

Abstract
Salmonella typhimurium AhpC is a founding member of the peroxiredoxin family, a ubiquitous group of cysteine-based peroxidases with high reactivity toward hydrogen peroxide, organic hydroperoxides and peroxynitrite. For all of the peroxiredoxins, the catalytic cysteine, referred to as the peroxidatic cysteine (CP), acts as a nucleophile in attacking the peroxide substrate, forming a cysteine sulfenic acid at the active site. Because thiolates are far stronger nucleophiles than thiol groups, it is generally accepted that cysteine-based peroxidases should exhibit pKa values lower than an unperturbed value of 8.3 – 8.5. In this investigation, several independent approaches were used to assess the pKa of the two cysteinyl residues of AhpC. Methods using two different iodoacetamide derivatives yielded unperturbed pKa values (7.9 – 8.7) for both cysteines, apparently due to reactivity with the “wrong” conformation of CP (i.e. locally unfolded and flipped out of the active site), as supported by X-ray crystallographic analyses. A functional pKa of 5.94 ± 0.10 presumably reflecting titration of CP within the fully folded active site was obtained by measuring AhpC competition with horseradish peroxidase for hydrogen peroxide; this value is quite similar to that obtained by analyzing the pH dependence of the ε240 of wild-type AhpC (5.84 ± 0.02), and similar to those obtained for two typical 2-cysteine peroxiredoxins from Saccharomyces cerevisiae (5.4 and 6.0). Thus, the pKa value of AhpC balances the need for a deprotonated thiol (at pH 7, ∼90% of the CP would be deprotonated) with the fact that thiolates with higher pKa values are stronger nucleophiles.
Keywords: peroxiredoxins, alkyl hydroperoxide reductase, peroxidases, disulfide redox centers, antioxidants, thiolate
Salmonella typhimurium AhpC (StAhpC)1 is a member of the peroxiredoxin (Prx) family, a ubiquitous group of cysteine based peroxidases with high reactivity toward H2O2 [Km for H2O2 of 1.4 μM; kcat/Km of 4 x 107 M-1 s-1 for StAhpC] (1). For all of the Prxs, the catalytic cysteine, referred to as the peroxidatic cysteine (CP) acts as a nucleophile to attack and reduce the peroxide substrate, and in doing so becomes oxidized to form a cysteine sulfenic acid. The fate of the sulfenic acid differs in various Prx enzymes, forming a disulfide bond either with another cysteine in the Prx, or with an external thiol in redox donors such as thioredoxin, glutaredoxin, or reduced glutathione (2-Cys and 1-Cys Prx mechanisms, respectively). The typical 2-Cys Prxs, which form an intersubunit disulfide bond during turnover, include StAhpC and are the most widely distributed and abundant type of Prxs. In mammalian cells they are on the order of 1% of the soluble protein (2, 3). AhpC in Escherichia coli has been shown to have a primary role as a peroxide scavenger (4), but since the discovery that many eukaryotic Prxs have an evolutionarily selected sensitivity to inactivation by peroxide (5), evidence has been accumulating supporting the hypothesis that the primary role of these enzymes in higher organisms is to regulate peroxide signaling (2, 6, 7).
Crystal structures of StAhpC have shown that the cysteines residues are oriented in very different ways depending upon the redox state of the protein. The structure of StAhpC with the CP mutated to Ser mimics the reduced conformation and represents a fully folded structure with the thiol(ate) form of the CP (C46) present in the first turn of an α-helix protruding into a highly conserved active site pocket, interacting with conserved Arg and Thr residues that likely serve to enhance its nucleophilicity (5). The CR (C165) is in the C-terminal end of the protein and is ∼13 Å apart from and pointing away from the CP. In contrast, the structure of the disulfide-bonded protein exhibits a local unfolding of the α-helix containing the CP, flipping the thiol group out of the active site pocket (8). The CR is also reoriented in this structure and the C-terminus beyond C165 (21 residues) is disordered. Analysis of the subunit composition of StAhpC by analytical ultracentrifugation has shown that there is an intimate link between redox state and oligomeric arrangement for StAhpC; the reduced protein is a strong decamer in solution while the oxidized protein tends to dissociate into dimers (8). Recent studies have shown that the decameric arrangement of StAhpC enhances the peroxide reductase activity of StAhpC, presumably through the stabilization of the fully folded active site by the dimer-dimer interface (1). If the local unfolding is unfavorable, as is the case for some eukaryotic enzymes, then the enzyme is highly sensitive to inactivation via further oxidation of CP by a second molecule of peroxide, leading to formation of a sulfinic acid (CP-SO2H). While StAhpC is resistant to inactivation, this pathway appears to be important for peroxide signaling in eukaryotes (5, 9).
It is generally accepted that, because thiolates are far stronger nucleophiles than thiol groups, cysteine-based peroxidases should exhibit pKa values that are significantly lower than the value of 8.3 – 8.5 observed for free cysteine to promote thiolate formation at the active site (10). There is a trade-off with lowering the cysteine pKa, however, in that the intrinsic nucleophilicity of a thiolate is lowered as the pKa is decreased (11, 12). In spite of the importance of this property for chemical reactivity of Prxs, pKa values for the CP in Prxs have been assessed in only a very few cases. The reported values range from pH 6.0 and 5.4 for yeast Tsa1 and Tsa2 (also known as cTPx1 and cTPx2) (13) to <5 reported for StAhpC (14), although a rigorous experimental determination of the pKa for the active site cysteine has not been carried out for StAhpC or most other Prxs. Here we present data from multiple different approaches used to assess the pKa of the cysteinyl residues of StAhpC. The pH dependence of alkylation rates using two different iodoacetamide derivatives yielded unperturbed pKa values between 7.9 and 8.7 for both cysteines, apparently due to reactivity with the “wrong” conformation of CP (i.e. flipped out of the active site), as supported by crystallographic analyses. Two other independent methods yielded perturbed pKa values of about 5.8 and 5.9 for C46 of wild type AhpC. The latter is, in fact, a functional pKa based on peroxide reactivity, supporting the interpretation that this measured pKa corresponds to the cysteine thiol(ate) within the active site.
Experimental Procedures
Materials
Horseradish peroxidase, type VI (HRP), NADH, iodoacetamide, trichloroacetic acid, calcium chloride, ammonium sulfate, sodium citrate, boric acid, and sodium phosphate were purchased from Sigma. 5-Iodoacetamidofluorescein was purchased from Molecular Probes (Invitrogen). TPCK-treated trypsin was from Worthington Biochemicals, and 1,4-dithiothreitol (DTT) was from Anatrace. Diethylenetriamine pentaacetic acid (DTPA) and 2,5-dihydroxybenzoic acid were purchased from Acros Organics. Guanidine hydrochloride (GuHCl) was from Lancaster Synthesis (Ward Hill, MA). Ethylenediamine tetraacetic acid (EDTA), 2-mercaptoethanol, ammonium bicarbonate, and 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) were purchased from Research Organics. Sodium citrate, HCl, acetonitrile, and hydrogen peroxide were from Fisher, and PD-10 desalting columns were from GE Healthcare. Deuterated (d5) and protiated (d0) N-phenyl iodoacetamide (iodoacetanilide, abbreviated d5-IAAn and d0-IAAn, respectively) were synthesized as described previously (15).
