

Abstract
Targeted delivery can potentially improve the pharmacological effects of antisense and siRNA oligonucleotides. Here we describe a novel bioconjugation approach to the delivery of splice-shifting antisense oligonucleotides (SSOs). The SSOs are linked to albumin via reversible S-S bonds. The albumin is also conjugated with polyethylene glycol (PEG) chains that terminate in an RGD ligand that selectively binds the αvβ3 integrin. As a test system we utilized human melanoma cells that express the αvβ3 integrin and that also contain a luciferase reporter gene that can be induced by delivery of SSOs to the cell nucleus. The RGD-PEG-SSO-albumin conjugates were endocytosed by the cells in an RGD-dependent manner; using confocal fluorescence microscopy evidence was obtained that the SSOs accumulate in the nucleus. The conjugates were able to robustly induce luciferase expression at concentrations in the 25−200nM range. At these levels little short-term or long-term toxicity was observed. Thus the RGD-PEG-Albumin conjugates may provide an effective tool for targeted delivery of oligonucleotides to certain cells and tissues.
INTRODUCTION
Antisense and siRNA oligonucleotides exhibit considerable promise as therapeutic agents (1, 2) While there are numerous examples of in vivo biological effects of oligonucleotides administered in free form, that is without use of a delivery system (3, 4), some investigators have suggested that oligonucleotide-based therapeutics could be enhanced via effective delivery strategies (5, 6). In this context, a variety of approaches have been pursued that generally fall into two broad categories. First, there are many reports dealing with the use of various types of nanoparticles for oligonucleotide delivery, including liposomes, lipoplexes, micelles, polymeric nanocarriers and metallic particles, as discussed in several recent reviews and articles (7-12). A second strategy has been to synthesize conjugates of oligonucleotides with various peptides including so-called cell-penetrating peptides (13, 14), or peptides able to bind to specific cell surface receptors and thus promote targeting of the conjugate (15, 16).
Both of these general approaches have advantages and liabilities. Even relatively small nanoparticles (∼100nm diameter) are quite large in molecular terms, with masses in the millions of daltons. Nanoparticles cannot readily pass across the capillary endothelium in most normal tissues, and thus their biodistribution is limited to tissues that have extensive fenestrations in the endothelium such as liver, spleen and some types of tumors (17-19). Nanoparticles are also scavenged by the phagocytes of the reticuloendothelial system (20) thus reducing their delivery to desired sites in the body, although this can be partially attenuated by surface modifications such as conjugation with polyethylene glycol (PEG) (21). In contrast, oligonucleotides themselves, being relatively small with masses of several thousand daltons, are rapidly cleared from the bloodstream via renal excretion but nonetheless have broad distribution in many tissues (22-24). The pharmacokinetics and biodistribution of peptide-oligonucleotide conjugates has only been studied to a limited degree; however, a recent analysis of a peptide-morpholino oligonucleotide conjugate indicated broad distribution to various tissues (25).
With these considerations in mind, it seemed desirable to develop intermediate-sized delivery agents for oligonucleotides that might avoid renal clearance and thus have a longer circulation time than free oligonucleotides, but not have the same size constraints on biodistribution as typical nanoparticles. As an initial step in that direction, we report on the synthesis and characterization of macromolecular-scale oligonucleotide carriers based on human serum albumin (HSA). Some of the surface amino groups of albumin are conjugated with PEG chains that terminate in a targeting ligand, namely a cyclic RGD (Arg-Gly-Asp) peptide. This ligand provides high affinity binding to the αvβ3 integrin which is strongly expressed in angiogenic endothelia and in some tumor cells (26). Additional surface amino groups are used to link antisense oligonucleotides to the albumin via bioreversible S-S bridges. In this study the antisense molecule used was a 2’-O-Me-phosphorothioate splice switching oligonucleotide (SSO) (27) that was designed to correct splicing of an aberrant luciferase reporter gene. Effective delivery of the SSO to the cell nucleus results in increased luciferase activity and thus provides an easily monitored positive readout.
