

Abstract
Hepatic lipid overloading mainly in the form of triglycerides is considered a prerequisite for the development of nonalcoholic fatty liver disease (NAFLD). However, triglyceride accumulation in the liver in response to lipid overflow may represent a protective mechanism against lipotoxicity. Our aims were to assess the fundamental cellular mechanisms that link lipid compartmentation in hepatocytes to liver damage and disease progression in NAFLD by using both in vivo dietary models of NAFLD and in vitro cell models of lipid overloading. Exposure of murine or human hepatocytes to monounsaturated fatty acids (MUFAs) resulted in lipid accumulation without changes in cell viability. In contrast, cell incubation with saturated fatty acids (SFAs) significantly decreased cell viability and increased caspase activation and apoptosis, with only minor lipid droplet accumulation. Genetic or pharmacological inhibition of stearoyl-CoA desaturase-1 (SCD1), the enzyme that converts SFA to MUFA, sensitized cells to SFA-induced apoptosis. Hepatic SCD1 expression increased in experimental steatosis resulting from high fat diet and decreased in a methionine-choline-deficient (MCD) dietary model of steatohepatitis resulting in the latter situation in significantly increased hepatic SFA levels. SCD1–/– mice on the MCD diet had decreased steatosis and markedly increased hepatocellular apoptosis, liver injury, and fibrosis compared with the SCD1+/+, whereas MUFA feeding prevents the MCD-induced injury. In conclusion, this study suggests hepatic SCD1 plays a key role in prevention of steatohepatitis by partitioning excess lipid into MUFA that can be safely stored. This concept has important implications for the development of novel treatment strategies for patients with this condition.
Nonalcoholic fatty liver disease (NAFLD)2 is currently the most common form of chronic liver disease affecting both adults and children and is strongly associated with obesity and insulin resistance (1, 2). One in three adults and one in ten children or adolescents in the United States have hepatic steatosis, a stage within the spectrum of NAFLD that is characterized by triglyceride accumulation in liver cells and follows a benign non-progressive clinical course. Nonalcoholic steatohepatitis (NASH) is defined as lipid accumulation with evidence of cellular damage, inflammation, and different degrees of scarring or fibrosis (3). NASH is a serious condition as ∼25% of these patients progress to cirrhosis and its feared complications of portal hypertension, liver failure and hepatocellular carcinoma (4–6).
The pathogenesis of NAFLD and NASH, and in particular the mechanisms responsible for liver injury and disease progression, remains poorly understood, but hepatic steatosis has been historically considered an initial event or the “first hit” in NAFLD development (7–9). This condition is postulated to increase sensitivity to a “second or multiple hits” that then trigger a cascade of events leading to cell death, inflammation, fibrogenesis, and fibrosis (8). However, this linear progression “two hit” model suggests any second stimulus must be infrequent and variably interact with steatotic cells. These implications arise firstly from extensive evidence from natural history studies in humans showing that only a small proportion of individuals with hepatic steatosis demonstrate disease progression or develop liver-related complications (4, 10, 11), and secondly, from recent epidemiological studies with serial liver biopsies on patients with NASH demonstrating that the degree of hepatic steatosis actually decreases in patients progressing to cirrhosis (12).
Steatosis and steatohepatitis can be modeled in mice by a high fat diet, or a diet deficient in methionine and choline, respectively. We noticed that in addition to the defining increase in triglyceride content in steatotic livers that there was an accompanying increase in free fatty acids (FFAs) and that the nature of FFA differed between the two models. The enzyme stearoyl-CoA desaturase-1 (SCD1) regulates the partitioning of incoming saturated free fatty acids (SFAs) between the monounsaturated free fatty acids (MUFAs) found in steatotic livers and the SFA present in inflamed and fibrotic livers. Here we tested the hypothesis that the relative abundance of cytotoxic SFA, rather than the extent of steatosis reflecting triglyceride accumulation (13, 14), determines how liver homeostasis is affected.
EXPERIMENTAL PROCEDURES
Cell Lines and Culture—Hepatocytes were isolated from wild-type C57BL/6 male mice (Jackson Laboratories, Bar Harbor, ME), purified by Percoll gradient centrifugation, cultured as previously described (15, 16), and used 4 h after isolation. Cells from a human, well differentiated hepatoblastoma cell line, HepG2, were cultured as previously described (17, 18). Previous studies from our laboratory showed that these cells are sensitive to FFA-induced mitochondrial permeabilization and cytochrome c release in a dose- and time-dependent manner with kinetics comparable to those observed in primary mouse hepatocytes (19). Cultured cells were treated with various concentrations (0.05–0.5 mm) of long chain FFA with different degrees of saturation, including palmitate and stearate (saturated), palmitoleate and oleate (one double bond), and linoleic (two double bonds) (Sigma) in media containing 1% bovine serum albumin for times ranging up to 24 h (0.5, 1, 2, 4, 8, 16, and 24 h). In selected experiments, cells were incubated with the fatty acid mixture in the presence or absence of the known SCD1 inhibitor trans-10,cis-12 isomer of conjugated linoleic acid (CLA, 45 μm, Matreya, LLC Biochemical, Pleasant Gap, PA) or a negative control (cis-9,trans-11 CLA, 45 μm, Matreya, LLC Biochemical) (20).
