

Abstract
The protective antigen (PA) component of the anthrax toxin (ATx) plays an essential role in the pathogenesis of the bioterrorism bacterium Bacillus anthracis. After oligomerization on the cell surface and docking of lethal factor and/or edema factor, PA is internalized and undergoes a conformational change when exposed to the low pH of the endosome to form a membrane-penetrating pore. While the structure of the PA prepore has been determined, precise structural information regarding the pore state remains lacking. Oxidative protein footprinting (OPF) can provide dynamic structural information about a protein complex through analysis of amino acid oxidation both before and after a conformational change. In this study, PA at pH 7.5 and 5.5 was exposed to hydroxyl radicals generated by ionizing radiation. Mass spectrometry was then used to both identify and quantitate the extent of oxidation of differentially modified residues. Several residues were found to be more readily oxidized at pH 7.5, most of which clustered toward the bottom plane of the prepore heptamer. Two amino acids had greater oxidation rates at pH 5.5, both found on the outer periphery of the prepore. When the OPF results were mapped to a current computational model of the pore, the accessibilities of some residues were consistent with their modeled positions in the pore (i.e., Y688 and V619/I620), while data for other residues (W346 and M350) appeared to conflict with the model. The results from this study illustrate the utility of OPF in generating empirical structural information for yet undetermined structures and offering opportunities for refinement for models thereof.
A keystone of the pathogenesis of the bioterrorism agent Bacillus anthracis is the anthrax toxin (ATx),1 a complex comprised of three distinct polypeptides. Each ATx complex contains seven copies of protective antigen (PA) and up to three copies of lethal factor (LF) and/or edema factor (EF) (1). The mechanism of action of ATx begins with the recruitment of 83 kDa PA subunits to the cell surface via binding to the ATx receptor capillary morphogenesis protein 2 (CMG2) (2) or anthrax toxin receptor/tumor endothelial marker 8 (3). Furin-like proteases then cleave a 20 kDa fragment from the N-terminus of PA, leaving activated PA63 subunits (4, 5) free to oligomerize on the surface of the cell (6). After PA63 oligomerization and clustering to lipid rafts (7), the cytotoxic LF and EF components dock onto the PA63 heptamers (1, 8). These membrane-associated toxin complexes are internalized into the cell in clathrin-coated pits, after which they are shuttled to early endosomes (7).
The low pH of the endosomal compartment has been shown to induce critical changes to the structures of all three ATx components. While the pH threshold for PA63 conformational changes can vary depending on the receptor to which the PA63 subunits are bound (9, 10), the acidic environment causes structural rearrangements of the PA63 heptamer, allowing penetration of the endosomal membrane (6, 11). This effect can also be seen in the absence of membranes in vitro, as the PA63 heptamer acquires SDS resistance at pH <7.5 (6, 11). In addition, LF and EF undergo partial unfolding at low pH values, allowing passage through the PA63 heptameric pore into the cytosol (reviewed in ref 12). LF is a metalloprotease that cleaves MAP-kinase-kinases, inactivating this pathway in the cell (13, 14). EF is a calmodulin- and calcium-dependent adenylyl cyclase which acts to increase intracellular cAMP levels (15, 16).
The three-dimensional structures of the PA monomer and the PA63 prepore heptamer have been determined by X-ray crystallography (17, 18). Typical of toxins in the β-barrel pore-forming toxin family (19), PA consists mostly of antiparallel β-sheets and contains four domains (Figure 1). The N-terminal furin cleavage site exists within calcium-binding domain 1. A large disordered loop exists within domain 2 (2β2–2β33) and is involved in the formation of the transmembrane channel. Domain 3 is regarded as being important for oligomerization of the protein, and domain 4 mediates binding to cell surface receptors (18). As mentioned, the PA63 prepore is ring-shaped and contains seven PA63 subunits, each with domains 1 and 2 facing inward and domains 3 and 4 outside of the cavity.
Figure 1.
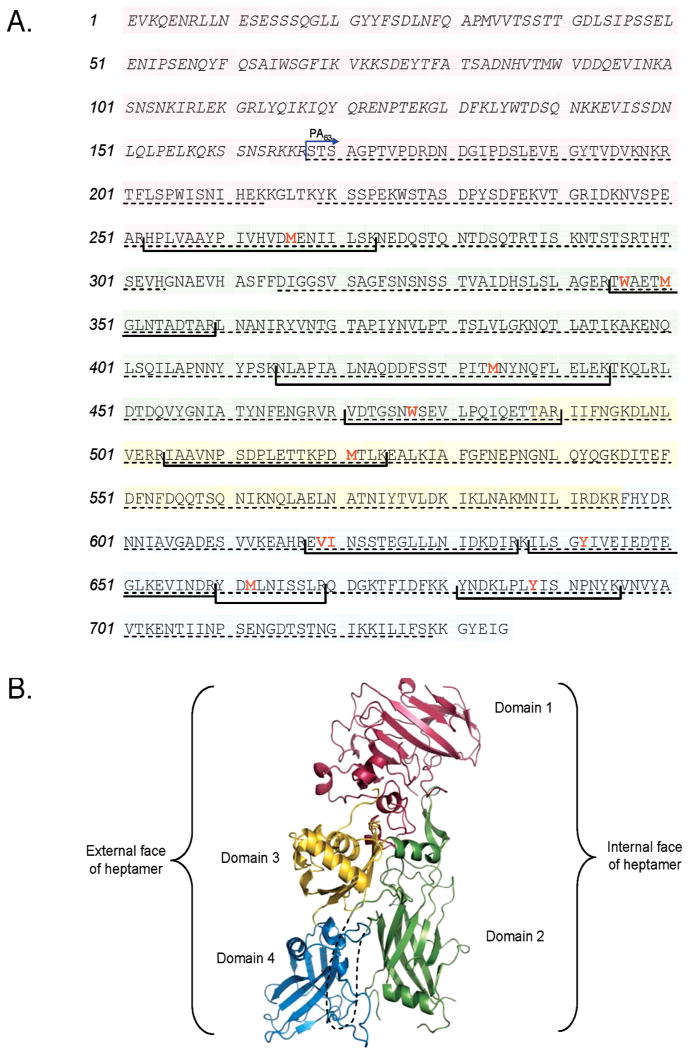
Structural map of PA. (A) Primary amino acid structure of PA highlighting domains 1 (pink), 2 (green), 3 (yellow), and 4 (blue). Proteolytic fragment PA20 consists of amino acids 1–167 and is italicized. Residues found to be oxidized in this study are in red and bold type, and tryptic fragments containing those residues are shown in brackets. Amino acid coverage by MS/MS analysis is shown with a dashed underline. (B) The 2.1 Å three-dimensional crystal structure of PA [PDB entry 1ACC (18)] with each of its four domains colored as in panel A. The disordered loop that forms part of the transmembrane stem domain (residues 302–325) is shown as a dashed line.
When the PA63 prepore heptamer is exposed to the aforementioned pH shift, major conformational changes that permit the 2β2–2β3 loops in domain 2 from each monomer to reach inside the oligomer and form the β-barrel transmembrane stem domain must take place. The disordered loop between amino acids 300 and 328 forms the tip of each β-hairpin, while two opposing β-sheets consisting of residues 288–300 and 328–335 must move away from the rest of domain 2 and insert down to create the stem structure. This restructuring would involve significant changes in both domains 2 and 4. Recent studies utilizing cysteine scanning and labeling techniques have provided evidence of this extended β-barrel structure (20, 21). Unfortunately, the structure of PA63 in its completed heptameric pore form has not been determined, likely due to the transmembrane configuration adopted by the toxin and the inherit difficulty in crystallizing such proteins and complexes.
