

Abstract
In this study, the potential role of Stat3 in UVB-induced skin carcinogenesis was examined using skin-specific gain and loss of function transgenic mice, i.e., K5.Stat3C and K5Cre.Stat3fl/fl mice, respectively. The epidermis of Stat3-deficient mice was highly sensitive to UVB-induced apoptosis, whereas the epidermis of K5.Stat3C mice was more resistant to UVB-induced apoptosis. In particular, the status of Stat3 influenced the survival of UV-photoproduct cells, including those located in the bulge region of hair follicles. K5.Stat3C mice exhibited significantly increased epidermal proliferation and hyperplasia in response to UVB irradiation, whereas Stat3-deficient mice showed reduced epidermal proliferation and hyperplasia. Expression of target genes regulated by Stat3, such as cyclin D1 and Bcl-xL, was increased in epidermis of both control and UVB-irradiated K5.Stat3C mice, and downregulated in epidermis of both control and UVB-irradiated K5Cre.Stat3fl/fl mice. Following UVB irradiation, the formation of skin tumors in K5.Stat3C mice was accelerated and both the incidence and multiplicity of skin tumors was significantly greater than wild-type controls. In contrast, Stat3-deficient mice were resistant to UVB skin carcinogenesis. These results demonstrate that Stat3 plays an important role in the development of UVB-induced skin tumors through its effects on both survival and proliferation of keratinocytes during carcinogenesis.
Keywords: Stat3, skin carcinogenesis, UVB
INTRODUCTION
Exposure to sunlight in the ultraviolet (UV) range causes DNA damage and other cellular responses that contribute to the development of all three major types of skin cancer, i.e., squamous cell carcinomas (SCCs), basal cell carcinomas, and cutaneous melanomas. UV radiation is divided into three categories: UVA (315-400 nm), UVB (280-315 nm), and UVC (190-280 nm) (Ichihashi et al., 2003; Kraemer, 1997; Sarasin, 1999). Among these, UVB is the most important for induction of skin cancer and can induce mutations in critical target genes as well as modulate cellular signal transduction pathways (Daya-Grosjean and Sarasin, 2005; Sarasin, 1999). Upon UVB irradiation, DNA-damaged keratinocytes may undergo apoptosis through several apoptotic pathways including p53 dependent pathways (de Gruijl et al., 2001; Melnikova and Ananthaswamy, 2005). Alternatively, UV-induced DNA damage may be repaired. In both cases, the risk of UV-induced skin cancer is reduced. In contrast, dysfunction of these pathways may contribute to the development of UVB-induced skin cancer (de Gruijl et al., 2001; Melnikova and Ananthaswamy, 2005).
Signal transducer and activator of transcription 3 (Stat3) is one of a family of cytoplasmic proteins that participate in normal cellular responses to cytokines and growth factors as transcription factors (Bowman et al., 2000; Bromberg, 2002; Levy and Darnell, 2002; Levy and Lee, 2002). Upon activation by a wide variety of cell surface receptors, tyrosine phosphorylated Stat3 dimerizes and translocates to the nucleus and modulates the expression of target genes that are involved in various physiological functions including apoptosis (e.g., Bcl-xL and Survivin), cell cycle regulation (e.g., Cyclin D1 and c-Myc), and angiogenesis (e.g., VEGF) (Darnell, 1997; Levy and Darnell, 2002). Studies have shown that constitutive activation of Stat3 is associated with a number of human tumors and cancer cell lines, including prostate, breast, lung, head and neck, brain, and pancreas (Alvarez et al., 2005; Frank, 2003; Li and Shaw, 2002; Masuda et al., 2002). Furthermore, inhibition of Stat3 can suppress growth of cancer cells by promoting apoptosis and inhibiting cell proliferation (Bromberg et al., 1999; Turkson and Jove, 2000), suggesting that Stat3 plays a critical role in both proliferation and survival of cancer cells. The primary mechanism for constitutive activation of Stat3 in human tumors and cancer cell lines appears to be aberrant growth factor signaling rather than genetic alterations (Bromberg, 2002).
Recent studies from our laboratory have demonstrated that Stat3 plays critical roles in epithelial carcinogenesis during the initiation, promotion and progression stages (Chan et al., 2004a; Chan et al., 2008; Chan et al., 2004b; Kataoka et al., 2008; Kim et al., 2007; Sano et al., 2007). In this regard, Stat3 is required for both the initiation and promotion stages of carcinogenesis by maintaining survival of DNA-damaged keratinocyte stem cells and by mediating cell proliferation necessary for the clonal expansion of initiated keratinocyte stem cells (Chan et al., 2004b). More recent studies using mice in which a constitutively active/dimerized form of Stat3 (Stat3C) was targeted to the proliferative compartment of the epidermis via the bovine keratin 5 promoter (referred to as K5.Stat3C transgenic mice) demonstrated a role in skin tumor progression through modulation of genes associated with epithelial-mesenchymal transition (Chan et al., 2008). In addition to its roles in multistage chemical carcinogenesis, several recent studies have shown that Stat3 plays a role in the response of keratinocytes and other cell types to UVB exposure (Ahsan et al., 2005; Aziz et al., 2007; Li et al., 2001; Sano et al., 2005b; Shen et al., 2001; Wheeler et al., 2004).
In the present study, we directly demonstrate an important role for Stat3 in UVB-induced skin carcinogenesis using both skin-specific gain and loss of function Stat3 transgenic mice. In this regard, K5.Stat3C mice showed accelerated development of skin tumors and increased numbers of skin tumors in response to a UVB skin carcinogenesis protocol compared to wild-type mice. In contrast, Stat3-deficient mice were resistant to UVB-induced skin carcinogenesis compared to wild-type controls. Further analyses indicate that Stat3 regulates keratinocyte apoptosis and proliferation in vivo in response to UVB during carcinogenesis. Collectively, these results demonstrate that Stat3 plays a similar role in both chemically-induced and UVB-induced skin cancer.
RESULTS
Constitutive Stat3 activation protects whereas Stat3 deficiency sensitizes keratinocytes to UVB-induced apoptosis
Recently, we reported that the number of sunburn cells 24 h after UVB exposure was significantly increased in the epidermis of Stat3-deficient mice, whereas the number of sunburn cells was dramatically reduced in the epidermis by overexpression of Stat3 (Sano et al., 2005b). In this study, we further examined the effect of Stat3 activation or deficiency on UVB-induced epidermal apoptosis by examining a longer timecourse and assessing the location of apoptotic cells. Wild-type, K5.Stat3C and Stat3-deficient mice were irradiated with a single dose of UVB (1200 J/m2), and apoptotic cells (caspase-3-positive cells) were measured 1, 2, 5 and 10 days later. Consistent with previous results, the number of apoptotic cells was significantly increased in the epidermis of Stat3-deficient mice, whereas the number of apoptotic cells was significantly reduced in the epidermis of K5.Stat3C mice compared with wild-type mice 24 hours after UVB irradiation (Figure 1a). This difference in apoptotic response was maintained throughout the time course examined, although the overall number of apoptotic cells in all three genotypes decreased with time (Figure 1a). Thus, the effects of Stat3 activation or deficiency on UVB-induced epidermal apoptosis persisted for at least 10 days after a single exposure. Further examination of caspase-3-positive cells revealed that the majority of UVB-induced apoptotic cells in Stat3-deficient mice were initially localized in the interfollicular epidermis although a significant number were also observed in the bulge region of the hair follicles (see Figure 1b). Thus, the data shown in Figure 1 indicate that Stat3 is a critical factor for maintaining the survival of DNA-damaged keratinocytes, including bulge region keratinocytes, following UVB irradiation.
