

Abstract
Human carbamoyl phosphate synthetase (hCPS) has evolved critical features that allow it to remove excess and potentially neurotoxic ammonia via the urea cycle, including use of only free ammonia as a nitrogen donor, a Km for ammonia 100-fold lower than for CPSs that also use glutamine as a nitrogen donor, and required allosteric activation by N-acetylglutamate (AGA), a sensor of excess amino acids. The recent availability of a Schizosaccharomyces pombe expression system for hCPS allowed us to utilize protein engineering approaches to elucidate the distinctive hCPS properties. Although the site of AGA interaction is not defined, it is known that the binding of AGA to CPS leads to a conformational change in which a pair of cysteine side chains become proximate and can then be selectively induced to undergo disulfide bonding. We analyzed the response of hCPS cysteine mutants to thiol-specific reagents and identified Cys-1327 and Cys-1337 as the AGA-responsive proximate cysteines. Possibly two of the features unique to urea-specific CPSs, relative to other CPSs (the conserved Cys-1327/Cys-1337 pair and the occurrence at very high concentrations in the liver mitochondrial matrix) co-evolved to provide buffering against reactive oxygen species. Reciprocal mutation analysis of Escherichia coli CPS (eCPS), creating P909C and G919C and establishing the ability of these engineered cysteine residues to share a disulfide bond, indicated an eCPS conformational change at least partly similar to the hCPS conformational change induced by AGA. These findings strongly suggested an alternative eCPS conformation relative to the single crystal conformation thus far identified.
When life adapted to a terrestrial habitat, removal of excess and potentially neurotoxic ammonia by the diffusion that occurred in an aquatic habitat was no longer possible (1). Arginine biosynthetic pathways almost certainly served as the precursors for the urea cycle, the present day metabolic pathway for removal of excess ammonia, with surprisingly few changes needed for the pathway evolution (2). Carbamoyl phosphate synthetase (CPS),2 the enzyme that catalyzes the entry and rate-limiting step of the urea cycle, was the site of four of these critical evolutionary changes: (a) gain of communication with a sensor of excess amino acids, N-acetylglutamate (AGA, which serves as a required allosteric activator only for urea-synthesizing CPSs), (b) a decrease in Km for ammonia to ∼1 mm from the ∼100 mm value for other CPSs, (c) loss of interaction with glutamine to avoid competition with the preferred substrate ammonia, and (d) localization to the hepatic mitochondrial matrix to allow independent regulation relative to CPSs with other metabolic roles. Structural changes have been identified in hCPS that correlate with the latter two functional changes (3-5); the cysteine residue required for releasing ammonia from glutamine has been replaced by serine, and hCPS is synthesized with 39 N-terminal residues that serve as a mitochondrial matrix targeting signal and that are cleaved as the hCPS precursor crosses the inner mitochondrial membrane. The structural basis for the first two critical functional features, AGA interaction and high affinity for ammonia, however, have not yet been elucidated nor has it been determined whether these two features are linked.
CPSs from all species with varied metabolic roles share strong sequence identity, and all appear to have the same overall domain organization (6) (Fig. 1) observed in the crystal structure of E. coli CPS (eCPS), the only solved CPS structure (7). All known CPSs appear to utilize a common mechanism (Fig. 1) to catalyze the formation of carbamoyl phosphate (CP), Pi, and two molecules of ADP from ammonia (either free in the cell or generated from the hydrolysis of glutamine), bicarbonate, and two molecules of ATP (6). A variety of studies have yielded evidence for a cycle of conformational changes accompanying the catalytic cycle (6-8). Vertebrate urea-specific CPSs, including hCPS, incorporate an additional type of conformational control in the form of dependence on the allosteric activator AGA. In the absence of AGA, a very small fraction of urea-specific CPS is in a conformation capable of catalytic activity, whereas binding of AGA yields CPS with activity equivalent to that of eCPS and other non-AGA-dependent CPSs (6). Although AGA functions as an intermediate in arginine biosynthesis in prokaryotes and lower eukaryotes, the only known function for AGA in vertebrates is activation of urea-specific CPSs (9). Co-localization of AGA synthetase in the hepatic mitochondrial matrix is consistent with this dedicated role, as is the fact that increases in the substrate glutamate signal increased degradation of proteins to yield both free amino acids and free ammonia. Allosteric activation of vertebrate AGA synthetase by arginine provides a further link to the presence of excess amino acids and the accompanying excess of free ammonia requiring detoxification.
FIGURE 1.

Domain and oligomeric structure of eCPS. Top panel, ribbon representation of one heterodimer of eCPS. The 42-kDa glutaminase subunit contains the glutamine binding domain (A2; violet) and domain A1 (purple) that is involved in communicating active site occupancy between the glutaminase and synthetase subunits. The 120-kDa synthetase subunit contains 4 domains. Domains B (green) and C (red) are regions of internal duplication, and each contains an ATP grasp fold. The oligomerization domain (D′, yellow) and allosteric domain (D, orange) are involved in side-by-side and end-on-end interactions, respectively. The ligands glutamine and ADP are shown in space-fill representations. The intramolecular tunnel connecting the three active sites is illustrated with arrows. Also shown are the individual chemical reactions at each of the active sites that are synchronized in the overall reaction. The domains of hCPS occur as a single polypeptide, with 1462 amino acid residues in the mature protein (from which the mitochondrial targeting signal has been removed) in contrast to the heterodimeric structure of eCPS. Bottom panel, the tetramer of the glutaminase + synthetase heterodimers. Created from PDB file 1a9x.
One reporter for the AGA-activated conformation of CPS is a pair of cysteine side chains that become proximate upon AGA binding and that can then be selectively induced to undergo disulfide bonding (10, 11). In the absence of AGA, only a small fraction of CPS is in a conformation with the cysteine pair proximate. This single pair of proximate sulfhydryl groups is uniquely modified when the AGA·CPS complex is exposed to a variety of disulfide-inducing reagents, and reversible activity loss accompanies disulfide formation. The recent availability (12) of a Schizosaccharomyces pombe expression system for hCPS allowed us to utilize protein engineering approaches for elucidation of the distinctive hCPS properties. To identify the residues of the cysteine reporter group, we have analyzed the response of hCPS cysteine mutants to thiol-specific reagents. We also have examined the potential for a conformational change in eCPS that parallels the AGA-induced conformational change of hCPS. Thus far only a single conformation of eCPS has been revealed from x-ray structural analysis. Defining the cycle of conformational changes that accompany the catalytic cycle and that allow synchronization of three active sites (glutamine amidotransferase site and two distinct ATP sites) to produce CP, therefore, remains a major challenge.
