

Abstract
Oxidative stress and aggregation of the protein α-synuclein are thought to be key factors in Parkinson’s disease. Previous work shows that cytochrome c plus H2O2 causes tyrosine-dependent in vitro peroxidative aggregation of proteins, including α-synuclein. Here, we examine the role of each of α-synuclein’s four tyrosine residues and how the protein’s conformation affects covalent oxidative aggregation. When α-synuclein adopts a collapsed conformation, tyrosine 39 is essential for wild-type-like covalent aggregation. This lone N-terminal tyrosine, however, is not required for wild type-like covalent aggregation in the presence of a denaturant or when α-synuclein is present in non-covalent fibrils. We also show that pre-formed oxidative aggregates are not incorporated into non-covalent fibrils. These data provide insight as to how dityrosine may be formed in Lewy bodies seen in Parkinson’s disease.
Parkinson’s disease, the second most common neurodegenerative disorder (1), affects over 1.5 million people in the United States, with over 60,000 new diagnoses each year (2). The cellular hallmark of Parkinson’s disease is the presence of Lewy bodies, intracellular aggregates of α-synuclein (3), cytochrome c (4) and other proteins (5–7). α-Synuclein is a 140-amino acid, intrinsically disordered protein (8) with three distinct regions (Fig. 1A). The N-terminal region is positively charged, the hydrophobic core (also known as the non-amyloid component) comprises residues 61–90, and the C-terminal region is negatively charged. Tyrosines are essential for α-synuclein oxidative aggregation (9). As shown in Fig. 1A, the protein has four unevenly distributed tyrosine residues, one (Y39) near the N-terminus and three (Y125, Y133, and Y136) near the C-terminus (the protein has no tryptophan residues). Although disordered, the protein adopts a compact state (10–13) wherein the charged termini collapse around the hydrophobic core (Fig. 1B). Little is known about the exact mechanism of Parkinson’s disease at the molecular level, but both covalent and non-covalent aggregation of α-synuclein are thought to play a key role (14).
Figure 1.

∝-Synuclein structure and conformation. A schematic representation of α-synuclein’s primary structure showing relevant regions, net charges, and position numbers (A), and a cartoon of its collapsed conformation (B). The positions of the tyrosine residues are indicated in green.
The oxidative stress marker dityrosine is detected in brain hydrolysates of murine models of Parkinson’s disease (15). Fenton-chemistry based oxidation systems (i.e., a transition metal plus H2O2) have been used extensively in vitro in attempts to reproduce in vivo covalent aggregation of α-synuclein, but these systems are probably not accurate mimics (16, 17). For instance, although dityrosine (tyrosines with a covalent bond between the 3/5 carbon atoms) is observed in Parkinson’s disease models, the covalent aggregation induced by several Fenton chemistry systems is tyrosine-independent (17). In fact, the HO• generated by Cu2+ plus H2O2 impedes dityrosine formation in the Alzheimer’s disease protein, Aβ (16). The cytochrome c/H2O2 oxidation system (4), which we call the peroxidative system, is a better model for α-synuclein covalent aggregation for two reasons. First, the peroxidative system causes tyrosine-dependent covalent aggregation of α-synuclein (9), similar to the dityrosine seen in murine models of Parkinson’s disease. Second, the peroxidative system causes the direct transfer of a free radical from cytochrome c to the acceptor protein (18, 19) without an HO• intermediate.
In addition to covalent aggregation, α-synuclein aggregates non-covalently into fibrils. The α-synuclein fibrils observed in patients with Parkinson’s are linear rods, 5–10 nm in diameter, much like those seen in other amyloid diseases (20). The fibrils comprise insoluble cross-β-sheets, and their growth in vitro exhibits a sigmoidal time dependence (21). Prior to fibril growth there is a lag, the length of which depends on factors such as protein concentration and pH (22). It is likely that structured intermediates, called nuclei, are required for fibril growth (23, 24). Following the lag is a period of elongation in which fibril concentration increases exponentially and plateaus (20). Fibril growth can be monitored by using thioflavin-T, which experiences a shift in its excitation spectrum when bound to β-sheets in fibrils, allowing it to be selectively excited at 442 nm (25).
