hmg/11.13.1517. [DOI] [PubMed] [Google Scholar]
- 117.Cheifetz S, Bellon T, Cales C, Vera S, Bernabeu C, Massague J, Letarte M. Endoglin is a component of the transforming growth factor-beta receptor system in human endothelial cells. J Biol Chem. 1992;267:19027–19030. [PubMed] [Google Scholar]
- 118.You HJ, Bruinsma MW, How T, Ostrander JH, Blobe GC. The type III TGF-beta receptor signals through both Smad3 and the p38 MAP kinase pathways to contribute to inhibition of cell proliferation. Carcinogenesis. 2007;28:2491–2500. doi: 10.1093/carcin/bgm195. [DOI] [PubMed] [Google Scholar]
- 119.Johnson DW, Berg JN, Baldwin MA, Gallione CJ, Marondel I, Yoon SJ, Stenzel TT, Speer M, Pericak-Vance MA, Diamond A, Guttmacher AE, Jackson CE, Attisano L, Kucherlapati R, Porteous ME, Marchuk DA. Mutations in the activin receptor-like kinase 1 gene in hereditary haemorrhagic telangiectasia type 2. Nat Genet. 1996;13:189–195. doi: 10.1038/ng0696-189. [DOI] [PubMed] [Google Scholar]
- 120.ten Dijke P, Yamashita H, Ichijo H, Franzen P, Laiho M, Miyazono K, Heldin CH. Characterization of type I receptors for transforming growth factor-beta and activin. Science. 1994;264:101–104. doi: 10.1126/science.8140412. [DOI] [PubMed] [Google Scholar]
- 121.Oh SP, Seki T, Goss KA, Imamura T, Yi Y, Donahoe PK, Li L, Miyazono K, ten Dijke P, Kim S, Li E. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proc Natl Acad Sci U S A. 2000;97:2626–2631. doi: 10.1073/pnas.97.6.2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Choke E, Cockerill GW, Dawson J, Wilson RW, Jones A, Loftus IM, Thompson MM. Increased angiogenesis at the site of abdominal aortic aneurysm rupture. Ann N Y Acad Sci. 2006;1085:315–319. doi: 10.1196/annals.1383.007. [DOI] [PubMed] [Google Scholar]
- 123.Miwa K, Nakashima H, Aoki M, Miyake T, Kawasaki T, Iwai M, Oishi M, Kataoka K, Ohgi S, Ogihara T, Kaneda Y, Morishita R. Inhibition of ets, an essential transcription factor for angiogenesis, to prevent the development of abdominal aortic aneurysm in a rat model. Gene Ther. 2005;12:1109–1118. doi: 10.1038/sj.gt.3302496. [DOI] [PubMed] [Google Scholar]
- 124.Paik DC, Fu C, Bhattacharya J, Tilson MD. Ongoing angiogenesis in blood vessels of the abdominal aortic aneurysm. Exp Mol Med. 2004;36:524–533. doi: 10.1038/emm.2004.67. [DOI] [PubMed] [Google Scholar]
- 125.Dietz HC, Loeys B, Carta L, Ramirez F. Recent progress towards a molecular understanding of Marfan syndrome. Am J Med Genet C Semin Med Genet. 2005;139:4–9. doi: 10.1002/ajmg.c.30068. [DOI] [PubMed] [Google Scholar]
- 126.Lee B, Godfrey M, Vitale E, Hori H, Mattei MG, Sarfarazi M, Tsipouras P, Ramirez F, Hollister DW. Linkage of Marfan syndrome and a phenotypically related disorder to two different fibrillin genes. Nature. 1991;352:330–334. doi: 10.1038/352330a0. [DOI] [PubMed] [Google Scholar]
- 127.Silverman DI, Gray J, Roman MJ, Bridges A, Burton K, Boxer M, Devereux RB, Tsipouras P. Family history of severe cardiovascular disease in Marfan syndrome is associated with increased aortic diameter and decreased survival. J Am Coll Cardiol. 1995;26:1062–1067. doi: 10.1016/0735-1097(95)00258-0. [DOI] [PubMed] [Google Scholar]
- 128.van Karnebeek CD, Naeff MS, Mulder BJ, Hennekam RC, Offringa M. Natural history of cardiovascular manifestations in Marfan syndrome. Arch Dis Child. 2001;84:129–137. doi: 10.1136/adc.84.2.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Ramirez F. Fibrillln mutations in Marfan syndrome and related phenotypes. Curr Opin Genet Dev. 1996;6:309–315. doi: 10.1016/s0959-437x(96)80007-4. [DOI] [PubMed] [Google Scholar]