Figure 1. UVB-induced apoptosis in wild-type, K5.Stat3C and Stat3-deficient mice.

Groups of mice (n = 3) were irradiated with UVB at 1,200 J/m2 and sacrificed 1, 2, 5 and 10 days after irradiation. (a) Number of caspase-3-positive cells per centimeter of epidermis from wild-type, K5.Stat3C, and Stat3-deficient mice irradiated with UVB. *P < 0.05 and **P < 0.01 by Mann-Whitney U test. (b) Representative Caspase-3 staining of epidermis from Stat3-deficient mice irradiated with UVB after 2 d. Note that caspase-3-positive cells are found in the bulge region of hair follicles (arrows) in addition to interfollicular epidermis (arrow-head). Scale bar: 100 μm.
Stat3 affects the survival of photoproduct-containing keratinocytes following UVB irradiation
UVB exposure causes DNA damage and mutations in runs of tandemly located pyrimidine residues of DNA and induces primarily CPDs and 6-4 PPs (Lippke et al., 1981; Mitchell and Nairn, 1989; Setlow and Carrier, 1966). These DNA lesions can be repaired or DNA-damaged cells may be removed by apoptosis (Friedberg, 2001; Hill et al., 1999). Previous studies revealed that there were no differences in the kinetics of disappearance of 6-4 PPs among wild-type, Stat3-deficient, and Stat3-overexpressing keratinocytes up to 8 hours after UVB exposure, suggesting that Stat3 is likely not involved in the repair of UV photoproducts (Sano et al., 2005b). However, as shown in Figure 2, the status of epidermal Stat3 impacts the number of UV-photoproduct-positive cells that persist over time following UVB irradiation. Wild-type, K5.Stat3C and Stat3-deficient mice were irradiated with UVB at 1,200 J/m2 and the number of 6-4 PP- and CPD-positive cells in the epidermis was compared among the genotypes for up to 10 days after UVB exposure. The number of 6-4 PP-positive cells in the epidermis of all three genotypes disappeared rapidly and could not be detected by immunohistochemistry 5 d after UVB exposure. As shown in Figure 2a, at one and two days after UVB irradiation the number of 6-4 PP-positive cells was significantly higher in epidermis of K5.Stat3C mice compared to epidermis of Stat3-deficient mice. In addition, 6-4 PP containing cells were observed in both the interfollicular epidermis as well as in the bulge region of hair follicles. UVB-induced 6-4 PP containing keratinocytes localized in the bulge region were particularly prominent in skin of K5.Stat3C mice (Figure 2b).
Figure 2. Induction of CPD and 6-4 PP after UVB irradiation in wild-type, K5.Stat3C and Stat3-deficient mice.

Groups of mice (n = 3) were irradiated with UVB at 1,200 J/m2 and sacrificed 1, 2, 5 and 10 days after irradiation. (a) The percentage of 6-4 PP-positive cells per centimeter of epidermis is shown for wild-type, K5.Stat3C, and Stat3-deficient mice irradiated with UVB. *P < 0.05 by Mann-Whitney U test. (b) Representative 6-4 PP staining of epidermis from K5.Stat3C mice irradiated with UVB. (c) The percentage of CPD-positive cells per centimeter of epidermis is shown for wild-type, K5.Stat3C, and Stat3-deficient mice irradiated with UVB. *P < 0.05 by Mann-Whitney U test. (d) Representative CPD staining of epidermis from K5.Stat3C mice irradiated with UVB.
In contrast to 6-4 PPs, CPDs persisted in epidermis of all three genotypes for a longer period. Significant differences in the number of CPD containing keratinocytes between K5.Stat3C and Stat3-deficient mice were apparent by one day after UVB exposure (Figure 2c). Five days after UVB exposure, significant differences were observed among all three of the genotypes. The number of CPD-positive cells was greater in epidermis of both wild-type and K5.Stat3C mice compared to epidermis from Stat3-deficient mice at 5 and 10 d after exposure. K5.Stat3C mice had the highest number whereas Stat3 deficient mice had the lowest number of CPD-positive cells at these two time points. Similar to 6-4 PPs, CPD containing keratinocytes were observed in both the interfollicular epidermis as well as in the bulge-region of hair follicles. In addition, CPD containing bulge-region keratinocytes appeared to persist longer than those in the interfollicular epidermis, especially in K5.Stat3C mice (Figure 2d). Collectively, these data demonstrate that Stat3 plays a specific role in the survival of keratinocytes (including bulge region keratinocytes) that have acquired UVB-induced DNA damage, especially CPDs, and that in the absence of Stat3 (i.e., K5Cre.Stat3fl/fl mice) these cells are lost more rapidly from both epidermal compartments.
UVB-induced keratinocyte proliferation and epidermal hyperplasia is dependent on Stat3
Keratinocyte proliferation and epidermal hyperplasia are induced in response to UVB irradiation following an initial increase in apoptosis in mouse epidermis. UVB-mediated epidermal hyperproliferation is thought to replenish damaged keratinocytes with “new” basal cells through activation of signal transduction pathways (Melnikova and Ananthaswamy, 2005). Our previous studies indicated that Stat3 is required for UVB-induced keratinocyte proliferation and epidermal hyperplasia at early times after exposure (Sano et al., 2005b). To further examine the effect of Stat3 activation in UVB-induced keratinocyte proliferation and epidermal hyperplasia over an extended time period, wild-type, K5.Stat3C and Stat3-deficient mice were irradiated with UVB at a single dose of 1,200 J/m2. Epidermal proliferation and epidermal thickness were then measured 1, 2, 5 and 10 days after UVB exposure. Following UVB exposure, the number of BrdU positive cells in the epidermis increased at 24 and 48 h in wild-type and K5.Stat3C transgenic mice. In contrast, there was only a slight increase in the Stat3-deficient mice at 48 h (Figure 3a). Similar to the previous results, the percentage of BrdU-positive cells was significantly increased in the epidermis of K5.Stat3C mice 24 and 48 hours after UVB irradiation compared with wild-type and Stat3-deficient mice (Figure 3a). Although BrdU-positive cells were reduced in all three groups of mice at the later time points (5 and 10 days) post UVB irradiation, the number of BrdU-positive cells in K5.Stat3C mice remained significantly greater than those in Stat3-deficient mice. Figure 3b shows representative BrdU-stained sections from wild-type, K5.Stat3C and Stat3-deficient mice at 24 and 48 h post UVB treatment. Epidermal thickness showed a pattern similar to BrdU labeling and again was higher in K5.Stat3C mice compared with wild-type or Stat3-deficient mice 5 and 10 days after UVB irradiation (Figure 3c and d). Collectively, these results indicate that Stat3 plays an important role in epidermal hyperproliferation and regeneration following UVB irradiation.
