

Abstract
Rapamycin (or sirolimus), the prototypical inhibitor of the mammalian target of rapamycin (mTOR) and an immunosuppressant used for the prevention of renal transplant rejection, has recently emerged as an effective treatment for Kaposi's sarcoma (KS), an enigmatic vascular tumor and a model for pathological angiogenesis. Indeed, recent work supports a role for mTOR as a central player in the transformation of endothelial cells by the KS-associated herpesvirus (KSHV)-encoded G protein-coupled receptor (vGPCR), the viral oncogene believed to be responsible for causing KS. However, emerging evidence that rapamycin may transiently promote the activation of Akt may limit its use as an anti-KS therapy. Here we demonstrate that activation of Akt in endothelial cells expressing vGPCR is augmented by treatment with rapamycin, resulting in the upregulation of several Akt proliferative and survival pathways. However, use of a novel dual PI3Kα/mTOR inhibitor, PI-103, effectively and independently blocked activation of both PI3K and mTOR in vGPCR-expressing endothelial cells. This resulted in more effective inhibition of endothelial cell proliferation and survival in vitro and tumor growth in vivo. Our results suggest that PI-103 may be an effective therapeutic option for the treatment of patients with KS. Moreover, as KS may serve as a model for pathological angiogenesis, our results further provide the basis for the early assessment of PI-103 as an anti-angiogenic chemotherapeutic.
Keywords: endothelial cell, Kaposi's sarcoma, rapamycin, sirolimus, PI-103, mTOR, Akt, PI3 kinase, Kaposi's sarcoma associated herpesvirus, KSHV, Human herpesvirus-8, HHV-8, G protein-coupled receptor, vGPCR
Introduction
Kaposi's sarcoma (KS) is a multifocal vascular neoplasm that affects immunosuppresed individuals (1, 2). Indeed, as the most common AIDS-related malignancy worldwide and the most frequent cancer among children and adult men in sub-Saharan Africa, KS has emerged as a major cause of morbidity and mortality amongst the AIDS population. Despite decades of investigation into the etiology of this enigmatic tumor, clinical management of KS has proven to be challenging.
Invariably associated with infection by the KS-associated herpesvirus (KSHV or HHV8), current research efforts have focused on the study of the relative contribution of KSHV-encoded genes to Kaposi's sarcomagenesis, to identify novel mechanism-based therapies for patients suffering from this neoplasm. Although several viral genes bear potential for KS pathogenesis, compelling data point to the KSHV-encoded G protein-coupled receptor (vGPCR) as a leading candidate viral gene for the initiation of KS (3, 4). Expression of KSHV vGPCR in mice by endothelial cell specific retroviral transduction (5) or in traditional transgenic models (6, 7) has revealed the remarkable sarcomagenic potential of this viral receptor.
Of interest, emerging evidence has implicated the Akt downstream effectors TSC/mTOR as a key intracellular route regulating endothelial cell biological responses, including endothelial tumor formation and pathological angiogenesis (8-11). Indeed, we have recently demonstrated that KSHV vGPCR activation of the PI3K/Akt/mTOR pathway plays a fundamental role in KS development and that rapamycin (sirolimus) is able to block vGPCR oncogenesis in vitro and in vivo (12). This drug has been shown to be an efficient therapy for transplant recipients with (iatrogenic) KS (13, 14) as well as for patients with the classic form of the disease (15, 16). However, treatment with rapamycin has not been successful in all KS patients (17-20). Why this treatment is successful in some patients – but not in others – remains unclear; the recent observation that rapamycin causes transient upregulation of Akt and Akt-mediated survival in some normal and tumor cells may provide one explanation (21-23). Indeed, as Akt activation is a recurring theme in oncogenesis, this transient upregulation of Akt may expose a potential Achilles' heel of rapamycin as a chemotherapeutic agent (24, 25).
Of note, a novel chemotherapeutic drug, PI-103, has recently been shown to independently inhibit both PI3Kα and mTOR (26), thereby overcoming a potential disadvantage of rapamycin in the treatment of Akt-dependent tumors. We therefore set out to assess the efficacy of PI-103 for the treatment of KS. Our results show that PI-103 blocks endothelial cell proliferation and survival more efficiently than rapamycin and demonstrate the potential of inhibiting both PI3Kα and mTOR as an effective anti-angiogenic approach.
Materials and methods
Expression plasmids and reagents
The expression plasmids for KSHV vGPCR, vGPCR R143A, Rheb and EGFP have been described elsewhere (11, 12). LY294002 and rapamycin were purchased from Calbiochem. The dual PI3Kα/mTOR inhibitor PI-103 has been previously described (26). For in vitro studies, rapamycin, LY294002 and PI-103 were reconstituted in DMSO as 1000x stock solutions, and were further diluted to the working concentration in culture medium. For in vivo studies, rapamycin (LC Laboratories) was dissolved in 100% ethanol as 20 mg/ml stock solutions, and further diluted in an aqueous solution of 5.2% Tween-80 and 5.2% PEG immediately before use (27). PI-103 was dissolved in 100% DMSO (5 mg/ml) and subsequently diluted to 50% DMSO in water before use.