- 130.Ramirez F, Pereira L. The fibrillins. Int J Biochem Cell Biol. 1999;31:255–259. doi: 10.1016/s1357-2725(98)00109-5. [DOI] [PubMed] [Google Scholar]
- 131.Ramirez F, Sakai LY, Dietz HC, Rifkin DB. Fibrillin microfibrils: multipurpose extracellular networks in organismal physiology. Physiol Genomics. 2004;19:151–154. doi: 10.1152/physiolgenomics.00092.2004. [DOI] [PubMed] [Google Scholar]
- 132.Bunton TE, Biery NJ, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. 2001;88:37–43. doi: 10.1161/01.res.88.1.37. [DOI] [PubMed] [Google Scholar]
- 133.Pereira L, Lee SY, Gayraud B, Andrikopoulos K, Shapiro SD, Bunton T, Biery NJ, Dietz HC, Sakai LY, Ramirez F. Pathogenetic sequence for aneurysm revealed in mice underexpressing fibrillin-1. Proc Natl Acad Sci U S A. 1999;96:3819–3823. doi: 10.1073/pnas.96.7.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kaartinen V, Warburton D. Fibrillin controls TGF-beta activation. Nat Genet. 2003;33:331–332. doi: 10.1038/ng0303-331. [DOI] [PubMed] [Google Scholar]
- 135.Annes JP, Munger JS, Rifkin DB. Making sense of latent TGFbeta activation. J Cell Sci. 2003;116:217–224. doi: 10.1242/jcs.00229. [DOI] [PubMed] [Google Scholar]
- 136.Chaudhry SS, Cain SA, Morgan A, Dallas SL, Shuttleworth CA, Kielty CM. Fibrillin-1 regulates the bioavailability of TGFbeta1. J Cell Biol. 2007;176:355–367. doi: 10.1083/jcb.200608167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Charbonneau NL, Ono RN, Corson GM, Keene DR, Sakai LY. Fine tuning of growth factor signals depends on fibrillin microfibril networks. Birth Defects Res C Embryo Today. 2004;72:37–50. doi: 10.1002/bdrc.20000. [DOI] [PubMed] [Google Scholar]
- 138.Isogai Z, Ono RN, Ushiro S, Keene DR, Chen Y, Mazzieri R, Charbonneau NL, Reinhardt DP, Rifkin DB, Sakai LY. Latent transforming growth factor beta-binding protein 1 interacts with fibrillin and is a microfibril-associated protein. J Biol Chem. 2003;278:2750–2757. doi: 10.1074/jbc.M209256200. [DOI] [PubMed] [Google Scholar]
- 139.Koli K, Saharinen J, Hyytiainen M, Penttinen C, Keski-Oja J. Latency, activation, and binding proteins of TGF-beta. Microsc Res Tech. 2001;52:354–362. doi: 10.1002/1097-0029(20010215)52:4<354::AID-JEMT1020>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 140.Neptune ER, Frischmeyer PA, Arking DE, Myers L, Bunton TE, Gayraud B, Ramirez F, Sakai LY, Dietz HC. Dysregulation of TGF-beta activation contributes to pathogenesis in Marfan syndrome. Nat Genet. 2003;33:407–411. doi: 10.1038/ng1116. [DOI] [PubMed] [Google Scholar]
- 141.Habashi JP, Judge DP, Holm TM, Cohn RD, Loeys BL, Cooper TK, Myers L, Klein EC, Liu G, Calvi C, Podowski M, Neptune ER, Halushka MK, Bedja D, Gabrielson K, Rifkin DB, Carta L, Ramirez F, Huso DL, Dietz HC. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science. 2006;312:117–121. doi: 10.1126/science.1124287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beighton P, de Paepe A, Danks D, Finidori G, Gedde-Dahl T, Goodman R, Hall JG, Hollister DW, Horton W, McKusick VA, et al. International Nosology of Heritable Disorders of Connective Tissue, Berlin, 1986. Am J Med Genet. 1988;29:581–594. doi: 10.1002/ajmg.1320290316. [DOI] [PubMed] [Google Scholar]
- 143.De Paepe A, Devereux RB, Dietz HC, Hennekam RC, Pyeritz RE. Revised diagnostic criteria for the Marfan syndrome. Am J Med Genet. 1996;62:417–426. doi: 10.1002/(SICI)1096-8628(19960424)62:4<417::AID-AJMG15>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- 144.Rose PS, Levy HP, Ahn NU, Sponseller PD, Magyari T, Davis J, Francomano CA. A comparison of the Berlin and Ghent nosologies and the influence of dural ectasia in the diagnosis of Marfan syndrome. Genet Med. 2000;2:278–282. doi: 10.1097/00125817-200009000-00002. [DOI] [PubMed] [Google Scholar]