Figure 3. UVB-induced epidermal cell proliferation in wild-type, K5.Stat3C and Stat3-deficient mice.

Groups of mice (n = 3) were irradiated with UVB at 1,200 J/m2 and sacrificed 1, 2, 5 and 10 days after irradiation. (a) Quantification of BrdU-labelled keratinocytes from the epidermis of wild-type, K5.Stat3C, and Stat3-deficient mice irradiated with UVB. *P < 0.05 and **P < 0.01 by Mann-Whitney U test. (b) BrdU stained skin section from wild-type and K5.Stat3C mice. (c) H&E staining of epidermis from wild-type, K5.Stat3C, and Stat3-deficient mice. Scale bar: 50 μm. (d) Quantification of epidermal thickness from wild-type, K5.Stat3C, and Stat3-deficient mice irradiated with UVB.
Status of selected Stat3-regulated target proteins in epidermis following UVB irradiation
The levels of both antiapoptotic (Bcl-xL) and cell cycle (cyclin D1 and E) target proteins that are known to be regulated by Stat3 were examined by Western blot analyses (Figure 4a). Quantitation of the levels of these three proteins is shown in Figure 4b – d (average ± SEM from 3 different blots). Relative to wild-type mice, the levels of Stat3 and phospho-Stat3 were higher in epidermis of K5.Stat3C mice while significantly lower in K5Cre.Stat3fl/fl mice both in the absence and presence of UVB exposure. The levels of cyclin D1, cyclin E and Bcl-xL were either higher (K5.Stat3C mice) or lower (K5Cre.Stat3fl/fl mice) compared to wild-type mice at all time points following UVB exposure. Furthermore, the levels of these three Stat3 target proteins in wild type and K5.Stat3C mice were also higher compared with Stat3-deficient mice even in the absence of UVB irradiation. Thus, the levels of these Stat3 target proteins correlated with the responses seen in terms of both epidermal apoptosis and proliferation following UVB exposure in the different genotypes.
Figure 4. Expression of target proteins of Stat3 in epidermis of wild-type, K5.Stat3C and Stat3-deficient mice after UVB exposure.

(a) Western blot analysis of Stat3 and phosphorylated Stat3 protein levels in relation to levels of its target proteins after 24 and 48 hours of UVB irradiation in wild-type, BK5.Stat3C, and Stat3-deficient mice. Relative expression of (b) cyclin D1, (c) cyclin E, and (d) Bcl-xL were quantified by densitometry.
Constitutive activation of Stat3 enhances susceptibility to UVB-induced skin carcinogenesis
To further investigate the potential role of Stat3 in UVB-induced skin carcinogenesis, K5.Stat3C and wild-type mice (12 per group) were subjected to the UVB skin carcinogenesis regimen described in the Material and Methods section. K5.Stat3C mice were significantly more sensitive to UVB skin carcinogenesis. In this regard, 100% of K5.Stat3C mice developed tumors on the ear skin and dorsal skin by the end of the experiment, whereas only 20% of wild-type mice developed tumors, primarily on the ears (Figure 5a; p < 0.05, χ2-test). The majority of the tumors that developed in both wild-type and K5.Stat3C mice were diagnosed as SCCs. For the data shown in Figure 5, only SCCs are tabulated. The average number of SCCs per mouse was also significantly greater in the K5.Stat3C mice compared to wild-type mice (Figure 5b; p < 0.05, Mann Whitney U Test). No differences were seen in the distribution of skin tumors between dorsal skin and ears between the two genotypes of mice in this experiment.
Figure 5. UVB-induced skin carcinogenesis in wild-type and BK5.Stat3C mice.
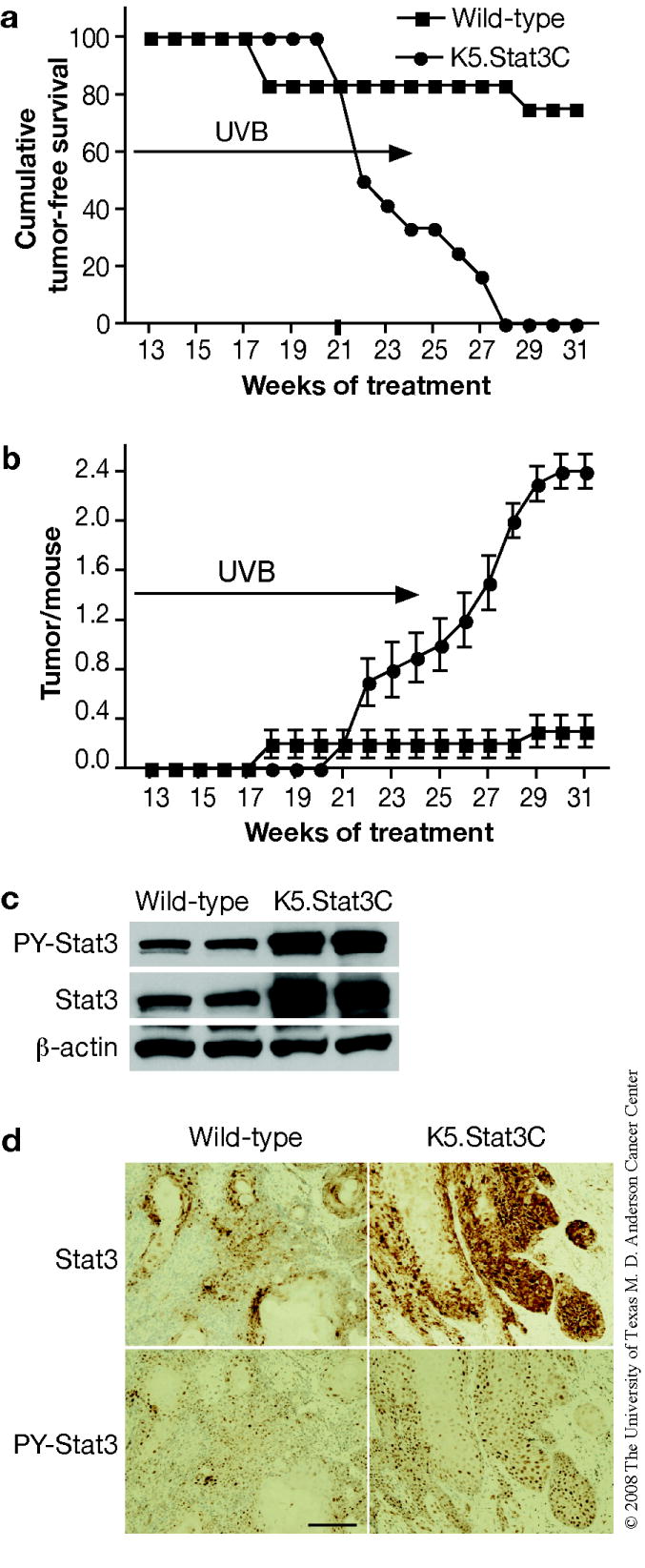
Groups of mice (n=12) were exposed to 1,200 J/m2 of UVB three times per week. The dose was increased gradually by increments of 25% weekly until 4,500 J/m2 of UVB was reached. UVB irradiation was stopped at 25 weeks and mice were kept without treatment until 31 weeks. (a) Percentage of mice without skin tumors. (b) Average number of skin tumors per mouse (the mean ± SEM). (c) Western blot analysis of Stat3 and phosphorylated Stat3 in skin tumors from both genotypes. (d) Immunohistochemical staining of Stat3 and phosphorylated Stat3 in a representative section of skin tumors. Scale bar: 100 μm.