Methods
Protein Expression and Purification
Wild type Salmonella typhimurium AhpC and two mutant forms, C46S and C165S, were expressed in a non-His-tagged form from a vector, pTHCm-ahpC, derived from Invitrogen's pTrcHisA in which the beta-lactamase gene was replaced by a chloramphenicol resistance gene. E. coli strain TA4315 (16) was used for expression of all AhpC proteins. The purification procedure for all proteins was essentially the same as described previously (17, 18); DTT was maintained at 5 mM in all buffers used during the purification of C165S in order to prevent hyperoxidation of the CP. AhpC concentrations (in terms of monomers) were determined by absorbance at 280 nm with ε = 24,300 M-1 cm-1 (17).
Stability of AhpC Toward Denaturation By GuHCl or Prolonged Incubation in Various pH Buffers
AhpC (20 μg) in 1.5 mL of buffer containing 10 mM sodium phosphate, 10 mM boric acid, 10 mM sodium citrate, 1 mM EDTA, and 100 mM ammonium sulfate (pH 7.0) including varying concentrations of GuHCl (0-6 M) at 25 °C was incubated for 2 h followed by measurement of the intrinsic tryptophan fluorescence at λex = 280 nm and λem = 350 nm using an Aminco-Bowman Series 2 luminescence spectrofluorometer. The photomultiplier tube voltage was set so that a fresh sample without GuHCl gave a fluorescence intensity that was 50% of the maximal fluorescence value.
To assess structural stability of the native protein in various pH buffers, AhpC (20 μg) was incubated at 23 °C for 24 h in a final volume of 1.5 mL at various pH values from 3 to 10 in a buffer containing 10 mM sodium phosphate, 10 mM sodium citrate, 10 mM boric acid, 1 mM EDTA, and 100 mM ammonium sulfate (BPACE buffer), with the pH adjusted with either ammonium hydroxide or sulfuric acid. The intrinsic tryptophan fluorescence for each sample was measured as described above. After recording the data at each pH, the fluorescence intensity for each sample was measured under denaturing conditions by the addition of 10 μL concentrated HCl.
Analysis of pKa Using pH Dependence of 240 nm Absorbance
AhpC (wild type and mutants) was reduced with 10 mM DTT for 10 min at room temperature, then separated from the DTT using a PD-10 size exclusion column. The absorbance of 3 to 10 μM protein was measured in 1X BPACE buffer on an Agilent HP8453 diode array spectrophotometer at a variety of pH values. The amount of protein in solution was determined spectrophotometrically at 280 nm as described above and used to calculate the ε240 at each pH. This value was plotted against pH and the pKa was determined by direct fit to equation 1,
| (1) |
where y = ε 240, A = the upper plateau at high pH (ε 240 for the deprotonated form), and B = the lower plateau at low pH (ε240 for the protonated form).
Determination of Cysteine pKa By Reactivity With Fluorescein Iodoacetamide Across a Range of pH Values
Wild type AhpC was reduced and reisolated from the DTT as described above. The protein was diluted to a final concentration of 0.5 mg/mL (24.3 μM) in 1X BPACE buffer at various pH values and incubated with 180 μM fluorescein iodoacetamide; after various incubation times, 20 μL aliquots were removed and quenched with 10 μL of 600 mM 2-mercaptoethanol. Samples were then separated by 12% SDS-PAGE, and the fluorescence associated with the protein and with the dye front (unreacted reagent) was measured on a STORM 840 fluorescence imager. Fluorescence intensity was determined using ImageQuant 5.2 gel analysis software. The percent of fluorescence in the protein fraction was plotted against time and fit to a single exponential equation to determine the kobs. The kobs at each pH was then plotted against pH and the pKa was determined by direct fit to equation 1, where y = kobs, A = the upper plateau at high pH (the rate of reaction of the deprotonated form), and B = the lower plateau at low pH (the rate of reaction of the protonated form).
Determination of Cysteine pKa By Reactivity With Iodoacetanilide (IAAn) Across a Range of pH Values Using Isotope Coded Reagents and MALDI-TOF Mass Spectrometry Analyses
As an internal standard for quantitation, reduced AhpC was incubated with a 5-fold excess of d5-IAAn at 24 °C for 21 h. After confirming complete reaction by MALDI-TOF MS, the resulting protein was exchanged into 25 mM ammonium bicarbonate at pH 8.5 and stored at -20 °C until needed.
AhpC (0.5 mM) was reduced with 10 mM DTT for 10-15 min, then reisolated using a PD10 column. The reaction mixture was prepared with a final concentration of 40 μM AhpC in a total volume of 1.055 mL using 1X BPACE buffer at the desired pH. The reaction was initiated by the addition of 400 μM (final concentration) d0-IAAn, and aliquots of 2.5 nmol AhpC were removed at appropriate time points and quenched with 500 mM 2-mercaptoethanol. A standard amount of AhpC labeled with d5-IAAn (1.25 nmol) was added to each sample prior to precipitation on ice with cold trichloroacetic acid at 10% final concentration. After centrifugation, the protein pellet was washed with 0.5 mL of 1:1 ether:ethanol, recentrifuged and resuspended in 40 mM ammonium bicarbonate, 1 mM CaCl2, and 10% acetonitrile at pH 8.5 and digested overnight at 37 °C with 2.5 ng (50 ng/mL) trypsin. The two cysteines of AhpC are located on different tryptic peptides; C46 was monitored at 3836 or 3841 Da (representing the d0- and d5-labeled peptides, respectively; WSVFFFYPADFTFVCPTELGDVADHYEELQK), and C165 was monitored at 1745 or 1750 Da (representing the d0- and d5-labeled peptides, respectively; AAQYVAAHPGEVCPAK) on a Brüker Autoflex MALDI-TOF mass spectrometer using dihydroxybenzoic acid as the matrix. Data were collected three times for every sample using the Autorun feature on the instrument. Peak intensities were used to determine the ratio of light to heavy peptides, and the ratios were plotted against time and fit to a single exponential equation to determine kobs. The actual pH of each reaction mixture was measured for the remaining sample. The kobs at each pH was plotted against pH and the pKa was determined by direct fit to equation 1.
