

Abstract
The relative simplicity and high specificity of peptide therapeutics has fueled recent interest. However, peptide and protein drugs generally require injection and suffer from low metabolic stability. We report here the design, synthesis and characterization of fluorinated analogues of the gut hormone peptide, GLP-1. Overall, fluorinated GLP-1 analogues displayed higher proteolytic stability with simultaneous retention of biological activity (efficacy). Fluorinated amino acids are useful for engineering peptide drug candidates and probing ligand-receptor interactions.
Keywords: GLP-1 peptide, fluorinated amino acids, anti-diabetics, ligand-receptor interaction
Type 2 diabetes is a debilitating disease and has become a worldwide epidemic.1 The unmet challenges in disease management are the lack of long-term efficacy in reducing hyperglycemia and the inability to stop progression. Glucagon-like peptide-1 (GLP-1) is viewed as the basis for a new class of therapeutics to meet such needs.2-4 GLP-1 has multifaceted actions: it stimulates insulin secretion in a glucose dependent manner increases β-cell mass and function and suppresses glucagon secretion and appetite.3,4 These properties illuminate its pharmacological potential in regulating blood glucose homeostasis. The concept of using GLP-1 to treat type 2 diabetes has been validated in human subjects,5 as administration of exogenous GLP-1 lowers blood sugar levels. However, the clinical utility of native GLP-1 is severely hampered by its rapid deactivation by the serine protease, dipeptidyl peptidase IV (DPP IV, EC 3.4.14.5).3,4 GLP-1 has a half-life of less than 2 minutes after intravenous administration.6 There is great interest in developing long-acting agents that emulate the functions of GLP-1.3,7 Indeed, a 39-residue peptide that mimics the functional attributes of GLP-1, exenatide, with a half-life of 26 min after i.v. injection,2 is currently on the market with a dosing frequency of twice a day.
Fluorine substitution has proven useful for improving the therapeutic index of medicinal agents. While the first examples may have been serendipitous, nevertheless, roughly one-fifth of the pharmaceuticals on the market contain fluorine.8 Introduction of fluorine into small molecules often results in increased hydrophobicity and metabolic stability, eventually leading to improved bioactivity and bioavailability,9 as evidenced in the case of the anti-hyperglycemic sitagliptin10 and the anti-depressant fluoxetine.11 However, this strategy has rarely been explored in peptide/protein therapeutics, a class of promising agents with high specificity and low metabolic stability.12
In limited studies, myriad effects of fluorination on hormonal and antimicrobial peptides13-19 have been documented. For example, replacement of Tyr4 and Phe8 with 4F–Phe resulted in two angiotensin II analogues with distinct functional profiles. The first of these analogues resulted in generating a competitive inhibitor of angiotensin receptors, while the latter is an equipotent agonist13 in both rat blood pressure and oxytocic assays. Introduction of trifluoromethyl isoleucine and leucine into the cell lytic peptide melittin14 and cell penetrating buforin17 led to enhanced membrane binding affinity and increased bacteriostatic activity, respectively. In addition, introduction of hexafluorovaline15 and hexafluoroleucine17,18 into hormonal and antimicrobial peptides, renders them more stable to certain hydrolytic enzymes.
We envisioned that incorporation of hexafluoroleucine at strategic sites in therapeutic peptides could modulate peptide-receptor interactions and provide improved resistance to proteolytic cleavage. We report here the synthesis of GLP-1 and selectively fluorinated analogues, their binding affinities to the cognate human receptor (GLP-1R) and signal transduction efficacy, and their protease stability against DPP IV. A fluorinated analogue was also tested for its ability to regulate the blood glucose level in vivo (see Sup Info).
Experimental Procedures
Cell Culture and Receptor Transfection
COS-7 cells were cultured in DME supplemented with 10% FBS, penicillin G sodium (100 units/mL) and streptomycin sulfate (100 μg/mL), 26 mM sodium bicarbonate, pH 7.4 at 37 °C in 5% CO2 humidified air. Cells (0.8 × 106 cells/plate) were seeded in 10 cm dishes. The following day, cells were transiently transfected with 5 μg of pcDNA1 vector using diethylaminoethyl-dextran (DEAE-dextran). The vector contains cDNA encoding the human GLP-1 receptor (hGLP1-R)20 and has been sequenced for the identity21.