Here, we shed light on the process of both peroxidative and non-covalent aggregation of α-synuclein and how these forms of aggregation may be related. Specifically, we address the role of each tyrosine residue and show how the protein’s conformation affects aggregation.
EXPERIMENTAL PROCEDURES
Preparation of Wild-Type and Mutant/Variant Recombinant Human α-Synucleins and Cytochromes
c-Twenty-four human α-synuclein mutants in the pT7-7 vector were created by using a site-directed mutagenesis kit (QuickChange, Stratagene, La Jolla, CA). The wild-type construct and the mutants with one, two, or three tyrosine codons converted to phenylalanine codons were created with and without the valine 3 codon converted to a cysteine codon for labeling purposes. Additionally, the wild-type construct was created with the valine 66 codon converted to cysteine codon, also for labeling purposes. Wild-type and variant α-synucleins were purified as previously described (26) except that the freeze/thaw step was eliminated. Protein concentration was determined with the Lowry method (27) (Modified Lowry Protein Assay Kit, Pierce, Rockford, IL) using human recombinant cytochrome c (28) as a standard. After purification, proteins were aliquoted and lyophilized for storage at −80°C.
Wild-type human cytochrome c in the pBTR vector (28) was produced with and without lysine 39 converted to cysteine for labeling purposes. The cytochrome c variants were purified as previously described (28). Protein concentration was determined from the absorbance at 410 nm by using a molar extinction coefficient of 106.1 mM−1cm−1 (29). After purification, proteins were aliquoted and lyophilized for storage at −80°C.
Alexa Fluor Labeling
Sixty nmol of V3C α-synuclein were reacted with 300 nmol of Alexa Fluor 633 C5 maleimide (Invitrogen, Carlsbad, CA) in 20 mM Tris, pH 7.5 for 2 h at room temperature. The reaction was allowed to proceed for 18–66 h at 4°C. Unreacted cysteines were blocked with 3 mmol of iodoacetamide (Sigma, St. Louis, MO). Unreacted dye was removed with a two-step gel filtration chromatography process. First, the bulk of the free dye was removed by using a HiTrap desalting column (GE Healthcare, Uppsala, Sweden) in 20-mM phosphate buffer, pH 7.4, containing 150-mM NaCl and 30% v/v acetonitrile. A subsequent, more rigorous, purification was performed by using a Superdex200 10/300 column (GE Healthcare) in the same buffer. Fractions with no remaining unreacted dye were dialyzed (7000 MWCO cassette, Pierce) into water, aliquoted and lyophilized for storage at −80°C. Alexa Fluor labeling efficiencies ranged from 31 to 85%, with most variants showing efficiencies between 45 and 55%.
The K39C variant of cytochrome c was labeled and purified in the same way by using Alexa Fluor 488 C5 maleimide (Invitrogen).
Covalent Aggregation Assays
α-Synuclein (100 μM, 9:1 unlabeled:Alexa Fluor labeled for wild-type), cytochrome c (10 μM, 1:3 unlabeled:Alexa Fluor labeled) and H2O2 (10 mM) were reacted in phosphate-buffered saline (PBS: 20-mM Na2HPO4, pH 7.4, 150-mM NaCl) at 37°C for 90 min. To compensate for the varying Alexa Fluor labeling efficiencies of α-synuclein variants, the amount of labeled α-synuclein was adjusted so that each variant had the same total fluorescence as the wild-type control sample. Control experiments show that this adjustment does not affect our conclusions. For the cytochrome c control reaction, 100-μM cytochrome c (100% AlexaFluor labeled) and 10-mM H2O2 were reacted under the same conditions as above. Samples were resolved by electrophoresis on 10–20% gradient sodium dodecyl sulfate (SDS1) polyacrylamide gels (PAG) (Criterion, BioRad, Hercules, CA) for 75 min at 200 V. Gels were analyzed for fluorescence with a VersaDoc MP imager (BioRad) and for total protein content with Coomassie Blue staining.