Determination of Reaction Rates of AhpC With Iodoacetamide, Fluorescein Iodoacetamide and IAAn at pH 7
Wild type AhpC was reduced and exchanged into BPACE pH 7.0 as described above. AhpC was diluted to 40 μM and the reaction was initiated by the addition of iodoacetamide, fluorescein iodoacetamide or IAAn. For fluorescein iodoacetamide modification, the reaction progress was monitored by fluorescence after SDS-PAGE as described above. To measure reaction progress with iodoacetamide or IAAn, an aliquot of each sample at various times was quenched by addition of 100 mM DTT, then applied to a Bio-Gel P6 spin column to remove small molecules and exchange the protein into 25 mM potassium phosphate buffer with 1 mM EDTA, pH 7.2. The loss of free thiol groups was monitored by addition of 0.25 mM DTNB (final concentration) to a cuvette containing 170 μL of the spin column flowthrough and 360 μL of buffer, and measurement of the 412 nm absorbance. The concentration of free thiols was then determined based on the release of 2-nitro-5-thiobenzoate (ε412 = 14,150 M-1 cm-1) (19). Protein concentration was determined by measurement of the 280 nm absorbance before DTNB addition.
Crystal Structure Determination of the IAAn Adduct of C165S AhpC
Purified C165S AhpC was separated from the DTT in the storage buffer using a PD10 column, then incubated with a 4-fold excess of d0-IAAn for 17 h at 4 °C in 25 mM phosphate, 1 mM EDTA, pH 7.0. MALDI-TOF MS analysis confirmed that the protein was completely alkylated. The protein was then exchanged into 25 mM phosphate, 1 mM EDTA (pH 7) using a G25 size exclusion column to remove excess reagent and concentrated to 14.6 mg/mL using 20 kDa cutoff Apollo ultrafiltration devices (Orbital Biosciences, Topsfield, MA).
Crystals of the S-acetanilide modified form of C165S (AhpC-C46AAn) were grown at 4 °C in hanging drops using a 0.4 mL reservoir solution with drops containing a 2:1 ratio of protein stock solution to reservoir solution. Commercial Wizard II screen condition #45 [Emerald Biosystems, 1.26 M (NH4)2SO4, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) pH 6.0] produced the best crystals. The rectangular prism crystals appeared after two weeks, growing out of a precipitate to a final size of 0.25 x 0.25 x 0.1 mm3. Crystals were harvested into 1.2 M (NH4)2SO4, 0.1 M MES, pH 6.0, and for data collection, crystals were placed in the same buffer plus 25% glycerol for 1 min, mounted in loops and flash frozen in liquid nitrogen.
Oscillation data were collected in house using Cu-Kα radiation, an R-axis IV detector and Δφ = 1.0°. The crystals belong to trigonal space group P3121 with one half-decamer in the asymmetric unit and unit cell dimensions a = b = 136.91 Å, c = 145.42 Å. AhpC-C46AAn data were merged from two isomorphous crystals, one with 60 images (20 min/image) and the other with 90 images (30 min/image). Useable data extended to 4.0 Å resolution (Table 1).
Table 1.
Data Collection and Refinement Statistics for C165S AhpC Alkylated With Iodoacetanilide (AhpC-C46AAn).
| Data Collection1 | |
| Resolution (Å) | 100 - 4.0 (4.14 – 4.0) |
| No. of unique observations | 13503 |
| Multiplicity | 8.8 (8.9) |
| Completeness (%) | 98.4 |
| I/σ | 12.2 (5.3) |
| Mosacity | 0.87 |
| Rmeas2 (%) | 14.3 (51.9) |
| Rmrdg-F2 (%) | 12.7 (38.5) |
| Refinement | |
| Fully-Folded Rfactor / Rfree | 0.366 / 0.371 |
| Locally Unfolded Rfactor / Rfree | 0.378 / 0.3903 |
Numbers in parentheses correspond to the highest resolution bin.
Rmeas is the multiplicity weighted merging R-factor and Rmrgd-F is an indicator of the quality of reduced data (42).
Final rigid body refinement of the locally unfolded model after manual rebuilding to minimize clases gives an Rfactor / Rfree of 0.301/0.308.
All crystallographic calculations were performed using ccp4 (20) version 5.99.5 and model viewing was done using Coot (21) version 0.2. The structure of AhpC-C46AAn was determined by molecular replacement using AmoRe (22). Two search models were used, an AhpC structure with a fully folded active site (PDB entry 1N8J) and an AhpC structure with a locally unfolded active site (the T77V mutant of AhpC, PDB entry 1YF1). For the search models, only the protein atoms for one half-decamer (chains A-E) were used. For the fully folded model, all chains were truncated after residue 166; for the locally unfolded model no chains extended beyond residue 165 so no truncation was needed. Five percent of the data were randomly selected for cross-validation. Using data from 15 to 4 Å resolution, both search models gave a unique solution that packed well in the unit cell. After rigid body refinement, no additional refinement was carried out before selection of the correct model due to the low resolution of the data. Electron density maps were calculated from both the fully folded and locally unfolded models. Omit maps were also generated from both models by removing residues 41 through 48 from each subunit prior to rigid body refinement. Non-crystallographic symmetry averaging was performed using DM (23) to improve the signal to noise ratio for each of the four maps. The locally unfolded model was further refined as a rigid body after removing significant clashes in the model by truncating all chains at residue 162 and using the “rigid body fit zone” option of Coot (18) to shift segments 28-31, 56-64, 125-129, 136-140, and 148-162 of chain A. The final model had R/Rfree=0.301/0.308 and has been deposited in the Protein Data Bank with pdb code 3EMP.
Determination of Functional pKa By pH-Dependence of Competition With HRP For Hydrogen Peroxide
The pH dependence of the AhpC reaction with H2O2 was determined by monitoring the ability of AhpC to compete with HRP based on the method of Ogusucu et al. (13). Briefly, HRP was dissolved in 5 mM potassium phosphate buffer with 0.1 mM DTPA, pH 7.0, and the concentration of the HRP stock was determined by measuring the absorbance at 403 nm (ε 403 = 1.02 × 105 M-1 cm-1) (24, 25). Wild-type AhpC was reduced with 10 mM DTT for 1 h at room temperature, then separated from the DTT using a PD-10 column and exchanged into 5 mM phosphate buffer containing 0.1 mM DTPA at pH 7.0. Stock solutions containing HRP and AhpC in the same buffer (75 μL) were aliquoted into a 96 well microplate, then mixed with 1 volume of buffer at various pH values between 4 and 9.5 to give a
| (2) |
final concentration of 10 mM phosphate, 10 mM boric acid, 10 mM sodium citrate, 100 mM sodium chloride, 0.1 mM DTPA, 7.5 μM HRP, and 0, 2, 4, 8, 12, or 16 μM AhpC in a final volume of 150 μL. The starting HRP absorbance for each sample was determined at 403 nm in a Tecan Safire 2 microplate reader. The reaction was started by the addition of 10 μL of 45 μM H2O2 in deionized H2O (final concentration of H2O2 = 3 μM). The extent of HRP oxidation by H2O2 was determined by monitoring the change in absorbance at 403 nm within 90 s of peroxide addition. The actual pH was determined by making a 1:1 dilution of each pH buffer into 5 mM phosphate containing 0.1 mM DTPA, mimicking the reaction mixtures. The percentage of inhibition of HRP oxidation (F/1–F) at each AhpC concentration (see Supplemental Information for details) was plotted against [AhpC] in order to obtain the second-order rate constant for AhpC (kAhpC) as established by equation 2 (26). Above pH 5, a second order rate constant of 1.8 × 107 M-1 s-1 (24) for kHRP was used. Below pH 5, the second order rate constant for HRP was calculated for each pH using equation 3 and the ionization constants determined in the previous study (24) (K1 = 5.64 × 10-4 and K2 = 1.29 × 10-4):
| (3) |
The kAhpC at each pH was plotted versus pH and the pKa was calculated by direct fit to equation 1, where y = kAhpC.
