

Abstract
The amygdala is a medial forebrain structure with an established role in nociceptive modulation, including the expression of stress-induced hypoalgesia (SIH). Projections from the locus coeruleus increase levels of noradrenaline in the amygdala during acute stress. α2-Noradrenergic receptor agonists have significant clinical utility as analgesic agents. We therefore hypothesized that α2-noradrenergic activation of the amygdala may result in behaviorally measurable hypoalgesia. Lightly anesthetized rats underwent microinjection of the α2-noradrenergic agonist clonidine into the amygdala and intermittent measurement of thermal nociception using the tail-flick latency. Bilateral microinjection of clonidine into the central nucleus of the amygdala (CeA) resulted in a significant, dose-dependent increase in tail-flick latency. This effect was blocked by systemic pre-treatment with the α2-antagonist yohimbine or by local pre-injection of the α2-antagonist idazoxan but not by local pre-injection of the α1-antagonist WB-4101. When injected alone, no antagonist resulted in a significant change in tail-flick latency compared to baseline. Clonidine injection into the amygdala but outside the CeA, including the BLA, did not significantly alter tail-flick latency. These results demonstrate that anatomically and pharmacologically specific activation of α2-receptors in the CeA in lightly anesthetized rats results in behaviorally measurable antinociception.
Keywords: hypoalgesia, clonidine, microinjection, α2-agonist, idazoxan, yohimbine
The amygdala integrates multiple sensory, including nociceptive, inputs leading to adaptive autonomic, endocrine, and behavioral responses (Bernard and Besson, 1990, Neugebauer et al., 2003, Li and Neugebauer, 2004, Neugebauer et al., 2004). It is comprised of several nuclei with differing cytoarchitecture, connectivity, and function (Davis and Whalen, 2001, Sah et al., 2003). The central nucleus of the amygdala (CeA), has been called the ‘nociceptive amygdala’ and also serves as the major output nucleus of the amygdala (Freedman and Aghajanian, 1985, Rizvi et al., 1991, Pitkanen et al., 1997, Bourgeais et al., 2001, Neugebauer et al., 2003). A majority (80%) of CeA neurons are excited or inhibited exclusively or preferentially by noxious stimuli including pinch, squeeze, and hot water bath (Bernard et al., 1992). The amygdala receives nociceptive input from the parabrachial nucleus and has reciprocal connections with the periaquductal gray, a midbrain structure with a role in descending nociceptive modulation (Beitz, 1982, Bernard and Besson, 1990, Rizvi et al., 1991, Bourgeais et al., 2001, Neugebauer et al., 2003).
Consistent with these anatomical and physiological observations, the amygdala contributes to nociceptive modulatory function. For example, electrical or chemical stimulation of various nuclei within the amygdala, including the CeA, produce antinociception (Helmstetter et al., 1993, Mena et al., 1995, Manning et al., 2003). The amygdala is also necessary for the expression of stress-induced hypoalgesia (SIH) (antinociception resulting from exposure to a stressful stimulus such as electric shock or immobilization) and conditioned hypoalgesia (antinociception resulting from exposure to a conditioned stimulus paired with a stressor). Furthermore, amygdala lesions block both SIH and conditioned hypoalgesia (Helmstetter and Bellgowan, 1993, Fox and Sorenson, 1994, Pavlovic et al., 1996, Shavit et al., 2005).
Noradrenaline is released within the amygdala in response to acute stress such as that which occurs during SIH (Tanaka et al., 1991, Fendt et al., 1994, Quirarte et al., 1998, Khoshbouei et al., 2002). In general, noradrenergic projections are activated by presentation of acute stressors and exert various complementary modulatory effects on target structures (Selden et al., 1991, Berridge and Waterhouse, 2003, Morilak et al., 2005). The locus coeruleus (LC) is a key component of this system, which becomes active in response to stressors including electric shock, loud noise, tail-pinch, and immobilization (U'Prichard et al., 1980, Aston-Jones et al., 1991, Passerin et al., 2000, Pardon et al., 2002, Ma and Morilak, 2004). The LC has efferent projections to the CeA via the dorsal noradrenergic bundle and noradrenaline levels increase in the CeA in response to the presentation of acute stressors (U'Prichard et al., 1980, Tanaka et al., 1991, Fendt et al., 1994, Quirarte et al., 1998, Khoshbouei et al., 2002). Lesion of the dorsal noradrenergic bundle attenuates the expected elevation of noradrenaline levels in the amygdala after acute stress (U'Prichard et al., 1980). Thus, noradrenergic input may influence amygdala function, including nociceptive modulation, particularly during stressful environmental contingencies.
Noradrenaline subserves many functions in response to acute stressors, including antinociception via the α2-adrenoceptor subtype. Activation of α2-adrenoceptors leads to neuronal inhibition through a G-protein (Gi) coupled mechanism (Summers and McMartin, 1993, Boyd, 2001, Calzada and De Artinano, 2001). Systemic, intrathecal, intracerebroventricular, and intramedullary administration of the α2-noradrenergic receptor agonist clonidine results in decreased nociceptive responsiveness (Hammond et al., 1980, Yasuoka and Taksh, 1983, Sagen and Proudfit, 1985, Haws et al., 1990, Calzada and De Artinano, 2001, Quartilho et al., 2004). This finding has been exploited clinically by utilizing clonidine to potentiate the intrathecal administration of analgesics (Schug et al., 2006). The antinociceptive effect of clonidine is blocked in gene knock-out and knock-down mouse models targeting the α2a receptor subtype. (Lakhlani et al., 1997, Wang et al., 2002, Lahdesmaki et al., 2003). α2-Adrenoceptors are also implicated in the expression of SIH. Systemic treatment with the α2-adrenoceptor antagonists yohimbine or idazoxan blocks the expression of SIH in rodents (Coderre and Rollman, 1984, Oluyomi and Hart, 1990, Tokuyama et al., 1991). α2-Adrenoceptors are present throughout the central nervous system, including within the CeA (U'Prichard et al., 1980, Freedman and Aghajanian, 1985, Nicholas et al., 1996).