Cell Viability and Apoptosis Assessment—The number of viable cells was determined using a fluorometric assay (CellTiter-Blue, Promega, Madison, WI) following the manufacturer's instructions. Caspase activation and apoptosis were quantified using biochemical and fluorescent microscopic techniques: Caspase-Glo 3/7 Assay (Promega), the cell death ELISA kit (Roche Diagnostics), and assessing the characteristic nuclear changes of apoptosis (i.e. chromatin condensation and nuclear fragmentation) using the nuclear binding dye 4′,6-diamidino-2-phenylindole dihydrochloride (Molecular Probes Inc., Eugene, OR) and fluorescence microscopy.
siRNA and Transfection—HepG2 cells were grown in 6-well plates and transiently transfected with 30 nm of pre-design SCD1 siRNA (Ambion) by using 8 μl of the siPORT NeoFXT-MTransfection Agent (Ambion) in a total transfection volume of 0.5 ml of Opti-MEM (Invitrogen). After incubation at 37 °C for 5 h, 1.5 ml of normal growth medium was added. Cells were used for experiments 48 h after transfection. Inhibition of SCD1 protein expression was assessed by immunoblot analysis.
Nile Red Staining and Immunofluorescence—Isolated hepatocytes from C57BL/6 male mice were incubated with FFA (0.05–0.5 mm) in media with bovine serum albumin as a carrier for up to 4 h. Following fixation, cells were incubated with 0.1 μmol/ml of Nile Red in phosphate-buffered saline and examined by digitized fluorescence microscopy. Nile Red staining was expressed as an increase in total cellular fluorescence (pixel number per average fluorescence intensity) per cell.
Immunoblot Analysis—Immunoblot analysis was performed using whole cell lysates. Samples were resolved by 12% SDS-PAGE, transferred to nitrocellulose membrane, and blotted with appropriate primary antibodies. The membrane was incubated with peroxidase-conjugated secondary antibody (1:10,000 dilution, BIOSOURCE International, Camarillo, CA), and the bound antibody was visualized using a chemiluminescent substrate (ECL, Amersham Biosciences) and Kodak X-Omat film (Eastman Kodak, Rochester, NY). Primary anti-SCD1 antibody (1:2,000 dilution, Abcam) and goat anti-glyceraldehyde-3-phosphate dehydrogenase (1:2,000) were also used.
Animal Studies—These experimental protocols were approved by the Institutional Animal Care and Use Committee at the Cleveland Clinic. Male C57BL/6 mice, 20–25 g of body weight, were purchased from Jackson Laboratory. C57BL/6 SCD1 knockout (SCD1–/–, Jackson Laboratories) mice were described previously (21). Mice were placed on two different diets that allow us to study the spectrum of human NAFLD. First, mice were fed a high fat (HFAT) diet (42% of kcal from fat, TD 88137, Teklad Mills, Madison, WI), which results in steatosis mainly due to increase delivery of exogenous fatty acids to the liver (22, 23). The second diet is a methionine and choline-deficient (MCD) diet (TD 90262, Teklad Mills), which has been extensively shown to result in steatosis associated with significant inflammation and progressive fibrosis pathologically similar to human severe steatohepatitis (24, 25). In this later model steatosis results from both decreased mitochondrial oxidation of fatty acids and decreased export of fatty acids in the form of very low density lipoprotein (26). Identical groups of animals (n = 6 in each group) received standard rodent chow to act as controls. In selective experiments, mice (n = 6) were fed a customized MCD diet in which the fat was supplemented entirely by oleate. Total body weight was measured weekly. Animals in each group were sacrificed after 6 weeks on respective diets.
Determination of Liver Fibrosis—Liver fibrosis was quantified with Sirius Red. Direct Red 80 and Fast Green FCF (color index 42053) were provided by Sigma-Aldrich. Liver sections were incubated in the dark for 2 h at room temperature with an aqueous solution of saturated picric acid containing 0.1% Fast Green FCF and 0.1% Direct Red (27). Stained slides were washed slowly under running distilled water for 6 min, dehydrated (3 min for each step), mounted, and examined by light microscopy. Red-stained collagen fibers were quantitated by digital image analysis.
TUNEL Assay, Histopathology, and Serum Assays—Blood samples and liver tissue were collected under deep anesthesia after a 5-h fast as previously described in detail (17). Liver tissue was fixed in 4% paraformaldehyde and embedded in Tissue Path (Fisher Scientific, Pittsburgh, PA). Tissue sections (4 μm) were prepared, and terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay was performed following manufacturer's instructions (in situ cell death detection kit; Roche Molecular Biochemicals, Mannheim, Germany). Hepatocyte apoptosis in liver sections was quantitated by counting the number of TUNEL-positive cells in 10 random microscopic fields (20×), as previously described (28). Hematoxylin and eosin as well as Oil Red O-stained liver specimens were evaluated by light microscopy. The presence of 4-hydroxynonenal, a representative lipid peroxide product of oxidative stress was assessed by immunohistochemistry (4-hydroxynonenal, Cayman Chemicals). Serum alanine aminotransferase determinations were performed using a commercial kit (Sigma Diagnostics).