Albumin has been extensively studied as a non-toxic carrier for conventional drugs (28, 29) and thus we felt that it might have merits as a macromolecular carrier for oligonucleotides as well. The albumin-RGD-SSO conjugates we developed were tested in a cell culture model and were found to provide highly effective receptor-specific delivery. The endocytotic pathway of the conjugates involved trafficking via smooth vesicles rather than clathin-coated pits, and ultimately led to substantial nuclear accumulation of the splice switching oligonucleotide. This resulted a marked increase in luciferase activity, an effect that was fully inhibited by excess RGD peptide, indicating receptor-dependent delivery. Thus, at the cell culture level, this approach has led to a highly specific and effective delivery system in the macromolecular or sub-nanoscale size range.
EXPERIMENTAL PROCEDURES
Materials
Human serum albumin (HSA) was purchased from Sigma-Aldrich (St. Louise, MO, USA). Alexa Fluor 488 C5 maleimide and a CBQCA amino assay kit were obtained from Invitrogen (Carlsbad, CA, USA). Dual functionalized polyethylene glycol, Malhex-NH-PEG-O-C3H6-CONHS (MW 5,000), was from Rapp Polymere (Tubingen, Germany). Cyclo[RGDfK(Ac-SCH2CO)] peptide was purchased from Peptide International (Louisville, KY, USA). Sulfosuccinimidyl 6-(3’-[2-pyridyldithio]-propionamido) hexanoate (Sulfo-LC-SPDP) was from Thermo Fisher Scientific (Rockford, IL, USA). Glass bottom tissue culture plates were obtained from MatTek (Ashland, MA, USA). Splice shifting oligonucleotide 623, or a mis-match version, were synthesized by an oligonucleotide core facility at University of North Carolina at Chapel Hill. These oligonucleotides were 2’-O-Me phosphorothioates functionalized with thiol groups at the 5’ position; in some cases a Tamra fluorophore was included at the 3’ position (15).
Cells
A375SM melanoma cells that express the αvβ3 integrin were stably transfected with a plasmid, pLuc/705, containing an aberrant intron inserted into the firefly luciferase coding sequence (15). The resulting A375SM-Luc705-B cells were grown in Dulbecco's Modified Eagle's Medium (DMEM) (Gibco/Invitrogen, Carlsbad, CA, USA) supplemented with 10% feta bovine serum (FBS).
Preparation of albumin conjugates with cyclic RGD PEG or cysteine PEG
Human serum albumin (denoted as A) (5 mg, 7.4 × 10−8 mole) was reacted with either L-cysteine (0.05 mg, 3.67 × 10−7 mole) or Alexa Fluor 488 C5 maleimide (0.27 mg, 3.67× 10−7 mole) in phosphate buffered saline (PBS) supplemented with 1 mM EDTA (pH 7.4) for 4 h at room temperature to conjugate the single surface thiol group on albumin; this was followed by dialysis (MW cut off 3,500). The amino groups of the albumin were then reacted with Malhex-NH-PEG-O-C3H6-CONHS (11 mg, 2.21 × 10−6 mole) in PBS/1 mM EDTA (pH 7.4) for 4 h at room temperature. The product, a human albumin derivative with PEG-maleimide groups on the surface (termed PA), was purified from unincorporated PEG materials by dialysis (MW cut off 100,000). The average number of PEG groups conjugated to albumin was determined by using the CBQCA assay, according to the manufacturer's instruction, to measure residual free amino groups. The thiol group on cyclic RGDfK needed for the conjugation with the maleimide group on albumin was freshly generated by 1 h incubation of cyclo[RGDfK(Ac-SCH2CO)] (5 mg, 7.35 × 10−6 mole) in pH 7.0 deprotection buffer (HEPES (50 mM), NH2OH (50 mM) and EDTA (30 mM)) at room temperature. The maleimide groups on the albumin derivative were then reacted with the thiol group of cyclic RGDfK (or cysteine as a control) in PBS with 1 mM EDTA (pH 7.4) overnight at room temperature and purified by dialysis (MW cut off 100,000).
Conjugation of RGD-PEG-albumin with oligonucleotides
PEG-albumin conjugates, derivatized with either cyclo RGD (termed RPA, 9.2 mg, 7.35 × 10−8 mole) or cysteine (termed CPA, 8.7 mg, 7.35 × 10−8 mole), were reacted with dual functional sulfosuccinimidyl 6-(3’-[2-pyridyldithio]-propionamido)hexanoate (1.2 mg, 2.21 × 10−6 mole) linker in PBS with 1 mM EDTA (pH 7.4) for 4 h at room temperature, and purified by dialysis (MW cut off 100,000). Thiol derivatized 623 oligonucleotide (or a mis-match version) (see Table 1) was then added to the intermediate in PBS with 1 mM EDTA (pH 7.4), and the reaction was maintained at room temperature for 24 h and purified by dialysis (MW cut off 100,000). The average number of oligonucleotides linked to albumin was determined as 9.8 by observing the release of pyridine-2-thione (λmax = 343 nm) from the reaction intermediate. This was also confirmed by monitoring the increase in OD260 subsequent to the oligonucleotide conjugation.