In sum, several lines of evidence suggest that the stress-related neurotransmitter noradrenaline, via its α2-adrenoceptor subtype, may play a role in the expression of nocifensive behavior mediated by the amygdala. We therefore tested the hypothesis that activation of α2-noradrenergic receptors in the amygdala would result in behaviorally measurable antinociception.
EXPERIMENTAL PROCEDURES
Animals and surgical preparation
Experimental protocols were approved by the Institutional Animal Care and Use Committee of Oregon Health & Science University. All experiments conformed to the guidelines of the International Association for the Study of Pain (IASP, 1983). Efforts were made throughout experiments to minimize animal discomfort and to reduce the number of animals used. Male Sprague–Dawley rats (Taconic, Indianapolis, IN, USA; 250−300 g) were anesthetized with pentobarbital (60 mg/kg, i.p.), and a catheter inserted into an external jugular vein for administration of anesthetic. The rats were placed in a stereotaxic apparatus, holes were drilled in the skull over the amygdala bilaterally, and the dura removed to allow for placement of a microinjection pipette. Body temperature was maintained at approximately 37 °C by a circulating water pad. Following surgery, the anesthetic level was allowed to lighten until a tail-flick reflex could be elicited by application of noxious heat using a feedback-controlled projector lamp focused on the blackened ventral surface of the tail. Following surgical preparation, the animals were then maintained in a lightly anesthetized state using a continuous infusion of methohexital at a rate (15−30 mg/kg per h, i.v.) that allowed a stable tail-flick latency (TFL) and that prevented any signs of discomfort. The animals did not move spontaneously, vocalize, or produce vigorous or prolonged withdrawal reflexes following noxious pinch. The protocol was begun after a stabilization period of at least 30 min, and the infusion rate was not altered during the protocol.
Nociceptive testing
Latency to tail-flick to heat was used as a measure of nociceptive responsiveness. Each trial consisted of a linear increase in temperature at approximately 1.8 °C/s from a holding temperature of 34 °C until the tail-flick occurred or to a maximum of 52 °C at 10.2 s. The holding temperature compensates for any potential variation in skin temperature of the subject during testing. Trials were carried out at 5 min intervals throughout the experiment. Tail-flick latency was considered stable after six successive trials with less than 1.5 sec variation between trials. Temperature and tail-flicks were monitored and recorded on a computer using commercially available software (Spike 2, Cambridge Electronic Design, Cambridge, England)
Microinfusion protocol
After achieving a stable TFL, a microinfusion pipette (60−80μm outer diameter beveled tip), secured in the carrier arm of the stereotaxic apparatus, was positioned over the amygdala to be infused first (right and left sided initial infusion sites were alternated in successive experiments). The micropipette tip was then lowered into the amygdala according to the coordinates of Paxinos & Watson (Paxinos and Watson, 1986) (relative to bregma, CeA: A/P −2.8mm, M/L ± 4.2mm, D/V −8.1mm; BLA: A/P −2.8mm, M/L ±4.8mm, D/V −8.5mm). Drug or vehicle was then injected through the micropipette which was attached via a length of polyethylene tubing (PE-50) to a glass microsyringe (1μL, Hamilton, Reno, NV). Infusions (0.2μL infusion volumes were used in all experiments) were performed over 1.5 min, and the micropipette tip was left in place for an additional 1 min. After completion of one amygdala, the micropipette was withdrawn and moved to the contralateral side. A small amount of solution was expressed from the tip to ensure free flow of solution, and the micropipette tip was lowered into the contralateral amygdala for an identical infusion. Beginning 5 min after the second infusion, tail-flick trials were resumed at 5 min intervals for 30 min.
For experiments in which systemic drug administration occurred, the following modifications were made. After achieving a stable TFL, an intra-peritoneal (i.p.) drug injection was made. Beginning 5 min after injection, three tail-flick trials were performed. Intra-amygdala microinfusion was then carried out in a manner identical to that described above and was followed by six tail-flick trials at 5 min intervals.
For experiments in which successive intra-amygdala microinfusions were performed, the following modifications were made. The initial microinfusion proceeded as described above. A single tail-flick was then recorded 5 min after completing the infusion. The drug solution was evacuated from the micropipette and tubing, which were then flushed with saline vehicle, and the next drug (or vehicle) to be tested was loaded for infusion. The bilateral sequential infusion procedure was then repeated using the second drug, and the final infusion was followed by 6 successive tail-flick trials at 5 min intervals.
Drugs
All drugs used were commercially available (Sigma/Aldrich, St. Louis, MO) and were dissolved in physiological saline. Drug solutions were prepared daily, and the pH adjusted to 7.4.