Receptor Binding Assay
Receptor binding of peptides was analyzed as previously described21. Briefly, COS-7 cells (104 cells/well) were subcultured onto 24-well tissue culture plates (Falcon, Primaria®, BD sciences) a day after transfection. Competitive binding experiments were carried out the next day at 25 °C for 100 min using 17 pM [125I]-exendin (9-39) amide as the radioligand. The peptides had a final concentration ranging from 3 × 10−6 to 3 × 10−11 M in 270 μL binding buffer freshly prepared in HBSS containing 0.2% BSA, 0.15 mM phenylmethylsulfonyl fluoride (PMSF), 25 mM HEPES, pH 7.3. Non-specific binding was determined in the presence of 1 μM unlabeled peptides. Cells were carefully washed before (1 × 1 mL) and after (3 × 1 mL) incubation with binding buffer. Cells were lysed in 1 N NaOH, washed with 1 N HCl, and transferred to polypropylene tubes (Sigma) for γ counting (Beckman Gamma counter 5500B).
cAMP Assay
COS-7 (105 cells/well) cells were transferred 24 h after transfection to 24-well plates, and cultured for another 24 h. Cells were stimulated with GLP-1 and analogues for 1 h at 25°C in DME (without phenol red) supplemented with 1% BSA, 1 mM 3-isobutyl-1-methylxanthine (IBMX), 0.4 μM Pro-boroPro, and 25 mM HEPES, pH 7.4. Pro-boroPro ([1-(2-pyrrolidinylcarbonyl)-2-pyrrolidinyl] boronic acid) is a potent DPP IV inhibitor22 (provided by Dr. W. W. Bachovchin, Tufts University). The final peptide concentrations ranged from 1 × 10−6 to 1 × 10−11 M in 10-fold increments in 270 μL of buffer. Upon removal of incubation buffer, cells were lysed by a freeze-thaw cycle in N2 (l), followed by the addition of 200 μL M–Per to ensure the complete lysis of cells. The concentrations of cAMP in its acetylated form were determined using a FlashPlate® kit (PerkinElmer Life Sciences) and [125I]-cAMP as the radioligand. Plate-bound radioactivity was measured using a Packard Topcount® proximity scintillation counter.
Degradation of Peptides by DPP IV
The proteolytic stability of peptides towards DPP IV was determined by an analytical RP–HPLC assay. The chromogenic Gly-Pro-p-nitroanilide was used to calibrate the specific activity of DPP IV (Δε410nm = 8800 M−1 ·cm−1) in 100 mM Tris•HCl, pH 8.0. Peptides (10 μM) were separately incubated with DPP IV (20 units/L) in 50 mM Tris•HCl, 1 mM EDTA, pH 7.6 at 37 °C over 200 min.23 Reactions were quenched with 600 μL of 0.2% TFA at time intervals and stored at −20°C. An analytical column [J.T.Baker C18, 5 μm, 4 mm × 250 mm] was used for separation and quantitation of intact and digested peptides with a binary solvent system of CH3CN/H2O/0.1%TFA (detection at 230 nm). First order rate constants were obtained as the fitted value ± standard deviation using equation (1):
| (1) |
where A is the concentration of peptides; k the first order rate constant; t the reaction time in min; and [A]0 the initial concentration of peptides. The fragments derived from the full-length peptides were manually collected and identified by ESI–MS.
Data Analysis
Data from radioligand binding and cAMP asssays were analyzed by nonlinear regression (GraphPad Prism v4.0, San Diego, CA) and are reported as mean ± s.e.m. Data were normalized relative to GLP-1 for both receptor binding and receptor activation assays.
Results
Peptide Design and Characterization
GLP-1 binds to its cognate seven transmembrane G protein-coupled receptor (GLP-1R) through noncovalent interactions.24 GLP-1 consists of an N-terminal random coil segment25 (7-13), two helical segments (13-20 and 24-36), and a less well-defined region (21-23). The C-terminal helix of GLP-1 is conformationally more stable than the N-terminal helix as judged by amide proton exchange experiments25 and is essential for receptor binding.24 Replacements of Phe28 and Ile29 with alanine in GLP-1 induced a dramatic loss of binding affinity to GLP-1R.24 These two residues along with Trp31 and Leu32 are conserved between GLP-1 and exendin 4, a synthetic GLP-1R agonist with high binding affinity,26 and are located on the C-terminal hydrophobic surface. In an attempt to manipulate the binding affinity of GLP-1 to GLP-1R, Phe28, Ile29, and Leu32 were selectively substituted with hexafluoroleucine assuming that increased hydrophobicity of hexafluoroleucine27 would lead to an enhanced binding affinity. Trp31 was retained because of its utility in concentration determination and the flat geometry and large volume of the aromatic side chain.