EXPERIMENTAL PROCEDURES
Recombinant DNA Methods—Bacterial transformations and recombinant DNA techniques were carried out as described in Sambrook et al. (13). Site-directed mutants were created using nested PCR. Each mutagenesis cassette was sequenced to verify that no undesired changes were incorporated into the nucleotide sequence. Mutagenesis primers (mutated base pairs in bold) and outside non-mutagenic primers corresponding to the coding strand sequence were as follows. Primers used for eCPS mutants (5′-3′) were: NP909CF1, CCGCTGTTAGGGTGTGAAATGCGATCGACCGGG; NP909CR1, CCCGGTCGATCGCATTTCACACCCTAACAGCGG; NG919CF1, GCGATCGACCGGGGAAGTCATGTGCGTGGG; NG919CR1, CCCACGCACATGACTTCCCCGGTCGATCGC; MOPF1, CCGGTTATCGGCACCAGCCCGG; MOPR1, GCCTCTCCCCGCGCGTTGGCCG; MOPF2, CGTGAACGCTTCCAGCATGCGGTTGAGCG; MOPR2, CACGACAGGTTTCCCGACTGGAAAGCGG. Primers used for hCPS mutants (5′-3′) were: NC1327AF1, CCCATTCTGAGAGCTGAGATGGCGTCGACTGG; NC1327AR1, CCAGTCGACGCCATCTCAGCTCTCAGAATGGG; NC1337AF1, GCGTCGACTGGAGAGGTGGCTGCCTTTGG; NC1337AR1, CCAAAGGCAGCCACCTCTCCAGTCGACGC; hMOPF1, GGGCACAAGCCCCCTGCAGATCGACAGGGC; hMOPR1, CCCTTGGAACAAGACCCGCATGCG; hMOPFi, GGCCTTCCTATGTTTTGAGTGGGTCTGC.
CPS Expression and Purification—The protocol for expression and purification of recombinant wild type eCPS and for its site-directed mutants was as previously described (14). The expressed eCPS was comprised of both subunits. The expression/purification protocol for wild type and mutant hCPSs was as described (12). The expressed hCPS was the mature protein (1462 amino acid residues) with the first 39 amino acids of the precursor protein replaced by a methionine and an N-terminal His6-FLAG® tag (yielding the N-terminal sequence HHHHHHDYKDDDKMSVKAQ, with the tag residues italicized). The fusion tag was not removed from the purified hCPS because (a) it appeared to be functionally neutral (12) and (b) in previous studies with frog CPS expressed as a fusion protein (14), attempts to remove the fusion tag resulted in cleavage of CPS, apparently at the links between domains that are extremely susceptible to proteolysis. Recombinant hCPS corresponded to gene ID 1373 (gene symbol CPSI, RefSeq transcript variant 2, NM_001875; threonine-1406 variant). The purity of all protein preparations was at least 95%, as assessed by Coomassie Blue staining of SDS-PAGE gels (15). Protein concentration was estimated using the dye binding assay of Bradford with bovine serum albumin as the standard (16) or by A280 (0.685/mg eCPS) (17).
CPS Activity Assays and Data Analysis—CP synthesis was determined in a two-step assay by coupling the CPS reaction to that of ornithine transcarbamoylase and then quantitating the resulting citrulline (14, 18). The reaction mixtures contained 50 mm HEPES, 100 mm KCl, 10 mm ATP, 20 mm MgCl2, 20 mm NaHCO3, 5 mm ornithine, 0.2 units ornithine transcarbamoylase, and either 300 mm NH4Cl or 10 mm glutamine and were initiated by the addition of CPS (final volume 100 μl, pH 7.6). For hCPS assay, 5 mm AGA was included, KCl was reduced to 10 mm, NH4Cl was reduced to 30 mm, and ornithine was increased to 20 mm. Standard incubation was at 37 °C for 20 min.
ATPase activities were determined in a pyruvate kinase/lactate
dehydrogenase-coupled assay
(18), where the formation of
ADP (with 2 ADP produced per CP synthesized) is coupled to NADH oxidation as
monitored continuously at 340 nm. The reaction mixtures contained variable
ATP, 50 mm HEPES, 100 mm KCl, 20 mm
MgCl2, 40 mm NaHCO3, 10 mm
ornithine, 1 mm sodium phosphoenolpyruvate, 0.2 mm NADH,
18 units pyruvate kinase, and 24 units of lactate dehydrogenase. For hCPS
assay, 5 mm AGA was included, KCl was reduced to 10 mm,
and ornithine was omitted. To determine ammonium-dependent ATPase activity,
300 mm NH4Cl (30 mm for hCPS) was included in
the reaction mixture, and to determine glutamine-dependent ATPase activity, 10
mm glutamine was included. Whereas only the unprotonated form of
ammonia (NH3) is a substrate for CPS
(19), the data are presented
as the total of  + NH3
because it was the level of NH4Cl that was varied during the
experiments. Under the assay conditions, NH3 represented about 4%
of the NH4Cl (pKa 9.25) added to the solution
(19). For determination of
bicarbonate-dependent ATPase activity in domain B, the assay was carried out
in the absence of both NH4Cl and glutamine. For determination of
allosteric effects, the assay was conducted in the absence of ornithine or in
the presence of 0.1 mm UMP. Assays were monitored for 10-20 min at
25 °C after the addition of 5-50 μg of CPS.
+ NH3
because it was the level of NH4Cl that was varied during the
experiments. Under the assay conditions, NH3 represented about 4%
of the NH4Cl (pKa 9.25) added to the solution
(19). For determination of
bicarbonate-dependent ATPase activity in domain B, the assay was carried out
in the absence of both NH4Cl and glutamine. For determination of
allosteric effects, the assay was conducted in the absence of ornithine or in
the presence of 0.1 mm UMP. Assays were monitored for 10-20 min at
25 °C after the addition of 5-50 μg of CPS.