Lipid Analyses—Fatty acid composition of liver tissues was measured by gas chromatography as described previously (29, 30).
Data Analysis—All data are expressed as the mean ± S.D. unless otherwise indicated. Differences between groups were compared by an analysis of variance followed by a post hoc Bonferroni test to correct for multiple comparisons. Differences were considered to be statistically significant at p < 0.05.
RESULTS
Viability and Lipid Storage Are Inversely Affected by Exogenous FFA—Lipid accumulation in hepatocytes is a critical event in NAFLD and is thought to be mainly a result of increased uptake of FFA from the circulation (31, 32). We first recapitulated this environment by using isolated hepatocytes from C57BL/6 mice incubated with varied concentrations of specific FFA for up to 24 h. Triglyceride accumulation was assessed by Nile Red staining coupled to digitized fluorescence microscopy, whereas cell viability, caspase activation, and apoptosis were assessed using various biochemical and fluorescence microscopic approaches. Exposure of hepatocytes to the MUFA oleic acid resulted in significant accumulation of triglycerides (Fig. 1, A and B) without changes in cell viability, and with no detectable caspase 3 activation or apoptotic cell death (Fig. 1, C–F). In contrast, cells incubated with the SFA palmitate showed only a minor increase in lipid content that was accompanied by significant decrease in cell viability, increased caspase 3 activation, and apoptotic cell death (Fig. 1, A–F). Co-incubation with oleate completely rescued cells from palmitate-induced caspase activation, apoptosis, and restored triglyceride accumulation (Fig. 1, A–F). Similar results were found with other long-chain SFAs and MUFAs, such as stearate and palmitoleate (data not shown).
FIGURE 1.

Triglyceride accumulation and cell viability in FFA-treated hepatocytes. Isolated hepatocytes from C57BL/6 mice were incubated in the presence or absence of various concentrations of FFA with different degrees of saturation for up 24 h. A, triglyceride accumulation was determined by staining with Nile Red in fixed cells and (B) quantitated by digitized fluorescence microscopy. C, cell viability and caspase activation were assessed by CellTiter Blue and Apo-ONE Homogeneous Caspase 3/7 fluorometric assays, respectively. Finally, apoptotic cell death was quantified by an ELISA cell death assay (D) and using the DNA-binding dye 4′,6-diamidino-2-phenylindole (DAPI) coupled to fluorescence microscopy (E). Results are expressed as mean ± S.D. from three independent experiments. *, p < 0.05 compared with controls. #, p < 0.05 compared with palmitate-treated cells.
SCD1 Is Rate-limiting in Detoxification of Exogenous SFA—SCD1 initiates SFA metabolism by catalyzing the conversion to MUFA. We next tested whether endogenous metabolism to oleate recapitulates the protection of exogenous oleate by either pharmacological or genetic inhibition of SCD1. We first evaluated apoptosis in FFA-treated cells in the presence or absence of a potent inhibitor of SCD1, 10,12-CLA. SCD1 inhibition significantly increased hepatocyte sensitivity to palmitate-induced apoptosis (Fig. 2A). To determine whether specific suppression of SCD1 expression had a similar effect, we used siRNA to silence SCD1 expression in HepG2 cells. By this approach, we reduced SCD1 expression to background levels (Fig. 2B). Cells transfected with the SCD1 siRNA showed a significant increase in apoptotic cell death after treatment with palmitate (Fig. 2C). Collectively, these data strongly suggest that SCD1 and the MUFA to SFA ratio in hepatocytes play a key role in protection from lipotoxicity in the context of FFA overloading.
FIGURE 2.

SCD1 inhibition sensitizes hepatocytes to FFA induce apoptotic cell death. A, cells were incubated with or without FFA in the presence or absence of a known SCD1 inhibitor trans-10,cis-12 isomer of conjugated linoleic acid (CLA, 45 μm) or a negative control (cis-9,trans-11 CLA, 45 μm). In A, apoptosis was quantified using the DNA-binding dye 4′,6-diamidino-2-phenylindole dihydrochloride and fluorescence microscopy. Next, SCD1 expression in HepG2 cells was silenced by siRNA as detailed under “Experimental Procedures.” After transfection of HepG2 cells with SCD1 siRNA, an immunoblot for SCD1 and glyceraldehyde-3-phosphate dehydrogenase was performed (B) and apoptosis was quantified as before (C). CTL, control; PA, palmitate; Scr., scramble; and siRNA, small interfering RNA.