Table 1.
Oligonucleotides used in this study
| 623 | 5'-GTTATTCTTTAGAATGGTGC-3' |
| 623-Tamra | 5'-GTTATTCTTTAGAATGGTGC-Tamra-3' |
| 623-SH | 5'-HS-(CH2)6-GTTATTCTTTAGAATGGTGC-3' |
| Tamra-623-SH | 5'-HS-(CH2)6-GTTATTCTTTAGAATGGTGC-Tamra-3' |
| MM | 5'-GTAATTATTTATAATCGTCC-3' |
| MM-Tamra | 5'-GTAATTATTTATAATCGTCC-Tamra-3' |
| MM-SH | 5'-HS-(CH2)6-GTAATTATTTATAATCGTCC-3' |
| Tamra-MM-SH | 5'-HS-(CH2)6-GTAATTATTTATAATCGTCC-Tamra-3' |
All oligonucleotides consist of 2'-OMe ribose residues with a phosphorothioate backbone.
Physical characterization of the RGD-PEG-Oligonucleotide albumin conjugates
The cyclic RGD derivatized albumin conjugate of oligonucleotide (termed RPAO) was analyzed by gel filtration fast protein chromatography system (FPLC) (GE Healthcare) using a Superose 6 10/300 size exclusion column. The size and polydispersity of the conjugates were determined by a quasi-elastic dynamic light scattering (QELS) method as they eluted from the column (Wyatt, Santa Barbara, CA, USA).
Oligonucleotide treatment of cells and luciferase assay
A375SM-Luc705-B cells were seeded onto 12 well plates at 1 × 105 cells per well in medium containing 10% FBS. After 24 h, cells were rinsed, placed in OPTI-MEM (Gibco/Invitrogen, Carlsbad, CA, USA) and treated with either free 623 oligonucleotide, various versions of the albumin-PEG-623 oligonucleotide conjugates, or 623 oligonucleotide complexed with Lipofectamine 2000 as per manufacturer's instruction. After 4 h of treatment, FBS was added to each well to 1%. After 24 h, cells were washed with DMEM medium containing 10 % FBS and incubated in DMEM supplemented with 1% FBS for 48 h prior to harvest. Activation of luciferase gene expression due to correction of splicing by oligonucleotide 623 was determined using a Luciferase assay kit (Promega, Madison, WI, USA) on a Monolight 2010 instrument (Analytical Luminescence Laboratory, San Diego, CA, USA).
Cellular uptake and confocal microscopy
Cells were seeded onto either 12 well plates at 1 × 105 cells per well for cellular uptake measurements or in 12 well glass bottom plates at 2 × 104 cells per well for live cell analysis by confocal microscopy. After treatment of cells with Tamra labeled oligonucleotide derivatives for 24 h, the cells were either lysed for measurement of Tamra fluorescence using a Nanodrop microfluorimeter (Nanodrop Technologies, Wilmington, DE, USA), or washed with DMEM containing 10% FBS and then placed in DMEM without phenol red, supplemented with 1% FBS, for confocal microscopy analysis. Co-localization of Tamra labeled oligonucleotides with Alexa 488 labeled Transferrin or Dextran (Molecular Probes, Beaverton, OR, USA) in live cells was also examined by confocal microscopy. An Olympus confocal microscope with a 60X objective lens was used and the data were processed using Fluoview software (Olympus, Center valley, PA, USA) as described previously (15).
Toxicity studies
Cells were treated with various concentrations of oligonucleotides or conjugates as described above. After 48 h of treatment, cells were trypsinized and viable cells were counted using a Cellzone electric particle counter (Elmhurst, IL, USA) for short term toxicity studies, or replated at low density in 6 well plates containing a mixture of 1% low gelling temperature agarose (SeaKem, Rockland, ME, USA) supplemented with DMEM-H/10% FBS for 14 day studies of colony forming ability as described previously (15).