- 145.Collod G, Babron MC, Jondeau G, Coulon M, Weissenbach J, Dubourg O, Bourdarias JP, Bonaiti-Pellie C, Junien C, Boileau C. A second locus for Marfan syndrome maps to chromosome 3p24.2-p25. Nat Genet. 1994;8:264–268. doi: 10.1038/ng1194-264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Dietz H, Francke U, Furthmayr H, Francomano C, De Paepe A, Devereux R, Ramirez F, Pyeritz R. The question of heterogeneity in Marfan syndrome. Nat Genet. 1995;9:228–231. doi: 10.1038/ng0395-228. [DOI] [PubMed] [Google Scholar]
- 147.Gilchrist DM. Marfan syndrome or Marfan-like connective-tissue disorder. Am J Hum Genet. 1994;54:553–554. [PMC free article] [PubMed] [Google Scholar]
- 148.Boileau C, Jondeau G, Babron MC, Coulon M, Alexandre JA, Sakai L, Melki J, Delorme G, Dubourg O, Bonaiti-Pellie C, et al. Autosomal dominant Marfan-like connective-tissue disorder with aortic dilation and skeletal anomalies not linked to the fibrillin genes. Am J Hum Genet. 1993;53:46–54. [PMC free article] [PubMed] [Google Scholar]
- 149.Mizuguchi T, Collod-Beroud G, Akiyama T, Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M, Morisaki H, Ihara M, Kinoshita A, Yoshiura K, Junien C, Kajii T, Jondeau G, Ohta T, Kishino T, Furukawa Y, Nakamura Y, Niikawa N, Boileau C, Matsumoto N. Heterozygous TGFBR2 mutations in Marfan syndrome. Nat Genet. 2004;36:855–860. doi: 10.1038/ng1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Biddinger A, Rocklin M, Coselli J, Milewicz DM. Familial thoracic aortic dilatations and dissections: a case control study. J Vasc Surg. 1997;25:506–511. doi: 10.1016/s0741-5214(97)70261-1. [DOI] [PubMed] [Google Scholar]
- 151.Loeys BL, Schwarze U, Holm T, Callewaert BL, Thomas GH, Pannu H, De Backer JF, Oswald GL, Symoens S, Manouvrier S, Roberts AE, Faravelli F, Greco MA, Pyeritz RE, Milewicz DM, Coucke PJ, Cameron DE, Braverman AC, Byers PH, De Paepe AM, Dietz HC. Aneurysm syndromes caused by mutations in the TGF-beta receptor. N Engl J Med. 2006;355:788–798. doi: 10.1056/NEJMoa055695. [DOI] [PubMed] [Google Scholar]
- 152.Pannu H, Avidan N, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and dissections: potential relevance to abdominal aortic aneurysms. Ann N Y Acad Sci. 2006;1085:242–255. doi: 10.1196/annals.1383.024. [DOI] [PubMed] [Google Scholar]
- 153.Zhu L, Vranckx R, Khau Van Kien P, Lalande A, Boisset N, Mathieu F, Wegman M, Glancy L, Gasc JM, Brunotte F, Bruneval P, Wolf JE, Michel JB, Jeunemaitre X. Mutations in myosin heavy chain 11 cause a syndrome associating thoracic aortic aneurysm/aortic dissection and patent ductus arteriosus. Nat Genet. 2006;38:343–349. doi: 10.1038/ng1721. [DOI] [PubMed] [Google Scholar]
- 154.Guo DC, Pannu H, Tran-Fadulu V, Papke CL, Yu RK, Avidan N, Bourgeois S, Estrera AL, Safi HJ, Sparks E, Amor D, Ades L, McConnell V, Willoughby CE, Abuelo D, Willing M, Lewis RA, Kim DH, Scherer S, Tung PP, Ahn C, Buja LM, Raman CS, Shete SS, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) lead to thoracic aortic aneurysms and dissections. Nat Genet. 2007;39:1488–1493. doi: 10.1038/ng.2007.6. [DOI] [PubMed] [Google Scholar]
- 155.Hasham SN, Willing MC, Guo DC, Muilenburg A, He R, Tran VT, Scherer SE, Shete SS, Milewicz DM. Mapping a locus for familial thoracic aortic aneurysms and dissections (TAAD2) to 3p24-25. Circulation. 2003;107:3184–3190. doi: 10.1161/01.CIR.0000078634.33124.95. [DOI] [PubMed] [Google Scholar]