Hepatic SCD1 Expression and FFA Profiles Markedly Differ in Experimental Models of Hepatic Steatosis and Steatohepatitis—To investigate the role of SCD1 and lipid partitioning in liver injury and steatohepatitis development, we initially placed C57BL/6 mice on either a HFAT or a MCD diet. The first diet results in hepatic steatosis and several features of the human metabolic syndrome, including obesity, insulin resistance and dyslipidemia, but no evidence of any significant liver damage or inflammation (22, 23). The second diet results in progressive fibrosing steatohepatitis pathologically similar to human severe steatohepatitis (24, 25). After 6 weeks on the respective diets we observed a significant increase in hepatic SCD1 expression in mice on the HFAT diet compared with those on the control diet (Fig. 3, A and B). In sharp contrast, mice on the MCD diet showed that SCD1 expression was significantly attenuated (Fig. 3, A and B). Both groups of animals on the diets developed hepatic steatosis, however they showed profound differences in their fatty acids profiles, with levels of SFA (palmitate (16:0) and stearate (18:0)) being significantly higher and those of MUFA (palmitoleate (16:1) and oleate (18:1)) being significantly lower in those mice on the MCD diet compared with those on the HFAT (Fig. 3, C and D). Thus, these data suggest a possible link between SCD1 activity and steatohepatitis development in vivo models of hepatic lipid overloading.
FIGURE 3.

Changes in hepatic SCD1 expression and FFA profiles in experimental models of NAFLD. C57BL/6 mice were placed on two different dietary models of NAFLD or a control diet for 6 weeks (n = 6 in each group). The first one resembles isolated hepatic steatosis (HFAT diet), and the second one results in steatohepatitis similar to severe human NASH (MCD diet). A, representative immunoblot analysis for SCD1 in livers from mice on the three different diets. β-Actin served as a control for protein loading. SCD1 subcellular localization in the livers was further investigated by immunohistochemistry. Liver tissue was fixed and incubated with anti-SCD1 antibody (1:100 dilution). B, representative microphotograph of SCD1 staining (magnification, 40×). Fatty acid composition of livers from animals on the three different diets was assessed as detailed under “Experimental Procedures.” C, the levels of SFA, including palmitate (16:0) and stearate (18:0), were determined in livers from mice on three different diets and expressed as moles of fatty acid as a percentage of total moles of fatty acid. Results are represented as mean ± S.D. *, p < 0.05 compared with HFAT and control diet group. #, p < 0.01 compared with HFAT and control diet. D, the levels of MUFA, including palmitoleate (16:1) and oleate (18:1), were determined in livers from mice on three different diets and expressed as moles of fatty acid as a percentage of total moles of fatty acid. Results are represented as mean ± S.D. *, p < 0.01 compared with HFAT. #, p < 0.01 compared with HFAT.
Hepatic Lipid Partitioning and Hepatocellular Apoptosis in SCD1 Knockout Mice—The findings of a key role of SCD1 in maintaining hepatocyte viability in the context of FFA overflow and the strong correlation of hepatic SCD1 expression with liver injury led us to further examine the role of SCD1 in steatohepatitis by using SCD1-null mice. SCD1–/– mice and their wild-type controls were placed on the three different diets (HFAT, MCD, or control diets) for 6 weeks. SCD1 knockouts were resistant to HFAT diet-induced obesity consistent with previous reports (21) and showed no evidence of hepatomegaly (Table 1) or liver histological changes (data not shown) compared with animals on the control diet. We next compared the two groups of animals on the MCD diet. SCD1–/– mice on the MCD diet had a significant reduction in hepatic steatosis compared with the wild-type animals on the same diet (Fig. 4). To determine if these changes in lipid partitioning in the liver were associated with an increase in liver cell apoptosis as we observed in cultured hepatocytes, we next quantified the amount of hepatocellular apoptosis present in the four groups of mice. TUNEL-positive hepatocytes were >2-fold greater in SCD1–/– on the MCD diet compared with SCD1+/+ mice on the same diet (Fig. 5, A and B).
TABLE 1.
Body and liver weight in SCD1-/- and wild-type littermates after 6 weeks on the respective diets
Values are mean ± S.D.
| Body weight | Liver weight | Liver/body weight | |
|---|---|---|---|
| g | g | % | |
| SCD1+/+ | |||
| Control diet | 28 ± 1.2 | 1.1 ± 0.2 | 3.9 |
| HFAT diet | 33 ± 1.5 | 2.7 ± 0.1 | 8 |
| p value | <0.01 | <0.01 | <0.01 |
| MCD diet | 15 ± 1.1 | 0.9 ± 0.3 | 6 |
| p value | <0.01 | NSa | <0.05 |
| SCD1-/- | |||
| Control diet | 24 ± 1.3 | 1.2 ± 0.2 | 4.6 |
| HFAT diet | 26 ± 1.1 | 1.4 ± 0.2 | 5.4 |
| p value | NS | NS | NS |
| MCD diet | 13 ± 1.2 | 0.5 ± 0.3 | 3.9 |
| p value | <0.01 | <0.05 | NS |
NS, not significant.