Histology
At the conclusion of the experiments, infusion sites were marked by injection of Pontamine Sky Blue dye. Animals were killed with an overdose of methohexital, and perfused intracardially with physiological saline followed by 10% formalin. Tissue was counterstained with Cresyl Violet, and infusion sites were histologically verified and plotted on standardized sections (Paxinos and Watson, 1986). A representative section through the amygdala is shown in Fig. 1 and the microinfusion sites are shown in Fig. 2.
Fig. 1.

Representative section through the left amygdala showing the CeA (thin arrow) and BLA (open arrow), the microinjector tip placement is seen within the CeA (bold arrow).
Fig. 2.
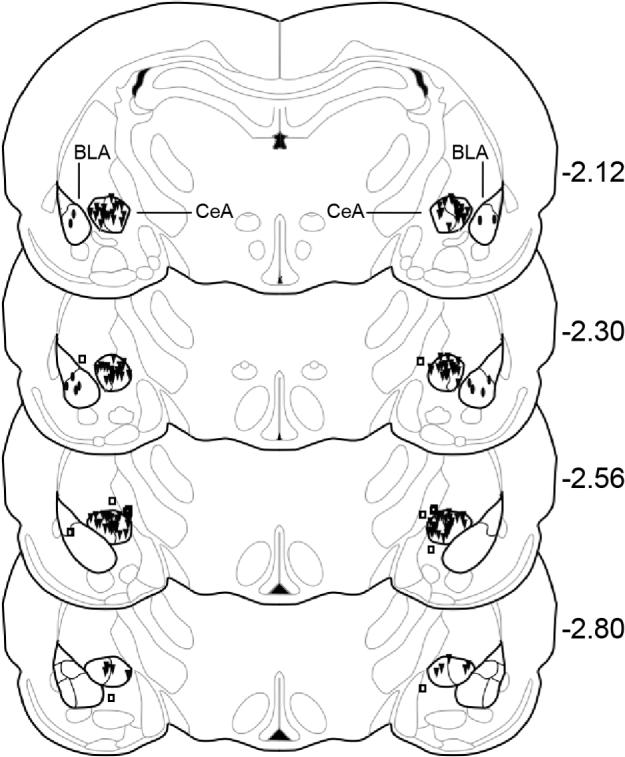
Serial coronal sections of the rat brain showing placement of microinjections (N = 73). Filled triangles = CeA injections, filled ovals = BLA injections, open squares = placement controls. CeA = central nucleus of the amygdala, BLA = basolateral nucleus of the amygdala, numbers to the right of sections represent millimeters relative to bregma, adapted from the atlas of Paxinos and Watson (1986).
Data analysis
Data are presented as percent maximal possible effect (% MPE), which is calculated according to the formula: [(average post injection TFL – average baseline TFL)/(cut-off TFL – average baseline TFL)] x 100. Student's t-test for correlated means was used for comparing baseline and post-injection tail-flick latencies. ANOVA was used for between group comparisons. P < 0.05 was considered significant.
RESULTS
Microinfusion of the α2-adrenoceptor agonist clonidine into the CeA significantly increased the TFL in a dose-responsive manner (Fig. 3). Systemic yohimbine (an α2-noradrenergic receptor antagonist; 2mg/kg i.p.) blocked the antinociceptive effect of intra-CeA clonidine (1.25μg) (TFL: −21.1 ± 3% MPE, p = NS compared to baseline). Local microinjection of idazoxan (an α2-adrenoceptor antagonist from a separate structural class than yohimbine) also blocked the effect of microinjected clonidine (Fig. 4). By contrast, microinjection of the α1-adrenoceptor antagonist WB-4101 did not block the effect of subsequently microinjected clonidine.
Fig. 3.

Microinjection of clonidine into the CeA results in a dose-dependent increase in the TFL to radiant heat. Saline microinjection did not result in a significant change in the TFL as compared to baseline (p > 0.05, Student's t-test for correlated means, N = 8−10 per group). There were no differences among the groups in baseline latencies (ANOVA). All groups differed significantly in post-treatment latencies (ANOVA). (*p<0.05, **p<0.01, values averaged over 30 min post-injection time period compared with baseline, Student's t-test for correlated means)
Fig. 4.
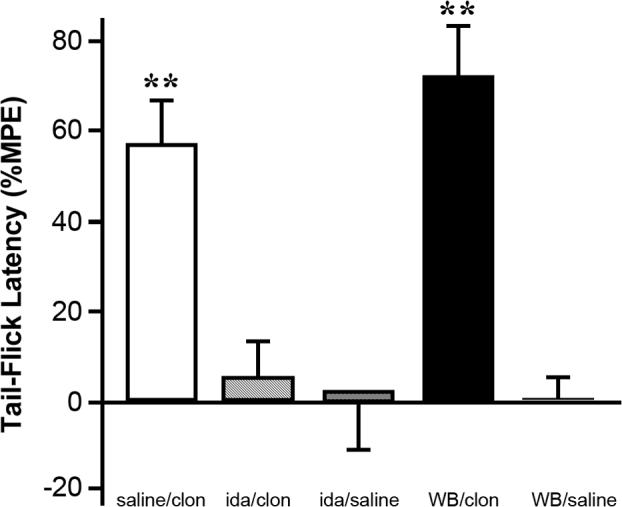
The increase in TFL to intra-CeA clonidine microinfusion is dependent upon the α2-noradrenergic receptor subtype. Saline microinjection followed by clonidine (1.25μg), an α2-noradrenergic receptor agonist, resulted in a significant increase in TFL. Microinjection of the α2-noradrenergic receptor antagonist idazoxan (9μg) blocked the effect of subsequently microinjected clonidine (1.25μg). Microinjection of idazoxan followed by saline microinjection resulted in a TFL that did not significantly differ from baseline. Microinjection of the α1-noradrenergic receptor antagonist WB-4101 (12μg) did not block the effect of subsequently microinjected clonidine (1.25μg). Microinjection of WB-4101 (12μg) followed by saline injection did not result in a significant alteration of baseline TFL, N = 5−8 per group. There were no differences among the groups in baseline latencies (ANOVA). There was no significant difference in post-treatment TFL between the saline/clonidine and WB-4101/clonidine groups (ANOVA). (**p<0.01, values averaged over 30 min post-injection time period compared with baseline, Student's t-test for correlated means). clon = clonidine, ida = idazoxan, WB = WB-4101.