ATP synthesis was assayed by coupling ATP formation from ADP and CP to the hexokinase and glucose-6-phosphate dehydrogenase reaction (18). The reaction mixture contained 50 mm HEPES, 20 mm MgCl2, 100 mm KCl, 10 mm ornithine, 1 mm NADP+, 20 mm glucose, 10 mm CP, 5 units of hexokinase, 2.5 units of glucose-6-phosphate dehydrogenase, and 0.005-1 mm ADP in a final volume of 500 μl, pH 7.6. For the hCPS assay, 5 mm AGA was included, KCl was reduced to 10 mm, and ornithine was omitted. Assays were conducted at 25 °C, and after the addition of 5 μg of CPS, the formation of NADPH was followed continuously at 340 nm.
Glutamine hydrolysis was determined as previously described (14). Kinetic data were collected on a Beckman DU 640 spectrophotometer and were fit by nonlinear regression (Grafit, Version 5.1) to the equation v = VmaxS/(Km + S), where v is the initial velocity, Vmax is the maximal velocity, S is the substrate concentration, and Km is the Michaelis-Menten constant.
Thiol-specific Reactions—hCPS (2.5 μm) was incubated with or without 2 mm AGA and/or 7 mm ATP plus 12 mm MgCl2 at 30 °C for 10 min before 5.5 μm phenylarsine oxide (PAO) or 5.18 μm 4,4′-dithiodipyridine (PySSPy) was added. Rates of inactivation were determined by adding 10-μl aliquots of the reaction mixture to the CP synthesis activity assay mixture (final reaction volume of 100 μl, pH 7.6) at the indicated times and incubating at 30 °C for 20 min. To test reversal of sulfhydryl modification, 10 mm dithiothreitol (DTT) was added after ∼66% inactivation, and aliquots were removed at intervals for determination of CP synthesis activity. eCPS (2.5 μm) was modified only with 5.5 μm PAO, and incubations were carried out at 37 °C; for protection assays, 10 mm ATP plus 20 mm MgCl2 was used.
Visualization of eCPS Crystal Structure—Figures which include representations of the eCPS crystal structure were created using the University of California, San Francisco Chimera (20) and Friend (21) programs.
RESULTS AND DISCUSSION
Effects of C1327A and C1337A Substitutions on the Function of hCPS—Previous studies have established that a pair of cysteine side chains become proximate upon AGA binding to urea-specific CPS and that the cysteine residues can then be selectively induced to undergo disulfide bonding (10, 11). This single pair of proximate sulfhydryl groups is uniquely modified when the AGA·CPS complex is exposed to a variety of disulfide-inducing reagents, and reversible activity loss accompanies disulfide formation. The specificity of these reactions must depend on the relative unreactivity or inaccessibility of the other 18 hCPS cysteinyl residues. Earlier studies (11, 22) have identified one of the cysteine pair in hCPS as either Cys-1327 or Cys-1337. To identify both residues of the cysteine reporter group, we first constructed the hCPS mutants C1327A and C1337A and analyzed their responses to thiol-specific reagents.
The effect of the mutations themselves on the function of hCPS was determined by analyzing the overall and partial reactions catalyzed by the enzyme. The synthesis of CP requires coordination of two hCPS active sites with ATP cleavage occurring at duplicated ATP grasp domains B and C (Fig. 1). Individual contributions of these active sites to the overall reaction are reflected in two partial reactions, 1) at ATPB, a bicarbonate-dependent ATPase that reflects formation of the intermediate carboxyl phosphate and its nonproductive reaction with water in the absence of ammonia and 2) at ATPC, the formation of ATP from CP and ADP that appears to reflect reversal of carbamate formation that occurs at ATPC during the synthesis of CP (7, 23). The allosteric activator AGA must be bound for hCPS to provide significant catalysis for any of these reactions.
Both C1327A and C1337A were able to effectively synthesize CP under standard assay conditions with all substrates in excess. The observed specific activities in μmol CP/min/mg were as follows: 0.86 for wild type hCPS, 0.43 for C1327A, and 0.47 for C1337A. Kinetic parameters for the WT and mutant hCPSs are shown in Table 1. There were only minor changes in the parameters for the bicarbonate-dependent ATPase reaction, the partial reaction occurring at the domain B ATP site. In contrast, both mutations significantly altered activity at the domain C ATP site, as gauged by the CP-dependent ATP synthesis partial reaction. The ADP Km values were 9- and 14-fold higher than that of WT for C1337A and C1327A, respectively, and kcat was decreased in both mutants. C1327A and C1337A displayed 20- and 28-fold lower kcat/Km values for ATP formation at domain C than wild type hCPS. These effects on the domain C ATP site were consistent with the previous report that Cys-1327 and/or Cys-1337 reacts with the ATP analog 5′-fluorosulfonylbenzoyladenosine (FSBA) and the resulting prediction that one or both residues are localized near the ATP site (11).
TABLE 1.
Kinetic parameters for hCPS WT, C1327A, and C1337A
Reaction conditions were as described under “Experimental Procedures.” ATP was varied from 0.05 to 5 mm, NH4Cl from 0.01 to 2.5 mm, AGA from 0.01 to 1 mm, and ADP from 0.001 to 1 mm. S.E. of the kinetic parameters was determined from nonlinear regression curve-fitting (GraFit, Version 5.01). kcat values were within ± 10%.
|
Ammonia-dependent ATPase
|
Bicarbonate-dependent ATPase
|
CP-dependent ATP synthesis
|
|||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Km ATP | kcat | kcat/Km ATP |
Km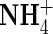
|
kcat |
kcat/Km
|
Km AGAa | kcat | kcat/Km AGA | Km ATP | kcat | kcat/Km | Km ADP | kcat | kcat/Km | |
| mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | |
| hCPS WT | 0.55 ± 0.04 | 5.73 | 10.42 | 0.35 ± 0.02 | 5.73 | 16.37 | 0.29 ± 0.01 | 5.73 | 19.76 | 0.56 ± 0.03 | 0.24 | 0.43 | 0.016 ± 0.002 | 0.28 | 17.5 |
| C1327A | 1.17 ± 0.29 | 1.92 | 1.64 | 0.11 ± 0.08 | 3.19 | 29.00 | 0.03 ± 0.01 | 1.18 | 39.33 | 0.52 ± 0.01 | 0.58 | 1.12 | 0.28 ± 0.06 | 0.20 | 0.71 |
| C1337A | 0.54 ± 0.10 | 1.50 | 2.80 | 0.35 ± 0.19 | 5.11 | 14.60 | 0.04 ± 0.01 | 1.18 | 29.50 | 0.18 ± 0.03 | 0.58 | 3.22 | 0.18 ± 0.03 | 0.09 | 0.50 |
Interaction of AGA with CPS is respresented as the apparent Km value.