Results
Stability of StAhpC After Prolonged Incubation in Buffers at Various pH Values
Changes in protein conformation often result in alterations in the fluorescence properties of a protein due to changes in the local environment of the tryptophan residues. AhpC has three tryptophan residues (W32, W81 and W169), with one, W81, located near C46. While no difference can be observed in the fluorescence properties of reduced and oxidized AhpC, protein denaturation in the presence of 3 M GuHCl or 80 mM HCl results in a 70% decrease in the fluorescence intensity of AhpC with excitation at 280 nm and emission at 350 nm (see Supplemental Information, Figure S1). Because this serves as a sensitive probe of the folding status of this protein, AhpC fluorescence was measured after 24 h incubations in buffers between pH 3 and 11. No significant change was observed in the fluorescence intensity of either reduced or oxidized AhpC between pH 4.5 and 10, indicating that both are stable across the pH range required for pKa determinations. In further experiments, AhpC fluorescence did not change over 20-30 min in buffers at pH values down to 4.14, allowing relatively short experiments to be conducted without stability problems within this limited range.
Determination of Cysteine pKa By Measuring Absorbance at 240 nm Across a Range of pH Values
Absorbance at 240 nm has been used previously to monitor the protonation state of cysteine residues in proteins (27-29). Given the stability results described above, the ε240 was determined for both oxidized and reduced wild-type AhpC at a variety of pH values between 4.5 and 9.5. While the oxidized protein did not exhibit a significant pH-dependent change in ε240, the reduced protein exhibited a single apparent pKa value of 5.84 ± 0.02 (Figure 1a). The difference in ε240 between reduced AhpC at high pH and the oxidized protein was ∼9,700 M–1 cm–1. If a standard range of ε240 for a single thiol group of 4,000 – 6,000 M–1 cm–1 (27) were used, this would suggest that two cysteine residues with similar pKa values were undergoing changes in protonation state during the course of this titration. This interpretation is not clearcut, however, since thiolate extinction coefficients may be quite variable, with reported values as low 2,300 and as high (at 229 nm) as 7,500 M–1 cm–1 (30, 31). Furthermore, the presence of nearby aromatic residues may alter the spectral signature of this ionization. Given data presented below, we can be relatively confident that C46 is centrally involved in the extinction coefficient change observed at 240 nm; whether or not C165 also titrates through this pH range with a very similar pKa is not clear.
FIGURE 1. Monitoring cysteine thiolate absorption of wild type AhpC at 240 nm over a pH range yields a single pKa value of 5.84 ± 0.02.

(a) The A240 and A280 values for oxidized (o) and reduced (●) AhpC (3-10 μM) were measured over a range of pH values and converted to ε240 assuming an ε280 value of 24,300 M-1 cm-1. The pKa was determined from the ε240 versus pH plot by direct fit to Equation 1 as described in Methods. (b) As described for panel a, the pH-dependent change in absorbance was measured for C46S (o), C165S (●) and the C46S, C165S double mutant of AhpC (Δ). These data were not able to be fit to equation 1.
In order to distinguish the pKa values for each individual cysteine and provide for an additional control titration in an AhpC lacking both cysteine residues, the experiment was repeated using the single cysteine mutants, C165S and C46S, and the double cysteine mutant C46S/C165S. Not surprisingly, the C165S/C46S mutant did not exhibit a significant pH-dependent change in the ε240 (Figure 1b). While there were some pH-dependent changes observed in ε240 for both the C165S and C46S mutants (Figure 1b), these changes were neither large enough nor in the expected direction to permit an evaluation of the cysteine pKa values, nor did they help in the interpretation of the pKa value obtained for the titration of the wild type protein. These complexities suggested that this type of analysis for AhpC may be confounded by additional conformational changes occurring during the pH titration, at least in the case of the mutant proteins.
Determination of Apparent Cysteine pKa By Monitoring Reactivity With Iodoacetamide Derivatives Across a Range of pH Values
Although a reasonable pKa value was obtained for the wild type protein by monitoring the ε240, the single apparent pKa value obtained for the two cysteine residues in wild type AhpC and the inconsistencies with the mutant proteins made it imperative to evaluate the pKa values using alternative methods. Nuclear magnetic resonance approaches, which in favorable cases also allow monitoring of the protonation/deprotonation behavior of cysteine residues in their native environment across a pH range, did not turn out to work for this protein. Even when 2-13C-cysteine was incorporated, neither cysteine signal was readily distinguished by 13C NMR; this is not surprising since the decameric form of AhpC is very large and would result in a significant broadening of peaks due to slow tumbling of the protein in solution (K. J. Nelson, D. A. Horita and L. B. Poole, unpublished results).
Because the protonated form of cysteine is unreactive with iodoacetamide, the increase in reaction rate with this reagent as pH increases reflects titration of the target cysteine residue (in the absence of changes in environment and/or accessibility of the target cysteine residue), and this technique has frequently been used to determine cysteine pKa values (32-35). In order to obtain the pKa value for both the peroxidatic (C46) and resolving (C165) cysteines, fluorescein iodoacetamide was incubated with either the C165S or C46S mutant of AhpC, and the rate of alkylation was determined by measuring the fluorescence intensity associated with the protein band after SDS-PAGE (Fig. 2a). Using this technique, the pKa values for C46 and C165 were 7.87 ± 0.06 and 8.64 ± 0.05, respectively. Both of these values are much higher than the value determined for wild type AhpC by measuring the pH dependence of ε240 and are approaching the pKa value of 8.44 measured for free cysteine (10), suggesting that fluorescein iodoacetamide might be reacting with both cysteine residues of AhpC in the locally unfolded rather than fully folded conformation.
FIGURE 2.