The pH-dependent conformational change in PA63 is a potential target for structural analysis by oxidative protein footprinting (OPF). This technique uses mass spectrometry to monitor the amount of oxidation at multiple sites in a protein simultaneously. Previous foundational work showed that amino acid side chains are oxidized at a rate dependent primarily on two factors: the inherent reactivity of the oxidation site (22, 23) and the solvent accessibility of that residue in the protein structure. OPF has been previously used to study protein structure (24–31), protein folding and unfolding (32–35), and conformational changes in proteins (36–38). Additionally, OPF has been utilized in evaluating computational models of protein structure and providing experimental constraints during the construction of such models (39, 40).
In this study, we used OPF coupled with mass spectrometry to detect changes in the rate of oxidation of different targets in the PA63 heptamers as a function of decreasing pH. Sites exhibiting differential oxidation were then mapped to the known prepore structure of PA63 to improve our understanding of its pH-dependent conformational change, and comparisons are made to existing biochemical data and computational models of the PA63 pore.
Experimental Procedures
Materials
PA63 was purchased from List Biological Laboratories, Inc. (Campbell, CA). These preparations of PA63 were preactivated by the manufacturer by limited trypsinization of PA83 and subsequent removal of the 20 kDa cleavage product. Slide-a-lyzer MINI dialysis units were obtained from Pierce (Rockford, IL), and sequencing-grade trypsin was purchased from Promega (Madison, WI). Acetonitrile (ACN) was supplied by Caledon Laboratory Chemicals (Georgetown, ON), while formic acid (96%), NH4HCO3, catalase tetramer, and renin substrate tetradecapeptide were products from Sigma-Aldrich (St. Louis, MO). Purified deionized H2O was produced by a Hydro Picopure 2 water purification system.
Oxidation of PA63
One hundred micrograms of lyophilized PA63 was reconstituted in 110 μL of deionized H2O by incubation for 2 min at RT and intermittent gentle mixing. Two 50 μL aliquots of reconstituted PA63 were transferred to two separate Slide-a-lyzer MINI dialysis cups (10K molecular weight cutoff). One cassette was dialyzed against 1.1 mL of 10 mM NH4HCO3 (pH 7.5) and the second against 1.1 mL of 10 mM NH4HCO3 (pH 5.5), both in 1.5 mL microfuge tubes. After 1 h at RT with gentle mixing, the dialysis cups were placed in fresh buffer, and this step was repeated twice more to allow buffer exchange for a total of 4 h. After collection of the dialysate, exchange to the appropriate pH was verified with pH test strips. Where indicated, catalase tetramer was included in the NH4HCO3 buffers at a final concentration of 10 nM. Both low- and high-pH samples were split into equal volumes in 0.5 mL microfuge tubes, and one tube from each set was irradiated while the other set aside as a control. Tubes destined for irradiation were exposed to a dual point source 137Cs γ-irradiator and rotated on a turntable at ∼60 rpm for 40 min (dosage rate of 21.42 Gy/min). After irradiation, PA63 was thermally denatured by incubation at 65 °C for 90 min, and then 5 volumes of 10 mM NH4HCO3 (pH 7.5) was added to all tubes to restore pH neutrality for digestion. Samples were then digested with trypsin (at a 1:50 trypsin:PA63 ratio) at 37 °C overnight with gentle shaking. Digested samples were then lyophilized and stored at −80 °C until they were analyzed by mass spectrometry. In total, there were four conditions in each experiment (nonirradiated PA63 at pH 7.5, irradiated PA63 at pH 7.5, nonirradiated PA63 at pH 5.5, and irradiated PA63 at pH 5.5), and this experiment was performed three independent times.
Reversed Phase High-Performance Liquid Chromatography–Nanoelectrospray Ion Trap Tandem Mass Spectrometry
Trypsin-digested and lyophilized samples were reconstituted in 60 μL of deionized H2O, 10 μL of which was loaded onto an Agilent (Foster City, CA) 1200 Series HPLC chip, consisting of a 40 nL enrichment column and a 43 mm × 75 μm analytical column packed with ZORBAX 300SB C18 (5 μm), connected online to an Agilent MSD XCT Ultra series ion trap mass spectrometer. After enrichment, peptides were eluted from the LC chip by applying a linear gradient from 3 to 40% ACN with 0.1% FA over 95 min. A second gradient followed from 40 to 95% ACN with 0.1% FA over 20 min, followed by a hold at 95% ACN for 5 min. Analyses of eluted peptides were performed in enhanced positive ion mode. The spray voltage for all analyses was set at 1.8 kV, and the ICC smart target was set to accumulate 125000 ions before scan-out. MS/MS analyses were performed in a data-dependent acquisition mode that preferentially chose multiply charged ions and had a threshold of 5000 counts and/or a 5% relative abundance. The fragmentation voltage was set to 1.3 V for all MS/MS acquisitions. Mass resolution for the Agilent ion trap was typically ∼2500 full width at half-maximum, while the mass accuracy was better than 100 ppm. Using the Spectrum Mill MS Proteomics Workbench (Agilent), MS/MS fragmentation data were searched against a database consisting of only the PA63 primary sequence. Oxidized peptides were found by using a modification search for peptides with mass shifts in multiples of 16 Da. Other oxidative modifications [decarboxylation, His ring opening, etc. (23)] were not typically seen and, when detectable, were at levels at which the confirmation of sequence could not be verified. All sites of oxidation on modified peptides were validated manually with Data Analysis, which accompanied the ion trap mass spectrometer (Agilent). The matched peptides for the MS/MS analysis covered 547 of the 568 amino acids (96%) of PA63 shown in Figure 1.
Flow Injection Analysis–Nanoelectrospray Quadrupole Time-of-Flight Mass Spectrometry
Peptides found to be oxidized in RP-HPLC–nanoESI-IT MS/MS experiments were targeted for quantitation of oxidation. For this analysis, 10 μL of electrospray solvent (50:50 mixture of deionized H2O and an ACN/0.1% FA mixture) was added to a 10 μL aliquot of the analyte. These samples were directly infused from a pressurized bomb (41) into the orthogonal nanoelectrospray source of a Micromass Q-TOF Ultima Global mass spectrometer (Waters, Milford, MA). Settings for MS acquisition were as follows: capillary potential of 3.5 kV, cone potential of 100 V, collision energy of 5.0 V, RF set to 100 V, cone gas flow of 50 L/h. Blanks consisting of electrospray solvent alone were run between each sample until the signal stabilized to baseline abundance. Mass resolution for these experiments was typically ∼8000 full width at half-maximum, while mass accuracy was better than 40 ppm using external calibration with renin substrate tetradecapeptide.
For each of the four experimental conditions, three separate FIA-nanoESI–Q-TOF MS acquisitions were performed, combining 10 min of acquisition for each sample to increase the signal-to-noise ratio. Resultant spectra were manually searched for ion packets representing the unoxidized peptides as well as for ion clusters that had multiples of +8 or +5.3 m/z shifts for doubly or triply charged ions, respectively, corresponding to single, double, or triple oxidation events. Ions with poor signal-to-noise ratios were avoided, and monoisotopic peaks were used for measurement of signal abundance where possible. When multiple charge states of the same ion were observed, the charge state with the smallest amount of variation in oxidation across all experimental conditions was chosen for quantitation. The extent of oxidation was not observed to change between the different charge states of the same ion.