Impaired functioning at domain C was also reflected in 3.7-6.4-fold decreases in kcat/Km for coupled utilization of both ATP molecules in the overall reaction, as gauged by ammonia-dependent ATPase activity. The mutations appeared to additionally affect interaction with AGA, with mutant apparent Km values that were 7-10-fold lower than for wild type hCPS. This observation was consistent with the previous reports that the C-terminal region of CPS is involved in binding AGA (24, 25). Interaction with ammonia was not significantly affected by the mutations. Whereas the specific effects of these mutations on the kinetic parameters provided insight into the structure/function relationship for hCPS, the kinetic analysis established that neither Cys-1327 nor Cys-1337 was critical for enzymatic activity. Thus, the Cys-1327 and Cys-1337 mutants could serve as effective probes for thiol reactivity.
Effects of C1327A and C1337A Substitutions on Reactivity with Thiol-selective Reagents—Previous studies on native rat liver CPS have established that two cysteine side chains become proximate in the AGA complex, as evidenced by the ability to form a disulfide bond when treated with PAO, a compound that preferentially reacts with dithiols that are capable of forming a cyclic dithioarsenite complex, PySSPy, a thiol-specific reagent that can induce disulfide bond formation, or with the ATP analog FSBA (10). FSBA reacts with a cysteinyl side chain to form a thiosulfonate, and subsequent attack by a second proximate cysteinyl side chain can displace the sulfonyl-benzoyladenosine moiety to yield a disulfide bond. Formation of the disulfide bond in the CPS·AGA complex could be monitored by the accompanying enzymatic inactivation as these two sulfhydryls were shown to be uniquely reactive. A later study (11) used a peptide chemistry approach to establish that FSBA initially reacts with either Cys-1327 or Cys-1337 at one of the CPS sites labeled by this analog but was not able to identify which of these cysteine residues was involved or which of the 20 CPS cysteine residues was the partner in the disulfide bond.
To determine whether Cys-1327 and/or Cys-1337 are the residues that become proximate in the CPS·AGA complex, we analyzed the response of wild type hCPS, C1327A, and C1337A to treatment with PAO. Replacing either of the two proximate cysteinyl residues with an alanine would remove the potential for the formation of a cyclic dithioarsenite complex and, thus, prevent the accompanying hCPS inactivation. As shown in Table 2, treatment of the wild type hCPS·AGA complex with a 2.2:1 molar ratio of PAO to hCPS led to 90% inactivation in 20 min. When AGA was omitted from the PAO treatment mixture, hCPS was only 27% inactivated. This inactivation of free hCPS could have two components; (a) PAO could react with the small fraction of hCPS that exists in the active conformation in the absence of AGA, with the PAO reaction pulling the equilibrium mixture of active and inactive conformations toward the active conformation as it is removed from the pool as the inactivated disulfide species, and (b) there could be some reaction with another cysteine pair that is at least transiently proximate, possibly via random intermolecular interactions.
TABLE 2.
Inactivation of hCPSI WT, C1327A, and C1337A by thiol selective reagents
The reaction mixtures contained 2.5 μm enzyme, 5.5 μm PAO, or 5.2 μm PySSPy and where indicated 2 mm AGA or 7 mm ATP + 12 mm MgCl2. Aliquots were removed at the indicated times of incubation at 30 °C and assayed for CP synthesis activity.

Treatment of the AGA complexes of C1327A and C1337A yielded 36 and 42% inactivation, respectively. This significant reduction in PAO inactivation relative to the 90% observed with WT suggested that Cys-1327 and Cys-1337 are the residues that become proximate in the AGA·hCPS complex and that then can be induced to form a disulfide bond. As was the case for wild type hCPS, both mutants showed significantly slower loss of activity in the absence of AGA (Table 2). Inactivation of native CPS by PAO can be prevented including ATP in the reaction mixture, presumably because binding of ATP at the domain C active site sterically blocks access to reagents such as PAO and FSBA (10, 11). Our present studies (Table 2) showed similar ATP protection against PAO inactivation of the AGA·hCPS complex, with activities of 88, 102 and 94% after 20 min of PAO treatment for WT, C1327A, and C1337A, respectively. The reducing agent DTT has been shown to reverse the inactivation of native CPS caused by PAO (10). Significant reversal of PAO inactivation was also observed in the present study. When enzyme with ∼66% remaining activity was treated for 10 s with 10 mm DTT, activities of 93, 92, and 90% were observed for WT, C1327A and C1337A, respectively. Taken together, these findings indicated that Cys-1327 and Cys-1337 constitute the AGA-responsive proximate cysteine pair. The findings also suggested that the residual ability of C1327A and C1337A to react with PAO in the absence of ATP protection resulted from the interaction of the remaining authentic disulfide pair member, i.e. Cys-1327 or C1337, with one of the other 18 cysteine residues in hCPS. Presumably this additional propinquity of cysteine side chains resulted from effects of the alanine substitution on polarity experienced by nearby residues leading to a local conformational change.