Cell lines and transfections and cell proliferation assays
Immortalized murine endothelial cells (SVEC), EC-vGPCR, EC-R143A and COS-7 cells were cultured as previously described (5). Transfection of COS-7 was performed using Polyfect (QIAGEN) according to the manufacturer's protocol. Cell proliferation was determined using the crystal violet staining assay (28).
5-Bromo-2′-deoxy-uridine (BrdU) uptake and flow cytometry
BrdU uptake was determined using the BrdU kit I from Roche Applied Sciences (Mannheim, Germany). Briefly, cells were plated on coverslips and serum starved for 24 hours. BrdU (10 mmol/L) was added and cells were then treated with LY294002, rapamycin or PI-103 for 6 hours. Cells were fixed with ethanol fixative (50mM glycine solution into EtOH), and incubated with anti-BrdU working solution and anti-mouse-Igfluorescein working solution. Cells were finally covered with Vectashield mounting medium containing propidium iodide (Vector Laboratories, Inc., California, USA). The samples were analyzed under a fluorescence microscope with a detection range of 515 to 565 nm. For apoptosis detection, 0.5 × 106 cells were seeded, serum starved for 24 hours and treated as indicated with LY294002, rapamycin or PI-103. Samples were analyzed by flow cytometry, using the Annexin V-FITC apoptosis detection kit (BD Biosciences, California, USA) and propidium iodide staining.
Establishment and treatment of tumor allografts in athymic nu/nu mice
SVEC, EC- vGPCR or EC-R143A (106) cells were used to induce allografts in 8-week-old athymic (nu/nu) nude females mice as described (11). Briefly, early passage, exponentially growing cells were harvested after stable selection with G418, washed with PBS, and resuspended in DMEM. Viable cells (106) were then transplanted subcutaneously in the right flank of the mouse. The animals were monitored three times weekly for tumor formation. Tumor's longest length (L) and shortest width (W) were measured using a caliper at different time points throughout the experiment. Tumor volume was then converted into tumor weight using the formula LW2/2, as described previously (29). PI-103 treatment was commenced when estimated tumor weight reached ∼0.25 g. For this procedure, tumor-bearing animals were randomly grouped (control, n = 5; PI-103 treated group, n = 5) and treated with PI-103 (10 mg/kg/d) or an equal volume of vehicle. Treatment schedule was a single injection per animal given intraperitoneally for 18 consecutive days (26). Results of animal experiments were expressed as mean estimated tumor weight ± SD. When appropriate, animals were euthanized, and tissue was fixed in 4% paraformaldehyde and embedded in paraffin or lysed for further analysis. For BrdU studies, mice were first given an intraperitoneal injection of BrdU (100mg/kg) 2 hours prior to sacrifice. Detection of apoptotic cells was performed by TUNEL assay using TdT end labeling with DIG. All procedures involving animals were approved by the Institutional Animal Care and Use Committee.
Western Blots and Immunohistochemistry
Western Blots and immunohistochemical analysis were performed as previously described (5, 11). Antibodies recognizing P-Akt, Akt, P-S6 ribosomal protein, S6 ribosomal protein, P-p38, p38, P-GSK3, GSK3, P-BAD, BAD, P-Mdm2, p27, P-p70 S6K and p70 S6K were obtained from Cell Signaling (Massachusetts, USA). Antibodies recognizing P-p27 and Mdm2 were obtained from R&D Systems (Minnesota, USA) and BD Biosciences (California, USA), respectively.
Results
Rapamycin inhibits mTOR but upregulates Akt signaling in vGPCR-expressing endothelial cells
Kaposi's sarcoma associated herpesvirus (KSHV), the human herpesvirus that causes KS, encodes a G protein-coupled receptor (vGPCR) that has been shown to reproduce KS-like tumors when expressed in transgenic mice (5-7). vGPCR is a homologue of the human chemokine receptors CXCR1 and CXCR2 that exhibits constitutive-signaling to several oncogenic pathways (4, 30, 31). Immortalized murine vascular endothelial cells (SVECs) expressing vGPCR (EC-vGPCR) are highly tumorigenic in nude mice (Fig. 1A, top). A single point mutation in the highly conserved DRY sequence abrogates vGPCR constitutive activity (32); in turn, murine endothelial cells stably expressing this inactive vGPCR R143A mutant (EC- R143A) are not tumorigenic. Of interest, the oncogenic potential of vGPCR appears to correlate with its ability to signal to Akt and its effector kinase, mTOR (Fig. 1A, bottom). These results suggest that constitutive signaling to Akt and mTOR may play a fundamental role in vGPCR oncogenesis.
Figure 1. Rapamycin upregulates Akt signaling in KSHV vGPCR-expressing endothelial cells.

(A) Tumor allografts were established in athymic nu/nu mice by injecting immortalized murine endothelial cells (SVECs), or SVECs expressing vGPCR (EC-vGPCR) or the inactive mutant vGPCR R143A (EC-R143A). The results are expressed as mean estimated tumor weight (g) ± SD. Data are from a representative independent experiment that was repeated two times with similar results. Western blot analysis shows the correlation with P-Akt, Akt, P-S6 or S6 expression levels. (B) Immunodetection of the levels of P-Akt, Akt, P-S6, S6, P-p38 or p38 of EC-vGPCR cells treated with increasing doses of rapamycin (Rap), LY294002 (LY) or DMSO (control) for 6 hours. (C) Phosphorylation of Akt or Akt downstream molecules (GSK3, BAD, Mdm2, p27, P70-S6K or S6) in EC-vGPCR cells treated with 10 nM rapamycin (Rap), 10 μM LY294002 (LY), or DMSO (control) for 6 hours.