pKa determination using two different iodoacetamide-based compounds and single cysteine mutants of AhpC provide pKa values for C46 and C165 close to 8.5. (a) Reduced C165S (—●—) or C46S (- -○- -) AhpC (24 μM) in various pH buffers was incubated with 180 μM 5-iodoacetamidofluorescein for various amounts of time, quenched with excess 2-mercaptoethanol, then analyzed on a 12% SDS-polyacrylamide gel to determine the % fluorescence in the protein fraction; the data were fit to a single exponential equation to determine kobs at each pH. Data plotted as kobs versus pH were fit to equation 1 as described in Methods. (b) Reduced wild-type AhpC (40 μM) in various pH buffers was incubated with 400 μM d0-IAAn over a time course, and aliquots were quenched with excess 2-mercaptoethanol. A standard amount of AhpC labeled with d5-IAAn was added to each sample, and the protein was digested with trypsin overnight as described in Experimental Procedures. The extent of alkylation of C46 (—●—) and C165 (- -○- -) was determined by measuring the ratio of the peak intensities at 3836/3841 or 1745/1750 Da, respectively. The ratios were fit to a single exponential equation to determine the kobs and used to calculate the pKa of each residue as in panel (a).
A new isotope-coded method was recently developed in our laboratory to determine cysteine pKa values by monitoring the pH dependence of their reactivity with IAAn (15). The rate of reaction with the protiated version of this reagent (d0-IAAn) was determined using mass spectrometry to compare the intensity of the alkylated tryptic peptide to a standard amount of the same peptide labeled with the deuterated version of the reagent (d5-IAAn). This method was tested with E. coli thioredoxin and gave pKa values very similar to those previously published for chemical modification studies with this protein (15). This technique can be used to study wild type AhpC directly since both cysteine residues in this protein are on different tryptic peptides. As cumene hydroperoxide is similar in size and a reasonably good substrate for AhpC (36), IAAn, which also possesses a phenyl group, could be expected to access the active site pocket as a substrate analogue during modification of the protein. Using this technique, the pKa values obtained for C46 and C165 were similar to those obtained with fluorescein iodoacetamide, however, at 8.55 ± 0.21 and 8.74 ± 0.19, respectively (Figure 2b).
We previously showed that IAAn is about 3-fold more reactive with free cysteine than is iodoacetamide (15). Interestingly, AhpC reacted 8.9-fold more rapidly with IAAn than with iodoacetamide at pH 7, suggestive of an activating effect from the hydrophobic nature of the IAAn (Table 2). The reaction rates of fluorescein iodoacetamide, IAAn, and the smallest of the reagents, iodoacetamide, are relatively slow compared to turnover rates of about 3100 min-1 and 2400 min-1 with hydrogen peroxide and cumene hydroperoxide, respectively, at 400 μM. In addition, the reactions with both iodoacetamide and IAAn were slower than with fluorescein iodoacetamide, the bulkiest derivative which was expected to have difficulties accessing C46 within the fully folded active site of AhpC. These data along with the fact that the pKa values were so similar to free cysteine suggested that the peroxidatic cysteine was partially or totally inaccessible within the fully folded protein and that the predominant reaction of these alkylating agents was with the portion of the AhpC population that was in the locally unfolded conformation.
Table 2.
Reaction Rates of StAhpC With Various Iodoacetamide Derivatives and Hydroperoxides at pH7.
| Reaction rate with AhpC (min-1)a | |
|---|---|
| Fluorescein iodoacetamide | 0.67 |
| Iodoacetanilide | 0.031 |
| Iodoacetamide | 0.0035 |
| Hydrogen peroxide | 3130b |
| Cumene hydroperoxide | 2460b |
Prereduced AhpC (40 μM) was incubated in pH 7 buffer with 400 μM iodoacetamide, iodoacetanilide (IAAn), or fluorescein iodoacetamide. Rates of reaction with iodoacetamide and IAAn were determined by a subsequent thiol assay conducted at multiple time points, whereas modification by fluorescein iodoacetamide was assessed using a gel method to detect fluorescently-labeled protein as described in Methods.
X-Ray Crystal Structure of the IAAn-Generated Adduct of AhpC Confirms a Locally Unfolded Active Site
To determine the conformation of the alkylated AhpC product (AhpC-C46AAn), the crystal structure was obtained for C165S AhpC that had been fully modified with IAAn. Since the usable data only extended to 4.0 Å resolution, search models were used of both a fully folded and a locally unfolded active site in order to compare and control for possible model bias in the electron density maps. For the fully folded structure, the extra step was taken to remove the C-terminal residues from the search model so that any electron density showing up for the ordered C-terminal helix would provide unbiased and strong proof of both its presence and a fully folded active site. Finally, for model selection, model bias was minimized by only carrying out rigid body refinement, and the close match between R and Rfree values (Table 1) confirms that minimal overfitting has occurred.
The omit maps generated with the fully folded and locally unfolded models show similar electron density in the alkylated AhpC active site and both omit maps clearly follow the path of the locally unfolded model (Figures 3b and 3d). Even if omit maps are not used, the electron density still strongly follows the path of the locally unfolded conformation (Figures 3a and 3c); however, in the map generated using the fully folded model, a slight bias can be seen as the electron density is somewhat stronger along the path of the fully folded chain (Figure 3a). Thus, the active site density is strongly indicative of the structure of AhpC-C46AAn adopting the locally unfolded conformation. Furthermore, independent confirmation that the structure is locally unfolded is provided by the lack of electron density for the C-terminal helix in any of the maps. These results support our hypothesis that these bulky reagents are excluded from the active site and react with the CP only in its locally unfolded conformation.
FIGURE 3.
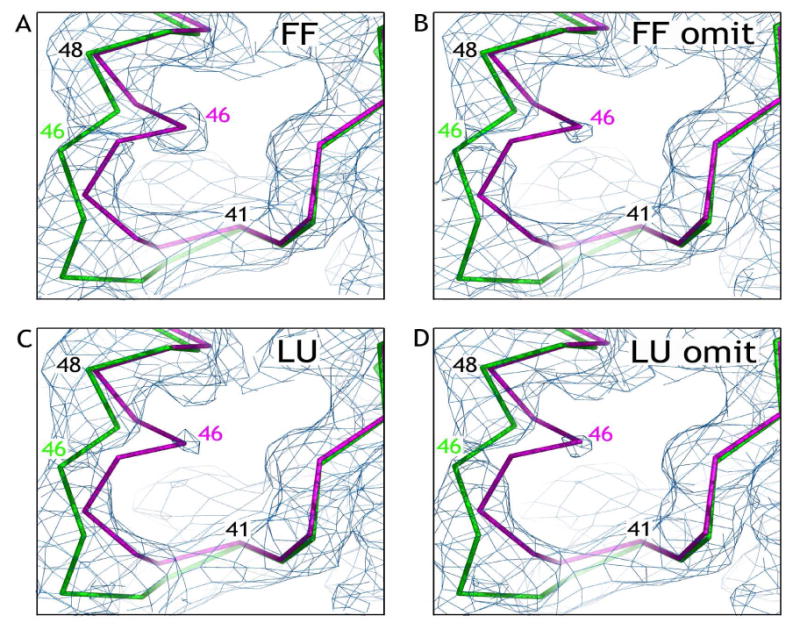
Crystal structure of C165S AhpC adduct with iodoacetanilide shows that the protein is in the locally unfolded conformation. In each panel, the Cα chain of residues 38 through 50 from the fully folded (FF) model (magenta) and the locally unfolded (LU) model (green) are shown along with 2Fo-Fc electron density contoured at 0.7·ρrms. (a and c) Electron density calculated using the FF or LU model, respectively; (b and d) Electron density calculated using the FF or LU model, respectively, with residues 41 through 48 omitted. Figures were prepared using Pymol.