Fibril Formation
α-Synuclein (200 μM) in PBS with 1-mM EDTA was shaken at 37°C and 225 rpm for 48 h. Fibril growth was quantified via thioflavin-T fluorescence in a 96-well plate by using the VersaDoc MP imager. Cytochrome c (final concentration 20 μM) and H2O2 (final concentration 20 mM) were added to the reaction either at the beginning or at the end of the shaking period. Fibrils were separated from α-synuclein monomers and smaller aggregates by centrifugation at 1.7 × 104 g for 10 min. Fibrils (pellet) and small aggregates (supernatant) were treated with SDS and boiled for 10 min before being analyzed by SDS-PAG electrophoresis as described above.
RESULTS
Alexa Fluor labeling and tyrosine-dependent α-synuclein covalent aggregation
Labeling α-synuclein and cytochrome c with different dyes, Alexa Fluor 633 (green) and 488 (red), respectively, allows us to follow each protein. Wild-type and the variant containing no tyrosines were combined with cytochrome c and H2O2. Fig. 2A shows the results of the reaction between cytochrome c and H2O2 as a standard. As shown in Fig. 2B, cytochrome c and H2O2, as well as the tyrosines in α-synuclein, are required for oxidative aggregation. This result agrees with our previous work with Coomassie Blue and anti-α-synuclein antibody detection (9). In all peroxidative aggregation reactions, a population of α-synuclein monomer is left unreacted, even after exposure to additional cytochrome c and H2O2. This result indicates that some portion of the α-synuclein is rendered incapable of covalent aggregation after exposure to cytochrome c and H2O2. This effect has also been seen when human neuroglobin is exposed to peroxide (30). Changing the position of the Alexa Fluor label in α-synuclein from position 3 (Fig. 2B, lanes 1–4) to 66 (data not shown) does not change aggregation. We conclude that Alexa Fluor labels on α-synuclein and cytochrome c allow facile detection of peroxidative covalent aggregation of α-synuclein.
Figure 2.

Alexa Fluor labeling detects peroxidative aggregation. (A) Cytochrome c was incubated with and without H2O2. (B) ∝-Synuclein, either the wild-type protein with the Alexa Fluor label at position 3 (WT, lanes 1–4), or a no tyrosine variant with the Alexa Fluor label at position 3 (noY, lanes 5–8) were combined with various combinations of cytochrome c and H2O2 for 90 min., separated on a 10–20% gradient polyacrylamide gel, and visualized by fluorescence (green, α-synuclein; red, cytochrome c).
Reactivity of the tyrosines in α-synuclein to covalent aggregation
To examine the chemical reactivity of each tyrosine, four variants, each containing only one tyrosine, were reacted with cytochrome c and H2O2. Our conditions do not promote non-covalent aggregation of α-synuclein. Fig. 3A shows the reaction between cytochrome c and H2O2 as a standard. Fig. 3B shows the differing ability of each tyrosine to form inter-molecular dityrosine bonds. Tyrosines 133 and 136 are the most reactive, as shown by distinct dimer formation and little α-synuclein degradation. Streaking of the α-synuclein because of random backbone cleavage and large amounts of degraded α-synuclein indicate that tyrosine 125 is less able to accept a radical from cytochrome c. Lack of α-synuclein dimer and large amounts of degradation indicate that tyrosine 39 is the least reactive. Tyrosine 39 is so unreactive that it does not form an intact heterodimer with cytochrome c as seen in the other variants. Instead, some of the free radicals generated cause degradation of the heterodimer. In summary, the chemical reactivities of the tyrosines in α-synuclein to form dityrosines increase in the following order: 39, 125, 133/136.
Figure 3.

Every tyrosine in α-synuclein is reactive, but to varying extents. (A) Cytochrome c was incubated with H2O2. (B) The wild-type protein (WT) and single tyrosine-containing variants of α-synuclein were reacted with cytochrome c and H2O2 for 90 minutes, and treated as described in the caption to Figure 2.