Of note, several studies have demonstrated the efficacy of the mTOR inhibitor, rapamycin (sirolimus), in the treatment of transplant recipients with (iatrogenic) KS (13, 14) as well as patients with classic KS (15, 16). However, recent reports documenting the therapeutic failure of rapamycin for some KS patients suggest that the relationship among vGPCR, mTOR, and rapamycin in KSHV-infected endothelial cells may be more complicated than previously thought (17-20). Indeed, emerging evidence points to a complex regulation of Akt/TSC/mTOR activity (24, 25). An intriguing recent finding is that Akt is upregulated in some cells treated with rapamycin, as a result of the loss of mTOR-mediated inhibitory feedback mechanisms (21-23, 25). We therefore set out to determine whether Akt was upregulated in vGPCR-expressing endothelial cells upon treatment with rapamycin. As shown in Figure 1B, treatment of EC-vGPCR cells with increasing doses of rapamycin for 6 hours resulted in an inhibition of mTOR; however, the levels of phosphorylated Akt increased in a dose-dependent manner. Conversely, EC-vGPCR cells exposed to increasing doses of the PI3K inhibitor, LY294002, showed inhibition of both Akt and mTOR under the same experimental conditions.
We then investigated whether the increase in the phosphorylation of Akt observed in EC-vGPCR cells treated with rapamycin was associated with an increased phosphorylation of endogenous Akt substrates. We found that treatment of vGPCR-expressing endothelial cells with rapamycin resulted in a marked increase in the phosphorylation of GSK3 and BAD (Fig. 1C). Phosphorylation of other Akt downstream effectors was unaffected (MDM2, p27) or downregulated (p70-S6K, S6 ribosomal protein). Collectively, these results suggest that, while inhibiting vGPCR-induced mTOR activity, rapamycin treatment may simultaneously lead to functional activation of Akt signaling in vGPCR-expressing endothelial cells.
Treatment of vGPCR-expressing endothelial cells with PI-103 inhibits activation of both Akt and mTOR, independently
These findings suggest that inhibiting Akt activation in rapamycin-treated KS tumors may improve the efficacy of rapamycin treatment for vGPCR-induced tumorigenesis. In this regard, a novel dual PI3Kα/mTOR inhibitor, PI-103, has shown to be highly effective in the inhibition of both Akt and mTOR activity in vitro and in vivo (26). We therefore set out to determine whether PI-103 could provide an alternative therapeutic option for the treatment of KS. We treated EC-vGPCR cells with increasing concentrations of PI-103 and found that this drug potently inhibited the phosphorylation of Akt as well as the phosphorylation of the mTOR substrate, p70-S6K, and its downstream effector, S6 ribosomal protein (Fig. 2A).
Figure 2. Treatment of KSHV vGPCR-expressing endothelial cells with PI-103 independently inhibits both PI3K/Akt and mTOR.

(A) Immunodetection of the levels of P-Akt, Akt, P-S6K, S6K, P-S6, S6, P-p38 or p38 of EC-vGPCR cells treated with increasing doses of PI-103 or DMSO (control) for 6 hours. (B) Immunodetection of the levels of P-Akt, Akt, P-S6 or S6 in cells expressing vGPCR or vGPCR and Rheb. Cells were treated with LY294002 (LY), rapamycin (Rap), PI-103 or DMSO (control) for 6 hours. Co-expression of vGPCR with Rheb partially rescued the inhibition of mTOR by LY294002, but not by PI-103 (or rapamycin). (C) Immunodetection of the levels of P-Akt, Akt, P-GSK3, GSK3, P-BAD BAD, P-Mdm2, Mdm2, P-p27 or p27 of EC-vGPCR cells treated with increasing doses of PI-103 or DMSO (control) for 6 hours.
To further explore the independent sensitivities of Akt and mTOR activation to treatment with PI-103, we co-expressed vGPCR with wild-type Rheb – the Ras-related small GTP binding protein that promotes the activation of mTOR. Rheb is unique among members of the small GTPase family in that the regulation of its function occurs predominately through its inactivation by Rheb GAPs (e.g., TSC2) rather than its activation by Rheb guanidine exchange factors (GEFs) (33); thus, overexpression of Rheb bypasses the requirement for PI3K activation of Akt to promote mTOR activity. Indeed, co-expression of Rheb was able to partially rescue the inhibition of mTOR by LY294002, but not by PI-103 (or rapamycin), confirming that PI-103 blocks S6 phosphorylation in these cells by direct inhibition of mTOR (Fig. 2B).
We next explored the effect of treating vGPCR-expressing endothelial cells with PI-103 on the phosphorylation of Akt substrates. Figure 2C shows that, unlike rapamycin, PI-103 potently inhibited the activation of GSK3, BAD, Mdm2 and p27. Collectively, these results demonstrate that PI-103 – unlike LY294002 or rapamycin – is capable of inhibiting vGPCR activation of both PI3K/Akt and mTOR, independently.