To perform the relative quantitative analysis of oxidation, the signal abundance for unoxidized and oxidized peptides was recorded for each of these acquisitions and converted a “percent oxidized” value: [oxidized peptide/(unoxidized peptide + oxidized peptide)] × 100. The average percent oxidized for control samples was subtracted from the average for the irradiated samples to control for any background oxidation. This entire analysis was repeated in three separate experiments, resulting in a total of nine analyses for each of the four experimental conditions. The background-corrected percent oxidized values for the prepore conformation (pH 7.5) were compared to those for the pore conformation (pH 5.5). A t test was then performed on these values using PROC GLM in SAS version 9. An oxidation site was considered increased, decreased, or unchanged in surface accessibility if it had a statistically significant (p ≤ 0.05) difference between prepore and pore conformations in two of the three trials (Table S2 of the Supporting Information). Note that this study provides relative quantitative information, not absolute.
Structural Analysis of the PA63 Prepore and Pore Model
Visualization, manipulation, coloring, and water accessibility renderings of PA63 were all performed with PyMOL Molecular Graphics System, version 0.99 (42). The fractional solvent accessible surface area was calculated with the GETAREA 1.1 online web service (43) with values in Table 1 representing the ratio of side chain surface area to random coil value per residue. Steric and geometric evaluation of the PA63 pore model was performed with MolProbity (44). MolProbity categories as listed in Table S1 are as follows: vdW clash, non-hydrogen-bonded atoms with a >0.4 Å (for example) van der Waals overlap; ϕ/ψ, ϕ and ψ angle outliers; rotamer, side chain angle outliers; Cβ, indicative of distortion of Cα bond angles; bond length, violations in lengths of peptide bonds; bond angle, violations in angles of peptide bonds.
Table 1.
Fractional Solvent Accessible Surface Areas for Selected Residues of the PA63 Prepore Structure and Pore Model Conformers Calculated by GETAREA (43)
| PA residue | prepore | Aa | Ba | Ca | Da | Ea | Fa | Ga |
|---|---|---|---|---|---|---|---|---|
| M266 | 0.0 | 19.3 | 11.2 | 6.3 | 8.9 | 13.3 | 1.9 | 30.3 |
| W346 | 10.9 | 52.3 | 52.7 | 57.1 | 48.4 | 35.9 | 56.3 | 49.8 |
| M350 | 0.0 | 47.0 | 33.3 | 15.5 | 39.9 | 79.9 | 12.9 | 16.6 |
| M434 | 0.0 | 15.3 | 0.0 | 12.4 | 3.4 | 2.8 | 32.2 | 1.7 |
| W477 | 0.0 | 0.2 | 0.0 | 1.5 | 0.4 | 0.0 | 0.0 | 0.0 |
| M521 | 2.8 | 6.8 | 38.6 | 6.4 | 0.7 | 12.4 | 73.1 | 27.9 |
| V619 | 34.8 | 36.3 | 7.2 | 5.8 | 33.3 | 6.1 | 54.1 | 39.2 |
| I620 | 42.8 | 42.6 | 60.0 | 73.8 | 57.8 | 76.8 | 53.2 | 39.3 |
| Y642 | 1.2 | 75.2 | 76.3 | 78.0 | 76.6 | 75.0 | 61.2 | 95.4 |
| M662 | 20.9 | 8.6 | 13.6 | 0.0 | 45.0 | 10.8 | 12.7 | 0.0 |
| Y688 | 83.3 | 60.6 | 56.3 | 55.7 | 56.1 | 0.5 | 29.9 | 55.3 |
Pore model conformer.
Results
Identification of Oxidized Residues by RP-HPLC–nanoESI-IT MS/MS
To mimic the prepore or pore conformational states, PA63 was dialyzed against NH4HCO3 buffer at either pH 7.5 or 5.5, respectively. After dialysis, the PA63 was exposed to hydroxyl radicals generated by the radiolysis of water. The PA63 samples were digested with trypsin after irradiation, and this digestion mixture was used for MS analysis. The liquid chromatography separation prior to MS/ MS analysis was used to allow greater sequence coverage and higher sensitivity for identifying lower-abundance peptides. Relatively long separations were performed (2 h; see Experimental Procedures); however, 70–80% sequence coverage was obtained in nearly all experiments.
Among the three repetitions of this experiment, several amino acids were consistently found to be oxidized at both pH values, with their locations appearing to be roughly every 40–70 amino acids (Figure 1). Consistent with previous studies that examined oxidation rates of each amino acid residue (23), sites of oxidation of PA63 were dominated by methionine, tryptophan, and tyrosine residues, with one example of valine or isoleucine being oxidized (Table 2 and Figure S1 of the Supporting Information). Most residues were modified with a single oxygen atom; however, there were two cases of doubly oxidized tryptophan residues. W477 was consistently identified by MS/MS analysis to have a +32 kDa mass shift. Also, the most abundant peptide containing W346 contained two oxygen atoms at this residue, and a third at M350 (Table 2 and Figure 1).
Table 2.
Sites of Oxidation Identified by RP-HPLC–nanoESI-IT MS/MS
| PA residues | amino acid sequence | oxidation | M + H |
|---|---|---|---|
| 253–273 | HPLVAAYPIVHVDmENIILSK | M266 + O | 2359.3 |
| 345–359 | TwAETmGLNTADTAR | W346/M350 + 3O | 1637.8 |
| 415–444 | NLAPIALNAQDDFSSTPITMNYmNYNQFLELEK | M434 + O | 3397.7 |
| 471–490 | VDTGSNwSEVLPQIQETTAR | W477 + 2O | 2231.1 |
| 505–524 | IAAVNPSDPLETTKPDmTLK | M521 + O | 2141.1 |
| 618–636 | EviNSSTEGLLLNIDKDIR | V619/I620 + O | 2145.6 |
| 638–659 | ILSGyIVEIEDTEGLKEVINDR | Y642 + O | 2505.3 |
| 660–669 | YDmLNISSLR | M662 + O | 1211.6 |
| 681–695 | YNDKLPLyISNPNYK | Y688 + O | 1841.9 |
A second peptide displayed dual valence with respect to oxidation. Tryptic peptide 618–636 contained a single oxidation modification, and MS/MS analysis of this peptide showed fragment ions reflecting oxidation at either V619 or I620 (data not shown). Double oxidations were not typically seen on this peptide, so this oxidized peptide was considered a mixture of that oxidized at V619 and that oxidized at I620. The oxidation sites of all consistently modified peptides were able to be identified from these MS/MS analyses with only one exception; tryptic peptide 638–659 contained a single oxidation modification which mapped to either S640, G641, or Y642. Given that the oxidation rate constants for tyrosine are more than 100-fold higher than that of serine and 1000-fold higher than that of glycine (23), it is highly likely that the oxidation site for this peptide is in fact at Y642.
Quantitative Analysis of Oxidation via FIA-nano-ESI–Q-TOF MS
While the LC separation and data-dependent MS/MS acquisition were important for the high-throughput identification of oxidative modifications of PA63 peptides, a more reliably quantitative method was required to measure the degree to which these peptides were oxidized. To this end, we used FIA-nanoESI combined with high-accuracy Q-TOF MS to provide a constant nanoliter scale flow of analyte. A representative example of oxidized ion quantitation can be found in Figure S2 of the Supporting Information. In these spectra, PA63 peptide 660–669 was detected as a doubly charged ion at m/z 606.38 ± 0.09. This peptide was identified by MS/MS analysis to have a single oxidation modification at M662 (Table 2 and Figure S1 of the Supporting Information). Consistent with this observation, a doubly charged ion cluster corresponding to an 8 m/z shift can be detected at m/z 614.36 ± 0.02 in the FIA-nanoESI–Q-TOF MS spectra (Figure S2 of the Supporting Information). Rate constants for the reaction of hydroxyl radicals in aqueous solution with amino acids have been previously reported, showing little change in reactivity as a function of pH (45). Nonetheless, to examine any effects of pH on oxidation chemistry, we performed our OPF analysis at pH 7.5 and 5.5 on peptides from trypsin-digested PA63. We saw no major changes in oxidation between PA63 at either pH (data not shown), indicating that any changes in oxidation seen with intact PA63 were due to changes in structure and conformation.