Tyrosine 39 is essential for wild-type-like covalent aggregation of native α-synuclein
To determine how each tyrosine contributes to covalent aggregation, seven variants, each containing two or three tyrosines, were reacted with cytochrome c and H2O2. The conditions used do not promote non-covalent aggregation of α-synuclein. Fig. 4A shows the reaction between cytochrome c and H2O2 as a standard. Fig. 4B shows the participation of each tyrosine in oxidative aggregation. When tyrosine 125, 133, or 136 is removed (Fig. 4B, lanes 3–5), covalently aggregated species are populated in the same manner as for the wild-type protein (lane 1). When the least reactive tyrosine, tyrosine 39, is removed (Fig. 4B, lane 2), even-numbered aggregates (dimer, tetramer, and perhaps hexamer and octamer) are favored over the trimer. In general, more uniform covalent aggregate populations are observed when α-synuclein has at least one tyrosine at each end. This observation is supported by the data in Figure 4C, where variants with tyrosine 39 and one other tyrosine are examined.
Figure 4.

At least one tyrosine on each end is required for wild-type-like covalent aggregation of collapsed α-synuclein. (A) Cytochrome c was incubated with H2O2. (B) The wild-type protein and variants containing three tyrosines or (C) two tyrosines were reacted with cytochrome c and H2O2 for 90 minutes, and treated as described in the caption to Figure 2. Legend at top indicates which tyrosines are present in each variant. α-Synuclein aggregate species are indicated on the right.
Changes in covalent aggregation induced by a denaturant
If α-synuclein were totally disordered, one would expect that wild-type-like covalent aggregation could be achieved if any two tyrosines are present. However, as shown in Fig. 4, tyrosine 39 is essential for wild-type-like covalent aggregation. To determine if protein conformation was the basis of this observation, wild-type α-synuclein or the variant with tyrosine 39 removed were reacted with cytochrome c and H2O2 in the presence and absence of the denaturant, guanidine hydrochloride (300 mM). A control experiment (not shown) illustrates that 300 mM guanidine hydrochloride does not affect radical formation as assessed by the bands formed in cytochrome c covalent aggregation with and without guanidine hydrochloride. Coomassie Blue staining was used because guanidine hydrochloride interferes with Alexa Fluor fluorescence. In the presence of guanidine hydrochloride (Fig. 5B–C, lane 2), large covalent aggregates are more heavily favored for both wild-type and Y39F α-synuclein compared to the reaction in the absence of guanidine hydrochloride (Fig. 5B–C, lane 1). In agreement with NMR-based experiments, these data show that α-synuclein must have some structure that can be destroyed by a denaturant (11).
Figure 5.
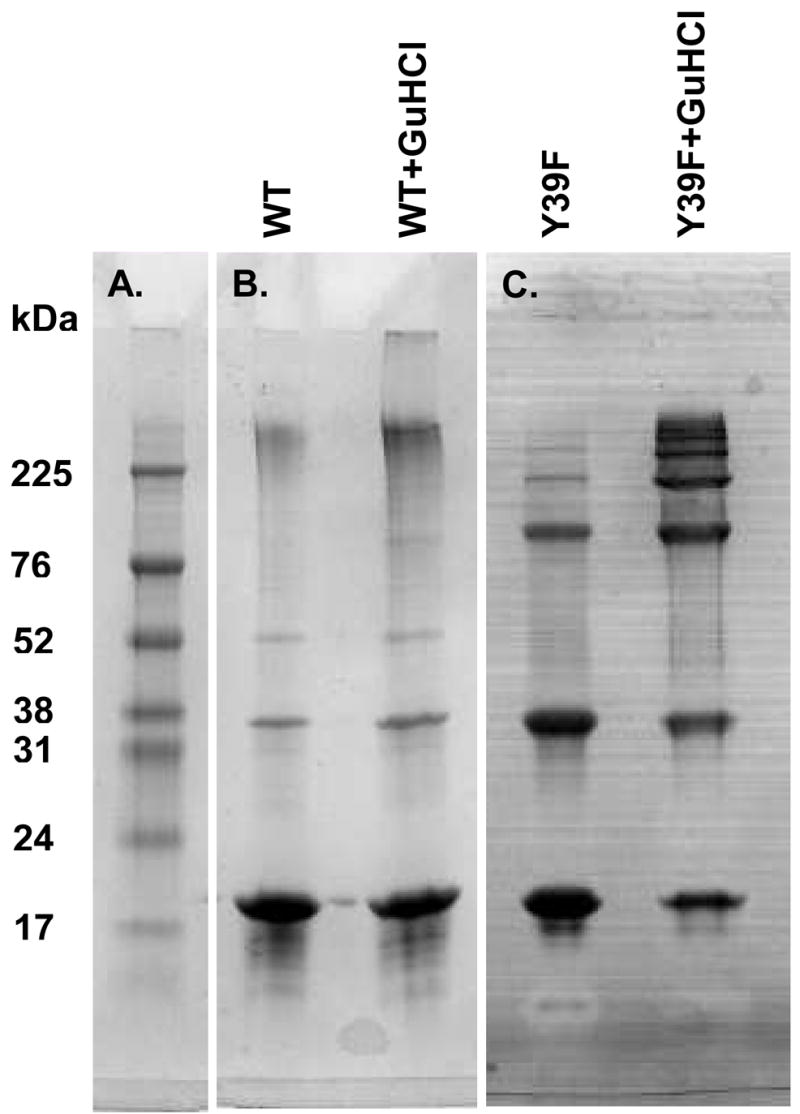
Tyrosine 39 is not required for full aggregation of denatured α-synuclein. (A) Molecular weight marker. (B) The wild-type protein (WT) and (C) the variant with tyrosine 39 removed (Y39F) were reacted with cytochrome c and H2O2 in the presence (lanes 2) or absence (lanes 1) of guanidine hydrochloride. Samples were separated on a 10–20% gradient polyacrylamide gel, and visualized by Coomassie Blue staining.