Several control experiments indicated that the various antagonist drugs used did not have independent effects on nociceptive responsiveness. Systemic yohimbine (2mg/kg i.p.) alone did not significantly alter the TFL ( −12.4 ± 12% MPE, p = NS, values averaged over 30 min post-injection time period compared with baseline, Student's t-test for correlated means, N = 6). Microinjection of neither idazoxan nor WB-4101 followed by saline vehicle microinjection significantly altered the TFL (Fig. 4). Furthermore, the principal antinociceptive effect of clonidine injection was replicated using the sequential local microinjection protocol employed for antagonist testing (saline vehicle microinfusion (0.2 μL) followed by clonidine microinfusion (1.25 μg)). Finally, high dose injections of clonidine (5.0 μg) within the BLA or within the amygdala but outside of both the CeA and BLA each failed to affect the TFL (Fig. 5).
Fig. 5.
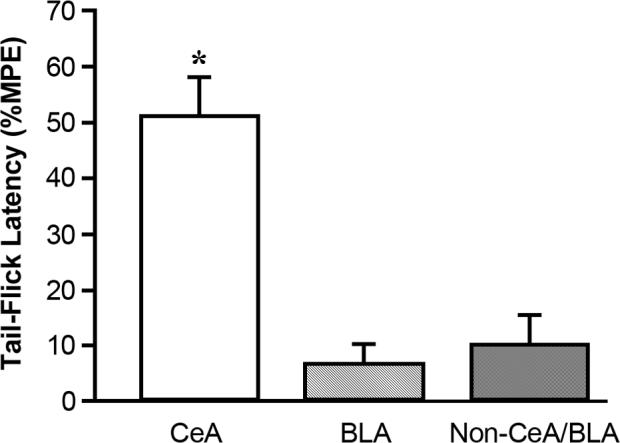
Placement of clonidine microinjections within the amygdala, but outside of the CeA did not result in TFLs that differed significantly from baseline. Intra-CeA microinjection of clonidine (1.25μg) is shown for comparison. Microinjections of clonidine (1.25μg) specifically targeted to the BLA did not result in significantly altered TFLs. Microinjections of clonidine (5μg) into the amygdala, but not within the CeA or BLA (either unilaterally or bilaterally) also did not result in significantly altered TFLs. (*p<0.05, values averaged over 30 min post-injection time period compared with baseline, Student's t-test for correlated means, N = 6−8 per group). CeA = central nucleus of the amygdala, BLA = basolateral nucleus of the amygdala
DISCUSSION
Injection of the α2-adrenoceptor agonist clonidine into the CeA resulted in dose-dependent thermal antinociception in lightly anesthetized rats. This effect was successfully antagonized by either peripheral or local administration of α2- but not α1-adrenoceptor antagonists. Intra-amygdalar injection of clonidine immediately adjacent to but outside of the CeA was ineffective in producing antinociception. These results demonstrate a pharmacologically and anatomically specific antinociceptive effect of α2-adrenoceptor activation in the CeA.
Modulation of nocifensive behavior by the amygdala
Microinjection of clonidine into the CeA resulted in a dose-dependent increase in TFL. This result is in agreement with previous studies demonstrating that pharmacological manipulation of the amygdala can influence nocifensive behavior. Microinfusion of opioids or endocannabinoids into the amygdala results in increased thresholds for nocifensive behaviors in both lightly anesthetized and awake behaving rats (Helmstetter et al., 1993, Manning and Mayer, 1995, McGaraughty and Heinricher, 2002, Hohmann et al., 2005). Similarly, electrical stimulation of the amygdala results in increased thresholds for nocifensive behavior (Mena et al., 1995). Noradrenergic agonists, opioids, endocannabinoids, and electrical stimulation all act on pain modulatory structures within the CNS (including the spinal cord dorsal horn, rostral ventromedial medulla, and periaqueductal gray) to increase the threshold for nocifensive behaviors (Fields et al., 1991, Hohmann and Suplita, 2006, Pertovaara, 2006). The current results therefore add to the evidence that the amygdala may be included among the CNS structures that exhibit nociceptive modulatory actions.
Blockade of α2-receptor function in the amygdala does not appear to alter baseline nociceptive function under the present conditions. Similarly, lesions or temporary inactivation of the amygdala globally fail to affect baseline nociceptive responsiveness (Helmstetter, 1992, Fox and Sorenson, 1994, Pavlovic et al., 1996, Manning et al., 2003). Instead, the amygdala appears to modulate nociception under particular environmental contingenices, such as SIH and conditioned hypoalgesia (Helmstetter, 1992, Helmstetter and Bellgowan, 1993, Fox and Sorenson, 1994). This distinction supports the hypothesis that the amygdala coordinates a set of species-specific behaviors in response to polymodal sensory input in the context of aversive environmental cues (Pitkanen et al., 1997, Berretta, 2005, Balleine and Killcross, 2006).