A second thiol selective reagent, PySSPy, was used to confirm the identification of Cys-1327 and Cys-1337 as the AGA-responsive proximate cysteines as well as to possibly identify which of the two residues is initially reactive. PySSPy forms a mixed disulfide with any reactive cysteine residue and can then be displaced by a proximate sulfhydryl group, forming an intramolecular disulfide. Thus, in contrast to PAO, PySSPy could react with only one of the proximate cysteine pair, although reaction with both to form the CPS disulfide bond was observed with native CPS (10). As previously observed for native enzyme, the WT/AGA complex rapidly lost the ability to synthesize CP when treated with PySSPy, with 62% of the original activity remaining after a 10-s treatment with a 2.07:1 molar ratio of PySSPy:hCPS (Table 2). The reaction was slower when AGA was omitted from the PySSPy incubation, with 92% activity remaining at 10 s. Both C1327A and C1337A displayed parallel behavior to WT, with an almost identical loss of activity for the AGA·CPS complex (58 and 64% activity at 10 s, respectively) and with much less activity loss when AGA was omitted from the reaction mixture. It was not possible to use our data to determine which residue is the initially reactive cysteine, but these data did confirm that Cys-1327 and Cys-1337 are the AGA-responsive pair.
The AGA complexes of all three constructions displayed complete protection from PySSPy inactivation when ATP was included in the incubation (Table 2). As also observed for PAO treatment, PySSPy inactivation of all three constructions was significantly reversed by DTT treatment. After a 10-s treatment with 10 mm DTT, WT activity increased to 81 from 57%, C1327A activity increased to 96 from 71%, and C1337A activity increased to 70 from 46%.
Implications for an hCPS Role in Buffering against Reactive Oxygen Species—Individual mutation of either Cys-1327 or Cys-1337 to alanine in hCPS resulted in specific activity values similar to that of the wild type enzyme, revealing that these residues were not critical for the function of the enzyme. The fact that the corresponding residues in eCPS are Pro-909 and Gly-919 also makes it is unlikely that either Cys-1327 or Cys-1337 is involved in any general feature of CP synthesis. Conservation of these residues in urea-specific CPSs could, therefore, indicate a critical role apart from enzymatic function.
Two earlier sets of experiments suggest physiological roles for the Cys-1327/Cys-1337 pair. First, studies on palmitoylation in mitochondria isolated from COS-7 cells and rat liver found that the majority of fatty-acylated protein in the mitochondrial matrix was urea-specific CPS (26). Although this study did not determine the site of palmitoylation, the findings that reaction with palmitoyl-CoA inactivated CPS and that the inactivation was prevented by ATP inclusion or prior FSBA treatment are consistent with Cys-1327 and/or Cys-1337 as the site(s) of palmitoylation. In the second set of earlier studies, Gautier et al. (27) reported that rat urea-specific CPS could be activated by thioredoxins isolated from rat liver. Without reduction of the thioredoxins by DTT, no activation of CPS was observed. Subsequent studies have revealed that thioredoxins constitute a family of 12-kDa proteins with thioredoxin 2 present in mitochondria. It is possible that thioredoxin 2 could serve as a reduction/oxidation sensitive regulator of hCPS activity under physiological conditions via interaction with the Cys-1327/Cys-1337 pair.
Urea-specific CPS is the major protein in the liver mitochondrial matrix, making up 15-20% that of the total protein in the mitochondrial matrix and occurring at an estimated in vivo concentration of 70 mg/ml (28). Thus, CPS has the potential ability to serve as a buffer against oxidative stress in the liver cell, especially under conditions of energy stress where ATP levels are low. In this situation AGA·CPS complexes could be oxidized to form the Cys-1327/Cys-1337 disulfide bond and thereby prevent interaction of the reactive oxygen species with other, less prevalent proteins, DNA or other molecules. Possibly two of the features unique to urea-specific CPSs, relative to other CPSs (the conserved Cys-1327/Cys-1337 pair and the occurrence at very high concentrations in the liver mitochondrial matrix) co-evolved to provide buffering against reactive oxygen species.
The hCPS inactivation accompanying formation of the Cys-1327/Cys-1337 disulfide bond also provides a rationale for one of the more puzzling features of Reye's syndrome; that is, the several days to several weeks that elapse between a viral infection that is treated with aspirin and the hyperammonemia that is the cause of mortality and morbidity of this disorder (29). Temporary deficiencies in mitochondrial ATP production and hCPS activity have been demonstrated in Reye's syndrome. It is possible that Cys-1327/Cys-1337 disulfide bond formation is the cause of the hCPSI deficiency and that the buildup of this oxidized hCPS as ATP concentrations become lower serves as the timer for Reye's syndrome.
Effects of P909C, G919C, and P909C/G919C Substitutions on the Function of eCPS—The residues in eCPS corresponding to the hCPS residues Cys-1327 and Cys-1337 are Pro-909 and Gly-919, respectively (Fig. 2). To examine the potential for an eCPS conformational change that parallels the AGA-induced conformational change of hCPS, we performed reciprocal mutagenesis to substitute cysteine residues both individually and simultaneously at these positions in eCPS. All three mutant enzymes were able to effectively catalyze the synthesis of CP under standard assay conditions with all substrates in excess. The specific activities of WT eCPS, P909C, G919C, and P909C/G919C in μmol of CP/min/mg were 0.855, 0.472, 0.524, and 0.348.
FIGURE 2.

CPS multiple sequence alignment. The sequences of urea-specific CPSs from Homo sapiens (human), Rattus norvegicus (rat), Rana catesbeiana (bullfrog), Xenopus laevis (toad), and glutamine-utilizing CPSs from Mesocricetus auratus (hamster), Saccharomyces cerevisiae (yeast), and E. coli are aligned to show correspondence of Cys-1327 in hCPS to Pro-909 in eCPS and Cys-1337 in hCPS to Gly-919 in eCPS (highlighted in blue). Residues conserved in all of the CPS sequences are highlighted in yellow.
Kinetic parameters for the three mutants as well as wild type eCPS are shown in Table 3. The most significant effects of mutations were on the interaction with ATP. When coupled use of both ATPs was assayed with ammonia and glutamine, respectively, as nitrogen donor, P909C/G919C displayed 13- and 34-fold lower kcat/Km and ATP values, and the two single mutants displayed smaller changes. There were relatively minor changes in the parameters for the bicarbonate-dependent ATPase reaction, the partial reaction occurring at the domain B ATP site. In contrast, P909C had an 8-fold lower Km for ATPC, as gauged by the CP-dependent ATP synthesis partial reaction, whereas G919C had an 8-fold higher Km for ATPC. Interestingly, the effects of the single mutants behaved in an apparently additive fashion to yield a double mutant with CP synthesis properties very similar to those of WT.