Simultaneous inhibition of mTOR and PI3K by PI-103 more effectively inhibits endothelial cell proliferation
We next set out to determine whether inhibition of both PI3K and mTOR would be more effective than treatment with rapamycin in preventing the proliferation of endothelial cells expressing vGPCR. To this end, we treated EC-vGPCR cells with increasing doses of PI-103, rapamycin, or LY294002. Figure 3A shows that treatment of cells with PI-103 was able to more efficiently block cell proliferation than rapamycin or LY294002 alone. The sensitivity to PI-103, as assessed by the IC50 (50% inhibitory concentration), was approximately 0.2 μM, achieving IC80 values with approximately 8 μM of compound. Furthermore, when we checked the incorporation of 5-Bromo-2′-deoxy-uridine (BrdU) by vGPCR-expressing endothelial cells treated with the different drugs, BrdU uptake was more significantly reduced in PI-103-treated EC-vGPCR cells (81%) than in rapamycin-treated EC-vGPCR cells (50%) (p <0.01; Fig. 3B and C). Of note, this remarkable sensitivity of the proliferative potential of EC-vGPCR was significantly higher than that of the parental SVEC endothelial cell line (Fig. 3C), emphasizing the importance of the Akt/mTOR pathway for the proliferation of vGPCR-expressing cells. Collectively, these results suggest that, compared to rapamycin, PI-103 is able to more effectively inhibit the proliferation of vGPCR-expressing endothelial cells.
Figure 3. PI-103 inhibits proliferation of KSHV vGPCR-expressing cells more effectively than rapamycin.
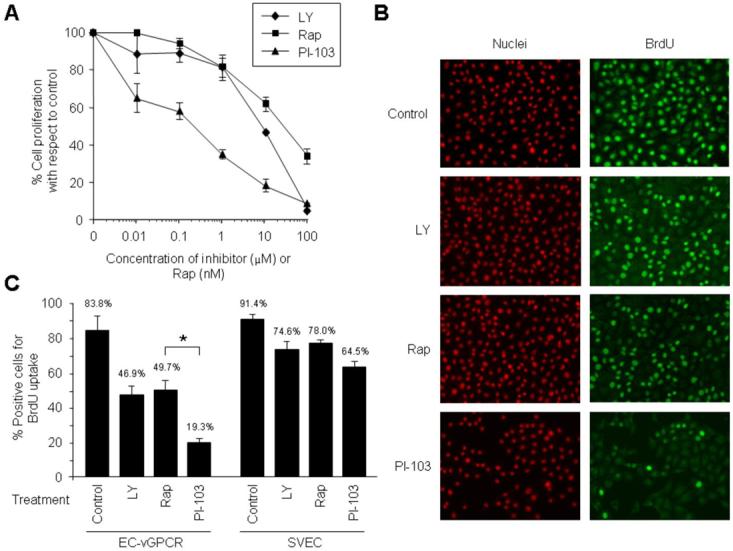
(A) Effect of the treatment with increasing doses of LY294002 (LY), rapamycin (Rap), or PI-103 on the proliferation of EC-vGPCR. Results are illustrated as percentage of cell proliferation relative to untreated control cells. The results are the mean ± SD of triplicate samples from a single representative experiment that was repeated three times with similar results. (B and C) BrdU uptake of EC-vGPCR cells treated with 10 μM LY294002 (LY), 10 nM rapamycin (Rap), 10 μM PI-103, or DMSO (control) for 6 hours. Samples were processed and analyzed under a fluorescence microscope as described in Materials and Methods (Magnification, 10 x) (B). Results are quantified as percentage of positive EC-vGPCR cells (or parental SVEC cells) for BrdU incorporation (C). Bars represent the mean percentage ± SD; * p <0.01 (C).
PI-103 promotes apoptosis in vGPCR-expressing endothelial cells
Close examination of PI-103- vs. rapamycin-treated EC-vGPCR cells revealed lower total cell numbers in the cells treated with the dual PI3Kα/mTOR inhibitor, suggesting that treatment with PI-103 may also more effectively promote cell death compared to rapamycin. We therefore set out to compare the efficacy of PI-103 in promoting apoptosis of endothelial cells expressing vGPCR. We cultured EC-vGPCR cells in the presence of LY294002, rapamycin or PI-103, and analyzed the samples by flow cytometry twenty-four hours following initiation of the treatment. Transient exposure (6 hour) to PI-103 induced apoptosis in 31% of the cells, compared to 20% in rapamycin treated cells (p <0.05; Fig 4A). Prolonged treatment (24 hours) with PI-103 was also associated with higher levels of apoptosis (55%) compared to rapamycin (38%) (p <0.05; Fig 4B). The sensitivity of EC-vGPCR survival was higher than that of the parental SVEC endothelial cell line (results not shown), again emphasizing the importance of the Akt/mTOR pathway for vGPCR-expressing cells. These results suggest that PI-103 is able to induce apoptosis of vGPCR-expressing endothelial cells more effectively than rapamycin.
Figure 4. PI-103 induces apoptosis of KSHV vGPCR-expressing cells more effectively than rapamycin.