Western blot and immunohistochemical analyses (Figure 5c and d, respectively) demonstrated that UVB-induced SCCs from K5.Stat3C mice had significantly higher levels of both total Stat3 and phospho (PY)-Stat3. Both total Stat3 and phospho-Stat3 also showed a high degree of nuclear localization in the tumors, especially those from K5.Stat3C mice.
Stat3 deficiency reduces susceptibility to UVB-induced skin carcinogenesis
To further confirm the role of Stat3 in UVB-induced skin carcinogenesis, groups of Stat3-deficient and wild-type mice (13 and 15 mice per group, respectively) were subjected to a UVB skin carcinogenesis regimen. For this experiment, mice were treated using a published protocol (Noonan et al., 2000). In contrast to K5.Stat3C mice, Stat3-deficient mice were resistant to UVB-induced skin carcinogenesis compared with wild-type controls (Figure 6a and b). In this regard, 93% of wild-type mice developed tumors on the ear and back skin at the end of the observation period (43 weeks), while only 31% of Stat3-deficient mice developed tumors (Figure 6a; p < 0.05, χ2-test). Again, the majority of these tumors were diagnosed as SCCs and only SCCs are tabulated in the figure. The average number of SCCs per mouse was significantly greater for the wild-type mice compared to Stat3-deficient mice (Figure 6b; p < 0.05, Mann Whitney U Test). Again, no differences were seen in the distribution of skin tumors between dorsal skin and ear skin between the two genotypes of mice in this experiment.
Figure 6. UVB-induced skin carcinogenesis in wild-type and Stat3-deficient mice.
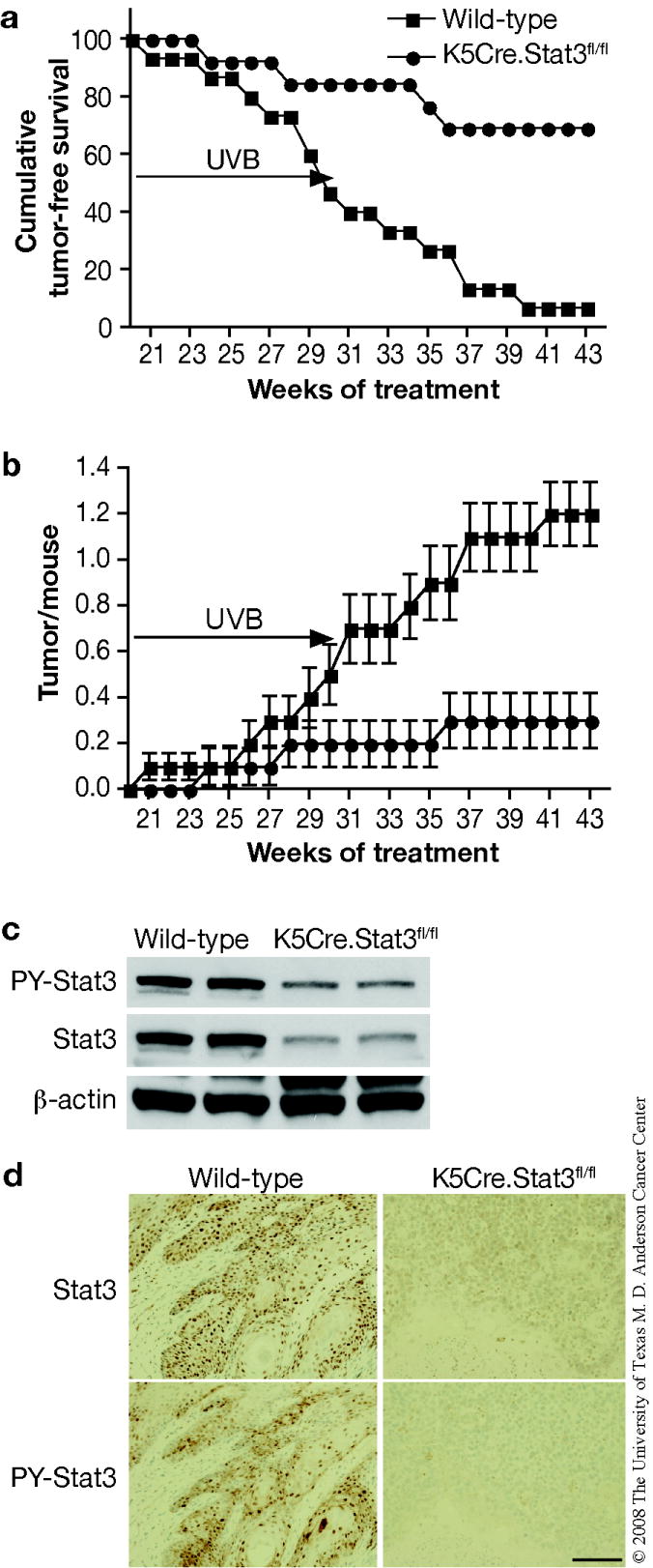
Groups of mice (wild-type, n=15; Stat3-deficient, n=13) were exposed to 2,200 J/m2 of UVB three times per week for weeks 1-6, 2,600 J/m2 of UVB for weeks 7-8, 3,000 J/m2 of UVB for weeks 9-10, 3,600 J/m2 of UVB for weeks 11-12, 4,050 J/m2 of UVB for weeks 13-14, 4,500 J/m2 of UVB for weeks 15-30. UVB irradiation was stopped at 31 weeks and mice were kept without treatment until 43 weeks. (a) Percentage of mice without skin tumors. (b) Average number of skin tumors per mouse (the mean ± SEM). (c) Western blot analysis of Stat3 and phosphorylated Stat3 in skin tumors from both genotypes. (d) Immunohistochemical staining of Stat3 and phosphorylated Stat3 in a representative section of skin tumors. Scale bar: 100 μm.
Western blot and immunohistochemical analyses (Figure 6c and d, respectively) demonstrated that UVB-induced tumors from K5Cre.Stat3fl/fl mice had lower levels of both total Stat3 and phospho-Stat3.