Determination of a Functional pKa For AhpC By Monitoring the pH Dependence of Competition With HRP For Hydrogen Peroxide
In order to directly target AhpC in the fully folded conformation which represents the peroxide-reactive structure, we turned to a technique by which we could monitor the pH dependence of the reaction of AhpC with hydrogen peroxide. The typical activity assay, which includes AhpF or a truncated form of this electron-donating protein (1, 17, 37), was not used due to potential complications resulting from pH-dependent changes in that protein. Instead, in the experiments undertaken here, the pH dependence of AhpC reduction of H2O2 was determined by monitoring the ability of AhpC to compete with HRP in a manner similar to the method used by Ogusucu et al. (13). At pH values of 5 or above, HRP reacts with hydrogen peroxide to produce compound I with a second order rate constant of 1.8 × 107 M-1 s-1 (24), a rate which is nearly identical with that for AhpC with hydrogen peroxide (3.7 × 107 M-1 s-1). As compound I forms, the 403 nm absorbance of HRP decreases (Δε403 = 5.4 × 104 M−1 s−1), allowing the amount of HRP oxidation to be determined spectroscopically. The rate of HRP reaction with hydrogen peroxide is independent of pH from 5 to 9.4 as confirmed again in our experiments (see below), which means any difference in rate as a function of pH is due to changes in AhpC reactivity (25). As long as the peroxide concentration is lower than the HRP concentration, the second order rate constant for AhpC reduction of hydrogen peroxide can be determined by plotting the percent inhibition of compound I formation (F/1-F) as a function of AhpC concentration using equation 2.
To test this approach, the amount of HRP (starting concentration = 7.5 μM) oxidized by 3 μM H2O2 at pH 7 was monitored in the presence of increasing concentrations of AhpC (0 - 16 μM) (Fig. S2a in Supplemental Information). AhpC inhibited the HRP reaction in a concentration dependent manner and the plot of (F/1-F)kHRP[HRP] versus [AhpC] was linear (Fig. S2b). At pH 7, the second order rate constant obtained using this technique was 3.2 × 107 M-1 s-1. This value is nearly identical to the published value for kcat/Km of 3.7 × 107 M-1 s-1 (1), confirming that we are measuring the reaction of AhpC with hydrogen peroxide. The rate of HRP oxidation was also determined in the absence of AhpC by stopped-flow spectroscopy and no change was observed in the rate of compound I formation between pH 5.43 and 9.05 (data not shown). Below pH 5, the rate decreased, consistent with the earlier report by Dolman et al. (24). We therefore corrected the kAhpC at pH 5 and below based on the decreased kHRP calculated from equation 3 and the pKa values reported earlier (24). The second order rate constants for AhpC reduction of H2O2 were determined at various pH values between 4 and 9, and the resulting rates were plotted versus pH to obtain a pKa value of 5.94 ± 0.10 (Figure 4). Because we are measuring the pH dependence of the catalytic activity of the protein, we attribute this pKa to C46 and can be confident that we are only measuring the conformational state of the protein in which the CP is activated for peroxide attack (the fully folded conformation).
FIGURE 4. pKa determination for wild type AhpC using a competition assay with horseradish peroxidase (HRP) yields a pKa of 5.94 ± 0.10.

Three μM H2O2 was added to HRP (7.5 μM) and reduced AhpC (0, 2, 4, 8, 12, or 16 μM) in various pH buffers. The extent of HRP oxidation by H2O2 was monitored using A403 (to measure complex I formation in HRP) before peroxide addition and again within 90 s after initiation of the reaction. The percentage of inhibition of HRP oxidation (F/1–F) versus [AhpC] was used to calculate the second-order rate constant for AhpC (kAhpC) using equation 2 (see Methods and Supplemental Figures S2 a and b). Data plotted as either kAhpC or relative rate versus pH were fit to equation 1 as described in Experimental Procedures.
Discussion
We report here a cysteine pKa value of 5.84 from ε240 measurements and a functional pKa of 5.94 that is based on reactivity with peroxide. Although the assignment of the spectroscopically determined pKa to one or both cysteine residues is problematic, the pKa from the peroxidase rate likely reflects only the protonation state of C46 in the fully folded, active conformation of the protein. Because the locally unfolded form would be essentially unreactive with peroxide at all pH values tested, this species would not be detected in the peroxide competition assay.
The functional pKa value we observe for AhpC (5.94 ± 0.10) is similar to the values reported by Ogusucu et al. for yeast Tsa1 and Tsa2 (5.4 and 6.0) (13) and the estimated pKa value of 5 to 6 for Prx2 suggested by Peskin et al. based upon the relative amount of disulfide bond formation across a range of pH values (38). It is also close to the value obtained by pH dependence of the ε240 of AhpC (5.84 ± 0.02), although the behavior of the corresponding mutant enzymes, and particularly C165S, does not firmly support assignment of this pKa to one or both cysteine residues. Our pKa values do not agree with an earlier report that suggests that the pKa for C46 in StAhpC is less than 5 (14). Given the difficulties we encountered using alkylating agents in the present study, the slow reaction of iodoacetamide with AhpC, and the use of mutant proteins and a single time point for generating the data in the previous study, our value of 5.9 using the HRP competition assay with wild type AhpC is a more reliable measurement.
The pKa value for AhpC is not as low as reported for some other proteins that use thiolates in nucleophilic redox reactions such as E. coli glutaredoxin-3 (pKa < 5.5) (39) and DsbA (pKa = 3.5) (34), but it is lower than the pKa for the nucleophilic cysteine (C32) of thioredoxin, which is 6.7 – 7.1 (35, 40). In fact, a pKa lower than 5.9 is unlikely to dramatically increase the efficiency of AhpC since a pKa of 6 means that 91% of the CP are deprotonated at pH 7 (based upon the Henderson-Hasselbach equation), compared to only 3% of cysteine being deprotonated with a pKa of 8.5. Also, once the thiolate is formed, its nucleophilicity actually decreases as its pKa is lowered (11, 12), a point that is often missed in the current “reactive cysteine” literature. Given the pKa of about 5.9, AhpC appears to balance these two factors which affect its enzymatic function.