Six peptides from intact PA63 were found to be significantly less oxidized at pH 5.5 than at pH 7.5. The most striking of these was the singly oxidized peptide containing residue M662, found to be nearly 100% oxidized after irradiation when at the high pH (Figure 2A). However, an only 12-fold increase in the level of oxidation was seen for the low-pH sample for this peptide compared to control (Figure 2B). The triply oxidized peptide containing W346 and M350 experienced a nearly 15-fold increase in the level of oxidation after PA63 samples at pH 7.5 were exposed to irradiation. A less impressive ∼2-fold increase in the level of detectable oxidation was observed for similar samples at pH 5.5 (Figure 2B). Residues M266 and Y688 each had lower extents of oxidation at the lower pH value, and Y642 and M521 had more modest reductions in the level of oxidation when the PA63 heptamer was in the pore conformation (Figure 2).
Figure 2.

Quantitative analysis of oxidation from ESI-MS. The amount of peptide oxidation was calculated for each PA63 amino acid found modified by MS/MS analysis. For each oxidized peptide under each experimental condition, the signal abundance of both unoxidized and oxidized ions was recorded, each in triplicate. (A) Data are expressed as the percent total oxidized peptide [oxidized/ (oxidized + unoxidized) × 100], and error bars represent the calculated standard deviation. (B) The increase in the level of oxidation for indicated PA63 residues was calculated by dividing the percent total oxidized peptide after irradiation by that of control samples. Shown are representative data for each of the amino acids in this study for which quantitation of oxidation was carried out.
Only a single peptide was shown to have a higher oxidation rate at pH 5.5. PA63 amino acids 618–636 contain the oxidation sites of V619 and I620, both found to be equivalently singly oxidized by MS/MS analysis. While no difference in the oxidation of this site was detected between control and irradiated samples at pH 7.5, a roughly 1.5-fold increase in the level of oxidation was seen when PA63 was in its pore conformation (Figure 2). In addition to differentially oxidized amino acids, there were also two residues for which no repeatable change was observed. Quantitation of oxidation for residues M434 and W477 generally demonstrated an increase in the abundance of oxidized peptides after irradiation, but no consistent trend was noted at these amino acids between PA63 samples at pH 7.5 or 5.5.
It has been reported previously that sulfur-containing amino acids can be subject to secondary oxidation reaction at rates independent of their surface accessibilities (46). Not only could such reactions obscure the interpretation of the surface accessibility data for Cys and Met residues themselves, but oxidation of sulfur-containing residues normally inaccessible to solvent could induce a conformational change that may potentially affect the solvent accessibility of other amino acids and complicate the data. Fortunately, the addition of trace amounts of catalase has been shown to mitigate these secondary oxidation reactions (46). Because several methionine residues were found to be differentially oxidized in this study, we performed a subset of our footprinting experiments in the presence or absence of 10 nM catalase. While a slight decrease in the amount of oxidation of Met residues was noted (as expected), we did not observe significant changes in the oxidation of non-sulfur-containing amino acids between the two pH values which were found to be differentially oxidized in our study (data not shown). Thus, we are confident that the changes in oxidation seen in PA63 residues at the two pH values result from conformational changes induced by the pH shift and not because of secondary oxidation of Met. Another possible source of false positives in OPF could be the fact that the abundance of more than one peptide is recorded from overlapping ion peaks during the manual quantitation of the MS runs; however, LC separation prior to quantitative MS analysis could overcome this challenge.
False negatives are inherently common in any mass spectrometric technique, given the frequent poor ionization and fragmentation efficiencies of peptides. This is perhaps best illustrated by our lack of detection of the highly accessible Y732, which resides in a peptide that was never detected by our analysis (Figure 1). It is possible that initial trypsin nicking to remove PA20 inadvertently also removed this fragment or that the multiply charged ions at the m/z values of this small peptide were too low to be effectively detected by our methodology. Another source of false negatives could be low levels of oxidation that may border on the detection limits of the instrumentation (i.e., inconsistent appearance of W206 and W226 oxidation).
Discussion
To assess the OPF of PA63 from a structural perspective, the results from the differential oxidation experiments were applied to the three-dimensional structure of the PA63 prepore heptamer [PDB entry 1TZO (17)]. We were encouraged to find that most of the residues found to be oxidized in the prepore in this study also appear to be generally accessible in the PA63 prepore (Figure 3). Most notable among these are several residues (W346, M350, M662, and Y688) that all cluster to the bottom plane of the prepore heptamer. These residues are found nearly aligned in any monomer (Figure 3D), and each amino acid is predicted to contact water (Figure S3 of the Supporting Information). M434 also maintains a location near the bottom plane but appears to be much more buried than other residues in this region (such as Y688). Water accessibility tracing of the heptamer reveals a very slight amount of contact with water for M434, despite the appreciable amounts of oxidation seen with OPF at this position (Figure S3 of the Supporting Information). Because Met residues are so readily oxidized (23), it is likely that the small amount of apparent surface accessibility is sufficient to allow ample oxidation of M434 in our experiments. M521 was also found by our OPF studies to be oxidized, and this appears consistent with its position in the prepore structure. M521 can be found in domain 3 near the top of the heptamer and appears at least partially buried (Figure 3), although a water accessibility tracing of the prepore shows some accessibility to solvent (Figure S3 of the Supporting Information). Another oxidation site, V619/I620, is located on the outer perimeter of the prepore and accessible according to the crystal structure. An R-helix formed by residues 608–617 does appear to shield the majority of V619 from water; however, I620 is visibly solvent accessible and is thus consistent with our OPF data.
Figure 3.
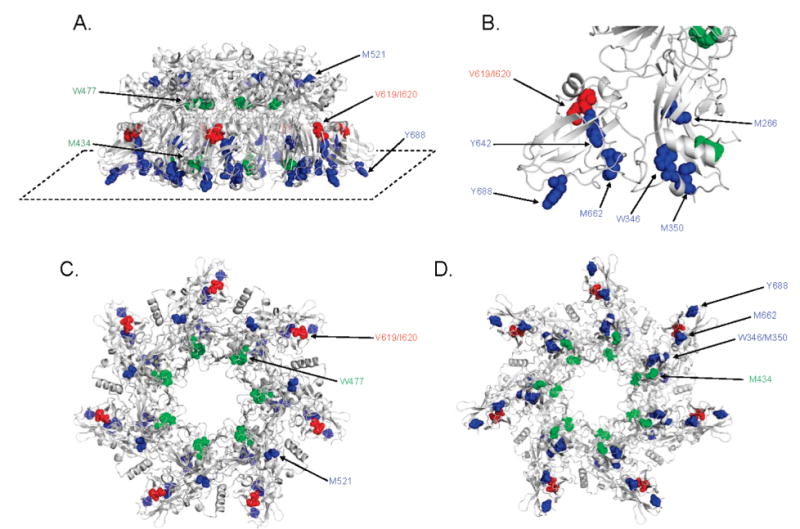
Mapping oxidized residues on the PA63 prepore. Amino acids found to be oxidized in this study are shown in colored spheres on a ribbon rendering of the 3.6 Å crystal structure of the PA63 prepore heptamer [PDB entry 1TZO (17)]. Residues observed to be oxidized more readily at pH 7.5 are colored blue, residues found to oxidized more readily at pH 5.5 red, and residues which demonstrated no change in oxidation between the two pH values green. Depicted in this figure are (A) a side view (with the “bottom plane” identified with a dashed line), (B) a zoomed view of the lower bottom plane, (C) a top-down view, and (D) a bottom-up view.