Fibril formation and oxidative aggregation
To determine if covalent aggregation affects the conformation required for non-covalent fibril formation, fibrils were grown from the wild-type protein. Coomassie Blue staining was used because the fibril growth conditions interfere with Alexa Fluor fluorescence. As a control, either cytochrome c or H2O2 were added either at the start or end of fibril growth. In each of these conditions, no covalent aggregates were detected in the supernatant or in the fibrils (data not shown). Fig. 6B shows the effects of exposing the fibrillization reaction to cytochrome c and H2O2 at the beginning or at the end of fibril growth. When peroxidation is introduced after 24 h of fibril growth (lanes 3 and 4), the covalent aggregates are observed in both the supernatant and the fibrils, and a large fibril pellet is observed. When peroxidation is introduced early, almost no fibrils are formed (as indicated by a miniscule pellet) and there is nearly a complete absence of covalent aggregates (lane 2). Instead, nearly all of the covalent aggregates are found in the supernatant (lane 1). Similar results are obtained for the variant containing only the tyrosine 39 to phenylalanine mutation (although the yield of fibrils is lower).
Figure 6.

Oxidative aggregation interferes with fibril formation. (A) Molecular weight marker. (B) The wild-type protein was reacted with cytochrome c and H2O2 before (lanes 1 and 2) or after (lanes 3 and 4) fibril formation. Fibrils were isolated by centrifugation. Fibrils (lanes 2 and 4) and the supernatant (lanes 1 and 3) were boiled with SDS, separated on 10–20% gradient polyacrylamide gels, and visualized with Coomassie Blue.
DISCUSSION
The present studies examine the role of α-synuclein conformation in its peroxidative aggregation. The conformations of this intrinsically disordered protein have been characterized by NMR, and the shape shown in Fig. 1B is just one of an ensemble of collapsed conformations adopted by α-synuclein (11). While the protein does not have a rigid secondary structure, these conformations have similar characteristics, namely that the charged termini collapse onto the hydrophobic core. In each conformation, the region surrounding tyrosine 39 abuts the core and C-teminus. This contact keeps tyrosine 39 partially protected from the solvent, which accounts for its low reactivity (Fig. 3). NMR-detected amide-proton exchange experiments also show that tyrosine 39 is protected from the solvent, as indicated by its low exchange rate (31). In each conformation, the region surrounding tyrosine 125 contacts three other regions (residues 11–13, 30–75, and 78–99) (12). Tyrosine 125 is in a region of increased rigidity compared to the rest of the protein (11, 12, 32), although the C-terminus as a whole is comparatively mobile. This synergy of rigidity and flexibility accounts for the decreased reactivity of tyrosine 125 compared to tyrosines 133 and 136, and its increased reactivity compared to tyrosine 39 (Fig. 3).