To confer resistance towards DPP IV, several N-terminal residues (at P1, P1′ and/or P2′ positions) were replaced with hexafluoroleucine, namely, Ala8, Glu9, Gly10 and both Ala8 and Glu9 to generate four fluorinated analogues. His7 was left unchanged due to its crucial role in receptor signaling.28 In short, selective hexafluoroleucine substitution was performed to probe the effects of fluorinating GLP-1 at sites that are crucial for the proteolytic stability and ligand-receptor interactions. The sequences of the fluorinated analogues, GLP-1, and exendin (9-39) amide are shown in Scheme 1.
Scheme 1.

The sequences of wild type GLP-1(7-36) amide, fluorinated analogues, Ex(9-39), and [125I]-Ex(9-39). The residues replaced with hexafluoroleucine in GLP-1 are underlined. The red arrow indicates the scissile bond that DPP IV cleaves. The conserved residues between GLP-1 and Ex(9-39) are colored blue. [125I]-Ex(9-39) amide served as the radioligand for the competition binding assay, in which 125I-labeled Bolton-Hunter reagent is conjugated to Lys12. L: 5,5,5,5′,5′,5′-2S-hexafluoroleucine. ORTEP drawing of the crystal structure of hexafluoroleucine methyl ester indicates the stereochemistry of α-carbon (S).
The crystal structure of hexafluoroleucine methyl ester hydrochloride salt (Scheme 1) validates the stereochemistry at α-carbon (S configuration). The secondary structures of all peptides were determined by CD in phosphate buffered saline (PBS), dodecylphosphocholine (DPC), and 35% (v/v) trifluoroethanol (TFE) in PBS. All fluorinated analogues were similar in structure to wt GLP-1 (representative spectra for GLP-1 and F8, see Fig. 1; other data not shown), suggesting no significant structural perturbation arose from fluorination in these buffered solutions, despite the lower α-helical propensity of hexafluoroleucine compared to leucine and alanine.29
Fig. 1.

CD spectra of GLP-1 (A) and F8 (B). Spectra in each panel correspond to peptides in 20 mM sodium phosphate (least helical), 40 mM dodecylphosphocholine, and 20 mM sodium phosphate containing 35% TFE (most helical) at 5 °C and pH 7.4. [Peptide] = 10 μM. Data represent the average of four scans.
Binding Assay
The binding affinity of fluorinated analogues was measured by a competition-binding assay using [125I]-Ex(9-39) amide as a radioligand. This Bolton-Hunter labeled peptide was assumed to have a similar affinity to hGLP-1R as Ex(9-39) amide since the modification at Lys12 side chain does not diminish receptor binding.30 The homologous antagonist competitive binding experiments showed that the binding of Ex(9-39) amide has a dissociation constant of 2.9 nM (three independent experiments in triplicate, see Sup Info), in good agreement with previously reported values.21,31 All 7 fluorinated analogues bound to the hGLP-1R expressed on COS-7 cells that lack endogenous GLP-1R. F9 had a binding affinity comparable to GLP-1 (IC50 5.1 nM vs 1.9 nM, Fig. 2 and Table 1), while F29 and F28 displayed 7-fold and 9.9-fold decreased affinity. F8, F89, F10, and F32 were weaker binders with a 27-60-fold increase in IC50 values (Figs. 2A and 2B). The side chain carboxylate of Glu9 has been shown to be important for receptor binding as substitution by Lys9 resulted in a significant loss in binding affinity.32 Substitution with Ala9 also led to relatively poor receptor binding (30-80-fold higher IC50),24,33 while substitution by Asp9 did not result in a significant change (about same IC50).33 These facts, together with the similar binding affinity showed by F9, in which Glu9 was replaced by hexafluoroleucine suggests that ‘polar hydrophobicity’34 of hexafluoroisopropyl group is likely responsible for the apparent retention in binding ability. Ligand-receptor binding interfaces are frequently devoid of solvent water. Therefore favorable polar interactions between CF3 groups and possible electropositive regions (or dipoles) could in principle mimic the interaction between the carboxylate and its binding partner. Multipolar C–F···C=O, C–F···H–X (X=O, N, S), C–F···H–Cα, and C–F···side chain (of Arg, Gln and Asn) interactions are commonly found in protein-ligand complexes in the PDB and CSD databases.8 An alternative explanation is that bulky hydrophobic side chains at this position are well tolerated. The latter argument is supported by the prior observation that substitution of Glu9 with Leu9 resulted in only a 1.5-fold decrease in binding affinity.35 The N-terminal modifications, except for F9, resulted in decreased binding affinity, while the C-terminal modifications were well tolerated indicated by only slightly diminished binding to GLP-1R.
Fig. 2.