DISCUSSION
In this study, we demonstrate that Stat3 activation contributes significantly to keratinocyte survival and proliferation as well as skin tumor development following UVB exposure. In this regard, increased levels of activated Stat3 in the epidermis of K5.Stat3C mice protected keratinocytes from UVB-induced apoptosis, whereas keratinocytes from Stat3-deficient mice were highly sensitive to UVB-induced apoptosis compared with control mice over an extended time course following UVB-exposure (Figure 1). Further analyses provided direct evidence that Stat3 was required for survival of keratinocytes, including those located in the bulge region of hair follicles. In addition, we obtained evidence showing that Stat3 regulated survival of UV-photoproduct (i.e., CPD and 6,4-PPs) containing keratinocytes, including those located in the bulge-region of hair follicles. Furthermore, we demonstrated that Stat3 regulates the long-term epidermal proliferative and epidermal regenerative response following UVB-exposure. Finally, we showed that Stat3 is required for UVB-induced skin carcinogenesis through evaluation of the susceptibility of both K5.Stat3C transgenic mice and skin specific Stat3-deficient mice. Collectively, these studies provide direct evidence for a critical role of Stat3 in UVB-induced skin cancer.
Previously, we found that Stat3 deficiency increased, whereas expression of Stat3C decreased responsiveness of mouse keratinocytes to UVB-induced apoptosis (Sano et al., 2005b). Furthermore, we also reported similar findings following treatment with 7,12-dimethylbenz[a]anthracene (DMBA) (Chan et al., 2004b). Following topical treatment with DMBA, the majority of apoptotic cells induced in Stat3-deficient mice were localized in the bulge region of hair follicles. The current data show that a significant number of apoptotic cells induced in Stat3 deficient mice following UVB treatment were located in the bulge-region of hair follicles. Stat3 is known to regulate anti-apoptotic genes, such as Bcl-xL (Grad et al., 2000) and Survivin (Gritsko et al., 2006), which play a role in Stat3-mediated cell survival. As shown in Figure 4, the level of Bcl-xL in wild-type, K5.Stat3C and K5.Cre.Stat3fl/fl mice both in the absence and presence of UVB treatment correlated with the susceptibility to UVB-induced apoptosis. Previous studies have also shown that modulating the levels of Bcl-xL in keratinocytes significantly altered susceptibility to UVB-induced apoptosis (Pena et al., 1997; Umeda et al., 2003). Recently we have found that skin specific Bcl-xL deficient mice (i.e., K5Cre.Bcl-xLfl/fl mice) have reduced sensitivity to both chemical and UVB-induced skin carcinogenesis (DJ Kim, K Kataoka, S Sano and J DiGiovanni, unpublished data). Collectively, these findings demonstrate that Stat3 plays a critical role in maintaining the survival of keratinocytes (including keratinocyte stem cells located in the bulge-region of hair follicles) following exposure to both DMBA and UVB irradiation and that Bcl-xL likely accounts for at least some of Stat3 actions in this regard. Additional studies will be required to determine exactly which additional survival genes regulated by Stat3 also contribute to these effects in keratinocytes and keratinocyte stem cells. Nevertheless, these data argue strongly that Stat3 plays an important role in the initiation of UVB-induced skin tumors.
An interesting aspect of the current study was the analysis of UV-photoproduct containing cells in epidermis of wild-type, K5.Stat3C and Stat3-deficient mice following UVB irradiation. A recent study (Nijhof et al., 2007) reported that UV-photoproducts (i.e., 6-4 PPs and CPDs) accumulated in epidermal basal cells, including putative stem and progenitor cells following chronic UVB exposure to skin of SKH-1 hairless mice. Our current results are consistent with these recent findings and show that both 6-4 PP-and CPD-containing keratinocytes can be found in hair follicles after only a single, large UVB exposure and that these cells appear to persist longer than those in the interfollicular epidermis. In human skin, NER repairs both CPD and 6-4 PP, but 6-4 PP repair is much faster that CPD repair (de Laat et al., 1999). Consistent with this observation, 6-4 PP-containing cells were eliminated from the interfollicular epidermis within 5 days after UVB irradiation, whereas CPD-containing cells were found as late as 10 days after UVB irradiation in mouse skin (Figure 2). Our previous studies indicated that the antiapoptotic function of Stat3 was not associated with altered NER following UVB irradiation (Sano et al., 2005b), however, the data in Figure 2 demonstrates that Stat3 is required for maintaining survival of UV-photoproduct bearing keratinocytes. Consistent with this statement, we found that UV-photoproduct containing cells were more prominent in hair follicles from K5.Stat3C mice. These data further support the conclusion that Stat3 plays a role in the initiation of UVB carcinogenesis through its affects on DNA damaged keratinocytes, including DNA-damaged bulge-region keratinocytes.
In addition to its antiapoptotic role, Stat3 activation promotes epidermal proliferation by regulating specific target genes, such as cyclin D1, cyclin E and c-myc (Chan et al., 2004b; Kim et al., 2007; Leslie et al., 2006). Previous studies showed that Stat3 deficiency resulted in a reduction of TPA-induced epidermal hyperproliferation (Chan et al., 2004b), whereas Stat3C expression increased TPA-induced epidermal hyperproliferation (Chan et al., 2008). Similarly, UVB-mediated epidermal hyperproliferation and regeneration were significantly increased in Stat3C mice and decreased in Stat3-deficient mice (see Figure 3). The levels of both cyclin D1 and cyclin E in the absence and presence of UVB-exposure correlated with Stat3 activation status and susceptibility to UVB-induced epidermal proliferation (see Figure 4). Cyclin D1 levels are known to increase throughout UVB-induced skin carcinogenesis in mice (Cooper et al., 2003; Kim et al., 2002a; Kim et al., 2002b). Furthermore, cyclin D1 has been shown to be important for chemically-mediated skin tumorigenesis. In this regard, cyclin D1 knockout mice display a significantly reduced sensitivity to two-stage chemical carcinogenesis (Robles et al., 1998). Thus, our current data, which are very similar to our earlier studies showing that TPA-induced epidermal hyperproliferation in K5.Stat3C and Stat3-deficient mice (Chan et al., 2008; Chan et al., 2004b) correlated with levels of cyclin D1, cyclin E and c-myc, suggest that cyclin D1 together with other Stat3 regulated genes that control G1→ S phase transition are responsible, in part, for its effects on UVB-induced skin carcinogenesis.
Finally, UVB skin carcinogenesis studies revealed that Stat3C mice were more susceptible to UVB-induced skin carcinogenesis with increased skin tumor development, whereas Stat3-deficient mice were more resistant to UVB-induced skin carcinogenesis with reduced skin tumor formation (Figures 5 and 6, respectively). These results obtained for UVB-induced skin carcinogenesis are remarkably similar to the results obtained from two-stage skin carcinogenesis studies using DMBA as the initiator and TPA as the promoter (Chan et al., 2008; Chan et al., 2004b). Interestingly, Stat3-deficient mice were completely resistant to skin tumor development induced by the DMBA/TPA, initiation-promotion protocol (Chan et al., 2004b), whereas about 30% of Stat3-deficient mice developed tumors induced by the UVB carcinogenesis protocol. Thus, under the current experimental conditions, Stat3-deficient mice, although highly resistant were not completely resistant to UVB-induced skin carcinogenesis. One possible explanation for this difference may be that during UVB carcinogenesis a subset of cells acquire additional genetic alterations that confer a growth advantage, even in the absence of Stat3. In support of this hypothesis are the data shown in Figure 6c and d. These data show that the UVB-induced tumors that grow in Stat3 deficient mice have low levels of Stat3 and phospho-Stat3. Further work will be required to verify this interesting hypothesis. Nevertheless, the function and underlying mechanisms for the role of Stat3 during UVB-induced skin carcinogenesis appear to be very similar to those observed for two-stage skin carcinogenesis (Chan et al., 2004a; Chan et al., 2008; Chan et al., 2004b; Kataoka et al., 2008; Kim et al., 2007; Sano et al., 2007).