The collapsed conformation of α-synuclein explains why at least one tyrosine on each end of the protein is required to maintain a distribution of covalent aggregates similar to that observed for the wild-type protein (Fig. 7A). When C-terminal tyrosines from two α-synuclein monomers react, the collapsed conformation sterically hinders the two remaining C-terminal tyrosines from reacting (Fig. 7B and 7C). Without a tyrosine near the N-terminus, i.e. tyrosine 39, dimers are favored (Fig. 7B), but once a sufficient population of dimers accumulates, they tend to form tetramers, hexamers, and higher-order even-numbered aggregates (Fig. 7C). The lower reactivities of tyrosines 39 and 125 (Fig. 3) explain why variants with only these tyrosines (Fig. 4C, lane 1) tend to form fewer higher-order covalent aggregates. When α-synuclein is denatured with guanidine hydrochloride, an N-terminal tyrosine is no longer needed for wild-type-like covalent aggregation (Fig. 7D) because the three C-terminal tyrosines are more accessible.
Figure 7.

α-Synuclein conformation and covalent aggregation. When α-synuclein adopts a collapsed conformation, various covalently aggregated species are more equally populated when at least one tyrosine is present on each end of the protein (A). Even-numbered aggregates are favored when tyrosine 39 is removed (B and C). When α-synuclein is completely disordered, the largest aggregate species are favored, even if tyrosine 39 is removed (D). Covalent α-synuclein aggregates are unable to fold into the beta-sheet aggregation nucleus, but once the folded monomers have assembled into protofibrils, the termini are stacked, allowing covalent aggregation of any of the tyrosines (E).
α-Synuclein must undergo a folding event to form an aggregation nucleus (23) before forming fibrils (Fig. 7E). Covalently cross-linking the protein’s tyrosines prevents it from folding properly, so that, as shown in Fig. 6, covalent aggregates are not incorporated into fibrils. In addition, when oxidation is introduced at the start of the fibrillization process, little to no fibril growth is observed because the concentration of monomer is too low to allow fibrillization in the time frame of the experiment. Once fibrils have assembled (Fig. 7E), however, the termini are stacked (33) so that the tyrosines are solvent-exposed and accessible for oxidative cross-linking2. While tyrosine 39 is essential for fibril formation (34), an N-terminal tyrosine is not required for covalent aggregation within fibrils. This observation is similar to that which is observed when the protein is denatured by guanidine hydrochloride. In summary, our data indicate that both covalent and non-covalent aggregation of α-synuclein depend on its conformation and suggest that covalent aggregation occurs in Lewy bodies after the formation of fibrils
Acknowledgments
We thank the members of the Pielak group for insightful comments. We thank Sandy An and Melanie Wiley for technical assistance.
This work was supported by a NIH Director’s Pioneer Award (5DP1OD783) to GJP and a Foundation for Aging Research GlaxoSmithKline Foundation Award to RASR.
Footnotes
Abbreviations: PAG, polyacrylamide gel; PBS, phosphate buffered saline; SDS, sodium dodecyl sulfate
Footnote: Studies are underway to determine if the covalent aggregation observed when H2O2 and cytochrome c are reacted with fibrils is tyrosine dependent.
References
- 1.Hague SM, Klaffke S, Bandmann O. Neurodegenerative disorders: Parkinson’s disease and Huntington’s disease. J Neurol Neurosurg Psychiatr. 2005;76:1058–1063. doi: 10.1136/jnnp.2004.060186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.National Parkinson’s foundation.