Some residues demonstrated by our OPF studies to be oxidized at both pH values did not appear to have obvious solvent accessibility in the prepore structure. Though present in the bottom plane, residue Y642 appears to be surrounded in boxlike fashion by two β-sheets formed by amino acids 651–735 (Figure 3B). Despite the apparent lack of surface accessibility of Y642, it was found to be oxidized only slightly less than Y688, a residue which appears to have exceptional exposure to solvent. It is possible that oxidation of Y642 results from some structural flexibility of PA63 in solution which is not represented in the crystal structure of this prepore. M266 is another residue which appears largely buried in the center of domain 2 but was still found readily oxidized after irradiation. As mentioned above, Met residues can be subject to secondary oxidation reactions and become oxidized without being exposed to solvent (25, 46). Our OPF with the inclusion of catalase to mitigate this effect, however, did not have a demonstrative effect on Met oxidation. Thus, it is possible that structural flexibility in this region may play a role in altering the accessibility of solvent to M266. A similar trend was seen for residue W477 in that, while some oxidation was seen at both pH values, the surface accessibility tracings of the prepore crystal structure show that it is obscured from contact (Figure S3B of the Supporting Information).
The somewhat low-resolution structure of PDB entry 1TZO (3.6 Å) has an inherently high overall anisotropic temperature factor (b-factor). However, analysis of relative atomic b-factors of the PA63 prepore structure with respect to oxidation illustrated some interesting points (Figure 4). The top of the PA63 heptamer harbored only a few oxidation sites and had relatively very low overall b-factor values. Conversely, the anisotropic temperatures were very high in residues in the bottom plane, a region to which seven of 11 total oxidation sites were mapped. Because this bottom plane region is involved with both receptor binding and structural rearrangements related to transmembrane pore formation, it is perhaps not surprising to find a large degree of flexibility and thermal motion among these residues. While oxidation of amino acids has been shown to be directly dependent on the solvent accessibility during irradiation (23), atomic thermal motion can also affect the accessibility of an amino acid to surrounding solvent if it fluctuates between buried and more accessible states. As shown here, anisotropic temperature can be predictive of apparent solvent accessibility and potential for oxidation.
Figure 4.
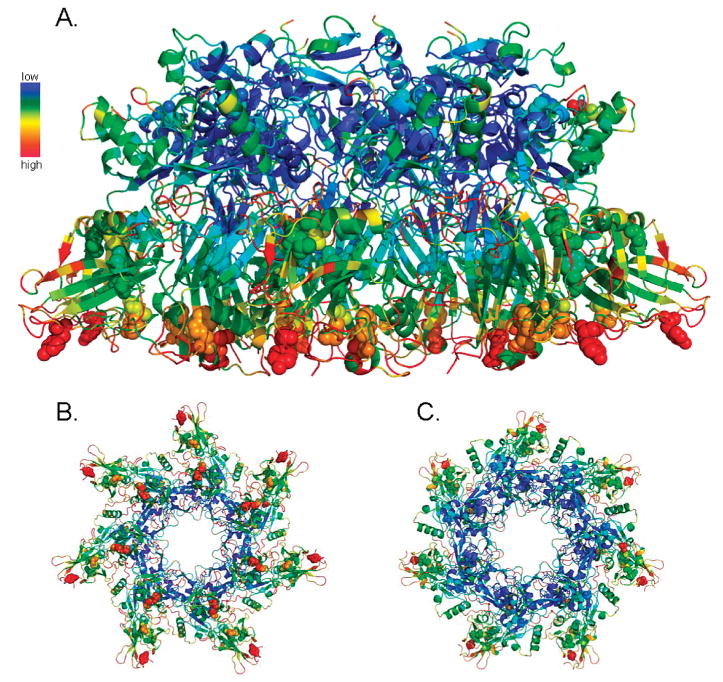
Anisotropic temperature factors of the PA63 prepore heptamer. The atoms of the PA63 prepore crystal structure [PDB entry 1TZO (17)] are colored on the basis of their anisotropic temperature factor. The spectrum from blue to red represents atoms with low to high b-factor values, respectively, relative to all atoms in the PA63 prepore structure. Sites identified to be oxidized in this study are represented as spheres. Depicted in this figure are side (A), bottom-up (B), and top-down (C) views of the prepore heptamer.
Structural Interpretation of Differentially Oxidized PA63 Residues
The majority of residues experienced a decrease in the level of oxidation when PA63 was at pH 5.5 and present in the heptameric pore conformation. While M521 was the exception, residues M266, W346, M350, Y642, M662, and Y688 can be found near the bottom of the prepore heptamer with W346, M350, M662, and Y688 each directly facing the bottom plane. It is interesting to note that the three regions in which W346/M350, M662, and Y688 are located have been shown by previous crystallographic studies to serve as contact points between PA63 and one of its receptors, CMG2. The PA63 prepore heptamer has been cocrystallized with CMG2, and that structure revealed that four loops contributed by PA domains 2 and 4 mediated the majority of the contact between PA63 and its receptor (17). These four loops contain residues spanning positions 340–348, 654–662, 681–688, and 714–716, thus inclusive of each of the bottom plane residues we have identified by OPF as being highly surface accessible. It also has been proposed that association of amino acids 340–348 with CMG2 residues 152–158 is in large part responsible for increasing the pH threshold of the conformational change by restricting unfolding of the transmembrane insertion loop (17). Our results demonstrating the higher surface accessibility of amino acids W346, M350, M662, and Y688 in the prepore state are consistent with these structural data and help to validate the use of OPF for probing protein structure. Experiments that aim to monitor the impact of receptor binding on the surface accessibility of these residues (at both prepore and pore states) are currently underway in our laboratory.
As mentioned above, only V619 and I620 were found to be more oxidized at pH 5.5 where PA63 undertakes its pore conformation. The limited water accessibility of these residues in the prepore state (Figure S3 of the Supporting Information) seems to be consistent with our OPF data, as these oxidation sites would be expected to gain a greater degree of surface accessibility after the pH-dependent conformational change. Several elegant biochemical studies have helped determine that, at acidic pH, amino acids 283–340 of the PA63 heptamer form an extended 14-strand β barrel structure (20, 21). In our oxidative analysis, we observed modification of two residues found to present in this stem domain, W346 and M3450 (as described above). Because we rarely detected oxidation of amino acids with hydroxyl radical reactivities lower than that of tyrosines in this study, we were not surprised to see additional oxidation of the stem domain which lacks any additional methionine, tryptophan, and tyrosine residues.
Application of OPF Data to a Computational PA63 Pore Model
The impetus for using nontraditional means to study the protein structure of PA63 in this study stems from the current lack of a determined structure of the final pore conformation. Though this transmembrane complex has been difficult for investigators to determine, a three-dimensional computational model for the PA63 pore has recently been published [PDB entry 1V36 (47)]. Although the Nguyen computational model is not without its caveats (i.e., each of the seven subunits of the heptameric PA pore has a slightly different conformation), this model structure does provide the opportunity to map our OPF data to a potential PA63 pore state.