Antinociceptive effect of the α2-adrenoceptor
Our results indicate that the α2-adrenoceptor mediates the increase in TFL observed after microinfusion of clonidine into the CeA. Microinjection of the α2-antagonist idazoxan or systemic injection of the α2-antagonist yohimbine, but not microinjection of the α1-antagonist WB-4101, blocked the expected increase in TFL after clonidine microinjection. Because both idazoxan (which is also active at imidazoline receptors) and yohimbine (which has no effect at imidazoline receptors) each effectively antagonized clonidine in these experiments, it is more likely that the antinociceptive effect of clonidine is specific to the α2-receptor and does not involve imidazoline receptors. None of the antagonists, when administered alone, significantly affected the TFL.
Clonidine induced antinociception is consistent with the known role of α2-adrenoceptors in nociceptive modulation. Noradrenaline is a key neurotransmitter in the modulation of nociception throughout the neuraxis (Pertovaara, 2006). Noradrenergic agonists induce antinociception when locally administered within CNS pain modulatory structures, including the locus coeruleus, periaqueductal gray, nucleus raphe magnus, and dorsal horn of the spinal cord (Hammond et al., 1980, Fleetwood-Walker et al., 1985, Haws et al., 1990, Guo et al., 1996). α2-Receptors mediate the antinociceptive effects of noradrenaline within these structures via coupling to an inhibitory G-protein (Gi) or direct modification of ion channels. Activation of the pre-synaptic receptor leads to membrane hyperpolarization and inhibition of further noradrenaline release (Summers and McMartin, 1993, Boyd, 2001, Calzada and De Artinano, 2001). The CeA contains both pre- and post-synaptic α2-adrenoceptors (U'Prichard et al., 1980, Glass et al., 2002, Khoshbouei et al., 2002). It is therefore likely that activation of α2-adrenoceptors by clonidine leads to inhibition of CeA neurons, which has a net antinociceptive effect. Consistent with this finding, intra-CeA administration of α2-adrenoceptor antagonists blocks the antinociceptive effect of clonidine, but does not affect baseline nociceptive responsiveness.
Contribution of Amygdalar Nuclei
Clonidine microinjection into the amygdala resulted in increased TFL when targeted to the CeA. Microinjection of clonidine into the BLA or within the amygdala, but outside the CeA or BLA, did not affect the TFL. This finding is consistent with identification of the CeA as a major output nucleus of the amygdala with a significant role in nociceptive processing (Freedman and Aghajanian, 1985, Rizvi et al., 1991, Pitkanen et al., 1997, Bourgeais et al., 2001, Neugebauer et al., 2003).
Nociceptive modulatory processing within the amygdala is specific to various individual nuclei and pharmacological systems (Helmstetter et al., 1993, Manning and Mayer, 1995, McGaraughty and Heinricher, 2002, Manning et al., 2003, Connell et al., 2006). For example, microinjections of μ-opioid agonists into the basolateral, but not the central nucleus of the amygdala results in thermal antinociception (McGaraughty and Heinricher, 2002). By contrast, morphine-induced antinociception after formalin hyperalgesia depends on the CeA (Manning and Mayer, 1995). The cannabinoid neurotransmitter system also mediates antinociception via either the basolateral or central nucleus depending on experimental conditions. CeA but not BLA lesions interfere with the thermal antinoceptive effects of systemic administration of cannabinoid receptor agonists (Manning et al., 2003). By contrast, local administration of a cannabinoid receptor antagonist into the BLA, but not the CeA, blocks the expression of footshock-induced SIH as measured by the tail-flick test (Connell et al., 2006). Taken together, these results suggest that nociceptive modulation by the amygdala may be dependent on the actions of particular neurotransmitters within individual amygdala nuclei under specific environmental conditions,
Microinfusion of clonidine into the amygdala results in dose-dependent increase in tail-flick latency to radiant heat in lightly anesthetized rats. This finding is anatomically and pharmacologically specific to α2-receptors in the CeA. The identification of non-opiate mechanisms of supraspinal nociceptive modulation may ultimately yield new therapeutic targets of clinical relevance.
Acknowledgements
Supported by grants from the National Institute of Neurological Disorders NS44255 (N.R.S) and the Cameron Foundation.
LIST OF ABBREVIATIONS
- A/P
Anterior/posterior
- ANOVA
Analysis of variance
- BLA
Basolateral nucleus of the amygdala
- CNS
Central nervous system
- CeA
Central nucleus of the amygdala
- D/V
Dorsal/ventral
- i.p.
Intra-peritoneal
- i.v.