FIGURE 4.

Hepatic lipid partitioning in SCD1–/– animals on the MCD diet. SCD1 knockout (SCD1–/–) and their wild-type controls (SCD1+/+) were placed on a methionine- and choline-deficient (MCD) diet or a control diet for 6 weeks (n = 6 in each group). Representative microphotograph of hematoxylin and eosin (H&E) staining, and Oil Red O staining from mice on each of the four groups.
FIGURE 5.

Hepatocellular apoptosis is increased in SCD1–/– mice on the MCD diet. To assess if the changes in lipid partitioning were associated with increase apoptotic cell death as we observed in cultured hepatocytes, hepatocellular apoptosis was quantified. A, representative microphotograph of TUNEL staining of liver section from SCD1–/– and SCD1+/+ mice on either the MCD or control diet. B, quantitation of TUNEL staining in liver sections from the four groups of mice by counting the number of TUNEL-positive cells in 10 random microscopic fields. Results are expressed as mean ± S.D.
Hepatic Inflammation and Collagen Deposition in SCD1 Knockout Mice—We next examined the effects of SCD1 ablation on liver inflammation, oxidative stress, and fibrosis induced by the MCD diet. Although, as expected after 6 weeks, on the MCD diet wild-type animals developed signs of liver injury as shown by an increase in serum alanine aminotransferase levels (Fig. 6B) and 4-hydroxynonenal expression in the liver (Fig. 6A), these changes were significantly enhanced in the SCD1–/– mice. More importantly, an almost 2-fold increase in collagen deposition as demonstrated by Sirius Red staining of liver tissue coupled to quantitation by digitized image analysis was present in SCD1–/– mice on the MCD diet compared with the wild-type animals on the same diet (Fig. 6, C and D). Taken together, these observations suggest that, in the context of hepatic lipid overloading, SCD1 activity and, as a consequence, the balance between MUFA and SFA accumulating in liver cells play a central role in whether isolated hepatic steatosis or progressive steatohepatitis and fibrosis occurs.
FIGURE 6.

SCD1–/– mice are sensitized to MCD diet-induced liver injury. A, immunohistochemistry for 4-hydroxynonenal (4-HNE), a representative lipid peroxide product of oxidative stress in liver sections from SCD1–/– and SCD1+/+ animals on a control diet or MCD diet, and (B) serum alanine aminotransferase levels in these four groups of mice (n = 4–6 in each group). Results are expressed as mean ± S.D. C, collagen fibers were stained with Sirius Red as described under “Experimental Procedures.” D, the surface area stained with Sirius Red was quantitated using digital image analysis. Results are expressed as mean ± S.D. (n = 4–6 in each group). *, p < 0.001 compared with SCD1–/– mice on a control diet. #, p < 0.01 compared with mice SCD1+/+ on MCD diet.
MCD Diet-induced Liver Injury Is Attenuated by MUFA Supplementation—To determined if exogenous supplementation of oleate could bypass the SCD-1 deficiency and in that way attenuate the MCD diet-induced apoptosis and liver damage, we generated a customized MCD diet in which the lipid component was completely replaced by this MUFA. The response of the high oleate MCD diet was compared with that of mice on the regular MCD diet or the control diet. After 6 weeks on the respective diets, MCD-induced apoptosis and liver injury were less severe in the high oleate MCD-fed animals compared with the animals on the regular MCD diet (Fig. 7, A–C).
FIGURE 7.

Oleic acid supplementation attenuates MCD diet-induced liver injury. To determine if exogenous supplementation of oleate could bypass the SCD-1 deficiency and attenuate the MCD diet-induced apoptosis and liver damage, mice were placed on either a standard MCD diet or a customized MCD diet in which the lipids were provided entirely by oleate. Representative microphotographs show hematoxylin & eosin staining (A) and TUNEL staining of liver sections from C57BL/6 mice on either the control diet, the MCD diet, or the MCD oleate diet (B). C, quantitation of TUNEL staining in liver sections from the three groups of mice by counting the number of TUNEL-positive cells in 10 random microscopic fields. Results are expressed as mean ± S.D.
DISCUSSION
The principal findings of this study relate to the mechanisms linking hepatocyte lipid overloading, to hepatocellular apoptosis and liver injury. The results demonstrate that the ratio of MUFA to SFA determines whether liver cells are damaged by the flux of exogenous FFA, that the nature rather than the quantity of FFA determines hepatic stress, and that hepatic triglyceride storage and MUFA in this context are protective.
Obesity and type 2 diabetes have reached epidemic proportions in most of the western world, and both conditions are strongly associated with NAFLD (33). NAFLD encompasses a wide spectrum of conditions associated with over-accumulation of lipids in the liver ranging from simple fatty liver or hepatic steatosis, in which there is evidence for fat accumulation without signs of liver cell injury or inflammation, to nonalcoholic steatohepatitis or NASH characterized by the accumulation of fat in the liver along with evidence of liver cell damage, inflammation, and different degrees of scarring or fibrosis (2). Although most patients with steatosis tend to have a benign nonprogressive clinical course, a significant proportion of those with NASH have a progressive disease with significant associated risk of developing cirrhosis and its feared complications of portal hypertension, liver failure, and hepatocellular carcinoma (4–6).