Binding of peptides to cloned human GLP-1R transiently expressed on COS-7 cells as examined by a competitive binding assay using [125I]-Ex(9-39) as the radioligand. (A) N-terminal analogues; (B) C-terminal analogues. Data represent five independent experiments in duplicate as mean ± s.e.m.
Table 1.
Summary of the receptor binding, cAMP production, and proteolytic stability of GLP-1 and fluorinated analogues.
| Binding to hGLP-1R | cAMP Production | Cleavage by DPP IV | ||||||
|---|---|---|---|---|---|---|---|---|
| IC50 (nM)a | Max.(CI95%)b | Ratio | EC50 (nM)c | Max.(CI95%) | Ratio | k (min−1) | Ratio | |
| GLP-1 | 1.9 | 99.1-104.9 | 1.0 | 1.0 | 95.6-103.8 | 1.0 | 0.0061 | 1 |
| F8 | 52.2 | 94.6-103.2 | 27.3 | 73.0 | 82.6-94.9 | 73.8 | 0c | |
| F9 | 5.1 | 96.5-103.9 | 2.7 | 2.0 | 95.9-106.0 | 2.1 | 0.0050 | 1.5 |
| F89 | 107.3 | 95.9-103.9 | 56.2 | 374.7 | 25.5-33.2 | 378.5 | 0c | |
| F10 | 113.1 | 95.8-104.2 | 59.2 | 67.3 | 82.0-98.1 | 68.0 | 0.0021 | 2.9 |
| F28 | 18.9 | 98.1-106.9 | 9.9 | 5.4 | 99.0-108.5 | 5.5 | 0.0046 | 1.3 |
| F29 | 13.4 | 92.4-101.7 | 7.0 | 3.6 | 91.0-111.0 | 3.6 | 0.0056 | 1.1 |
| F32 | 57.1 | 94.6-103.0 | 29.9 | 2.4 | 88.1-106.8 | 2.4 | – | – |
IC50: concentration required for 50% inhibition of the maximal binding.
CI95%: 95% confidence intervals.
EC50: concentration required for producing 50% of the maximal response.
No detectable hydrolysis after 24 h.
Formation of cAMP
In a radioimmunoassay, COS-7 cells expressing hGPL-1R were stimulated by peptides and the accumulation of intracellular secondary messenger cAMP was measured. All fluorinated peptides functioned as full agonists with the lone exception of F89 (Fig. 3). F9, F32, F29, and F28 had a 2 to 5-fold decreased potency while retaining the same efficacy as GLP-1 (Fig. 3 and Table 1). F8 and F10 displayed a moderate decrease in potency but efficacies were judged to equivalent, as judged by p-tests. A previous report24 found that substitution of Gly10 with Ala completely abolished receptor activation ability, suggesting the crucial role of Gly. Our study indicates that the bulky side chains can be accommodated at position 10. Replacement of Phe28 with Ala resulted in a 1000-fold decrease in receptor activation.24 In contrast, substitution with hexafluoroleucine was well tolerated in terms of both receptor binding and activation, suggesting hydrophobicity is a required element while aromaticity is dispensable at this position. Interestingly, F89 was a partial agonist with a maximal response being ∼30% of that elicited by natural GLP-1. This is different from the binding ability of F89 to GLP-1R, which is similar to F10 in the range of tested concentrations. This result suggests that the correct positioning of His7 into the receptor-binding pocket is critical for receptor activation. Comparing the potency of F89 to that of F9, it appears that the two adjacent side chains of hexafluoroleucine at positions 8 and 9 have enough steric bulk to perturb the spatial arrangement of His7. Overall, analogues with a lower receptor affinity, by and large, exhibited a higher EC50 value with respect to activation of adenylyl cyclase.
Fig. 3.

cAMP production stimulated by GLP-1 and fluorinated analogues using a radioimmunoassay with [125I]-cAMP as the tracer. All values are normalized to the maximal cAMP level induced by GLP-1. (A) N-terminal analogues; (B) C-terminal analogues. Data represent five independent experiments in duplicate as mean ± s.e.m.