TABLE 3.
Kinetic parameters for eCPS WT, P909C, G919C, and P909C/G919C
Reaction conditions were as described under “Experimental Procedures.” ATP was varied from 0.05 to 5 mM, NH4Cl from 50 to 250 mm, Gln from 0.01 to 1 mm, and ADP from 0.005 to 1 mm. To determine glutamine-dependent ATPase activity, 10 mm glutamine was included in the reaction mixture. To determine ammonia-dependent activity, 300 mm NH4Cl was in included in the reaction mixture. Ornithine (10 mm) was present except in reactions indicated as “no effector” or where substituted by 0.1 mm UMP. Ornithine (orn) is conventionally included in eCPS assays to yield Michaelis-Menten kinetic data. S.E. of the kinetic parameters was determined from nonlinear regression curve-fitting (GraFit, Version 5.01). kcat values were within ± 10%.
|
Ammonia-dependent ATPase
|
Bicarbonate-dependent ATPase
|
CP-dependent ATP synthesis
|
||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Km ATP | kcat | kcat/Km ATP |
Km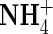
|
kcat |
kcat/Km
|
Km ATP | kcat | kcat/Km | Km ADP | kcat | kcat/Km | |
| mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | |
| eCPS WT | 0.03 ± 0.01 | 1.61 | 53.67 | 111 ± 11 | 5.10 | 0.046 | 0.10 ± 0.02 | 0.20 | 2.00 | 0.008 ± 0.001 | 0.14 | 17.5 |
| P909C | 0.07 ± 0.06 | 0.61 | 8.71 | 154 ± 27 | 1.99 | 0.013 | 0.03 ± 0.02 | 0.09 | 3.00 | 0.001 ± 0.002 | 0.17 | 170 |
| G919C | 0.02 ± 0.02 | 0.45 | 22.50 | 148 ± 30 | 1.53 | 0.010 | 0.15 ± 0.08 | 0.10 | 0.67 | 0.06 ± 0.01 | 0.27 | 4.50 |
| P909C/G919C | 0.14 ± 0.02 | 0.58 | 4.14 | 158 ± 52 | 1.60 | 0.010 | 0.14 ± 0.04 | 0.12 | 0.86 | 0.02 ± 0.02 | 0.42 | 21.0 |
|
Glutamine-dependent ATPase
|
||||||
|---|---|---|---|---|---|---|
| Km ATP | kcat | kcat/Km ATP | Km Gln | kcat | kcat/Km Gln | |
| mm | s−1 | mm−1 s−1 | mm | s−1 | mm−1 s−1 | |
| eCPS WT + orn | 0.04 ± 0.01 | 4.98 | 125 | 0.14 ± 0.02 | 5.81 | 41.5 |
| No effector | 0.47 ± 0.06 | 7.20 | 15.3 | |||
| + UMP | 1.60 ± 0.20 | 3.70 | 2.31 | |||
| P909C + orn | 0.09 ± 0.03 | 2.01 | 22.3 | 0.20 ± 0.10 | 3.15 | 15.8 |
| No effector | 2.33 ± 0.27 | 2.43 | 1.04 | |||
| + UMP | 0.91 ± 0.15 | 0.38 | 0.42 | |||
| G919C + orn | 0.16 ± 0.06 | 2.81 | 17.6 | 0.13 ± 0.03 | 3.35 | 25.8 |
| No effector | 4.00 ± 0.71 | 3.04 | 0.76 | |||
| + UMP | 2.25 ± 0.09 | 0.48 | 0.21 | |||
| P909C/G919C + orn | 0.54 ± 0.11 | 2.92 | 5.41 | 0.09 ± 0.01 | 3.16 | 35.1 |
| No effector | 5.29 ± 1.46 | 2.40 | 0.45 | |||
| + UMP | 1.47 ± 0.09 | 0.38 | 0.26 | |||
We also examined the effect of these mutations on the allosteric behavior of eCPS (Table 3). Ornithine, co-substrate with CP for the second step of arginine biosynthesis, acts as a positive allosteric effector for eCPS, the catalyst for the first pathway step. UMP, product of the pyrimidine nucleotide pathway, acts as a negative allosteric effector for eCPS, which also catalyzes the first step of this pathway. It should be noted that these effectors act as traditional allosteric effectors that alter the rate of an active unliganded enzyme rather then acting in the non-traditional manner of AGA, the essential allosteric activator for hCPS. Because allosteric effectors bind at domain D of eCPS and function primarily to alter the Km for ATP in domain C (7), the present mutations might provide insight into the molecular mechanism for allosteric communication. Comparison of the kinetic parameters for the mutants to those of WT indicated that the response to ornithine is unaffected but that the response to UMP occupancy was altered, with much less decrease in Km for all three mutants as well as an increased effect on kcat. These findings support the previous suggestion of independent routes of allosteric communication for ornithine and UMP within eCPS (7).
In sum, the kinetic analysis established that neither Pro-909 nor Gly-919 was critical for eCPS function and that activity was not significantly impaired in the P909C/G919C double mutant. Thus, P909C/G919C could be used to probe for disulfide bond formation that might occur if eCPS assumes a conformation that parallels the AGA-induced conformation of hCPS.
Response of P909C, G919C, and P909C/G919C to PAO Treatment—To determine whether the engineered cysteinyl residues P909C and G919C are located in eCPS positions that allow induction of a disulfide bond, we used PAO, a compound that preferentially reacts with dithiols capable of forming a cyclic dithioarsenite complex. We used loss of enzymatic activity to monitor this reaction, with the assumption that the ATP grasp folds of eCPS and hCPS are generally conserved. If this is the case, formation of a disulfide bond between P909C and G919C would be expected to result in sterically blocked access to ATP binding in domain C, as was observed for the analogous disulfide bond in hCPS. This expectation is supported by the occurrence of the Pro-909 oxygen 3.6 Å from C5 of the ADP-ribose in the solved eCPS structure (7). It should be noted that the expectation of disulfide bond formation itself is not supported by this structure, the only available solved eCPS conformation. The α carbons of Pro-909 and Gly-919 are 23.97 Å apart, far from the average disulfide bond length of 5.99 Å (Fig. 3). Thus, identification of a disulfide bond between the engineered cysteinyl residues would also serve to identify an alternative eCPS conformation.