Alkylation-based studies gave pKa values around 8.5 for both cysteine residues; the similarity to values for unperturbed free cysteines can be explained by the use of relatively bulky reagents that presumably are excluded from the fully folded active site pocket containing C46, and from the similarly buried C165. Localized unfolding in the region of these residues is well documented (41), and the dynamic effects which allow for transient accessibility of the cysteine residues may be responsible for the relatively slow alkylation observed (Table 2). This was further investigated by X-ray crystallography which indeed showed a locally unfolded conformation for the IAAn-modified protein. These results therefore support the idea that the reduced enzyme exists in dynamically fluctuating conformations in solution even without prior oxidation of the CP to sulfenic acid.
We were not able to obtain a clear pKa value for the CR in these studies. It is possible that the values of 8.6 – 8.7 obtained from the iodoacetamide-based alkylation experiments are correct for C165 in the fully folded conformation, but we expect from the crystal structure and data with C46 that these values also reflect the locally unfolded conformation of the CR. Previous studies by Bryk et al. using alkylation data also suggested a value of 8.7 for the pKa of C165 from StAhpC (14), but these results may similarly have reflected the locally unfolded rather than fully folded form of C46S.
With the present investigation, the pKa value for the CP of StAhpC joins those from two other studies of Prxs (13, 38) which suggest that the conserved active site environment of this broad family of peroxidases serves to lower the pKa of the critical cysteine by roughly 2.5 to 3 pH units to a value between 5.4 and 6. Such a pKa value allows for the CP within a large percentage of enzyme molecules to exist in its activated, deprotonated state at pH values of 7 or greater, yet helps promote the nucleophilicity of the CP thiolate which would suffer if the pKa were lowered even further.
Supplementary Material
Additional figures supporting the present work are included as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
The authors thank Amanda Day for technical assistance, Bu-Bing Zeng and S. Bruce King for providing the d5-IAAn and d0-IAAn used in these studies, and Dave Horita and Marcus Wright for advice and assistance in acquisition of NMR data for [2-13C]-cysteine labeled AhpC.
Footnotes
This study was supported by a grant from the National Institutes of Health to L.B.P. with a subcontract to P.A.K. (RO1 GM050389), a grant from the National Science Foundation to Jacquelyn S. Fetrow with L.B.P. as a co-investigator (MCB-0517343), and a Ruth L. Kirschstein Individual Fellowship from the National Institutes of Health to K.J.N (F32 GM074537)
The coordinates and structure factors have been deposited in the Protein Data Bank (PDB) with an ID code of 3EMP.
Abbreviations: Prx, peroxiredoxin; AhpC, alkyl hydroperoxide reductase C component (the cysteine-based peroxidase); StAhpC, AhpC from Salmonella typhimurium; AhpF, alkyl hydroperoxide reductase F component (the flavoprotein disulfide reductase); CP, peroxidatic cysteine (C46) of AhpC; CR, resolving cysteine (C165) of AhpC; HRP, horseradish peroxidase; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); SDS, sodium dodecyl sulfate; PAGE, polyacrylamide gel electrophoresis; DTT, 1,4-dithiothreitol; GuHCl, guanidine hydrochloride; DTPA, diethylenetriamine pentaacetic acid; EDTA, ethylenediamine tetraacetic acid; IAAn, iodoacetanilide; d5-IAAn, deuterated iodoacetanilide; d0-IAAn, protiated iodoacetanilide; MALDI-TOF, matrix-assisted laser desorption/ionization time-of-flight; MS, mass spectrometry; AhpC-C46AAn, S-acetanilide modified form of the C165S mutant of AhpC; MES, 2-(N-morpholino)ethanesulfonic acid.
References
- 1.Parsonage D, Youngblood DS, Sarma GN, Wood ZA, Karplus PA, Poole LB. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Veal EA, Day AM, Morgan BA. Hydrogen peroxide sensing and signaling. Mol Cell. 2007;26:1–14. doi: 10.1016/j.molcel.2007.03.016. [DOI] [PubMed] [Google Scholar]
- 3.Wood ZA, Schröder E, Harris JR, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 4.Seaver LC, Imlay JA. Alkyl hydroperoxide reductase is the primary scavenger of endogenous hydrogen peroxide in Escherichia coli. J Bacteriol. 2001;183:7173–7181. doi: 10.1128/JB.183.24.7173-7181.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 6.Poole LB, Karplus PA, Claiborne A. Protein sulfenic acids in redox signaling. Annu Rev Pharmacol Toxicol. 2004;44:325–347. doi: 10.1146/annurev.pharmtox.44.101802.121735. [DOI] [PubMed] [Google Scholar]
- 7.Rhee SG. Cell signaling H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 8.Wood ZA, Poole LB, Hantgan RR, Karplus PA. Dimers to doughnuts: redox-sensitive oligomerization of 2-cysteine peroxiredoxins. Biochemistry. 2002;41:5493–5504. doi: 10.1021/bi012173m. [DOI] [PubMed] [Google Scholar]
- 9.Georgiou G, Masip L. An overoxidation journey with a return ticket. Science. 2003;300:592–594. doi: 10.1126/science.1084976. [DOI] [PubMed] [Google Scholar]
- 10.Luo D, Smith SW, Anderson BD. Kinetics and mechanism of the reaction of cysteine and hydrogen peroxide in aqueous solution. Journal of pharmaceutical sciences. 2005;94:304–316. doi: 10.1002/jps.20253. [DOI] [PubMed] [Google Scholar]
- 11.Wilson JM, Bayer RJ, Hupe DJ. Structure-Reactivity Correlations for the Thiol-Disulfide Interchange Reaction. J Am Chem Soc. 1977;99:7922–7926. [Google Scholar]
- 12.Whitesides GM, Liburn JE, Szajewski RP. Rates of thiol-disulfide interchange reactions between mono- and dithiols and Ellman's reagent. J Org Chem. 1977;42:332–338. [Google Scholar]
- 13.Ogusucu R, Rettori D, Munhoz DC, Soares Netto LE, Augusto O. Reactions of yeast thioredoxin peroxidases I and II with hydrogen peroxide and peroxynitrite: Rate constants by competitive kinetics. Free Radic Biol Med. 2007;42:326–334. doi: 10.1016/j.freeradbiomed.2006.10.042. [DOI] [PubMed] [Google Scholar]
- 14.Bryk R, Griffin P, Nathan C. Peroxynitrite reductase activity of bacterial peroxiredoxins. Nature. 2000;407:211–215. doi: 10.1038/35025109. [DOI] [PubMed] [Google Scholar]