Residues found with less oxidation at pH 5.5 appear to line the bottom plane of the cap region and top of the stem region in a fashion somewhat similar to that of the prepore (Figure 5). Interestingly, the pore model depicts W346 and M350 oriented as distinctly facing outward from the transmembrane stem and highly accessible in all seven conformers. Despite this, the peptide containing W346 and M350 was found in our study to be nearly 2.5-fold less oxidized at pH 5.5 than at pH 7.5 (Figure 2B). When the fractional solvent accessible surface areas of the PA63 prepore structure and pore model were quantitated using GETAREA (43), W346 and M350 had values that disagreed with our OPF data (Table 1). According to these calculations, both of these residues are calculated to have an increase in solvent accessibility in the pore state. The robust amount of oxidation of the W346/M350-containing peptide at pH 7.5 was not observed at pH 5.5 in this study (Figure 2), and therefore, a discrepancy exists between our OPF data and the structural position assigned by the Nguyen pore model. It is interesting to note that a structural evaluation of the model using MolProbity (44) identified some unusual steric and geometric conformations at amino acids W346 and M350 and adjacent residues. Several overlapping van der Waals radii were noted, along with some abnormal bond lengths in this region (Table S1 of the Supporting Information). It remains possible, therefore, that additional structural rearrangements occur near W346 and M350 that are currently not represented in the model and that could affect solvent accessibility.
Figure 5.
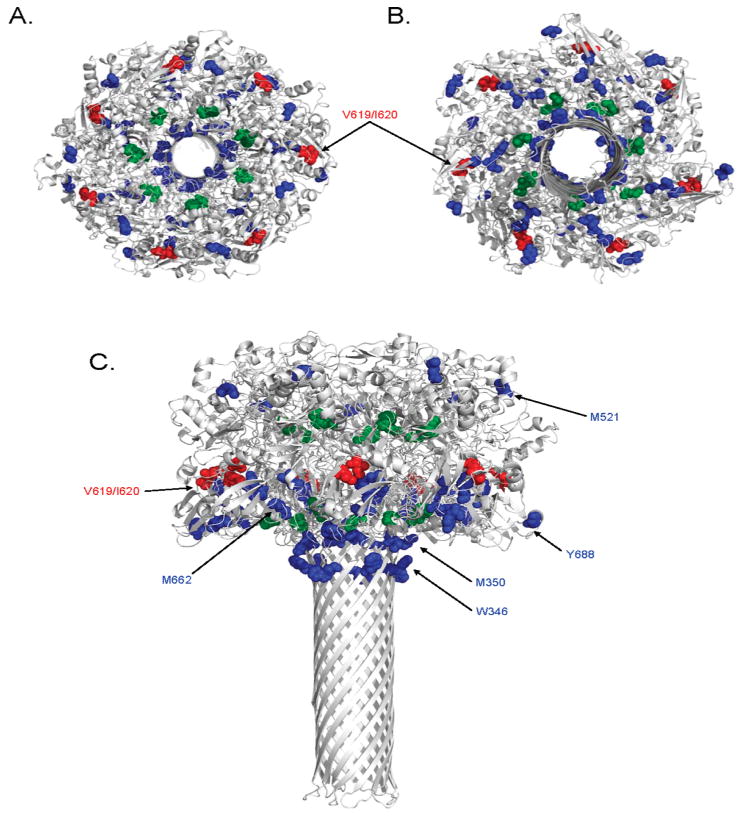
Mapping oxidized residues on the PA63 pore model. Amino acids found to be oxidized in this study are shown as colored spheres on a ribbon rendering of three-dimensional model of the PA63 pore heptamer [PDB entry 1V36 (47)]. Residues observed to be oxidized more readily at pH 7.5 are colored blue, residues found to oxidized more readily at pH 5.5 red, and residues which demonstrated no change in oxidation between the two pH values green. Depicted in this figure are top-down (A), bottom-up (B), and side (C) views of the pore heptamer.
The surface accessibility of Y688 in the pore model appears to be consistent with our OPF analysis. Oxidation of Y688 in the pore conformation was roughly 50% of that in the prepore (Figure 2), and fractional solvent accessible surface area calculations predict an ∼30% decrease in accessibility when PA transitions from the prepore to pore. M662, on the other hand, appears to be somewhat ambiguous in the pore model, since some of the conformers have this residue more buried than others (Figure 5 and Figure S4 of the Supporting Information). Our data would predict this residue to be less accessible in the pore conformation because a greater than 2-fold reduction in the level of oxidation was noted in the pore conformational state (Figure 2B). Interestingly, analysis of this region with MolProbity found several unusual steric conformations, including overlapping van der Waals radii in nearly all conformers of the pore model (Table S1 of the Supporting Information). Taken together, these observations support a conformer in which M662 is more buried from solvent than is predicted by any of the calculated structures. Conformer E of the Nguyen pore model would best represent the structural configuration most consistent with our findings for M662.
Y642 appears buried and inaccessible to solvent in the prepore structure, as mentioned above (Figure S3A of the Supporting Information), yet it still experienced appreciable amounts of oxidation after irradiation at pH 7.5 (Figure 2). The water accessibility of Y642 in the pore model appears similarly limited (Figure S4 of the Supporting Information), but quantitation of oxidation revealed a modest decrease in the level of oxidation at pH 5.5 (Figure 2). It is relevant to note that the PA monomers comprising the cap of the Nguyen pore model are more compactly folded than the prepore structure (47). This effect is most obvious when one examines the nearly 10 Å interdomain groove existing between domains 2 and 4 (Figure 1), a space which is nearly nonexistent in the pore model (Figure 5). If this more compact and closed configuration is representative of the actual pore structure, than this may explain why residues such as Y642 were observed to be less oxidized at pH 5.5. Fractional solvent accessible surface area calculations for Y642 appear to contradict this idea (Table 1); however, Y642 appears to be completely unable to access water in both conformations (Figures S3 and S4 of the Supporting Information). The surface accessibility of Y642 and its dynamics during the pH change will warrant further investigation.
Examination of the two residues found to be more oxidized at pH 5.5 was also mixed in consistency with the Nguyen model. The water accessibility rendering of both the PA63 prepore and pore depicts V619 and I620 as only slightly accessible to water (Figure S3A of the Supporting Information). While some of the conformers in the model do exhibit small increases in accessibility when compared to the prepore structure (Figure S4 of the Supporting Information), a large increase is not readily observable, especially in the water accessibility rendering of the pore model. There was also a mixed degree of change in the fractional solvent accessible surface area of V619 and I620 in the pore model conformers, with no clear trend between the two residues (Table 1). In addition, MolProbity found relatively few structural inconsistencies at or near these two residues, giving strength to the structural assignments in the Nguyen model for these residues (Table S1 of the Supporting Information). It should be noted that our OPF results showed an only small increase in the level of oxidation of these residues at pH 5.5, so perhaps the slight qualitative increase in accessibility seen in some of the conformers in Figure 5 is, in fact, representative of subtle structural alterations occurring in vivo.