A net retention of triglycerides in hepatocytes to form large cytosolic lipid droplets has been historically considered a prerequisite and the “first hit” in the development of NAFLD. The mechanisms that link hepatic steatosis to liver damage resulting in disease progression to NASH and NASH-cirrhosis have been the center of extensive investigation and remain incompletely understood. A central question is why most patients with hepatic steatosis have a benign nonprogressive disease while others develop steatohepatitis and end-stage liver disease.
Lipids accumulate in the form of triglycerides in the context of lipid overloading of the liver, but steatosis is associated with an excess of a variety of FFA and cholesterol (34, 35). Several studies have now demonstrated that SFAs are potentially cytotoxic in different cell lines by triggering of apoptotic cell death (14, 36, 37). SCD1 catalyzes the desaturation of SFA palmitate and stearate to the MUFA palmitoleate and oleate, respectively, and recently, has been implicated in the development of diet-induced obesity and insulin resistance (21, 38, 39). SCD1 knockout mice are resistant to high fat diet-induced obesity, whereas antisense oligonucleotide inhibition of SCD1 protected against diet-induced insulin resistance and obesity. However, other studies suggest hepatic SCD1 activity may maintain metabolic homeostasis and prevent inflammation (29, 30). Our in vitro studies showed that SCD1 protected hepatocytes from apoptotic cell death in the context of FFA overflow. We have previously demonstrated that exposing cultured hepatocytes to SFA, but not to mono- or polyunsaturated FFA, initiates a time- and dose-dependent permeabilization of the mitochondrial outer membrane, with release of cytochrome c, caspase 3 activation, and apoptosis. We hypothesize that SCD1 protection is through channeling these SFAs into MUFAs, which are then more easily incorporated into triglycerides and then safely stored in lipid droplets. This is supported by the fact that incubation with palmitate resulted in only minor increases in the triglyceride pool, whereas co-incubation with oleate completely prevented palmitate toxicity and increased the triglyceride pool to a similar extent to that seen in cells incubated with oleate alone. The in vitro observations can be extrapolated to in vivo hepatic lipid overloading because feeding mice a high fat diet, which results in extensive steatosis without apoptotic cell death, hepatocellular injury, or fibrosis, is associated with significantly increased hepatocyte SCD1 expression and a major increase in the MUFA to SFA ratio. In sharp contrast, feeding mice an MCD diet, that has been extensively associated with fibrosing steatohepatitis similar to that seen in patients with severe NASH, reversed the changes in the desaturation index. To further examine the role of SCD1 in lipid-mediated cytotoxicity in vivo we utilized SCD1-null mice. When we placed the SCD1–/– on an MCD diet, the mice accumulated less triglyceride in the liver compared with the wild-type animals on the same diet, the SCD1–/– mice also showed a pronounced decreased in the desaturation index that was accompanied by an increase in hepatocellular apoptosis and liver injury. Finally, the MCD diet-induced liver toxicity could by partially prevented by feeding mice a high oleate MCD diet. These results demonstrate that hepatic SCD1 deficiency and the desaturation index of hepatic FFA play a key role in development of steatohepatitis in the context of lipid overload in the liver, which is characteristic of patients with NAFLD. This concept has important implications for the pathogenesis and development of novel diagnostic and treatment strategies for NAFLD and at the same time alerts about the potential negative consequences to the liver of pharmacological inhibition of SCD1 for treatment of obesity and insulin resistance as a significant proportion of patients with these conditions also suffers from NAFLD.
In summary, the current studies uncover a pathogenic link between hepatic lipid metabolism and liver injury in the context of fat overloading of the liver. The results support a model (Fig. 8) in which, during the development of NAFLD, overflow of FFA to the liver is associated with an increase in SCD1 expression and activity resulting in a tilt of the balance toward MUFA formation, triglyceride storage, liver adaptation, and development of isolated hepatic steatosis. In contrast, SCD1 deficiency results in hepatic over-accumulation of SFA and a decreased desaturation index triggering hepatocellular apoptosis, liver damage, and development of steatohepatitis.
FIGURE 8.