- 156.Matyas G, Arnold E, Carrel T, Baumgartner D, Boileau C, Berger W, Steinmann B. Identification and in silico analyses of novel TGFBR1 and TGFBR2 mutations in Marfan syndrome-related disorders. Hum Mutat. 2006;27:760–769. doi: 10.1002/humu.20353. [DOI] [PubMed] [Google Scholar]
- 157.Pannu H, Tran-Fadulu V, Milewicz DM. Genetic basis of thoracic aortic aneurysms and aortic dissections. Am J Med Genet C Semin Med Genet. 2005;139:10–16. doi: 10.1002/ajmg.c.30069. [DOI] [PubMed] [Google Scholar]
- 158.Pannu H, Fadulu VT, Chang J, Lafont A, Hasham SN, Sparks E, Giampietro PF, Zaleski C, Estrera AL, Safi HJ, Shete S, Willing MC, Raman CS, Milewicz DM. Mutations in transforming growth factor-beta receptor type II cause familial thoracic aortic aneurysms and dissections. Circulation. 2005;112:513–520. doi: 10.1161/CIRCULATIONAHA.105.537340. [DOI] [PubMed] [Google Scholar]
- 159.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, Xu FL, Myers LA, Spevak PJ, Cameron DE, De Backer J, Hellemans J, Chen Y, Davis EC, Webb CL, Kress W, Coucke P, Rifkin DB, De Paepe AM, Dietz HC. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 160.Singh KK, Rommel K, Mishra A, Karck M, Haverich A, Schmidtke J, Arslan-Kirchner M. TGFBR1 and TGFBR2 mutations in patients with features of Marfan syndrome and Loeys-Dietz syndrome. Hum Mutat. 2006;27:770–777. doi: 10.1002/humu.20354. [DOI] [PubMed] [Google Scholar]
- 161.Superti-Furga A, Gugler E, Gitzelmann R, Steinmann B. Ehlers-Danlos syndrome type IV: a multi-exon deletion in one of the two COL3A1 alleles affecting structure, stability, and processing of type III procollagen. J Biol Chem. 1988;263:6226–6232. [PubMed] [Google Scholar]
- 162.Boutouyrie P, Germain DP, Fiessinger JN, Laloux B, Perdu J, Laurent S. Increased carotid wall stress in vascular Ehlers-Danlos syndrome. Circulation. 2004;109:1530–1535. doi: 10.1161/01.CIR.0000121741.50315.C2. [DOI] [PubMed] [Google Scholar]
- 163.Denton CP, Zheng B, Evans LA, Shi-wen X, Ong VH, Fisher I, Lazaridis K, Abraham DJ, Black CM, de Crombrugghe B. Fibroblast-specific expression of a kinase-deficient type II transforming growth factor beta (TGFbeta) receptor leads to paradoxical activation of TGFbeta signaling pathways with fibrosis in transgenic mice. J Biol Chem. 2003;278:25109–25119. doi: 10.1074/jbc.M300636200. [DOI] [PubMed] [Google Scholar]
- 164.Wong SH, Hamel L, Chevalier S, Philip A. Endoglin expression on human microvascular endothelial cells association with betaglycan and formation of higher order complexes with TGF-beta signalling receptors. Eur J Biochem. 2000;267:5550–5560. doi: 10.1046/j.1432-1327.2000.01621.x. [DOI] [PubMed] [Google Scholar]
- 165.Di Guglielmo GM, Le Roy C, Goodfellow AF, Wrana JL. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410–421. doi: 10.1038/ncb975. [DOI] [PubMed] [Google Scholar]
- 166.Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFbeta-inducible antagonist of TGF-beta signalling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- 167.Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor-beta-mediated transcription. J Biol Chem. 1999;274:37413–37420. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 168.Lee KS, Hong SH, Bae SC. Both the Smad and p38 MAPK pathways play a crucial role in Runx2 expression following induction by transforming growth factor-beta and bone morphogenetic protein. Oncogene. 2002;21:7156–7163. doi: 10.1038/sj.onc.1205937. [DOI] [PubMed] [Google Scholar]
- 169.Henderson EL, Geng YJ, Sukhova GK, Whittemore AD, Knox J, Libby P. Death of smooth muscle cells and expression of mediators of apoptosis by T lymphocytes in human abdominal aortic aneurysms. Circulation. 1999;99:96–104. doi: 10.1161/01.cir.99.1.96. [DOI] [PubMed] [Google Scholar]