In conclusion, using both skin-specific gain and loss of function Stat3 transgenic mice, we provide direct evidence that Stat3 is a critical regulator in UVB skin carcinogenesis by promoting cell survival and proliferation during UVB exposure. Further studies on specific Stat3 target genes involved in UVB-induced skin carcinogenesis will aid in understanding the role of Stat3 signaling in this process. Overall, these studies further support the suggestion that Stat3 is an important target for prevention of skin cancer.
MATERIALS AND METHODS
Animals
Development of transgenic mice whose keratinocytes express a constitutively active form of Stat3 (K5.Stat3C) and skin specific Stat3-deficient mice (K5Cre.Stat3fl/fl) have previously been described (Sano et al., 2005a; Sano et al., 1999). All K5.Stat3C and K5Cre.Stat3fl/fl mice were maintained on an FVB/N genetic background. Transgenic mice and non-transgenic littermates were used at 7-8 weeks of age and experiments were carried out with strict adherence to institutional guidelines for minimizing distress. The dorsal skin of each mouse was shaved 48 hours before UVB irradiation; only those mice in the resting phase of the hair cycle were used. For UVB irradiation, Westinghouse FS20 sun lamp bulbs with a peak emission at 313 nm were used. The fluence rate was measured with an IL1400A Radiometer/Photometer coupled to a SEL240/UVB-1/TD detector (International Light, Inc., Newburyport, MA). Each mouse was held in individual compartments of a plastic cage on a rotating base to prevent any differences in fluence. An UVB-transparent lid covering the radiation chamber was used to filter out the small amount of UVC radiation emitted.
Analysis of epidermal thickness, cell proliferation and apoptosis
For analysis of epidermal thickness, proliferation and apoptosis, groups of mice (n = 3) were irradiated with UVB at 1,200 J/m2 and sacrificed 1, 2, 5 and 10 days after irradiation. Epidermal cell proliferation, presented as the labeling index (LI), and epidermal thickness were determined as previously described (Chan et al., 2004b; Sano et al., 2005b). Epidermal apoptotic rates were also determined as previously described using caspase-3 staining (Chan et al., 2004b).
Immunohistochemistry of DNA photoproducts
Groups of mice (n = 3) were irradiated with UVB at 1,200 J/m2 and sacrificed 1, 2, 5 and 10 days after irradiation. After sacrifice, the dorsal skin was removed and fixed in 10% neutral-buffered formalin. To detect UVB-induced cyclobutane pyrimidine dimer (CPD) and pyrimidine (6-4) pyrimidone photoproduct (6-4 PP) formation, skin sections were deparaffinized, rehydrated in xylene followed by graded ethanol (100%, 95%) and endogenous peroxidase activity was blocked with 3% hydrogen peroxide in water for 10 min. Skin sections were then boiled in 1.0 mM EDTA buffer (pH 8.0) for 10 min for antigen retrieval. After cooling down slowly, skin sections were preincubated for 10 min at room temperature with Biocare blocking reagent (Biocare Medical Co., Concord, CA) to block non-specific antibody binding. To detect CPD formation, slides were incubated with a primary mouse monoclonal CPD antibody (Kamiya Biomedical Co., Seattle, WA) at 1:80,000 dilution for 1 hour at room temperature. To detect 6-4 PP formation, slides were incubated with a primary mouse monoclonal 6-4 PP antibody (MBL International Co., Woburn, MA) at 1:2,000 dilution overnight at 4°C. The slides were then incubated with biotinylated rabbit anti-mouse antibody, followed by streptavidin-horseradish peroxidase conjugate for detection and developed with diaminobenzidine as a substrate. Photoproduct-positive keratinocytes were counted microscopically in at least three nonoverlapping fields in sections from each mouse and calculated as the percent of photoproduct-positive cells/cm.
Western blot analysis
Epidermis was collected and protein lysates prepared as previously described (Kataoka et al., 2008). Western blots were incubated for 2 hours at room temperature with specific primary antibodies for Stat3, phospho-Stat3, and Bcl-xL (Cell Signaling Technology Inc., Beverly, MA), cyclin D1, cyclin E, (Santa Cruz Biotechnology Inc., Santa Cruz, CA), and β-actin (Sigma-Aldrich). Blots were then washed with TPBS and subjected to corresponding horseradish peroxidase-conjugated secondary antibodies against rabbit or mouse (Amersham Biosciences, Arlington Heights, IL), washed again with TPBS and protein expression was detected using ECL Western Blotting Substrate (Pierce Biotechnology Inc., Rockford, IL).
UV skin carcinogenesis bioassays
In the first UV skin carcinogenesis experiment, K5.Stat3C and control FVB mice (n=12 per group) were exposed to 1,200 J/m2 of UVB three times per week. The dose was increased gradually by increments of 25% weekly until 4,500 J/m2 of UVB was reached. A maximal dose of 4,500 J/m2 was used for weeks 7-24 and was stopped at 25 weeks and mice were kept without treatment until week 31. In the second UV skin carcinogenesis experiment, an incrementally graded UV protocol described previously (Noonan et al., 2000) was modified and used to reduce ear papilloma formation. Stat3-deficient (K5Cre.Stat3fl/fl, n=13) and control (K5Cre.Stat3wt/wt, n=15) mice were exposed to 2,200 J/m2 of UVB three times per week for weeks 1-6, 2,600 J/m2 of UVB for weeks 7-8, 3,000 J/m2 of UVB for weeks 9-10, 3,600 J/m2 of UVB for weeks 11-12, 4,050 J/m2 of UVB for weeks 13-14, 4,500 J/m2 of UVB for weeks 15-30. UVB irradiation was stopped at 31 weeks and mice were kept without treatment until 43 weeks. In both experiments, mice were monitored for tumor formation weekly. Skin tumors developed on dorsal skin and ear were counted following the first tumor appearance. After termination of experiments, mice were sacrificed and tumors were collected for histopathologic diagnosis.
Acknowledgments
This work was supported by NCI grants CA76520, CA105345, University of Texas M.D. Anderson Cancer Center support Grant CA16672, and National Institute of Environmental Health Sciences Center Grant ES07784. Funding as an Odyssey Fellow (to D. J. Kim) was supported by the Odyssey Program and The H-E-B Award for Scientific Achievement at The University of Texas M.D. Anderson Cancer Center.