(A) Effect of the treatment with 10 μM LY294002 (LY), 10 nM rapamycin (Rap), 10 μM PI-103, or DMSO (control) on EC-vGPCR apoptosis. Cells were cultured for 6 hours (A) or 24 hours (B) in the presence of the different compounds. Twenty-four hours following initiation of the treatment, cells were harvested and processed for flow cytometry analysis. Results are illustrated as percentage of apoptotic cells and are from a representative independent experiment that was repeated three times with similar results. * p <0.05.
PI-103 effectively inhibits tumor growth in an allograft model for KS
These observations prompted us to study the therapeutic potential of PI-103 as an anti-KS treatment. We therefore established KS tumor allografts by injecting EC-vGPCR cells in athymic nu/nu mice. Once lesions were established, tumor volume was measured and then converted into tumor weight at different time points as described in Materials and Methods. When estimated tumor weight reached ∼ 0.25 g (day 45), animals were treated with either PI-103 or vehicle (control) intraperitoneally, for 18 consecutive days (26). Tumor regression in PI-103-treated animals was observed within 2 days after the initiation of treatment, and inhibition of tumor growth was sustained for the duration of the experiment (Fig. 5A). At the end of the study (day 62), we observed that the average estimated weight of vehicle-treated tumors was 1312 mg, an almost 4 fold increase in 17 days. However, the PI-103-treated group demonstrated minimal growth over the same period, with an average estimated tumor weight of 460 mg on day 62 (Fig. 5A), representing only a 1.7 fold increase in tumor mass 17 days after the initiation of the treatment with the drug. Drug toxicity, as assessed by weight loss, was minimal in the treated group (reduction <5%) during the treatment period (results not shown).
Figure 5. PI-103 inhibits tumor growth in an allograft model for KS.
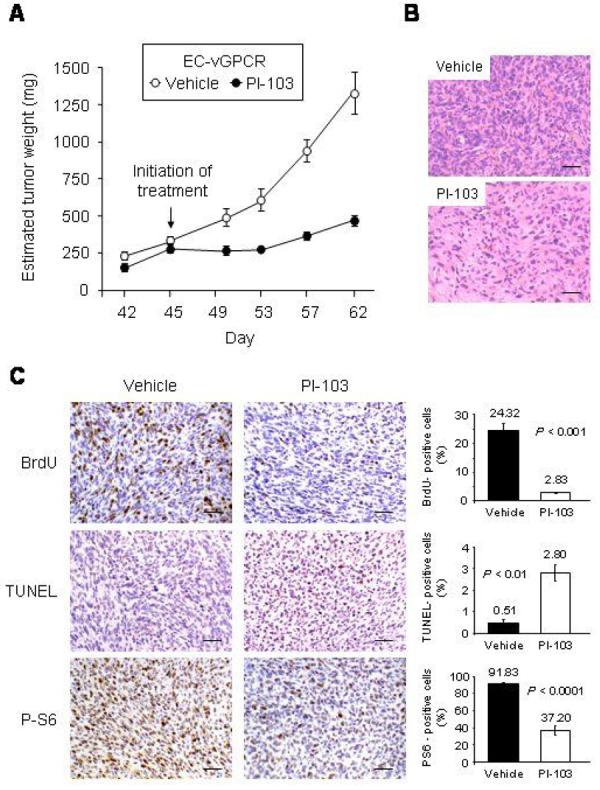
(A) Effect of the treatment of EC-vGPCR allografts established in athymic nu/nu females with the PI3Kα/mTOR inhibitor, PI-103. EC-vGPCR cells were injected s.c. into athymic nu/nu mice as described in Materials and Methods. When resulting allografts reached an estimated tumor weight of ∼ 0.25 g, mice were treated with PI-103 (10 mg/kg/d) or an equal volume of vehicle. Treatment schedule was a single injection per animal given intraperitoneally for 18 consecutive days. The results are expressed as mean estimated tumor weight (mg) ± SD. (B and C) H&E staining (B) and BrdU, TUNEL and phospho-S6 ribosomal protein staining (C) in representative sections of vehicle- and PI-103-treated EC-vGPCR tumor tissue. Scale bar 50 μm. Magnification, 20 x. Results are quantified as percentage of stained-positive cells; bars represent the mean percentage ± SD.
Tumors from PI-103-treated animals showed decreased cellularity (Fig 5B). Tumor cell proliferation, as assessed by the incorporation of BrdU, from PI-103-treated animals was also diminished compared to control animals (Fig 5C). In contrast, detection of apoptotic cells using TUNEL reaction showed increased cell death in PI-103-treated tumors. Immunohistochemical analysis of these tumors also demonstrated a dramatic reduction in the levels of phospho-S6 ribosomal protein compared to control animals (Fig. 5C). Of note, treatment of animals with PI-103 prior to establishment of tumors completely prevented tumor formation (results not shown). Taken together, our data provide the basis for the early assessment of molecules independently inhibiting the Akt and mTOR pathways, including PI-103, as an anti-KS therapy.