FIGURE 3.

Location of residues Pro-909 and Gly-919. The α carbons of residues Pro-909 and Gly-919 occur 23.97 Å apart in the crystal structure of eCPS. This figure was created from PDB file 1a9x.
As shown in Table 4, treatment of wild type eCPS, P909C, or G919C with a 2.2:1 molar ratio of PAO to CPS had essentially no effect on activity. These findings indicated the absence of vicinal cysteinyl residues in WT eCPS as well as the absence of a cysteine residue vicinal to P909C or G919C that could be induced to form a disulfide with the newly created cysteine. In dramatic contrast, treatment of P909C/G919C with a low concentration of PAO (2.2:1 molar ratio of PAO to CPS) led to rapid inactivation, with 21% activity loss at 10 s and a plateau of ∼40% activity loss. This significant and construct-specific inactivation indicated that eCPS can assume a conformation in which the engineered cysteinyl residues, P909C and G919C, are proximate and can be induced to form a disulfide bond. Because the essential allosteric activator AGA stabilizes the hCPS conformation where the analogous disulfide bond can be formed, we tested the effect of the positive allosteric effector ornithine on eCPS behavior. However, the addition of 10 mm ornithine to the PAO incubation mixture had essentially no effect on the PAO response of WT or any of the three mutants (data not shown). Inactivation of P909C/G919C by PAO could be prevented by the inclusion of ATP in the reaction mixture (Table 4), presumably because binding of ATP at the domain C active site sterically blocks access to PAO. When P909C/G919C that had been 40% inactivated by PAO treatment was subsequently treated with 10 mm DTT, 19% activity was recovered after 10 s of treatment. The failure to observe further DTT reactivation with treatment times up to 2 min suggested that the engineered disulfide bond was not fully accessible to reducing agent.
TABLE 4.
Inactivation of WT and mutant eCPSs by PAO
The reaction mixtures contained 2.5 μm enzyme and 5.5 μm PAO. Where indicated 10 mm ATP plus 20 mm MgCl2 was included in the reaction mixture. Aliquots were removed at the indicated times of incubation at 37 °C and assayed for CP synthesis activity.
| Time |
% Activity remaining after PAO treatment
|
% P909C/G919C activity remaining
|
|||
|---|---|---|---|---|---|
| WT eCPS | P909C | G919C | PAO only | +ATP/Mg++ | |
| s | |||||
| 10 | 98 ± 0.9 | 97 ± 4.2 | 97 ± 2.2 | 79 ± 1.3 | 95 ± 2.6 |
| 40 | 98 ± 2.9 | 95 ± 0.7 | 95 ± 4.4 | 73 ± 2.0 | 94 ± 3.3 |
| 120 | 98 ± 2.2 | 92 ± 0.3 | 94 ± 0.7 | 71 ± 0.5 | 93 ± 2.3 |
| 480 | 96 ± 2.7 | 90 ± 0.6 | 93 ± 1.9 | 64 ± 1.8 | 91 ± 0.1 |
| 840 | 93 ± 0.2 | 92 ± 1.5 | 96 ± 0.9 | 64 ± 0.2 | 93 ± 0.3 |
| 1200 | 98 ± 0.3 | 90 ± 2.1 | 94 ± 5.1 | 59 ± 3.8 | 94 ± 0.4 |
eCPS occurs almost entirely as a tetramer (four 42-kDa subunits plus four 118-kDa subunits; Fig. 1) under both physiological conditions and the present experimental conditions (7, 30). Thus, it is feasible that the disulfide bond observed for P909C/G919C was an intermolecular bond involving the P909C of one monomer and the G919C of another. The intermolecular bonding possibility was not a concern for the hCPS studies because that enzyme is known to exist as a monomer under the experimental conditions utilized (31) and because earlier gel analysis of the FSBA-induced disulfide ruled out formation of any species other than the monomer.3 To determine whether the PAO-induced disulfide bond of P909C/G919C was intramolecular or intermolecular, we carried out non-reducing SDS-PAGE analysis of PAO-treated WT and mutant constructs. None of the PAO treated eCPSs, including the double mutant, display shifts in mobility that would accompany introduction of an intermolecular disulfide bond (data not shown). Therefore, the engineered disulfide bond of P909C/G919C was intramolecular.
Implications for Parallel Conformational Changes in eCPS and hCPS—In sum, these findings indicated at least some conformational similarity between the AGA·hCPS complex and eCPS. Additionally, the fact that the α carbons of Pro-909 and Gly-919 are 23.97 Å apart, far from the average disulfide bond length of 5.99 Å, in the single solved conformation of eCPS (Fig. 3) indicated that the conformation with the engineered disulfide bond represents an alternative eCPS conformation. Pro-909 and Gly-919 occur near the boundary of domain C, one of the duplicated ATP grasp domains, and domain D, where allosteric effectors bind. As seen in Fig. 3, Pro-909 and Gly-919 do not occur within constrained secondary structural elements in the crystal structure. Gly-919 occurs at the end of a β-strand (913-921), and Pro-909 falls within an extended area without classical secondary structure (901-911). The fact that enzymatic activity was reasonably well maintained despite the drastic substitution of cysteine residues for Pro-909 and Gly-919 further indicated that the residues are not structurally critical. Additionally, both residues are located very near the internal molecular tunnel proposed to connect the active sites of domains B and C and to move the carbamate intermediate which is highly unstable and cannot be exposed to water (32); residues 911, 913, 914, 915, and 916 comprise part of the tunnel. Thus, conformational flexibility that would move Pro-909 and Gly-919 into more proximate positions is not inconsistent with the single available crystal structure. Further work is necessary to define this alternative conformation and its relationship to the cycle of conformational changes that accompany the catalytic cycle and that allow synchronization of three active sites (glutamine amidotransferase site and two distinct ATP sites) to produce CP.