Proteolytic Stability
The major obstacle of using natural GLP-1 as a therapeutic is its rapid inactivation by the ubiquitous protease DPP IV (t1/2 ≈ 2 min, i.v. in humans).4 DPP IV has a high substrate specificity at positions proximal to the scissile amide bond.36 Especially, Pro and Ala are highly favored at the P1 position.36 Other amino acids at this position enhanced the stability of peptides, as reported in the cases of GLP-1 derivatives with the substitutions Gly8, Aib8, Ser8, Thr8, Leu8.37,38 However, other positions such as P1′ and P2′ are relatively less studied. We found that the substitution by hexafluoroleucine conferred GLP-1 DPP IV resistance not only at position 8 but also at positions 9 and 10 (Fig. 4). F8 and F89 completely resisted enzymatic hydrolysis as no fragments were detected after 24 h incubation with DPP IV (Figs. 4B and 4C). F8 was further incubated with DPP IV at a 10-fold higher concentration; no digested fragments were detected after 1 h. F9 and F10 exhibited 1.5-fold and 2.9-fold increased stability relative to GLP-1. RP–HPLC analysis showed the formation of only one other major peak, which was identified by ESI–MS as corresponding peptide fragment GLP-1(9-36) or fluorinated GLP-1(9-36). The kinetic data reported here for the fluorinated GLP-1 analogues could be correlated to the prolonged metabolic stability in vivo, which has been established by Deacon and coworkers.38 Green et al39 have reported that intraperitoneal injection of Val8-GLP-1 into (ob/ob) mice resulted in increased insulin levels and reduced plasma glucose presumably due to its protracted lifetime. Indeed, Val8-GLP-1 is 220-fold and 3.5-fold less potent than GLP-1 with respect to in vitro receptor binding and cAMP stimulation.40 McIntosh and colleagues41 have recently demonstrated that subcutaneous administration of complete DPP IV resistant Ser8(P)2-GLP-1 into Wistar rats resulted in a more pronounced reduction in the glycemic profile, although it was deemed not to be statistically significant. Therefore, F8, F9, and F10 have the potential as candidates for further animal glucose tolerance study (see Sup Info).
Fig. 4.
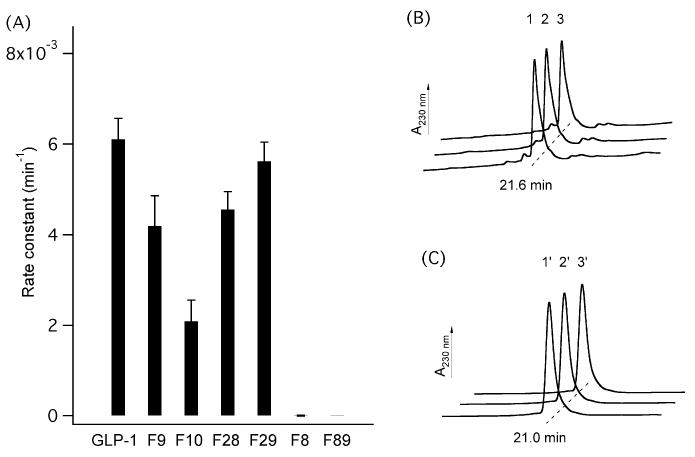
(A) Rate constants of peptide degradation by DPP IV. [Peptide] = 10 μM. [DPP IV] = 20 U/L. Error bars represent one standard deviation. (B) RP–HPLC traces of digestive mixtures of F8. Peaks 1, 2, and 3 represent F8 at 0, 48 h at [DPP IV] = 20 U/L, and 1 h at [DPP IV] = 200 U/L. (C) RP–HPLC traces of digestive mixtures of F89. Peaks 1′, 2′, and 3′ denote F89 at 0, 1, and 24 h at [DPP IV] = 20 U/L. No digestive products for either F8 or F89 by DPP IV were detectable. HPLC traces are offset along the time axis for clarity.
Conclusion and Outlook
In our continuing efforts to probe effects of fluorination on peptides/proteins, the results here are informative in that this is the first study of hexafluoroleucine containing hormonal ligand-receptor interaction investigated to date. Introduction of hexafluoroleucine into GLP-1 at strategic sites conferred resistance against its regulatory protease, DPP IV. Although the in vitro binding affinity and signal transduction activity decreased slightly, the all-important efficacy was retained in 6 out of 7 fluorinated analogues. Other interesting findings were that (1) the P2′ site had a significant impact on protease stability; (2) double substitutions at 8 and 9 positions turned GLP-1 into a partial agonist with retention of maximal binding ability; (3) F9 was similar to natural GLP-1 in every aspect, suggesting a large hydrophobic side chain is well tolerated at position 9, or that the CF3 group is able to make multipolar contacts in the binding interface. Considering the successful introduction of fluorine into small molecule drugs, incorporation of fluorinated amino acids into naturally bioactive peptides demands further studies. We are currently conducting further in vivo studies on fluorinated GLP-1s so that the integrated metabolic effects would be illuminated. We are also exploring suitable systems for further application of fluorinated amino acids in modulating biological functions.