Discussion
The Akt/TSC/mTOR pathway, a central player in endothelial cell biology (8, 10), has been identified as a critical signaling route in vGPCR-induced Kaposi's sarcomagenesis (9, 11, 12). We have previously demonstrated that this viral oncogene stimulates the phosphorylation and inactivation of tuberin (TSC2), promoting the activation of mTOR and its signaling effectors (12). mTOR activation by vGPCR is necessary and sufficient for the ability of this viral oncogene to render expressing endothelial cells oncogenic (12). Moreover, mice haploinsufficient for TSC2 are predisposed to vascular sarcomas remarkably similar to KS, suggesting that the vascular tumors induced by infection with KSHV may be a consequence of the profound sensitivity of endothelial cells to dysregulation of the mTOR pathway by vGPCR (12). Indeed, infection of endothelial cells by KSHV has recently been shown to induce the activation of the PI3K/Akt/mTOR pathway, thereby promoting endothelial cell survival (37). Collectively, these data suggest that drugs targeting this important signaling route may be an effective therapeutic approach for the treatment of patients with KS.
Of note, the mTOR-inhibitor, rapamycin (sirolimus), has recently emerged as an effective treatment for renal-transplant patients with KS; the switch from other immunosuppressive agents to rapamycin leads to remission of dermal KS lesions, while effective immunosuppresion is still provided (14, 38-40). More recently, classic KS has demonstrated a similarly remarkable sensitivity to treatment with sirolimus (15, 16). This argues against a reduction in immunosuppression with this drug compared to other immunosuppressive agents and further supports a central role for the Akt/TSC/mTOR pathway in KS pathogenesis.
However, emerging evidence suggests that rapamycin may have some limitations in its clinical use as an anti-cancer agent. Several laboratories have demonstrated that treatment of cancer cell lines with rapamycin results in a transient paradoxical increase in Akt phosphorylation, which appears to be due to the loss of negative feedback regulation in the PI3K/Akt pathway (21-23). This negative regulatory loop could explain the relative benign nature of tumors associated with TSC mutations, and also may provide a means for tumor resistance to mTOR inhibition (21, 41).
We demonstrate here that exposure of EC-vGPCR cells to increasing doses of rapamycin lead to an increase of Akt phosphorylation, suggesting that this drug also interferes with the regulation of PI3K/Akt activity in endothelial cells expressing vGPCR. As activation of Akt alone has been shown to be sufficient to promote endothelial cell transformation in vivo (11), this may prove to be an Achilles' heel for rapamycin in the treatment of KS; this may provide insight into why treatment with rapamycin has not been successful in some KS patients (17-20).
To overcome this limitation, we set out to assess the efficacy of a novel dual PI3Kα/mTOR inhibitor for the treatment of KS. Combinatorial inhibition of multiple targets has proven to be a very effective approach in the development of cancer inhibitors due to the complexity and redundancy of signaling networks underlying malignant transformation. We demonstrate here that PI-103 is able to effectively and independently block activation of both PI3K and mTOR by vGPCR in expressing endothelial cells. In turn, PI-103 inhibited the proliferation of endothelial cells expressing vGPCR in vitro, and efficiently inhibited the ability of this cell line to form tumors in vivo. Inhibition of tumor growth by PI-103 correlated with the induction of apoptosis through the inhibition of the PI3K/Akt and mTOR pathways. Collectively, our results demonstrate that PI-103 may prove to be a more effective treatment for patients with iatrogenic KS.
As drugs that inhibit mTOR have been shown to exhibit potent immunosuppressive activity, this may potentially discourage its use in patients with a chronic immunosuppressive state such as occurs in AIDS-KS patients. Indeed, how mTOR inhibitors affect viral proliferation (KSHV and HIV) and the immune responses to these viruses, remains unclear. However, the results of several small clinical studies suggest that immunosuppressive medications might be safe and effective for patients with stable HIV disease (42). Moreover, given the importance of immune activation for HIV disease progression, it has been suggested that pharmacologic modulation of immune activation may be of potential benefit in slowing down the rate of AIDS progression (42). In addition, it has been demonstrated that rapamycin may exert a direct antiretroviral effect by repressing HIV replication (43). The administration of rapamycin to non-human primates has also been shown to decrease CCR5 mRNA expression in vivo, further suggesting that rapamycin treatment may promote the prevention and treatment of HIV infection (44). Of note, it has also been hypothesized that co-receptor activation of the PI3K/AKT signaling pathway by HIV gp120 may promote the survival of infected T lymphocytes in HIV patients (4). In light of our results, this suggests that simultaneous inhibition of both mTOR and PI3K, as occurs with PI-103, may provide an effective therapeutic approach for patients with AIDS-KS.
Our results may further have more broad implications. Emerging appreciation for rapamycin as an anti-angiogenic treatment suggests that drugs targeting the mTOR pathway may prove to be an effective anti-angiogenic strategy for the treatment of a variety of diseases. Indeed, as KS may serve as a model for tumor induced- or pathologic angiogenesis, our results collectively provide the basis for the early assessment of PI-103 as an anti-KS and anti-angiogenic chemotherapeutic agent.
Acknowledgments
This work was supported by grant R01CA119911 (National Cancer Institute, NIH). We thank Histoserv, Inc for their assistance in the processing of the murine tissues. BCJ is a recipient of a predoctoral fellowship from the CNPq-Brazil.