Acknowledgments
We greatly appreciate the valuable discussions with Amna Saeed-Kothe, Michael Kothe, Vibha Ahuja, and Alexej Abysov.
This work was supported, in whole or in part, by National Institutes of Health Grant DK54423. The costs of publication of this article were defrayed in part by the payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Footnotes
The abbreviations used are: CPS, carbamoyl phosphate synthetase; eCPS, E. coli CPS; hCPS, urea-specific human liver CPS (also termed CPSI); AGA, N-acetylglutamate; CP, carbamoyl phosphate; DTT, dithiothreitol; FSBA, 5′-fluorosulfonylbenzoyladenosine; PAO, phenylarsine oxide; PySSPy, 4,4′-dithiodipyridine; WT, wild type.
M. D. Potter and S. G. Powers-Lee, unpublished observation.
References
- 1.Mommsen, T. P., and Walsh, P. J. (1989) Science 243 72-75 [DOI] [PubMed] [Google Scholar]
- 2.Paulus, H. (1983) Curr. Top. Cell. Regul. 22 177-200 [DOI] [PubMed] [Google Scholar]
- 3.Finckh, U., Kohlschutter, A., Schafer, H., Sperhake, K., Colombo, J. P., and Gal, A. (1998) Hum. Mutat. 12 206-211 [DOI] [PubMed] [Google Scholar]
- 4.Haraguchi, Y., Uchino, T., Takiguchi, M., Endo, F., Mori, M., and Matsuda, I. (1991) Gene (Amst.) 107 335-340 [DOI] [PubMed] [Google Scholar]
- 5.Nyunoya, H., and Lusty, C. J. (1983) Proc. Natl. Acad. Sci. U. S. A. 80 4629-4633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Anderson, P. M. (1995) Molecular Aspects of Carbamoyl Phosphate Synthetase in Nitrogen Metabolism and Excretion, pp. 33-49, CRC Press, Inc., Boca Raton, FL
- 7.Thoden, J. B., Raushel, F. M., Benning, M. M., Rayment, I., and Holden, H. M. (1999) Acta Crystallogr. D Biol. Crystallogr. 55 8-24 [DOI] [PubMed] [Google Scholar]
- 8.Miles, B. W., and Raushel, F. M. (2000) Biochemistry 39 5051-5056 [DOI] [PubMed] [Google Scholar]
- 9.Caldovic, L., and Tuchman, M. (2003) Biochem. J. 372 279-290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marshall, M., and Fahien, L. A. (1985) Arch. Biochem. Biophys. 241 200-214 [DOI] [PubMed] [Google Scholar]
- 11.Potter, M. D., and Powers-Lee, S. G. (1992) J. Biol. Chem. 267 2023-2031 [PubMed] [Google Scholar]
- 12.Ahuja, V., and Powers-Lee, S. (2008) J. Inherited Metab. Dis. 31 481-491 [DOI] [PubMed] [Google Scholar]
- 13.Sambrook, J., Fritsch, E. F., and Maniatis, T. (2001) Molecular Cloning: A Laboratory Manual, Cold Spring Harbor Laboratory Press, New York
- 14.Saeed-Kothe, A., and Powers-Lee, S. G. (2002) J. Biol. Chem. 277 7231-7238 [DOI] [PubMed] [Google Scholar]
- 15.Laemmli, U. K. (1970) Nature 227 680-685 [DOI] [PubMed] [Google Scholar]
- 16.McGivan, J. D., Bradford, N. M., and Mendes-Mourao, J. (1976) Biochem. J. 154 415-421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rubino, S. D., Nyunoya, H., and Lusty, C. J. (1986) J. Biol. Chem. 261 11320-11327 [PubMed] [Google Scholar]
- 18.Kothe, M., and Powers-Lee, S. G. (2004) Protein Sci. 13 466-475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen, N. S., Kyan, F. S., Kyan, S. S., Cheung, C. W., and Raijman, L. (1985) Biochem. J. 229 205-211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pettersen, E. F., Goddard, T. D., Huang, C. C., Couch, G. S., Greenblatt, D. M., Meng, E. C., and Ferrin, T. E. (2004) J. Comput. Chem. 25 1605-1612 [DOI] [PubMed] [Google Scholar]
- 21.Abyzov, A., Errami, M., Leslin, C. M., and V. A., I. (2005) Bioinformatics 21 3677-3678 [DOI] [PubMed] [Google Scholar]
- 22.Geschwill, K., and Lumper, L. (1989) Biochem. J. 260 573-576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kothe, M., Purcarea, C., Guy, H. I., Evans, D. R., and Powers-Lee, S. G. (2005) Protein Sci. 14 37-44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.McCudden, C. R., and Powers-Lee, S. G. (1996) J. Biol. Chem. 271 18285-18294 [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Aparicio, L. B., Guadalajara, A. M., and Rubio, V. (1989) Biochemistry 28 3070-3074 [DOI] [PubMed] [Google Scholar]
- 26.Corvi, M. M., Soltys, C. L., and Berthiaume, L. G. (2001) J. Biol. Chem. 276 45704-45712 [DOI] [PubMed] [Google Scholar]
- 27.Gautier, C., Habechi, Z., Balange, A. P., and Vaillant, R. (1984) C. R. Acad. Sci. III 299 849-852 [PubMed] [Google Scholar]
- 28.Clarke, S. (1976) J. Biol. Chem. 251 950-961 [PubMed] [Google Scholar]
- 29.Belay, E., Bresee, J., Holman, R., Khan, A., Shahriari, A., and LB, S. (1999) N. Engl. J. Med. 340 1377-1382 [DOI] [PubMed] [Google Scholar]
- 30.Kim, J., and Raushel, F. M. (2001) Biochemistry 40 11030-11036 [DOI] [PubMed] [Google Scholar]
- 31.Lusty, C. J. (1981) Biochemistry 20 3665-3674 [DOI] [PubMed] [Google Scholar]
- 32.Kim, J., and Raushel, F. M. (2004) Arch. Biochem. Biophys. 425 33-41 [DOI] [PubMed] [Google Scholar]