Summary
One of the most crucial steps in the mechanism of action of ATx is the translocation of the LF and EF cytotoxins into cytoplasm so they may act on their cellular targets. PA provides this function, but only after undergoing a significant conformational change to form a transmembrane pore from a membrane-surface prepore complex. In this study, an emerging technique known as OPF was utilized with PA63 heptamers under conditions that mimicked the prepore and pore conformational states. OPF of PA63 identified several amino acids that were differentially modified by oxidation, illustrating their differences in solvent accessibilities between the two states. Residues more surface accessible in the PA63 prepore were mostly confined to the bottom plane of the heptamer, a region of PA63 of high anisotropic temperature and structural significance, including receptor binding and pore formation. PA63 amino acids found to be more surface accessible in the pore state were found on the outer perimeter of the ringed heptamer, mostly buried in both conformations. Some of our results support aspects of the PA63 pore model, while some do not, especially where a structural evaluation of the computational pore model indicated unusual conformations. In addition, our results indicate that the anisotropy of the prepore crystal structure is consistent with solution state flexibility not represented in the determined structure but detected with OPF. While the field will ultimately benefit most from a determined three-dimensional structure of the PA63 pore, methods such as OPF combined with mass spectrometry can play important roles in providing empirical and dynamic structural information in the void of such a structure and adding experimental constraints to future models of the pore. The study presented here provides such information and can serve as a foundation for additional surface accessibility studies of ATx as well as other proteins and complexes.
Acknowledgments
We acknowledge helpful discussions with and instrument training from Drs. Leesa J. Deterding, Jason G. Williams, Roxana E. Iacob, Suraj Dhungana, and Allison N. Schorzman. We also thank Dr. Shyamal Peddada for help with statistical analysis and Drs. Lake N. Paul and Lars C. Pedersen for critical review of the manuscript.
Footnotes
This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences.
Abbreviations: ATx, anthrax toxin; PA, protective antigen; EF, edema factor; LF, lethal factor; OPF, oxidative protein footprinting; CMG2, capillary morphogenesis protein 2; RT, room temperature; ACN, acetonitrile; FA, formic acid; MS, mass spectrometry; MS/MS, tandem mass spectrometry; RP-HPLC–nanoESI-IT MS/MS, reversed phase high-performance liquid chromatography–nanoelectrospray ion trap tandem mass spectrometry; FIA-nanoESI–Q-TOF MS, flow injection analysis nanoelectrospray–quadrupole time-of-flight mass spectrometry; PDB, Protein Data Bank.
Supporting Information Available Sample MS/MS spectrum depicting oxidative modification (Figure S1), representative MS spectrum used for quantitative analysis (Figure S2), water accessibility renderings of the prepore and pore (Figures S3 and S4), SDS–PAGE analysis of PA63 prepore to pore conversion (Figure S5), MolProbity analysis of the pore model (Table S1), and statistical significance of oxidative analysis (Table S2). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Mogridge J, Cunningham K, Lacy DB, Mourez M, Collier RJ. The lethal and edema factors of anthrax toxin bind only to oligomeric forms of the protective antigen. Proc Natl Acad Sci USA. 2002;99:7045–7048. doi: 10.1073/pnas.052160199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bradley KA, Mogridge J, Mourez M, Collier RJ, Young JA. Identification of the cellular receptor for anthrax toxin. Nature. 2001;414:225–229. doi: 10.1038/n35101999. [DOI] [PubMed] [Google Scholar]
- 3.Scobie HM, Rainey GJ, Bradley KA, Young JA. Human capillary morphogenesis protein 2 functions as an anthrax toxin receptor. Proc Natl Acad Sci USA. 2003;100:5170–5174. doi: 10.1073/pnas.0431098100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Klimpel KR, Molloy SS, Thomas G, Leppla SH. Anthrax toxin protective antigen is activated by a cell surface protease with the sequence specificity and catalytic properties of furin. Proc Natl Acad Sci USA. 1992;89:10277–10281. doi: 10.1073/pnas.89.21.10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Molloy SS, Bresnahan PA, Leppla SH, Klimpel KR, Thomas G. Human furin is a calcium-dependent serine endoprotease that recognizes the sequence Arg-X-X-Arg and efficiently cleaves anthrax toxin protective antigen. J Biol Chem. 1992;267:16396–16402. [PubMed] [Google Scholar]
- 6.Milne JC, Furlong D, Hanna PC, Wall JS, Collier RJ. Anthrax protective antigen forms oligomers during intoxication of mammalian cells. J Biol Chem. 1994;269:20607–20612. [PubMed] [Google Scholar]
- 7.Abrami L, Liu S, Cosson P, Leppla SH, van der Goot FG. Anthrax toxin triggers endocytosis of its receptor via a lipid raft-mediated clathrin-dependent process. J Cell Biol. 2003;160:321–328. doi: 10.1083/jcb.200211018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cunningham K, Lacy DB, Mogridge J, Collier RJ. Mapping the lethal factor and edema factor binding sites on oligomeric anthrax protective antigen. Proc Natl Acad Sci USA. 2002;99:7049–7053. doi: 10.1073/pnas.062160399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rainey GJ, Wigelsworth DJ, Ryan PL, Scobie HM, Collier RJ, Young JA. Receptor-specific requirements for anthrax toxin delivery into cells. Proc Natl Acad Sci USA. 2005;102:13278–13283. doi: 10.1073/pnas.0505865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wolfe JT, Krantz BA, Rainey GJ, Young JA, Collier RJ. Whole-cell voltage clamp measurements of anthrax toxin pore current. J Biol Chem. 2005;280:39417–39422. doi: 10.1074/jbc.M509049200. [DOI] [PubMed] [Google Scholar]
- 11.Miller CJ, Elliott JL, Collier RJ. Anthrax protective antigen: Prepore-to-pore conversion. Biochemistry. 1999;38:10432–10441. doi: 10.1021/bi990792d. [DOI] [PubMed] [Google Scholar]
- 12.Young JA, Collier RJ. Anthrax toxin: Receptor binding, internalization, pore formation, and translocation. Annu Rev Biochem. 2007;76:243–265. doi: 10.1146/annurev.biochem.75.103004.142728. [DOI] [PubMed] [Google Scholar]
- 13.Duesbery NS, Webb CP, Leppla SH, Gordon VM, Klimpel KR, Copeland TD, Ahn NG, Oskarsson MK, Fukasawa K, Paull KD, Vande Woude GF. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science. 1998;280:734–737. doi: 10.1126/science.280.5364.734. [DOI] [PubMed] [Google Scholar]
- 14.Vitale G, Pellizzari R, Recchi C, Napolitani G, Mock M, Montecucco C. Anthrax lethal factor cleaves the N-terminus of MAPKKS and induces tyrosine/threonine phosphorylation of MAPKS in cultured macrophages. J Appl Microbiol. 1999;87:288. doi: 10.1046/j.1365-2672.1999.00893.x. [DOI] [PubMed] [Google Scholar]