Supplementary Material
Experimental procedures and analytical data. This material is available via the Internet at http://pubs.acs.org/.
Acknowledgments
We thank Dr. David Sanford (Tufts University School of Medicine) for help with OGTT experiments. This work was supported in part by the NIH (GM65500) and by the Massachusetts Technology Transfer Center. The ESI-MS facility at Tufts is supported by the NSF (0320783).
Abbreviations
- Boc
tert-butyloxycarbonyl
- CD
circular dichroism
- DPC
dodecylphosphocholine
- DPP IV
dipeptidyl peptidase IV
- GLP-1
glucagon-like peptide-1
- hGLP-1R
human glucagon-like peptide-1 receptor
- PMSF
phenylmethylsulfonyl fluoride
- TFE
trifluoroethanol
References
- 1.Yach D, Stuckler D, Brownell KD. Epidemiologic and economic consequences of the global epidemics of obesity and diabetes. Nat Med. 2006;12:62–66. doi: 10.1038/nm0106-62. [DOI] [PubMed] [Google Scholar]
- 2.Knudsen LB. Glucagon-like peptide-1: The basis of a new class of treatment for type 2 diabetes. J Med Chem. 2004;47:4128–4134. doi: 10.1021/jm030630m. [DOI] [PubMed] [Google Scholar]
- 3.Holst JJ, Deacon CF. Glucagon-like peptide 1 and inhibitors of dipeptidyl peptidase IV in the treatment of type 2 diabetes mellitus. Curr Opin Pharmacol. 2004;4:589–596. doi: 10.1016/j.coph.2004.08.005. [DOI] [PubMed] [Google Scholar]
- 4.Drucker DJ. Development of glucagon-like peptide-1-based pharmaceuticals as therapeutic agents for the treatment of diabetes. Curr Pharm Design. 2001;7:1399–1412. doi: 10.2174/1381612013397401. [DOI] [PubMed] [Google Scholar]
- 5.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet. 2002;359:824–830. doi: 10.1016/S0140-6736(02)07952-7. [DOI] [PubMed] [Google Scholar]
- 6.Deacon CF, Nauck MA, Toftnielsen M, Pridal L, et al. Both subcutaneously and intravenously administered Glucagon-Like Peptide-I are rapidly degraded from the NH2-terminus in type-II diabetic-patients and in healthy-subjects. Diabetes. 1995;44:1126–1131. doi: 10.2337/diab.44.9.1126. [DOI] [PubMed] [Google Scholar]
- 7.Knudsen LB, Nielsen PF, Huusfeldt PO, Johansen NL, et al. Potent derivatives of glucagon-like peptide-1 with pharmacokinetic properties suitable for once daily administration. J Med Chem. 2000;43:1664–1669. doi: 10.1021/jm9909645. [DOI] [PubMed] [Google Scholar]
- 8.Muller K, Faeh C, Diederich F. Fluorine in pharmaceuticals: Looking beyond intuition. Science. 2007;317:1881–1886. doi: 10.1126/science.1131943. [DOI] [PubMed] [Google Scholar]
- 9.Bohm HJ, Banner D, Bendels S, Kansy M, et al. Fluorine in medicinal chemistry. Chembiochem. 2004;5:637–643. doi: 10.1002/cbic.200301023. [DOI] [PubMed] [Google Scholar]
- 10.Kim D, Wang LP, Beconi M, Eiermann GJ, et al. (2R)-4-Oxo-4- 3-(trifluoromethyl)-5,6-dihydro 1,2,4 triazolo 4,3-alpha p yrazin-7(8H)-yl -1-(2,4,5-trifluorophenyl)butan-2-amine: A potent, orally active dipeptidyl peptidase IV inhibitor for the treatment of type 2 diabetes. J Med Chem. 2005;48:141–151. doi: 10.1021/jm0493156. [DOI] [PubMed] [Google Scholar]
- 11.Wong DT, Bymaster FP, Engleman EA. Prozac (Fluoxetine, Lilly-110140), the first selective serotonin uptake inhibitor and an antidepressant drug - 20 years since its first publication. Life Sci. 1995;57:411–441. doi: 10.1016/0024-3205(95)00209-o. [DOI] [PubMed] [Google Scholar]
- 12.Sato AK, Viswanathan M, Kent RB, Wood CR. Therapeutic peptides: technological advances driving peptides into development. Curr Opin Biotechnol. 2006;17:638–642. doi: 10.1016/j.copbio.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 13.Vine WH, Brueckne Da, Needlema P, Marshall GR. Synthesis, biological-activity, and F-19 Nuclear Magnetic Resonance spectral of angiotensin-II analogs containing fluorine. Biochemistry. 1973;12:1630–1637. doi: 10.1021/bi00732a026. [DOI] [PubMed] [Google Scholar]