Intravenous
- LC
Locus coeruleus
- M/L
Medial/lateral
- NS
Not significant
- % MPE
Percent maximal possible effect
- SIH
Stress-induced hypoalgesia
- TFL
Tail-flick latency
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Aston-Jones G, Chiang C, Alexinsky T. Discharge of noradrenergic locus coeruleus neurons in behaving rats and monkeys suggests a role in vigilance. Prog Brain Res. 1991;88:501–520. doi: 10.1016/s0079-6123(08)63830-3. [DOI] [PubMed] [Google Scholar]
- Balleine BW, Killcross S. Parallel incentive processing: an integrated view of amygdala function. Trends Neurosci. 2006;29:272–279. doi: 10.1016/j.tins.2006.03.002. [DOI] [PubMed] [Google Scholar]
- Beitz AJ. The organization of afferent projections to the midbrain periaqueductal gray of the rat. Neuroscience. 1982;7:133–159. doi: 10.1016/0306-4522(82)90157-9. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1990;63:473–490. doi: 10.1152/jn.1990.63.3.473. [DOI] [PubMed] [Google Scholar]
- Bernard JF, Huang GF, Besson JM. Nucleus Centralis of the Amygdala and the Globus Pallidus Ventralis: Electrophysiological Evidence for an Involvement in Pain Processes. J Neurophys. 1992;68:551–569. doi: 10.1152/jn.1992.68.2.551. [DOI] [PubMed] [Google Scholar]
- Berretta S. Cortico-amygdala circuits: role in the conditioned stress response. Stress. 2005;8:221–232. doi: 10.1080/10253890500489395. [DOI] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev. 2003;42:33–84. doi: 10.1016/s0165-0173(03)00143-7. [DOI] [PubMed] [Google Scholar]
- Bourgeais L, Gauriau C, Bernard JF. Projections from the nociceptive area of the central nucleus of the amygdala to the forebrain: a PHA-L study in the rat. Eur J Neurosci. 2001;14:229–255. doi: 10.1046/j.0953-816x.2001.01640.x. [DOI] [PubMed] [Google Scholar]
- Boyd RE. α2-Adrenergic Receptor Agonists as Analgesics. Curr Top Med Chem. 2001;1:193–197. doi: 10.2174/1568026013395182. [DOI] [PubMed] [Google Scholar]
- Calzada BC, De Artinano AA. Alpha-Adrenoceptor Subtypes. Pharmacol Res. 2001;44:195–208. doi: 10.1006/phrs.2001.0857. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Rollman GB. Stress analgesia: effects of PCPA, yohimbine, and naloxone. Pharmacol Biochem Behav. 1984;21:681–686. doi: 10.1016/s0091-3057(84)80002-7. [DOI] [PubMed] [Google Scholar]
- Connell K, Bolton N, Olsen D, Piomelli D, Hohmann AG. Role of the basolateral nucleus of the amygdala in endocannabinoid-mediated stress-induced analgesia. Neurosci Lett. 2006;397:180–184. doi: 10.1016/j.neulet.2005.12.008. [DOI] [PubMed] [Google Scholar]
- Davis M, Whalen P. The Amygdala: Vigilance and Emotion. Mol Psychiatr. 2001;6:13–34. doi: 10.1038/sj.mp.4000812. [DOI] [PubMed] [Google Scholar]
- Fendt M, Koch M, Schnitzler HU. Amygdaloid noradrenaline is involved in the sensitization of the acoustic startle response in rats. Pharmacol Biochem Behav. 1994;48:307–314. doi: 10.1016/0091-3057(94)90532-0. [DOI] [PubMed] [Google Scholar]
- Fields HL, Heinricher MM, Mason P. Neurotransmitters in nociceptive modulatory circuits. Annu Rev Neurosci. 1991;14:219–245. doi: 10.1146/annurev.ne.14.030191.001251. [DOI] [PubMed] [Google Scholar]
- Fleetwood-Walker SM, Mitchell R, Hope PJ, Molony V, Iggo A. An alpha 2 receptor mediates the selective inhibition by noradrenaline of nociceptive responses of identified dorsal horn neurones. Brain Res. 1985;334:243–254. doi: 10.1016/0006-8993(85)90216-1. [DOI] [PubMed] [Google Scholar]
- Fox RJ, Sorenson CA. Bilateral lesions of the amygdala attenuate analgesia induced by diverse environmental challenges. Brain Res. 1994;648:215–221. doi: 10.1016/0006-8993(94)91120-7. [DOI] [PubMed] [Google Scholar]
- Freedman JE, Aghajanian GK. Opiate and alpha 2-adrenoceptor responses of rat amygdaloid neurons: co-localization and interactions during withdrawal. J Neurosci. 1985;5:3016–3024. doi: 10.1523/JNEUROSCI.05-11-03016.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glass MJ, Colago EE, Pickel VM. Alpha-2A-adrenergic receptors are present on neurons in the central nucleus of the amygdala that project to the dorsal vagal complex in the rat. Synapse. 2002;46:258–268. doi: 10.1002/syn.10136. [DOI] [PubMed] [Google Scholar]
- Guo TZ, Jiang JY, Buttermann AE, Maze M. Dexmedetomidine injection into the locus ceruleus produces antinociception. Anesthesiology. 1996;84:873–881. doi: 10.1097/00000542-199604000-00015. [DOI] [PubMed] [Google Scholar]
- Hammond DL, Levy RA, Proudfit HK. Hypoalgesia following microinjection of noradrenergic antagonists in the nucleus raphe magnus. Pain. 1980;9:85–101. doi: 10.1016/0304-3959(80)90031-7. [DOI] [PubMed] [Google Scholar]