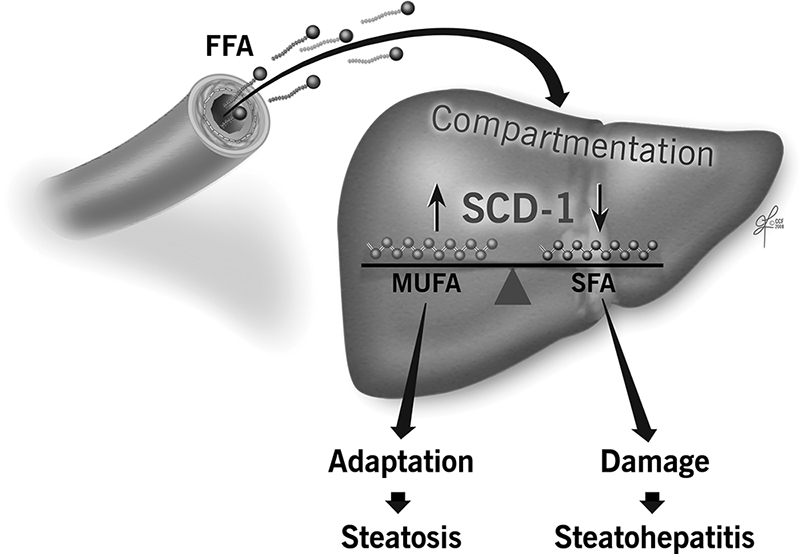
Lipotoxicity model in the context of lipid overloading of the liver. Hepatic steatosis, characteristic of patients with NAFLD, which is commonly associated with obesity and insulin resistance, results mainly from increased flow of free fatty acids from circulation. In this context, lipid compartmentation in liver cells and, in particular, the ratio of monounsaturated to saturated free fatty acids that is a function of SCD1 activity, play a central role in whether adaptation occurs, resulting in a benign clinical course, or there is ongoing hepatocellular apoptosis, and liver damage resulting in steatohepatitis and fibrosis development.
This work was supported, in whole or in part, by National Institutes of Health Grant DK076852. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: NAFLD, nonalcoholic fatty liver disease; NASH, nonalcoholic steatohepatitis; SCD, stearoyl-CoA desaturase; FFA, free fatty acid; SFA, saturated fatty acid; MUFA, monounsaturated fatty acid; CLA, conjugated linoleic acid; ELISA, enzyme-linked immunosorbent assay; siRNA, small interference RNA; HFAT, high fat diet; MCD, methionine-choline-deficient diet; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling.
References
- 1.Wieckowska, A., and Feldstein, A. E. (2005) Curr. Opin. Pediatr. 17 636–641 [DOI] [PubMed] [Google Scholar]
- 2.Angulo, P. (2002) N. Engl. J. Med. 346 1221–1231 [DOI] [PubMed] [Google Scholar]
- 3.Brunt, E. M., Neuschwander-Tetri, B. A., Oliver, D., Wehmeier, K. R., and Bacon, B. R. (2004) Hum. Pathol. 35 1070–1082 [DOI] [PubMed] [Google Scholar]
- 4.Adams, L. A., Lymp, J. F., St. Sauver, J., Sanderson, S. O., Lindor, K. D., Feldstein, A., and Angulo, P. (2005) Gastroenterology 129 113–121 [DOI] [PubMed] [Google Scholar]
- 5.Matteoni, C. A., Younossi, Z. M., Gramlich, T., Boparai, N., Liu, Y. C., and McCullough, A. J. (1999) Gastroenterology 116 1413–1419 [DOI] [PubMed] [Google Scholar]
- 6.Ekstedt, M., Franzen, L. E., Mathiesen, U. L., Thorelius, L., Holmqvist, M., Bodemar, G., and Kechagias, S. (2006) Hepatology 44 865–873 [DOI] [PubMed] [Google Scholar]
- 7.Diehl, A. M. (2002) Am. J. Physiol. 282 G1–G5 [DOI] [PubMed] [Google Scholar]
- 8.Browning, J. D., and Horton, J. D. (2004) J. Clin. Invest. 114 147–152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Day, C. P., and James, O. F. (1998) Gastroenterology 114 842–845 [DOI] [PubMed] [Google Scholar]
- 10.Dam-Larsen, S., Franzmann, M., Andersen, I. B., Christoffersen, P., Jensen, L. B., Sorensen, T. I., Becker, U., and Bendtsen, F. (2004) Gut 53 750–755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dam-Larsen, S., Franzmann, M. B., Christoffersen, P., Larsen, K., Becker, U., and Bendtsen, F. (2005) Scand. J. Gastroenterol. 40 460–467 [DOI] [PubMed] [Google Scholar]
- 12.Clark, J. M., and Diehl, A. M. (2003) J. Am. Med. Assoc. 289 3000–3004 [DOI] [PubMed] [Google Scholar]
- 13.Listenberger, L. L., Han, X., Lewis, S. E., Cases, S., Farese, R. V., Jr., Ory, D. S., and Schaffer, J. E. (2003) Proc. Natl. Acad. Sci. U. S. A. 100 3077–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Unger, R. H. (2003) Trends Endocrinol. Metab. 14 398–403 [DOI] [PubMed] [Google Scholar]
- 15.Feldstein, A. E., Werneburg, N. W., Li, Z., Bronk, S. F., and Gores, G. J. (2006) Am. J. Physiol. 290 G1339–G1346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canbay, A., Feldstein, A. E., Higuchi, H., Werneburg, N., Grambihler, A., Bronk, S. F., and Gores, G. J. (2003) Hepatology 38 1188–1198 [DOI] [PubMed] [Google Scholar]
- 17.Feldstein, A. E., Canbay, A., Guicciardi, M. E., Higuchi, H., Bronk, S. F., and Gores, G. J. (2003) J. Hepatol. 39 978–983 [DOI] [PubMed] [Google Scholar]