Contributor Information
Dae Joon Kim, Department of Carcinogenesis, The University of Texas M.D. Anderson Cancer Center, Science Park-Research Division, Smithville, Texas, USA.
Joe M. Angel, Department of Carcinogenesis, The University of Texas M.D. Anderson Cancer Center, Science Park-Research Division, Smithville, Texas, USA
Shigetoshi Sano, Department of Dermatology, Osaka University Graduate School of Medicine, Osaka, Japan.
John DiGiovanni, Department of Carcinogenesis, The University of Texas M.D. Anderson Cancer Center, Science Park-Research Division, Smithville, Texas, USA.
References
- Ahsan H, Aziz MH, Ahmad N. Ultraviolet B exposure activates Stat3 signaling via phosphorylation at tyrosine705 in skin of SKH1 hairless mouse: a target for the management of skin cancer? Biochem Biophys Res Commun. 2005;333:241–6. doi: 10.1016/j.bbrc.2005.05.106. [DOI] [PubMed] [Google Scholar]
- Alvarez JV, Febbo PG, Ramaswamy S, Loda M, Richardson A, Frank DA. Identification of a genetic signature of activated signal transducer and activator of transcription 3 in human tumors. Cancer Res. 2005;65:5054–62. doi: 10.1158/0008-5472.CAN-04-4281. [DOI] [PubMed] [Google Scholar]
- Aziz MH, Manoharan HT, Verma AK. Protein kinase C epsilon, which sensitizes skin to sun’s UV radiation-induced cutaneous damage and development of squamous cell carcinomas, associates with Stat3. Cancer Res. 2007;67:1385–94. doi: 10.1158/0008-5472.CAN-06-3350. [DOI] [PubMed] [Google Scholar]
- Bowman T, Garcia R, Turkson J, Jove R. STATs in oncogenesis. Oncogene. 2000;19:2474–88. doi: 10.1038/sj.onc.1203527. [DOI] [PubMed] [Google Scholar]
- Bromberg J. Stat proteins and oncogenesis. J Clin Invest. 2002;109:1139–42. doi: 10.1172/JCI15617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bromberg JF, Wrzeszczynska MH, Devgan G, Zhao Y, Pestell RG, Albanese C, et al. Stat3 as an oncogene. Cell. 1999;98:295–303. doi: 10.1016/s0092-8674(00)81959-5. [DOI] [PubMed] [Google Scholar]
- Chan KS, Carbajal S, Kiguchi K, Clifford J, Sano S, DiGiovanni J. Epidermal growth factor receptor-mediated activation of Stat3 during multistage skin carcinogenesis. Cancer Res. 2004a;64:2382–9. doi: 10.1158/0008-5472.can-03-3197. [DOI] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kataoka K, Abel E, Carbajal S, Beltran L, et al. Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene. 2008;27:1087–94. doi: 10.1038/sj.onc.1210726. [DOI] [PubMed] [Google Scholar]
- Chan KS, Sano S, Kiguchi K, Anders J, Komazawa N, Takeda J, et al. Disruption of Stat3 reveals a critical role in both the initiation and the promotion stages of epithelial carcinogenesis. J Clin Invest. 2004b;114:720–8. doi: 10.1172/JCI21032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper SJ, MacGowan J, Ranger-Moore J, Young MR, Colburn NH, Bowden GT. Expression of dominant negative c-jun inhibits ultraviolet B-induced squamous cell carcinoma number and size in an SKH-1 hairless mouse model. Mol Cancer Res. 2003;1:848–54. [PubMed] [Google Scholar]
- Darnell JE., Jr STATs and gene regulation. Science. 1997;277:1630–5. doi: 10.1126/science.277.5332.1630. [DOI] [PubMed] [Google Scholar]
- Daya-Grosjean L, Sarasin A. The role of UV induced lesions in skin carcinogenesis: an overview of oncogene and tumor suppressor gene modifications in xeroderma pigmentosum skin tumors. Mutat Res. 2005;571:43–56. doi: 10.1016/j.mrfmmm.2004.11.013. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR, van Kranen HJ, Mullenders LH. UV-induced DNA damage, repair, mutations and oncogenic pathways in skin cancer. J Photochem Photobiol B. 2001;63:19–27. doi: 10.1016/s1011-1344(01)00199-3. [DOI] [PubMed] [Google Scholar]
- de Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev. 1999;13:768–85. doi: 10.1101/gad.13.7.768. [DOI] [PubMed] [Google Scholar]
- Frank DA. StAT signaling in cancer: insights into pathogenesis and treatment strategies. Cancer Treat Res. 2003;115:267–91. doi: 10.1007/0-306-48158-8_11. [DOI] [PubMed] [Google Scholar]
- Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer. 2001;1:22–33. doi: 10.1038/35094000. [DOI] [PubMed] [Google Scholar]
- Grad JM, Zeng XR, Boise LH. Regulation of Bcl-xL: a little bit of this and a little bit of STAT. Curr Opin Oncol. 2000;12:543–9. doi: 10.1097/00001622-200011000-00006. [DOI] [PubMed] [Google Scholar]
- Gritsko T, Williams A, Turkson J, Kaneko S, Bowman T, Huang M, et al. Persistent activation of stat3 signaling induces survivin gene expression and confers resistance to apoptosis in human breast cancer cells. Clin Cancer Res. 2006;12:11–9. doi: 10.1158/1078-0432.CCR-04-1752. [DOI] [PubMed] [Google Scholar]
- Hill LL, Ouhtit A, Loughlin SM, Kripke ML, Ananthaswamy HN, Owen-Schaub LB. Fas ligand: a sensor for DNA damage critical in skin cancer etiology. Science. 1999;285:898–900. doi: 10.1126/science.285.5429.898. [DOI] [PubMed] [Google Scholar]
- Ichihashi M, Ueda M, Budiyanto A, Bito T, Oka M, Fukunaga M, et al. UV-induced skin damage. Toxicology. 2003;189:21–39. doi: 10.1016/s0300-483x(03)00150-1. [DOI] [PubMed] [Google Scholar]
- Kataoka K, Kim DJ, Carbajal S, Clifford J, DiGiovanni J. Stage-specific disruption of Stat3 demonstrates a direct requirement during both the initiation and promotion stages of mouse skin tumorigenesis. Carcinogenesis. 2008;29:1108–1114. doi: 10.1093/carcin/bgn061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim AL, Athar M, Bickers DR, Gautier J. Stage-specific alterations of cyclin expression during UVB-induced murine skin tumor development. Photochem Photobiol. 2002a;75:58–67. doi: 10.1562/0031-8655(2002)075<0058:ssaoce>2.0.co;2. [DOI] [PubMed] [Google Scholar]
- Kim AL, Gautier J, Bickers DR, Athar M. Reduced cyclin D1 ubiquitination in UVB-induced murine squamous cell carcinomas. Biochem Biophys Res Commun. 2002b;298:377–82. doi: 10.1016/s0006-291x(02)02435-x. [DOI] [PubMed] [Google Scholar]