Nuclease stability
Micrococcal nuclease (400 gel units) was added to solutions of 623-Tamra or RPA-623-Tamra in and the samples were incubated for various periods of time at 37 °C. The reactions were then stopped by the addition of EDTA (50 mM) and the fluorescent samples were analyzed on 10% polyacrylamide gels and examined under long wave length ultraviolet illumination to detect possible nucleolytic cleavage of the Tamra-labeled oligonucleotides.
RESULTS
Synthesis and Characterization of Albumin-PEG-Antisense Conjugates
The overall strategy for preparation of the various conjugates is outlined in Scheme 1. First, the single sulfhydryl group on human serum albumin was labeled with the green fluorophore Alexa 488 (in some cases the sulfhydryl was simply blocked by forming an s-s bridge with cysteine). Subsequently several surface amino groups were reacted with Mal-PEG-NHS to form a pegylated albumin (PA). The number of PEG chains conjugated to PA was determined by using a CBQCA assay to quantitate the number of exposed amino groups on albumin before and after the reaction with PEG; this indicated that approximately 10 PEG chains were conjugated per albumin molecule (Table 2). After purification of the PA conjugate, excess thiol-containing cyclic RGD peptide was reacted with the terminal maleimide groups on PA to form RGD-PEG-albumin (RPA). As a control, cysteine was used instead of RGD to react with the PA; this product was termed CPA. After purification, additional exposed amino groups were reacted with the bifunctional reagent Sulfo-LC-SPDP and then the 623 oligonucleotide (or mismatch version) was conjugated to form RPA-Oligonucleotide (RPAO) (also termed RPA-623). In some cases the 623 oligonucleotide included a 3’ Tamra (red) fluorophore (with the resulting product termed RPA-623-Tamra). The number of oligonucleotides linked to the conjugate was determined in two ways (Figure 1). First, formation of the colored product pyridine-2-thione (λmax = 343 nm) was monitored as the 5’-thiol oligonucleotide reacted with the SPDP-conjugated albumin. Second, the OD260 was determined before and after the conjugation reaction. Both of these methods led to close agreement with 8−11 oligonucleotides linked per albumin in various preparations.
Scheme 1. Preparation of RGD Targeted HSA Oligonucleotide Conjugates.

Alexa 488-Mal = Alexa Fluor 488 C5 maleimde; Mal-PEG-NHS = Malhex-NH-PEG-O-C3H6-CONHS; cRGD-SH = cyclo[RGDfK-COCH2SH]; SPDP = Sulfo-LC-SPDP; Oligo-SH = 623-SH, Tamra-623-SH, MM-SH or Tamra-MM-SH (see Table 1)
Table 2.
Measurement of Available Amino Groups on HSA by CBQCA.
| CBQCA fluorescence (%) | Number of estimated surface amino groups | |
|---|---|---|
| HSA | 100.00 ± 2.2 | 59 |
| HSA-PEG | 81.3 ± 3.3 | 48 |
Figure 1. Cleavable disulfide formation between RPA and oligonucleotide.
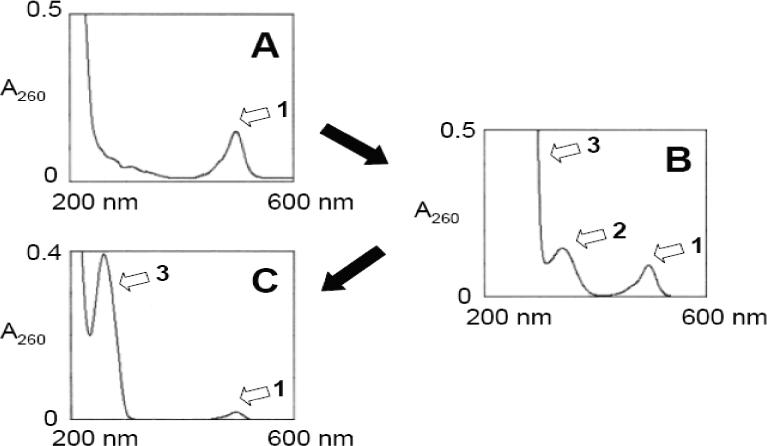
UV spectra of (A) RPA, (B) Reaction mixture of RPA-SPDP and 623-SH, (C) RPA-623. Peak 1: Alexa 488, Peak 2: Pyridine-2-thione, Peak 3: 623. Spectra of diluted samples are shown to include peak 2 and peak 3.