- 15.Nelson KJ, Day AE, Zeng BB, King SB, Poole LB. Isotope-coded, iodoacetamide-based reagent to determine individual cysteine pK(a) values by matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Anal Biochem. 2008;375:187–195. doi: 10.1016/j.ab.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Storz G, Jacobson FS, Tartaglia LA, Morgan RW, Silveira LA, Ames BN. An alkyl hydroperoxide reductase induced by oxidative stress in Salmonella typhimurium and Escherichia coli: genetic characterization and cloning of ahp. J Bacteriol. 1989;171:2049–2055. doi: 10.1128/jb.171.4.2049-2055.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poole LB, Ellis HR. Flavin-dependent alkyl hydroperoxide reductase from Salmonella typhimurium. 1. Purification and enzymatic activities of overexpressed AhpF and AhpC proteins. Biochemistry. 1996;35:56–64. doi: 10.1021/bi951887s. [DOI] [PubMed] [Google Scholar]
- 18.Ellis HR, Poole LB. Roles for the two cysteine residues of AhpC in catalysis of peroxide reduction by alkyl hydroperoxide reductase from Salmonella typhimurium. Biochemistry. 1997;36:13349–13356. doi: 10.1021/bi9713658. [DOI] [PubMed] [Google Scholar]
- 19.Riddles PW, Blakeley RL, Zerner B. Ellman's reagent: 5,5′-dithiobis(2-nitrobenzoic acid)--a reexamination. Anal Biochem. 1979;94:75–81. doi: 10.1016/0003-2697(79)90792-9. [DOI] [PubMed] [Google Scholar]
- 20.The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- 21.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 22.Navaza J. Acta Crystallogr. 1994;A50:157–163. [Google Scholar]
- 23.Cowtan K. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:34–38. [Google Scholar]
- 24.Dolman D, Newell GA, Thurlow MD, Dunford HB. A kinetic study of the reaction of horseradish peroxidase with hydrogen peroxide. Can J Biochem. 1975;53:495–501. doi: 10.1139/o75-069. [DOI] [PubMed] [Google Scholar]
- 25.Dunford HB. Spectroscopy of horseradish peroxidase. I. Optical, resonance raman, magnetic circular dichroism, x-ray absorption, and diffraction. In: Dunford HB, editor. Heme peroxidases. Wiley; New York: 1999. pp. 135–174. [Google Scholar]
- 26.Winterbourn CC. The ability of scavengers to distinguish OH production in the iron-catalyzed Haber-Weiss reaction: comparison of four assays for OH. Free Radic Biol Med. 1987;3:33–39. doi: 10.1016/0891-5849(87)90037-2. [DOI] [PubMed] [Google Scholar]
- 27.Benesch RE, Lardy HA, Benesch R. The sulfhydryl groups of crystalline proteins. I. Some albumins, enzymes, and hemoglobins. J Biol Chem. 1955;216:663–676. [PubMed] [Google Scholar]
- 28.Kortemme T, Darby NJ, Creighton TE. Electrostatic interactions in the active site of the N-terminal thioredoxin-like domain of protein disulfide isomerase. Biochemistry. 1996;35:14503–14511. doi: 10.1021/bi9617724. [DOI] [PubMed] [Google Scholar]
- 29.Roberts BR, Wood ZA, Jönsson TJ, Poole LB, Karplus PA. Oxidized and synchrotron cleaved structures of the disulfide redox center in the N-terminal domain of Salmonella typhimurium AhpF. Protein Sci. 2005;14:2414–2420. doi: 10.1110/ps.051459705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lo Bello M, Parker MW, Desideri A, Polticelli F, Falconi M, Del Boccio G, Pennelli A, Federici G, Ricci G. Peculiar spectroscopic and kinetic properties of Cys-47 in human placental glutathione transferase. Evidence for an atypical thiolate ion pair near the active site. J Biol Chem. 1993;268:19033–19038. [PubMed] [Google Scholar]
- 31.Sarkany Z, Szeltner Z, Polgar L. Thiolate-imidazolium ion pair is not an obligatory catalytic entity of cysteine peptidases: the active site of picornain 3C. Biochemistry. 2001;40:10601–10606. doi: 10.1021/bi010550p. [DOI] [PubMed] [Google Scholar]
- 32.Jocelyn PC. Biochemistry of the SH Group. Academic Press; New York: 1972. [Google Scholar]
- 33.Lindley H. A study of the kinetics of the reaction between thiol compounds and choloracetamide. Biochem J. 1960;74:577–584. doi: 10.1042/bj0740577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nelson JW, Creighton TE. Reactivity and ionization of the active site cysteine residues of DsbA, a protein required for disulfide bond formation in vivo. Biochemistry. 1994;33:5974–5983. doi: 10.1021/bi00185a039. [DOI] [PubMed] [Google Scholar]
- 35.Kallis GB, Holmgren A. Differential reactivity of the functional sulfhydryl groups of cysteine-32 and cysteine-35 present in the reduced form of thioredoxin from Escherichia coli. J Biol Chem. 1980;255:10261–10265. [PubMed] [Google Scholar]
- 36.Parsonage D, Karplus PA, Poole LB. Substrate specificity and redox potential of AhpC, a bacterial peroxiredoxin. Proc Natl Acad Sci U S A. 2008;105:8209–8214. doi: 10.1073/pnas.0708308105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Poole LB, Higuchi M, Shimada M, Calzi ML, Kamio Y. Streptococcus mutans H2O2-forming NADH oxidase is an alkyl hydroperoxide reductase protein. Free Radic Biol Med. 2000;28:108–120. doi: 10.1016/s0891-5849(99)00218-x. [DOI] [PubMed] [Google Scholar]
- 38.Peskin AV, Low FM, Paton LN, Maghzal GJ, Hampton MB, Winterbourn CC. The high reactivity of peroxiredoxin 2 with H2O2 is not reflected in its reaction with other oxidants and thiol reagents. J Biol Chem. 2007;282:11885–11892. doi: 10.1074/jbc.M700339200. [DOI] [PubMed] [Google Scholar]
- 39.Nordstrand K, Aslund F, Meunier S, Holmgren A, Otting G, Berndt KD. Direct NMR observation of the Cys-14 thiol proton of reduced Escherichia coli glutaredoxin-3 supports the presence of an active site thiol-thiolate hydrogen bond. FEBS Lett. 1999;449:196–200. doi: 10.1016/s0014-5793(99)00401-9. [DOI] [PubMed] [Google Scholar]
- 40.Mössner E, Huber-Wunderlich M, Glockshuber R. Characterization of Escherichia coli thioredoxin variants mimicking the active-sites of other thiol-disulfide oxidoreductases. Prot Sci. 1998;7:1233–1244. doi: 10.1002/pro.5560070519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Karplus PA, Hall A. Structural Survey of the Peroxiredoxins. In: Flohé L, Harris JR, editors. Peroxiredoxin Systems. Springer; New York: 2007. pp. 41–60. [DOI] [PubMed] [Google Scholar]
- 42.Diederichs K, Karplus PA. Improved R-factors for diffraction data analysis in macromolecular crystallography. Nat Struct Biol. 1997;4:269–275. doi: 10.1038/nsb0497-269. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional figures supporting the present work are included as Supporting Information. This material is available free of charge via the Internet at http://pubs.acs.org.