- 15.Leppla SH. Anthrax toxin edema factor: A bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc Natl Acad Sci USA. 1982;79:3162–3166. doi: 10.1073/pnas.79.10.3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leppla SH. Bacillus anthracis calmodulin-dependent adenylate cyclase: Chemical and enzymatic properties and interactions with eucaryotic cells. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1984;17:189–198. [PubMed] [Google Scholar]
- 17.Lacy DB, Wigelsworth DJ, Melnyk RA, Harrison SC, Collier RJ. Structure of heptameric protective antigen bound to an anthrax toxin receptor: A role for receptor in pH-dependent pore formation. Proc Natl Acad Sci USA. 2004;101:13147–13151. doi: 10.1073/pnas.0405405101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Petosa C, Collier RJ, Klimpel KR, Leppla SH, Liddington RC. Crystal structure of the anthrax toxin protective antigen. Nature. 1997;385:833–838. doi: 10.1038/385833a0. [DOI] [PubMed] [Google Scholar]
- 19.Parker MW, Feil SC. Pore-forming protein toxins: From structure to function. Prog Biophys Mol Biol. 2005;88:91–142. doi: 10.1016/j.pbiomolbio.2004.01.009. [DOI] [PubMed] [Google Scholar]
- 20.Nassi S, Collier RJ, Finkelstein A. PA63 channel of anthrax toxin: An extended β-barrel. Biochemistry. 2002;41:1445–1450. doi: 10.1021/bi0119518. [DOI] [PubMed] [Google Scholar]
- 21.Qa'dan M, Christensen KA, Zhang L, Roberts TM, Collier RJ. Membrane insertion by anthrax protective antigen in cultured cells. Mol Cell Biol. 2005;25:5492–5498. doi: 10.1128/MCB.25.13.5492-5498.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Buxton DB, Schwaiger M, Nguyen A, Phelps ME, Schelbert HR. Radiolabeled acetate as a tracer of myocardial tricarboxylic acid cycle flux. Circ Res. 1988;63:628–634. doi: 10.1161/01.res.63.3.628. [DOI] [PubMed] [Google Scholar]
- 23.Xu G, Chance MR. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal Chem. 2005;77:4549–4555. doi: 10.1021/ac050299+. [DOI] [PubMed] [Google Scholar]
- 24.Aye TT, Low TY, Sze SK. Nanosecond laser-induced photochemical oxidation method for protein surface mapping with mass spectrometry. Anal Chem. 2005;77:5814–822. doi: 10.1021/ac050353m. [DOI] [PubMed] [Google Scholar]
- 25.Kiselar JG, Maleknia SD, Sullivan M, Downard KM, Chance MR. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int J Radiat Biol. 2002;78:101–114. doi: 10.1080/09553000110094805. [DOI] [PubMed] [Google Scholar]
- 26.Maleknia SD, Kiselar JG, Downard KM. Hydroxyl radical probe of the surface of lysozyme by synchrotron radiolysis and mass spectrometry. Rapid Commun Mass Spectrom. 2002;16:53–61. doi: 10.1002/rcm.543. [DOI] [PubMed] [Google Scholar]
- 27.Guan JQ, Takamoto K, Almo SC, Reisler E, Chance MR. Structure and dynamics of the actin filament. Biochemistry. 2005;44:3166–3175. doi: 10.1021/bi048021j. [DOI] [PubMed] [Google Scholar]
- 28.Guan JQ, Vorobiev S, Almo SC, Chance MR. Mapping the G-actin binding surface of cofilin using synchrotron protein footprinting. Biochemistry. 2002;41:5765–5775. doi: 10.1021/bi0121104. [DOI] [PubMed] [Google Scholar]
- 29.Hambly DM, Gross ML. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J Am Soc Mass Spectrom. 2005;16:2057–2063. doi: 10.1016/j.jasms.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 30.Rashidzadeh H, Khrapunov S, Chance MR, Brenowitz M. Solution structure and interdomain interactions of the Saccharomyces cerevisiae “TATA binding protein” (TBP) probed by radiolytic protein footprinting. Biochemistry. 2003;42:3655–3665. doi: 10.1021/bi027203f. [DOI] [PubMed] [Google Scholar]
- 31.Sharp JS, Becker JM, Hettich RL. Protein surface mapping by chemical oxidation: Structural analysis by mass spectrometry. Anal Biochem. 2003;313:216–225. doi: 10.1016/s0003-2697(02)00612-7. [DOI] [PubMed] [Google Scholar]
- 32.Chance MR. Unfolding of apomyoglobin examined by synchrotron footprinting. Biochem Biophys Res Commun. 2001;287:614–621. doi: 10.1006/bbrc.2001.5628. [DOI] [PubMed] [Google Scholar]
- 33.Maleknia SD, Downard KM. Unfolding of apomyoglobin helices by synchrotron radiolysis and mass spectrometry. Eur J Biochem. 2001;268:5578–5588. doi: 10.1046/j.1432-1033.2001.02492.x. [DOI] [PubMed] [Google Scholar]
- 34.Maleknia SD, Ralston CY, Brenowitz MD, Downard KM, Chance MR. Determination of macromolecular folding and structure by synchrotron X-ray radiolysis techniques. Anal Biochem. 2001;289:103–115. doi: 10.1006/abio.2000.4910. [DOI] [PubMed] [Google Scholar]
- 35.Sharp JS, Sullivan DM, Cavanagh J, Tomer KB. Measurement of multisite oxidation kinetics reveals an active site conformational change in Spo0F as a result of protein oxidation. Biochemistry. 2006;45:6260–6266. doi: 10.1021/bi060470r. [DOI] [PubMed] [Google Scholar]
- 36.Guan JQ, Almo SC, Reisler E, Chance MR. Structural reorganization of proteins revealed by radiolysis and mass spectrometry: G-actin solution structure is divalent cation dependent. Biochemistry. 2003;42:11992–2000. doi: 10.1021/bi034914k. [DOI] [PubMed] [Google Scholar]
- 37.Kiselar JG, Janmey PA, Almo SC, Chance MR. Structural analysis of gelsolin using synchrotron protein footprinting. Mol Cell Proteomics. 2003;2:1120–1132. doi: 10.1074/mcp.M300068-MCP200. [DOI] [PubMed] [Google Scholar]
- 38.Liu R, Guan JQ, Zak O, Aisen P, Chance MR. Structural reorganization of the transferrin C-lobe and transferrin receptor upon complex formation: The C-lobe binds to the receptor helical domain. Biochemistry. 2003;42:12447–12454. doi: 10.1021/bi0352973. [DOI] [PubMed] [Google Scholar]
- 39.Sharp JS, Guo JT, Uchiki T, Xu Y, Dealwis C, Hettich RL. Photochemical surface mapping of C14S-Sml1p for constrained computational modeling of protein structure. Anal Biochem. 2005;340:201–212. doi: 10.1016/j.ab.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 40.Takamoto K, Chance MR. Radiolytic protein footprinting with mass spectrometry to probe the structure of macromolecular complexes. Annu ReV Biophys Biomol Struct. 2006;35:251–276. doi: 10.1146/annurev.biophys.35.040405.102050. [DOI] [PubMed] [Google Scholar]
- 41.Deterding LJ, Moseley MA, Tomer KB, Jorgenson JW. Coaxial continuous flow fast atom bombardment in conjunction with tandem mass spectrometry for the analysis of biomolecules. Anal Chem. 1989;61:2504–2511. doi: 10.1021/ac00197a011. [DOI] [PubMed] [Google Scholar]
- 42.DeLano WL. PyMOL. DeLano Scientific, LLC; Palo Alto, CA: 2002. [Google Scholar]
- 43.Fraczkiewicz R, Braun W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J Comput Chem. 1998;19:319–333. [Google Scholar]
- 44.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, III, Snoeyink J, Richardson JS, Richardson DC. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buxton GV, Greenstock CL, Helman WP, Ross AB. Critical Review of Rate Constants for Reactions of Hydrated Electrons, Hydrogen Atoms and Hydroxyl Radicals in Aqueous Solution. J Phys Chem Ref Data. 1988;17:513–886. [Google Scholar]
- 46.Xu G, Kiselar J, He Q, Chance MR. Secondary reactions and strategies to improve quantitative protein footprinting. Anal Chem. 2005;77:3029–3037. doi: 10.1021/ac048282z. [DOI] [PubMed] [Google Scholar]
- 47.Nguyen TL. Three-dimensional model of the pore form of anthrax protective antigen. Structure and biological implications. J Biomol Struct Dyn. 2004;22:253–265. doi: 10.1080/07391102.2004.10531226. [DOI] [PubMed] [Google Scholar]