The polyacrylamide gel migration behavior of the starting materials and the conjugates are illustrated in Figure 2A. The 623-Tamra and RPA-623-Tamra are detected by their red fluoresence while the RPA is detected by its green (Alexa 488) fluorescence. As seen, Alexa 488-modified human serum albumin migrated well into the gel consistent with its molecular weight of 68 kDa, while the unconjugated 623-Tamra oligonucleotide migrated near the dye front. Both RPA and RPA-623-Tamra failed to significantly enter the gel indicating molecular size greater than the largest molecular weight marker used (188 kilodaltons). We also used gel analysis to evaluate the relative nuclease stability of 623-Tamra versus the RPA-623-Tamra. As seen in Figure 2B, although 623-Tamra is a rather stable 2’-O-Me-phophorothioate oligonucleotide, incubation with micrococcal nuclease caused a gradual degradation of this material. In contrast, there was no loss of Tamra-labeled oligonucleotide from the RPA-623-Tamra conjugate. This suggests that the oligonucleotides linked to PEG-albumin are partially protected against nuclease degradation.
Figure 2. Polyacrylamide gel (10%) electrophoresis analysis of conjugates (A) and nuclease resistance (B).

Panel A.1) HSA-Alexa 488 2) PA-Alexa 488 3) RPA-Alexa 488 4) 623-Tamra 5) RPA-623-Tamra. Panel B. RPA-623-Tamra (1−5) and 623-Tamra (6−10) were digested with Micrococcal nuclease (400 gel units) for 0 h (1 and 6), 1 h (2 and 7), 2 h (3 and 8), 4 h (4 and 9) and 12 h (5 and 10).
The molecular size of the RPA-623 conjugate was estimated using size-exclusion chromatography and quasi-elastic laser light scattering. As indicated in Figure 3, the RPA-623 conjugate is heterodisperse, migrating as a broad peak while albumin has a sharper migration profile. The hydrodynamic radius of the RPA-623 conjugate was estimated at approximately 6 nm, while that of albumin was estimated at 2.2 nm. Thus the average radius of the conjugate is about 2.7 times that of the ummodified albumin carrier. The albumin-oligonucleotide conjugates were stable during several weeks’ storage in buffer at 4°C, with no indication of aggregation.
Figure 3. FPLC/QELS analysis.
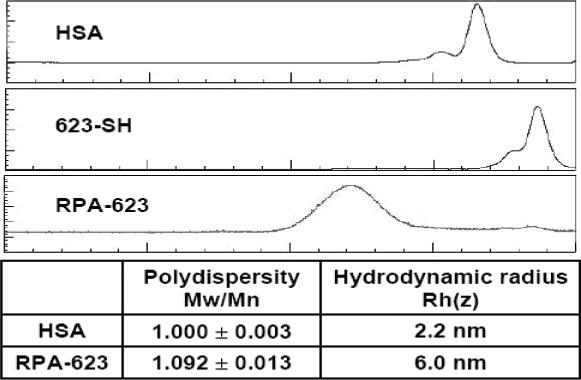
Starting materials (HSA and 623 oligonucleotide) and the final RPA-623 product were analyzed for relative molecular size using FPLC/QELS as described in experimental procedures. The faster migrating shoulder peaks seen in the HSA and 623-SH samples likely represent S-S bridged dimers.