- 3.Spillantini MG, Schmidt ML, Lee VMY, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–840. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 4.Hashimoto M, Takeda A, Hsu LJ, Takenouchi T, Masliah E. Role of cytochrome c as a stimulator of alpha-synuclein aggregation in Lewy body disease. J Biol Chem. 1999;274:28849–28852. doi: 10.1074/jbc.274.41.28849. [DOI] [PubMed] [Google Scholar]
- 5.D’Andrea MR, Ilyin S, Plata-Salaman CR. Abnormal patterns of microtubule-associated protein-2 (map-2) immunolabeling in neuronal nuclei and Lewy bodies in Parkinson’s disease substantia nigra brain tissues. Neuroscience Letters. 2001;306:137–140. doi: 10.1016/s0304-3940(01)01811-0. [DOI] [PubMed] [Google Scholar]
- 6.Gai WP, Power JHT, Blumbergs PC, Culvenor JG, Jensen PH. α-synuclein immunoisolation of glial inclusions from multiple system atrophy brain tissue reveals multiprotein components. J Neurochem. 1999;73:2093–2100. [PubMed] [Google Scholar]
- 7.Goldman JE, Yen SH, Chiu FC, Peress NS. Lewy bodies of Parkinson’s disease contain neurofilament antigens. Science. 1983;221:1082–1084. doi: 10.1126/science.6308771. [DOI] [PubMed] [Google Scholar]
- 8.Uversky VN. Neuropathology, biochemistry, and biophysics of α-synuclein aggregation. J Neurochem. 2007;103:17–37. doi: 10.1111/j.1471-4159.2007.04764.x. [DOI] [PubMed] [Google Scholar]
- 9.Olteanu A, Pielak GJ. Peroxidative aggregation of α-synuclein requires tyrosines. Protein Sci. 2004;13:2852–2856. doi: 10.1110/ps.04947204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Morar AS, Olteanu A, Young GB, Pielak GJ. Solvent-induced collapse of alpha synuclein and acid-denatured cytochrome c. Protein Sci. 2001;10:2195–2199. doi: 10.1110/ps.24301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bertoncini CW, Jung YS, Fernandez CO, Hoyer W, Greisinger C, Jovin TM, Zweckstetter M. Release of long range tertiary interactions potentiates aggregation of natively unstructured alpha-synuclein. Proc Natl Acad Sci U S A. 2005;102:1430–1435. doi: 10.1073/pnas.0407146102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dedmon MM, Lindorff-Larsen K, Christodoulou J, Vendruscolo M, Dobson CM. Mapping long-range interactions in alpha-synuclein using spin-label NMR and ensemble molecular dynamics simulations. J Am Chem Soc. 2004;127:476–477. doi: 10.1021/ja044834j. [DOI] [PubMed] [Google Scholar]
- 13.McNulty BC, Tripathy A, Young GB, Orans J, Pielak GJ. Temperature-induced reversible conformational change in the first 100 residues of alpha-synuclein. Protein Sci. 2005;15:602–608. doi: 10.1110/ps.051867106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schults CW. Lewy bodies. Proc Natl Acad Sci U S A. 2006;103:1661–1668. doi: 10.1073/pnas.0509567103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pannathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. Mass spectrometric quantification of 3-nitrotyrosine, ortho-tyrosine, and o,o’-dityrosine in brain tissue of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-treated mice, a model of oxidative stress in Parkinson’s disease. J Biol Chem. 1999;274:34621–34628. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 16.Atwood CS, Perry G, Zeng H, Kato Y, Jones WD, Ling K, Huang X, Moir RD, Wang D, Sayre LM, Smith MA, Chen SG, Bush AI. Copper mediates dityrosine cross-linking of Alzheimer’s amyloid beta. Biochemistry. 2004;43:560–568. doi: 10.1021/bi0358824. [DOI] [PubMed] [Google Scholar]
- 17.Norris EH, Giasson BI, Ischiropoulos H, Lee VM. Effects of oxidative and nitrative challeneges on α-synuclein fibrillogenesis involve distinct mechanisms of protein modificaions. J Biol Chem. 2003;278:27230–27240. doi: 10.1074/jbc.M212436200. [DOI] [PubMed] [Google Scholar]