- 14.Niemz A, Tirrell DA. Self-association and membrane-binding behavior of melittins containing trifluoroleucine. J Am Chem Soc. 2001;123:7407–7413. doi: 10.1021/ja004351p. [DOI] [PubMed] [Google Scholar]
- 15.Hsieh KH, Needleman P, Marshall GR. Long-acting angiotensin-II inhibitors containing hexafluorovaline in position-8. J Med Chem. 1987;30:1097–1100. doi: 10.1021/jm00389a021. [DOI] [PubMed] [Google Scholar]
- 16.Vine WH, Hsieh KH, Marshall GR. Synthesis of fluorine-containing peptides -Analogs of angiotensin-II containing hexafluorovaline. J Med Chem. 1981;24:1043–1047. doi: 10.1021/jm00141a005. [DOI] [PubMed] [Google Scholar]
- 17.Meng H, Kumar K. Antimicrobial activity and protease stability of peptides containing fluorinated amino acids. J Am Chem Soc. 2007;129:15615–15622. doi: 10.1021/ja075373f. [DOI] [PubMed] [Google Scholar]
- 18.Gottler LM, Lee HY, Shelburne CE, Ramamoorthy A, et al. Using fluorous amino acids to modulate the biological activity of an antimicrobial peptide. Chembiochem. 2008;9:370–373. doi: 10.1002/cbic.200700643. [DOI] [PubMed] [Google Scholar]
- 19.Gimenez D, Andreu C, del Olmo ML, Varea T, et al. The introduction of fluorine atoms or trifluoromethyl groups in short cationic peptides enhances their antimicrobial activity. Bioorg Med Chem. 2006;14:6971–6978. doi: 10.1016/j.bmc.2006.06.027. [DOI] [PubMed] [Google Scholar]
- 20.Dillon JS, Tanizawa Y, Wheeler MB, Leng XH, et al. Cloning and Functional Expression of the Human Glucagon-Like Peptide-1 (Glp-1) Receptor. Endocrinology. 1993;133:1907–1910. doi: 10.1210/endo.133.4.8404634. [DOI] [PubMed] [Google Scholar]
- 21.Tibaduiza EC, Chen C, Beinborn M. A small molecule ligand of the glucagon-like peptide 1 receptor targets its amino-terminal hormone binding domain. J Biol Chem. 2001;276:37787–37793. doi: 10.1074/jbc.M106692200. [DOI] [PubMed] [Google Scholar]
- 22.Flentke GR, Munoz E, Huber BT, Plaut AG, et al. Inhibition of dipeptidyl aminopeptidase-IV (DP-IV) by Xaa-Boropro dipeptides and use of these inhibitors to examine the role of DP-IV in T-Cell function. Proc Natl Acad Sci U S A. 1991;88:1556–1559. doi: 10.1073/pnas.88.4.1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lambeir AM, Proost P, Scharpe S, De Meester I. A kinetic study of glucagon-like peptide-1 and glucagon-like peptide-2 truncation by dipeptidyl peptidase IV, in vitro. Biochem Pharmacol. 2002;64:1753–1756. doi: 10.1016/s0006-2952(02)01415-6. [DOI] [PubMed] [Google Scholar]
- 24.Adelhorst K, Hedegaard BB, Knudsen LB, Kirk O. Structure-Activity Studies of Glucagon-Like Peptide-1. J Biol Chem. 1994;269:6275–6278. [PubMed] [Google Scholar]
- 25.Thornton K, Gorenstein DG. Structure of Glucagon-Like Peptide(7-36) Amide in a Dodecylphosphocholine Micelle as Determined by 2d NMR. Biochemistry. 1994;33:3532–3539. doi: 10.1021/bi00178a009. [DOI] [PubMed] [Google Scholar]
- 26.Goke R, Fehmann HC, Linn T, Schmidt H, et al. Exendin-4 Is a High Potency Agonist and Truncated Exendin-(9-39)-Amide an Antagonist at the Glucagon-Like Peptide 1-(7-36)-Amide Receptor of Insulin-Secreting Beta-Cells. J Biol Chem. 1993;268:19650–19655. [PubMed] [Google Scholar]
- 27.Lee KH, Lee HY, Slutsky MM, Anderson JT, et al. Fluorous effect in proteins: De novo design and characterization of a four-alpha-helix bundle protein containing hexafluoroleucine. Biochemistry. 2004;43:16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]
- 28.Hareter A, Hoffmann E, Bode HP, Goke B, et al. The positive charge of the imidazole side chain of histidine(7) is crucial for GLP-1 action. Endocr J. 1997;44:701–705. doi: 10.1507/endocrj.44.701. [DOI] [PubMed] [Google Scholar]