- Haws CM, Heinricher MM, Fields HL. Alpha-adrenergic receptor agonists, but not antagonists, alter the tail-flick latency when microinjected into the rostral ventromedial medulla of the lightly anesthetized rat. Brain Res. 1990;533:192–195. doi: 10.1016/0006-8993(90)91339-i. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ. The amygdala is essential for the expression of conditional hypoalgesia. Behav Neurosci. 1992;106:518–528. doi: 10.1037//0735-7044.106.3.518. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Bellgowan PS. Lesions of the amygdala block conditional hypoalgesia on the tail flick test. Brain Res. 1993;612:253–257. doi: 10.1016/0006-8993(93)91669-j. [DOI] [PubMed] [Google Scholar]
- Helmstetter FJ, Bellgowan PS, Tershner SA. Inhibition of the tail flick reflex following microinjection of morphine into the amygdala. Neuroreport. 1993;4:471–474. doi: 10.1097/00001756-199305000-00002. [DOI] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL., 2nd Endocannabinoid mechanisms of pain modulation. Aaps J. 2006;8:E693–708. doi: 10.1208/aapsj080479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hohmann AG, Suplita RL, Bolton NM, Neely MH, Fegley D, Mangieri R, Krey JF, Walker JM, Holmes PV, Crystal JD, Duranti A, Tontini A, Mor M, Tarzia G, Piomelli D. An endocannabinoid mechanism for stress-induced analgesia. Nature. 2005;435:1108–1112. doi: 10.1038/nature03658. [DOI] [PubMed] [Google Scholar]
- IASP Ethical guidelines for investigations of experimental pain in conscious animals [Guest editorial]. Pain. 1983;16:109–110. doi: 10.1016/0304-3959(83)90201-4. [DOI] [PubMed] [Google Scholar]
- Khoshbouei H, Cecchi M, Dove S, Javors M, Morilak DA. Behavioral reactivity to stress: amplification of stress-induced noradrenergic activation elicits a galanin-mediated anxiolytic effect in central amygdala. Pharmacol Biochem Behav. 2002;71:407–417. doi: 10.1016/s0091-3057(01)00683-9. [DOI] [PubMed] [Google Scholar]
- Lahdesmaki J, Scheinin M, Pertovaara A, Mansilla H. The α2a-Adrenoceptor Subtype is Not Involved in Inflammatory Hyperalgesia or Morphine-induced Antinociception. Eur J Pharmacol. 2003;468:183–189. doi: 10.1016/s0014-2999(03)01677-7. [DOI] [PubMed] [Google Scholar]
- Lakhlani P, MacMillan L, Guo T, McCool B, Lovinger D, Maze M, Limbird L. Substitution of a Mutant α2a-Adrenergic Receptor via “Hit and Run” Gene Targeting Reveals the Role of this Subtype in Sedative, Analgesic, and Anesthetic-sparing Responses in vivo. Proc Natl Acad Sci. 1997;94:9950–9955. doi: 10.1073/pnas.94.18.9950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Block of NMDA and non-NMDA receptor activation results in reduced background and evoked activity of central amygdala neurons in a model of arthritic pain. Pain. 2004;110:112–122. doi: 10.1016/j.pain.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Ma S, Morilak DA. Induction of FOS expression by acute immobilization stress is reduced in locus coeruleus and medial amygdala of Wistar-Kyoto rats compared to Sprague-Dawley rats. Neuroscience. 2004;124:963–972. doi: 10.1016/j.neuroscience.2003.12.028. [DOI] [PubMed] [Google Scholar]
- Manning BH, Martin WJ, Meng ID. The rodent amygdala contributes to the production of cannabinoid-induced antinociception. Neuroscience. 2003;120:1157–1170. doi: 10.1016/s0306-4522(03)00356-7. [DOI] [PubMed] [Google Scholar]
- Manning BH, Mayer DJ. The central nucleus of the amygdala contributes to the production of morphine antinociception in the formalin test. Pain. 1995;63:141–152. doi: 10.1016/0304-3959(95)00027-P. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Heinricher MM. Microinjection of morphine into various amygdaloid nuclei differentially affects nociceptive responsiveness and RVM neuronal activity. Pain. 2002;96:153–162. doi: 10.1016/s0304-3959(01)00440-7. [DOI] [PubMed] [Google Scholar]
- Mena NB, Mathur R, Nayar U. Amygdalar involvement in pain. Indian J Physiol Pharmacol. 1995;39:339–346. [PubMed] [Google Scholar]
- Morilak DA, Barrera G, Echevarria DJ, Garcia AS, Hernandez A, Ma S, Petre CO. Role of brain norepinephrine in the behavioral response to stress. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29:1214–1224. doi: 10.1016/j.pnpbp.2005.08.007. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RWt. Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Hokfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends Pharmacol Sci. 1996;17:245–255. doi: 10.1016/0165-6147(96)10022-5. [DOI] [PubMed] [Google Scholar]
- Oluyomi AO, Hart SL. Alpha-adrenoceptor involvement in swim stress-induced antinociception in the mouse. J Pharm Pharmacol. 1990;42:778–784. doi: 10.1111/j.2042-7158.1990.tb07020.x. [DOI] [PubMed] [Google Scholar]
- Pardon MC, Gould GG, Garcia A, Phillips L, Cook MC, Miller SA, Mason PA, Morilak DA. Stress reactivity of the brain noradrenergic system in three rat strains differing in their neuroendocrine and behavioral responses to stress: implications for susceptibility to stress-related neuropsychiatric disorders. Neuroscience. 2002;115:229–242. doi: 10.1016/s0306-4522(02)00364-0. [DOI] [PubMed] [Google Scholar]