References
- 1.Dourmishev LA, Dourmishev AL, Palmeri D, Schwartz RA, Lukac DM. Molecular genetics of Kaposi's sarcoma-associated herpesvirus (human herpesvirus-8) epidemiology and pathogenesis. Microbiol Mol Biol Rev. 2003;67:175–212. doi: 10.1128/MMBR.67.2.175-212.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Moore PS, Chang Y. Molecular virology of Kaposi's sarcoma-associated herpesvirus. Philos Trans R Soc Lond B Biol Sci. 2001;356:499–516. doi: 10.1098/rstb.2000.0777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sodhi A, Montaner S, Gutkind JS. Does dysregulated expression of a deregulated viral GPCR trigger Kaposi's sarcomagenesis? Faseb J. 2004;18:422–7. doi: 10.1096/fj.03-1035hyp. [DOI] [PubMed] [Google Scholar]
- 4.Sodhi A, Montaner S, Gutkind JS. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol. 2004;5:998–1012. doi: 10.1038/nrm1529. [DOI] [PubMed] [Google Scholar]
- 5.Montaner S, Sodhi A, Molinolo A, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–36. doi: 10.1016/s1535-6108(02)00237-4. [DOI] [PubMed] [Google Scholar]
- 6.Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G, Reitz M. Kaposi's sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse. J Virol. 2003;77:2631–9. doi: 10.1128/JVI.77.4.2631-2639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang TY, Chen SC, Leach MW, et al. Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi's sarcoma. J Exp Med. 2000;191:445–54. doi: 10.1084/jem.191.3.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Curatolo P. Tuberous Sclerosis Complex: from Basic Science to Clinical Phenotypes. 2003 [Google Scholar]
- 9.Montaner S, Sodhi A, Pece S, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B. Cancer Res. 2001;61:2641–8. [PubMed] [Google Scholar]
- 10.Phung TL, Ziv K, Dabydeen D, et al. Pathological angiogenesis is induced by sustained Akt signaling and inhibited by rapamycin. Cancer Cell. 2006;10:159–70. doi: 10.1016/j.ccr.2006.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sodhi A, Montaner S, Patel V, et al. Akt plays a central role in sarcomagenesis induced by Kaposi's sarcoma herpesvirus-encoded G protein-coupled receptor. Proc Natl Acad Sci U S A. 2004;101:4821–6. doi: 10.1073/pnas.0400835101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sodhi A, Chaisuparat R, Hu J, et al. The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. Cancer Cell. 2006;10:133–43. doi: 10.1016/j.ccr.2006.05.026. [DOI] [PubMed] [Google Scholar]
- 13.Campistol JM, Schena FP. Kaposi's sarcoma in renal transplant recipients--the impact of proliferation signal inhibitors. Nephrol Dial Transplant. 2007;22(Suppl 1):i17–22. doi: 10.1093/ndt/gfm089. [DOI] [PubMed] [Google Scholar]
- 14.Montaner S. Akt/TSC/mTOR activation by the KSHV G protein-coupled receptor: emerging insights into the molecular oncogenesis and treatment of Kaposi's sarcoma. Cell Cycle. 2007;6:438–43. doi: 10.4161/cc.6.4.3843. [DOI] [PubMed] [Google Scholar]
- 15.Guenova E, Metzler G, Hoetzenecker W, Berneburg M, Rocken M. Classic Mediterranean Kaposi's sarcoma regression with sirolimus treatment. Arch Dermatol. 2008;144:692–3. doi: 10.1001/archderm.144.5.692. [DOI] [PubMed] [Google Scholar]
- 16.Merimsky O, Jiveliouk I, Sagi-Eisenberg R. Targeting mTOR in HIV-Negative Classic Kaposi's Sarcoma. Sarcoma. 2008;2008:825093. doi: 10.1155/2008/825093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Boratynska M, Zmonarski SC, Klinger M. Reccurence of Kaposi's sarcoma after increased exposure to sirolimus. Int Immunopharmacol. 2006;6:2018–22. doi: 10.1016/j.intimp.2006.09.018. [DOI] [PubMed] [Google Scholar]
- 18.Descoeudres B, Giannini O, Graf T, Steiger J, Mayr M. No effect of sirolimus for kaposi sarcoma in a renal transplant recipient. Transplantation. 2006;81:1472–4. doi: 10.1097/01.tp.0000203322.99037.d2. [DOI] [PubMed] [Google Scholar]
- 19.Lebbe C, Euvrard S, Barrou B, et al. Sirolimus conversion for patients with posttransplant Kaposi's sarcoma. Am J Transplant. 2006;6:2164–8. doi: 10.1111/j.1600-6143.2006.01412.x. [DOI] [PubMed] [Google Scholar]
- 20.Wasywich CA, Croxson MC, van Doornum GJ, Coverdale HA, Ruygrok PN. Sirolimus for Kaposi's sarcoma. J Heart Lung Transplant. 2006;25:726–9. doi: 10.1016/j.healun.2006.01.010. [DOI] [PubMed] [Google Scholar]
- 21.Easton JB, Kurmasheva RT, Houghton PJ. IRS-1: auditing the effectiveness of mTOR inhibitors. Cancer Cell. 2006;9:153–5. doi: 10.1016/j.ccr.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 22.O'Reilly KE, Rojo F, She QB, et al. mTOR inhibition induces upstream receptor tyrosine kinase signaling and activates Akt. Cancer Res. 2006;66:1500–8. doi: 10.1158/0008-5472.CAN-05-2925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi Y, Yan H, Frost P, Gera J, Lichtenstein A. Mammalian target of rapamycin inhibitors activate the AKT kinase in multiple myeloma cells by up-regulating the insulin-like growth factor receptor/insulin receptor substrate-1/phosphatidylinositol 3-kinase cascade. Mol Cancer Ther. 2005;4:1533–40. doi: 10.1158/1535-7163.MCT-05-0068. [DOI] [PubMed] [Google Scholar]