- 29.Chiu HP, Suzuki Y, Gullickson D, Ahmad R, et al. Helix propensity of highly fluorinated amino acids. J Am Chem Soc. 2006;128:15556–15557. doi: 10.1021/ja0640445. [DOI] [PubMed] [Google Scholar]
- 30.Al-Sabah S, Donnelly D. A model for receptor-peptide binding at the glucagon-like peptide-1 (GLP-1) receptor through the analysis of truncated ligands and receptors. Br J Pharmacol. 2003;140:339–346. doi: 10.1038/sj.bjp.0705453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beinborn M, Worrall CI, McBride EW, Kopin AS. A human glucagon-like peptide-1 receptor polymorphism results in reduced agonist responsiveness. Regul Pept. 2005;130:1–6. doi: 10.1016/j.regpep.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 32.Green BD, Mooney MH, Gault VA, Irwin N, et al. Lys(9) for Glu(9) substitution in glucagon-like peptide-1(7-36)amide confers dipeptidylpeptidase IV resistance with cellular and metabolic actions similar to those of established antagonists glucagon-like peptide-1(9-36)amide and exendin (9-39) Metab Clin Exp. 2004;53:252–259. doi: 10.1016/j.metabol.2003.09.015. [DOI] [PubMed] [Google Scholar]
- 33.Xiao Q, Giguere J, Parisien M, Jeng W, et al. Biological activities of glucagon-like peptide-1 analogues in vitro and in vivo. Biochemistry. 2001;40:2860–2869. doi: 10.1021/bi0014498. [DOI] [PubMed] [Google Scholar]
- 34.Biffinger JC, Kim HW, DiMagno SG. The polar hydrophobicity of fluorinated compounds. Chembiochem. 2004;5:622–627. doi: 10.1002/cbic.200300910. [DOI] [PubMed] [Google Scholar]
- 35.de Menthiere CS, Chavanieu A, Grassy G, Dalle S, et al. Structural requirements of the N-terminal region of GLP-1- 7-37 -NH2 for receptor interaction and cAMP production. Eur J Med Chem. 2004;39:473–480. doi: 10.1016/j.ejmech.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 36.Mentlein R. Dipeptidyl-peptidase IV (CD26)-role in the inactivation of regulatory peptides. Regul Pept. 1999;85:9–24. doi: 10.1016/s0167-0115(99)00089-0. [DOI] [PubMed] [Google Scholar]
- 37.Ritzel U, Leonhardt U, Ottleben M, Ruhmann A, et al. A synthetic glucagon-like peptide-1 analog with improved plasma stability. J Endocrinol. 1998;159:93–102. doi: 10.1677/joe.0.1590093. [DOI] [PubMed] [Google Scholar]
- 38.Deacon CF, Knudsen LB, Madsen K, Wiberg FC, et al. Dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1 which have extended metabolic stability and improved biological activity. Diabetologia. 1998;41:271–278. doi: 10.1007/s001250050903. [DOI] [PubMed] [Google Scholar]
- 39.Green BD, Lavery KS, Irwin N, O'Harte FPM, et al. Novel glucagon-like peptide-1 (GLP-1) analog (Val(8))GLP-1 results in significant improvements of glucose tolerance and pancreatic beta-cell function after 3-week daily administration in obese diabetic (ob/ob) mice. J Pharmacol Exp Ther. 2006;318:914–921. doi: 10.1124/jpet.105.097824. [DOI] [PubMed] [Google Scholar]
- 40.Green BD, Gault VA, Mooney MH, Irwin N, et al. Novel dipeptidyl peptidase IV resistant analogues of glucagon-like peptide-1(7-36)amide have preserved biological activities in vitro conferring improved glucose-lowering action in vivo. J Mol Endocrinol. 2003;31:529–540. doi: 10.1677/jme.0.0310529. [DOI] [PubMed] [Google Scholar]
- 41.Hinke SA, Manhartll S, Kuhn-Wache K, Nian C, et al. Ser(2) - and Ser(P)(2) incretin analogs comparison of dipeptidyl peptidase IV resistance and biological activities in vitro and in vivo. J Biol Chem. 2004;279:3998–4006. doi: 10.1074/jbc.M311304200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental procedures and analytical data. This material is available via the Internet at http://pubs.acs.org/.