- Passerin AM, Cano G, Rabin BS, Delano BA, Napier JL, Sved AF. Role of locus coeruleus in foot shock-evoked Fos expression in rat brain. Neuroscience. 2000;101:1071–1082. doi: 10.1016/s0306-4522(00)00372-9. [DOI] [PubMed] [Google Scholar]
- Pavlovic ZW, Cooper ML, Bodnar RJ. Enhancements in swim stress-induced hypothermia, but not analgesia, following amygdala lesions in rats. Physiol Behav. 1996;59:77–82. doi: 10.1016/0031-9384(95)02038-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain and Stereotaxic Coordinates. 2nd Edition Academic Press; New York: 1986. [Google Scholar]
- Pertovaara A. Noradrenergic pain modulation. Prog Neurobiol. 2006;80:53–83. doi: 10.1016/j.pneurobio.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Pitkanen A, Savander V, LeDoux JE. Organization of intra-amygdaloid circuitries in the rat: an emerging framework for understanding functions of the amygdala. Trends Neurosci. 1997;20:517–523. doi: 10.1016/s0166-2236(97)01125-9. [DOI] [PubMed] [Google Scholar]
- Quartilho A, Mata HP, Ibrahim MM, Vanderah TW, Ossipov MH, Lai J, Porreca F, Malan TP., Jr. Production of paradoxical sensory hypersensitivity by alpha 2-adrenoreceptor agonists. Anesthesiology. 2004;100:1538–1544. doi: 10.1097/00000542-200406000-00029. [DOI] [PubMed] [Google Scholar]
- Quirarte GL, Galvez R, Roozendaal B, McGaugh JL. Norepinephrine release in the amygdala in response to footshock and opioid peptidergic drugs. Brain Res. 1998;808:134–140. doi: 10.1016/s0006-8993(98)00795-1. [DOI] [PubMed] [Google Scholar]
- Rizvi TA, Ennis M, Behbehani MM, Shipley MT. Connections between the central nucleus of the amygdala and the midbrain periaqueductal gray: topography and reciprocity. J Comp Neurol. 1991;303:121–131. doi: 10.1002/cne.903030111. [DOI] [PubMed] [Google Scholar]
- Sagen J, Proudfit HK. Evidence for pain modulation by pre- and postsynaptic noradrenergic receptors in the medulla oblongata. Brain Res. 1985;331:285–293. doi: 10.1016/0006-8993(85)91554-9. [DOI] [PubMed] [Google Scholar]
- Sah P, Faber ES, Lopez De Armentia M, Power J. The amygdaloid complex: anatomy and physiology. Physiol Rev. 2003;83:803–834. doi: 10.1152/physrev.00002.2003. [DOI] [PubMed] [Google Scholar]
- Schug SA, Saunders D, Kurowski I, Paech MJ. Neuraxial drug administration: a review of treatment options for anaesthesia and analgesia. CNS Drugs. 2006;20:917–933. doi: 10.2165/00023210-200620110-00005. [DOI] [PubMed] [Google Scholar]
- Selden NR, Everitt BJ, Jarrard LE, Robbins TW. Complementary roles for the amygdala and hippocampus in aversive conditioning to explicit and contextual cues. Neuroscience. 1991;42:335–350. doi: 10.1016/0306-4522(91)90379-3. [DOI] [PubMed] [Google Scholar]
- Shavit Y, Weidenfeld J, DeKeyser FG, Fish G, Wolf G, Mayburd E, Meerson Y, Beilin B. Effects of surgical stress on brain prostaglandin E2 production and on the pituitary-adrenal axis: attenuation by preemptive analgesia and by central amygdala lesion. Brain Res. 2005;1047:10–17. doi: 10.1016/j.brainres.2005.04.003. [DOI] [PubMed] [Google Scholar]
- Summers RJ, McMartin LR. Adrenoceptors and their second messenger systems. J Neurochem. 1993;60:10–23. doi: 10.1111/j.1471-4159.1993.tb05817.x. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Yokoo H, Mizoguchi K, Yoshida M, Tsuda A, Tanaka M. Noradrenaline release in the rat amygdala is increased by stress: studies with intracerebral microdialysis. Brain Res. 1991;544:174–176. doi: 10.1016/0006-8993(91)90902-8. [DOI] [PubMed] [Google Scholar]
- Tokuyama S, Takahashi M, Kaneto H. Participation of an alpha 2-mediated mechanism in the production of forced swimming-stress induced analgesia in mice. J Pharmacobiodyn. 1991;14:357–361. doi: 10.1248/bpb1978.14.357. [DOI] [PubMed] [Google Scholar]
- U'Prichard DC, Reisine TD, Mason ST, Fibiger HC, Yamamura HI. Modulation of rat brain alpha- and beta-adrenergic receptor populations by lesion of the dorsal noradrenergic bundle. Brain Res. 1980;187:143–154. doi: 10.1016/0006-8993(80)90500-4. [DOI] [PubMed] [Google Scholar]
- Wang XM, Zhang ZJ, Bains R, Mokha SS. Effect of antisense knock-down of alpha(2a)- and alpha(2c)-adrenoceptors on the antinociceptive action of clonidine on trigeminal nociception in the rat. Pain. 2002;98:27–35. doi: 10.1016/s0304-3959(01)00464-x. [DOI] [PubMed] [Google Scholar]
- Yasuoka S, Taksh T. Effects on Nociceptive Threshold and Blood Pressure of Intrathecally Administered Morphine and α-Adrenergic Agonists. Neuropharmacology. 1983;22:309–315. doi: 10.1016/0028-3908(83)90245-9. [DOI] [PubMed] [Google Scholar]