- 18.Barr DP, Gunther MR, Deterding LJ, Tomer KB, Mason RP. ESR spin-trapping of a protein-derived tyrosyl radical from the reaction of cytochrome c with hydrogen peroxide. J Biol Chem. 1996;271:15498–15503. doi: 10.1074/jbc.271.26.15498. [DOI] [PubMed] [Google Scholar]
- 19.Deterding LJ, Barr DP, Mason RP, Tomer KB. Characterization of cytochrome c free radical reaction with peptides by mass spectrometry. J Biol Chem. 1998;273:12963–112869. doi: 10.1074/jbc.273.21.12863. [DOI] [PubMed] [Google Scholar]
- 20.Fink AL. The aggregation and fibrillation of α-synuclein. Acc Chem Res. 2006;39:628–634. doi: 10.1021/ar050073t. [DOI] [PubMed] [Google Scholar]
- 21.Nielsen L, Khurana R, Coats A, Frokjaer S, Brange J, Vyas S, Uversky VN, Fink AL. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry. 2001;40:6036–6046. doi: 10.1021/bi002555c. [DOI] [PubMed] [Google Scholar]
- 22.Lee CC, Nayak A, Sethuraman A, Belfort G, McRae GJ. A three-stage kinetic model of amyloid fibrillation . Biophys J. 2007;92:3448–3458. doi: 10.1529/biophysj.106.098608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shahi P, Sharma R, Sanger S, Kumar I, Jolly RS. Formation of amyloid fibrils via longitudinal growth of oligomers. Biochemistry. 2007;46:7365–7373. doi: 10.1021/bi7001136. [DOI] [PubMed] [Google Scholar]
- 24.Lomakin A, Chung DS, Benedek GB, Kirschner DA, Teplow DB. On the nucleation and growth of amyloid β protein fibrils: Detection of nuclei and quantitation of rate constants. Proc Natl Acad Sci U S A. 1996;93:1125–1129. doi: 10.1073/pnas.93.3.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.LeVine H. Quantification of β-sheet amyloid fibril structures with thioflavin T. Method Enzymol. 1999;309:274–284. doi: 10.1016/s0076-6879(99)09020-5. [DOI] [PubMed] [Google Scholar]
- 26.Conway KA, Lee SJ, Rochet JC, Ding TT, Williamson RE, Lansbury PT., Jr Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: Implications for pathogenesis and therapy. Proc Natl Acad Sci U S A. 2000;97:571–576. doi: 10.1073/pnas.97.2.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 28.Olteanu A, Patel CN, Dedmon MM, Kennedy S, Linhoff MW, Minder CM, Potts PR, Deshmukh M, Pielak GJ. Stability and apoptotic activity of recombinant human cytochrome c. Biochem Biophys Res Commun. 2003;312:733–740. doi: 10.1016/j.bbrc.2003.10.182. [DOI] [PubMed] [Google Scholar]
- 29.Margoliash E, Frohwirt N. Spectrum of horse-heart cytochrome c. Biochem J. 1959;71:570–572. doi: 10.1042/bj0710570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lardinois OM, Tomer KB, Mason RP, Deterding LJ. Identification of protein radicals formed in the human neuroglobin—H2O2 reaction using immuno-spin trapping and mass spectrometry. Biochemistry. 2008;47:10440–10448. doi: 10.1021/bi800771k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Croke RL, Sallum CO, Watson E, Watt ED, Alexandrescu AT. Hydrogen exchange of monomeric α-synuclein shows unfolded structure persists at physiological temperature and is independent of molecular crowding in escherichia coli. Protein Sci. 2008;17:1434–1445. doi: 10.1110/ps.033803.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bussell R, Jr, Eliezer D. Residual structure and dynamics in Parkinson’s disease associated mutants of alpha synuclein. J Biol Chem. 2001;276:45996–46003. doi: 10.1074/jbc.M106777200. [DOI] [PubMed] [Google Scholar]
- 33.Vilar M, Chou HT, Luhrs T, Maji SK, Riek-Loher D, Verel R, Manning G, Stahlberg H, Riek R. The fold of α-synuclein fibrils. Proc Natl Acad Sci U S A. 2008;105:8637–8642. doi: 10.1073/pnas.0712179105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ulrih NP, Barry CH, Fink AL. Impact of Tyr to Ala mutations on α-synuclein fibrillation and structural properties. Biochim Biophys Acta. 2008;1782:581–585. doi: 10.1016/j.bbadis.2008.07.004. [DOI] [PubMed] [Google Scholar]