- Kim DJ, Chan KS, Sano S, Digiovanni J. Signal transducer and activator of transcription 3 (Stat3) in epithelial carcinogenesis. Mol Carcinog. 2007;46:725–31. doi: 10.1002/mc.20342. [DOI] [PubMed] [Google Scholar]
- Kraemer KH. Sunlight and skin cancer: another link revealed. Proc Natl Acad Sci U S A. 1997;94:11–4. doi: 10.1073/pnas.94.1.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie K, Lang C, Devgan G, Azare J, Berishaj M, Gerald W, et al. Cyclin D1 is transcriptionally regulated by and required for transformation by activated signal transducer and activator of transcription 3. Cancer Res. 2006;66:2544–52. doi: 10.1158/0008-5472.CAN-05-2203. [DOI] [PubMed] [Google Scholar]
- Levy DE, Darnell JE., Jr Stats: transcriptional control and biological impact. Nat Rev Mol Cell Biol. 2002;3:651–62. doi: 10.1038/nrm909. [DOI] [PubMed] [Google Scholar]
- Levy DE, Lee CK. What does Stat3 do? J Clin Invest. 2002;109:1143–8. doi: 10.1172/JCI15650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Turi TG, Schuck A, Freedberg IM, Khitrov G, Blumenberg M. Rays and arrays: the transcriptional program in the response of human epidermal keratinocytes to UVB illumination. Faseb J. 2001;15:2533–5. doi: 10.1096/fj.01-0172fje. [DOI] [PubMed] [Google Scholar]
- Li L, Shaw PE. Autocrine-mediated activation of STAT3 correlates with cell proliferation in breast carcinoma lines. J Biol Chem. 2002;277:17397–405. doi: 10.1074/jbc.M109962200. [DOI] [PubMed] [Google Scholar]
- Lippke JA, Gordon LK, Brash DE, Haseltine WA. Distribution of UV light-induced damage in a defined sequence of human DNA: detection of alkaline-sensitive lesions at pyrimidine nucleoside-cytidine sequences. Proc Natl Acad Sci U S A. 1981;78:3388–92. doi: 10.1073/pnas.78.6.3388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda M, Suzui M, Yasumatu R, Nakashima T, Kuratomi Y, Azuma K, et al. Constitutive activation of signal transducers and activators of transcription 3 correlates with cyclin D1 overexpression and may provide a novel prognostic marker in head and neck squamous cell carcinoma. Cancer Res. 2002;62:3351–5. [PubMed] [Google Scholar]
- Melnikova VO, Ananthaswamy HN. Cellular and molecular events leading to the development of skin cancer. Mutat Res. 2005;571:91–106. doi: 10.1016/j.mrfmmm.2004.11.015. [DOI] [PubMed] [Google Scholar]
- Mitchell DL, Nairn RS. The biology of the (6-4) photoproduct. Photochem Photobiol. 1989;49:805–19. doi: 10.1111/j.1751-1097.1989.tb05578.x. [DOI] [PubMed] [Google Scholar]
- Nijhof JG, van Pelt C, Mulder AA, Mitchell DL, Mullenders LH, de Gruijl FR. Epidermal stem and progenitor cells in murine epidermis accumulate UV damage despite NER proficiency. Carcinogenesis. 2007;28:792–800. doi: 10.1093/carcin/bgl213. [DOI] [PubMed] [Google Scholar]
- Noonan FP, Otsuka T, Bang S, Anver MR, Merlino G. Accelerated ultraviolet radiation-induced carcinogenesis in hepatocyte growth factor/scatter factor transgenic mice. Cancer Res. 2000;60:3738–43. [PubMed] [Google Scholar]
- Pena JC, Fuchs E, Thompson CB. Bcl-x expression influences keratinocyte cell survival but not terminal differentiation. Cell Growth Differ. 1997;8:619–29. [PubMed] [Google Scholar]
- Robles AI, Rodriguez-Puebla ML, Glick AB, Trempus C, Hansen L, Sicinski P, et al. Reduced skin tumor development in cyclin D1-deficient mice highlights the oncogenic ras pathway in vivo. Genes Dev. 1998;12:2469–74. doi: 10.1101/gad.12.16.2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano S, Chan KS, Carbajal S, Clifford J, Peavey M, Kiguchi K, et al. Stat3 links activated keratinocytes and immunocytes required for development of psoriasis in a novel transgenic mouse model. Nat Med. 2005a;11:43–9. doi: 10.1038/nm1162. [DOI] [PubMed] [Google Scholar]
- Sano S, Chan KS, Digiovanni J. Impact of Stat3 activation upon skin biology: A dichotomy of its role between homeostasis and diseases. J Dermatol Sci. 2007 doi: 10.1016/j.jdermsci.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Sano S, Chan KS, Kira M, Kataoka K, Takagi S, Tarutani M, et al. Signal transducer and activator of transcription 3 is a key regulator of keratinocyte survival and proliferation following UV irradiation. Cancer Res. 2005b;65:5720–9. doi: 10.1158/0008-5472.CAN-04-4359. [DOI] [PubMed] [Google Scholar]
- Sano S, Itami S, Takeda K, Tarutani M, Yamaguchi Y, Miura H, et al. Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. Embo J. 1999;18:4657–68. doi: 10.1093/emboj/18.17.4657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarasin A. The molecular pathways of ultraviolet-induced carcinogenesis. Mutat Res. 1999;428:5–10. doi: 10.1016/s1383-5742(99)00025-3. [DOI] [PubMed] [Google Scholar]
- Setlow RB, Carrier WL. Pyrimidine dimers in ultraviolet-irradiated DNA’s. J Mol Biol. 1966;17:237–54. doi: 10.1016/s0022-2836(66)80105-5. [DOI] [PubMed] [Google Scholar]
- Shen Y, Devgan G, Darnell JE, Jr, Bromberg JF. Constitutively activated Stat3 protects fibroblasts from serum withdrawal and UV-induced apoptosis and antagonizes the proapoptotic effects of activated Stat1. Proc Natl Acad Sci U S A. 2001;98:1543–8. doi: 10.1073/pnas.041588198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkson J, Jove R. STAT proteins: novel molecular targets for cancer drug discovery. Oncogene. 2000;19:6613–26. doi: 10.1038/sj.onc.1204086. [DOI] [PubMed] [Google Scholar]
- Umeda J, Sano S, Kogawa K, Motoyama N, Yoshikawa K, Itami S, et al. In vivo cooperation between Bcl-xL and the phosphoinositide 3-kinase-Akt signaling pathway for the protection of epidermal keratinocytes from apoptosis. Faseb J. 2003;17:610–20. doi: 10.1096/fj.02-0597com. [DOI] [PubMed] [Google Scholar]
- Wheeler DL, Martin KE, Ness KJ, Li Y, Dreckschmidt NE, Wartman M, et al. Protein kinase C epsilon is an endogenous photosensitizer that enhances ultraviolet radiation-induced cutaneous damage and development of squamous cell carcinomas. Cancer Res. 2004;64:7756–65. doi: 10.1158/0008-5472.CAN-04-1881. [DOI] [PubMed] [Google Scholar]