- 24.Chiang GG, Abraham RT. Targeting the mTOR signaling network in cancer. Trends Mol Med. 2007;13:433–42. doi: 10.1016/j.molmed.2007.08.001. [DOI] [PubMed] [Google Scholar]
- 25.Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12:9–22. doi: 10.1016/j.ccr.2007.05.008. [DOI] [PubMed] [Google Scholar]
- 26.Fan QW, Knight ZA, Goldenberg DD, et al. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell. 2006;9:341–9. doi: 10.1016/j.ccr.2006.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wendel HG, De Stanchina E, Fridman JS, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 28.Orsulic S, Li Y, Soslow RA, Vitale-Cross LA, Gutkind JS, Varmus HE. Induction of ovarian cancer by defined multiple genetic changes in a mouse model system. Cancer Cell. 2002;1:53–62. doi: 10.1016/s1535-6108(01)00002-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Patel V, Lahusen T, Leethanakul C, et al. Antitumor activity of UCN-01 in carcinomas of the head and neck is associated with altered expression of cyclin D3 and p27(KIP1) Clin Cancer Res. 2002;8:3549–60. [PubMed] [Google Scholar]
- 30.Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC, Cesarman E. Human herpesvirus KSHV encodes a constitutively active G-protein- coupled receptor linked to cell proliferation. Nature. 1997;385:347–50. doi: 10.1038/385347a0. [DOI] [PubMed] [Google Scholar]
- 31.Cesarman E, Nador RG, Bai F, et al. Kaposi's sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi's sarcoma and malignant lymphoma. J Virol. 1996;70:8218–23. doi: 10.1128/jvi.70.11.8218-8223.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ho HH, Ganeshalingam N, Rosenhouse-Dantsker A, Osman R, Gershengorn MC. Charged residues at the intracellular boundary of transmembrane helices 2 and 3 independently affect constitutive activity of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor. J Biol Chem. 2001;276:1376–82. doi: 10.1074/jbc.M007885200. [DOI] [PubMed] [Google Scholar]
- 33.Im E, von Lintig FC, Chen J, et al. Rheb is in a high activation state and inhibits BRaf kinase in mammalian cells. Oncogene. 2002;21:6356–65. doi: 10.1038/sj.onc.1205792. [DOI] [PubMed] [Google Scholar]
- 34.Folkman J. Angiogenesis. Annu Rev Med. 2006;57:1–18. doi: 10.1146/annurev.med.57.121304.131306. [DOI] [PubMed] [Google Scholar]
- 35.Ensoli B, Barillari G, Gallo RC. Cytokines and growth factors in the pathogenesis of AIDS-associated Kaposi's sarcoma. Immunol Rev. 1992;127:147–55. doi: 10.1111/j.1600-065x.1992.tb01412.x. [DOI] [PubMed] [Google Scholar]
- 36.Jenner RG, Boshoff C. The molecular pathology of Kaposi's sarcoma-associated herpesvirus. Biochim Biophys Acta. 2002;1602:1–22. doi: 10.1016/s0304-419x(01)00040-3. [DOI] [PubMed] [Google Scholar]
- 37.Wang L, Damania B. Kaposi's sarcoma-associated herpesvirus confers a survival advantage to endothelial cells. Cancer Res. 2008;68:4640–8. doi: 10.1158/0008-5472.CAN-07-5988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Campistol JM, Gutierrez-Dalmau A, Torregrosa JV. Conversion to sirolimus: a successful treatment for posttransplantation Kaposi's sarcoma. Transplantation. 2004;77:760–2. doi: 10.1097/01.tp.0000115344.18025.0b. [DOI] [PubMed] [Google Scholar]
- 39.McCaffrey P. Sirolimus does double duty after organ transplantation. Lancet Oncol. 2005;6:261. doi: 10.1016/s1470-2045(05)70146-7. [DOI] [PubMed] [Google Scholar]
- 40.Stallone G, Schena A, Infante B, et al. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–23. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 41.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19:1773–8. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Argyropoulos C, Mouzaki A. Immunosuppressive drugs in HIV disease. Curr Top Med Chem. 2006;6:1769–89. doi: 10.2174/156802606778194271. [DOI] [PubMed] [Google Scholar]
- 43.Roy J, Paquette JS, Fortin JF, Tremblay MJ. The immunosuppressant rapamycin represses human immunodeficiency virus type 1 replication. Antimicrob Agents Chemother. 2002;46:3447–55. doi: 10.1128/AAC.46.11.3447-3455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gilliam BL, Heredia A, Devico A, et al. Rapamycin reduces CCR5 mRNA levels in macaques: potential applications in HIV-1 prevention and treatment. Aids. 2007;21:2108–10. doi: 10.1097/QAD.0b013e3282f02a4f. [DOI] [PubMed] [Google Scholar]