- 170.Lopez-Candales A, Holmes DR, Liao S, Scott MJ, Wickline SA, Thompson RW. Decreased vascular smooth muscle cell density in medial degeneration of human abdominal aortic aneurysms. Am J Pathol. 1997;150:993–1007. [PMC free article] [PubMed] [Google Scholar]
- 171.Sho E, Sho M, Nanjo H, Kawamura K, Masuda H, Dalman RL. Comparison of cell-type-specific vs transmural aortic gene expression in experimental aneurysms. J Vasc Surg. 2005;41:844–852. doi: 10.1016/j.jvs.2005.02.027. [DOI] [PubMed] [Google Scholar]
- 172.Thompson RW, Liao S, Curci JA. Vascular smooth muscle cell apoptosis in abdominal aortic aneurysms. Coron Artery Dis. 1997;8:623–631. doi: 10.1097/00019501-199710000-00005. [DOI] [PubMed] [Google Scholar]
- 173.Fukui D, Miyagawa S, Soeda J, Tanaka K, Urayama H, Kawasaki S. Overexpression of transforming growth factor beta1 in smooth muscle cells of human abdominal aortic aneurysm. Eur J Vasc Endovasc Surg. 2003;25:540–545. doi: 10.1053/ejvs.2002.1857. [DOI] [PubMed] [Google Scholar]
- 174.Zalewski A, Shi Y. Vascular myofibroblasts. Lessons from coronary repair and remodeling. Arterioscler Thromb Vasc Biol. 1997;17:417–422. doi: 10.1161/01.atv.17.3.417. [DOI] [PubMed] [Google Scholar]
- 175.Willis BC, Borok Z. TGF-beta-induced EMT: mechanisms and implications for fibrotic lung disease. Am J Physiol Lung Cell Mol Physiol. 2007;293:L525–534. doi: 10.1152/ajplung.00163.2007. [DOI] [PubMed] [Google Scholar]
- 176.Shi Y, O’Brien JE, Jr, Fard A, Zalewski A. Transforming growth factor-beta 1 expression and myofibroblast formation during arterial repair. Arterioscler Thromb Vasc Biol. 1996;16:1298–1305. doi: 10.1161/01.atv.16.10.1298. [DOI] [PubMed] [Google Scholar]
- 177.Vaughan MB, Howard EW, Tomasek JJ. Transforming growth factor-beta1 promotes the morphological and functional differentiation of the myofibroblast. Exp Cell Res. 2000;257:180–189. doi: 10.1006/excr.2000.4869. [DOI] [PubMed] [Google Scholar]
- 178.Hu Y, Peng J, Feng D, Chu L, Li X, Jin Z, Lin Z, Zeng Q. Role of extracellular signal-regulated kinase, p38 kinase, and activator protein-1 in transforming growth factor-beta1-induced alpha smooth muscle actin expression in human fetal lung fibroblasts in vitro. Lung. 2006;184:33–42. doi: 10.1007/s00408-005-2560-5. [DOI] [PubMed] [Google Scholar]
- 179.Kapoun AM, Gaspar NJ, Wang Y, Damm D, Liu YW, O’Young G, Quon D, Lam A, Munson K, Tran TT, Ma JY, Murphy A, Dugar S, Chakravarty S, Protter AA, Wen FQ, Liu X, Rennard SI, Higgins LS. Transforming growth factor-beta receptor type 1 (TGFbetaRI) kinase activity but not p38 activation is required for TGFbetaRI-induced myofibroblast differentiation and profibrotic gene expression. Mol Pharmacol. 2006;70:518–531. doi: 10.1124/mol.105.021600. [DOI] [PubMed] [Google Scholar]
- 180.Shi Y, Patel S, Niculescu R, Chung W, Desrochers P, Zalewski A. Role of matrix metalloproteinases and their tissue inhibitors in the regulation of coronary cell migration. Arterioscler Thromb Vasc Biol. 1999;19:1150–1155. doi: 10.1161/01.atv.19.5.1150. [DOI] [PubMed] [Google Scholar]
- 181.Shi Y, Pieniek M, Fard A, O’Brien J, Mannion JD, Zalewski A. Adventitial remodeling after coronary arterial injury. Circulation. 1996;93:340–348. doi: 10.1161/01.cir.93.2.340. [DOI] [PubMed] [Google Scholar]
- 182.Sakata N, Nabeshima K, Iwasaki H, Tashiro T, Uesugi N, Nakashima O, Ito H, Kawanami T, Furuya K, Kojima M. Possible involvement of myofibroblast in the development of inflammatory aortic aneurysm. Pathol Res Pract. 2007;203:21–29. doi: 10.1016/j.prp.2006.08.008. [DOI] [PubMed] [Google Scholar]