- 18.Li, Z., Berk, M., McIntyre, T. M., Gores, G. J., and Feldstein, A. E. (2008) Hepatology 47 1495–1503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldstein, A. E., Werneburg, N. W., Canbay, A., Guicciardi, M. E., Bronk, S. F., Rydzewski, R., Burgart, L. J., and Gores, G. J. (2004) Hepatology 40 185–194 [DOI] [PubMed] [Google Scholar]
- 20.Ntambi, J. M., Choi, Y., Park, Y., Peters, J. M., and Pariza, M. W. (2002) Can. J. Appl. Physiol. 27 617–628 [DOI] [PubMed] [Google Scholar]
- 21.Ntambi, J. M., Miyazaki, M., Stoehr, J. P., Lan, H., Kendziorski, C. M., Yandell, B. S., Song, Y., Cohen, P., Friedman, J. M., and Attie, A. D. (2002) Proc. Natl. Acad. Sci. U. S. A. 99 11482–11486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Koteish, A., and Diehl, A. M. (2001) Semin. Liver Dis. 21 89–104 [DOI] [PubMed] [Google Scholar]
- 23.Nanji, A. A. (2004) Clin. Liver Dis. 8 559–574 [DOI] [PubMed] [Google Scholar]
- 24.Dela Pena, A., Leclercq, I., Field, J., George, J., Jones, B., and Farrell, G. (2005) Gastroenterology 129 1663–1674 [DOI] [PubMed] [Google Scholar]
- 25.den Boer, M., Voshol, P. J., Kuipers, F., Havekes, L. M., and Romijn, J. A. (2004) Arterioscler. Thromb. Vasc. Biol. 24 644–649 [DOI] [PubMed] [Google Scholar]
- 26.London, R. M., and George, J. (2007) Clin. Liver Dis. 11 55–74 [DOI] [PubMed] [Google Scholar]
- 27.Canbay, A., Guicciardi, M. E., Higuchi, H., Feldstein, A., Bronk, S. F., Rydzewski, R., Taniai, M., and Gores, G. J. (2003) J. Clin. Invest. 112 152–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Feldstein, A. E., Canbay, A., Angulo, P., Taniai, M., Burgart, L. J., Lindor, K. D., and Gores, G. J. (2003) Gastroenterology 125 437–443 [DOI] [PubMed] [Google Scholar]
- 29.Chen, C., Shah, Y. M., Morimura, K., Krausz, K. W., Miyazaki, M., Richardson, T. A., Morgan, E. T., Ntambi, J. M., Idle, J. R., and Gonzalez, F. J. (2008) Cell Metab. 7 135–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flowers, J. B., Rabaglia, M. E., Schueler, K. L., Flowers, M. T., Lan, H., Keller, M. P., Ntambi, J. M., and Attie, A. D. (2007) Diabetes 56 1228–1239 [DOI] [PubMed] [Google Scholar]
- 31.Donnelly, K. L., Smith, C. I., Schwarzenberg, S. J., Jessurun, J., Boldt, M. D., and Parks, E. J. (2005) J. Clin. Invest. 115 1343–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Postic, C., and Girard, J. (2008) J. Clin. Invest. 118 829–838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Clark, J. M., Brancati, F. L., and Diehl, A. M. (2002) Gastroenterology 122 1649–1657 [DOI] [PubMed] [Google Scholar]
- 34.McClain, C. J., Barve, S., and Deaciuc, I. (2007) Hepatology 45 1343–1346 [DOI] [PubMed] [Google Scholar]
- 35.Mari, M., Caballero, F., Colell, A., Morales, A., Caballeria, J., Fernandez, A., Enrich, C., Fernandez-Checa, J. C., and Garcia-Ruiz, C. (2006) Cell Metab. 4 185–198 [DOI] [PubMed] [Google Scholar]
- 36.Schaffer, J. E. (2003) Curr. Opin. Lipidol. 14 281–287 [DOI] [PubMed] [Google Scholar]
- 37.Shimabukuro, M., Zhou, Y. T., Levi, M., and Unger, R. H. (1998) Proc. Natl. Acad. Sci. U. S. A. 95 2498–2502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gutierrez-Juarez, R., Pocai, A., Mulas, C., Ono, H., Bhanot, S., Monia, B. P., and Rossetti, L. (2006) J. Clin. Invest. 116 1686–1695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang, G., Li, Z., Liu, F., Ellsworth, K., Dallas-Yang, Q., Wu, M., Ronan, J., Esau, C., Murphy, C., Szalkowski, D., Bergeron, R., Doebber, T., and Zhang, B. B. (2005) J. Clin. Invest. 115 1030–1038 [DOI] [PMC free article] [PubMed] [Google Scholar]