Pharmacological Effect of the Albumin-PEG-Antisense Oligonucleotide Conjugates
In order to evaluate the pharmacological effectiveness of the conjugates, A375SM-Luc705-B cells were incubated with various concentrations of ‘free’ 623 oligonucleotide, 623 complexed with Lipofectamine 2000, or several versions of the conjugates, and the increase in luciferase activity was quantitated. As indicated in Figure 4, treatment with the RPA-623 conjugate resulted in a concentration-dependent increase in luciferase activity over the range 25−200 nM, while use of a mis-matched oligonucleotide version (RPA-MM), or a version where the PEG chains were terminated with cysteine (CPA-623), failed to achieve an effect. Free 623 also did not cause a significant increase in luciferase activity, while 623 complexed with Lipofectamine 2000 had a strong effect. It is interesting to note that the magnitude of the effect achieved by the RPA-623 conjugate almost equaled that of the Lipofectamine complex; the latter is often considered as the ‘gold standard’ for delivery of oligonucleotides to cells in culture. The time course of luciferase activation differed markedly between RPA-623 and the 623-Lipofectamine complex. Thus, as seen in Figure 5, treatment with RPA-623 resulted in activity that rose gradually, attained a maximum at 72 h post treatment, and then gradually declined. In contrast, use of the 623-Lipofectamine complex resulted in a rapid rise of activity within 24 h of treatment followed by a monotonic decline. This pattern suggests that the Lipofectamine complex rapidly delivers olgonucleotide to the cytosol and nucleus, while delivery of oligonucleotide from the RPA-623 conjugate involves a more complex and protracted trafficking within the cell. The intracellular delivery mediated by the RPA-623 conjugate clearly involves the αvβ3 integrin since, as seen in Figure 6, incubation with excess cyclic RGD peptide completely blocks the ability of the conjugate to activate luciferase. At the concentrations used the RGD peptide does not cause cell detachment or other obvious toxicity.
Figure 4. Dose response and specificity studies.
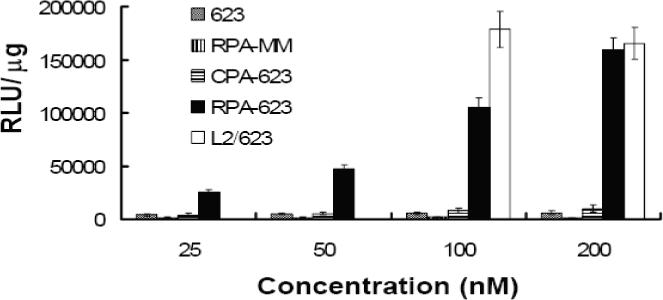
Cells were treated with either free 623, RPA-MM, CPA-623 (prepared by conjugating cysteine with the maleimide on PEG), RPA-623 at 25 to 200 nM, or 623 complexed with Lipofectamine 2000 (1.5 ug/ml) at 100 and 200 nM (L2/623) as described in experimental procedures. Luciferase activity was determined after 72 h from the cell lysates and expressed as relative luminescence units (RLUs) per ug of protein. Results are means and standard errors of three determinations.
Figure 5. Time response studies.
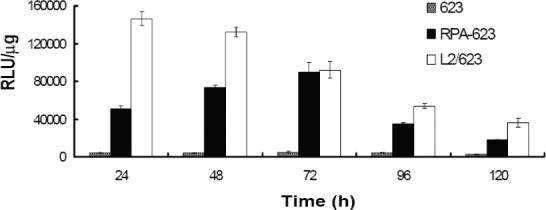
Cells were treated with 200 nM of either free 623, RPA-623 or 623 complexed with Lipofectamine 2000 (1.5 ug/ml) (L2/623). After 24 h, cells were washed and placed in DMEM/1% FBS. Luciferase activity was determined from cell lysates collected at various times and expressed as relative luminescence units (RLUs) per ug of cell protein as described in experimental procedures. Results are means and standard errors of three determinations.
Figure 6. Inhibition of 623 antisense effect by excess cRGD peptide.
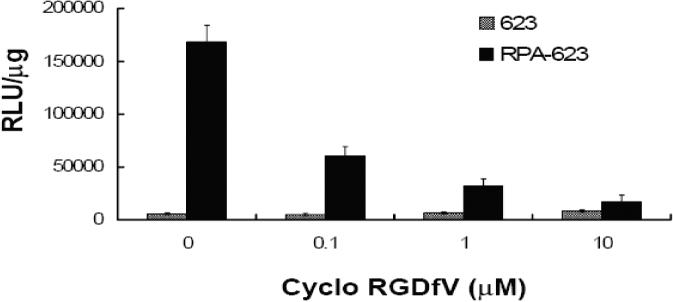
Free cyclo RGDfV peptide at the indicated concentrations was added to the cells 30 min prior to treatment with either free 623 (100 nM) or RPA-623 (100 nM). Luciferase activity was determined after 48 h from cell lysates and expressed as relative luminescence units (RLUs) per ug of cell protein. Results are means and standard errors of three determinations.
Uptake and Sub Cellular Distribution of the Conjugates
We examined the total cellular accumulation of the various materials using a Nanodrop fluorimeter assay, as seen in Figure 7. Total uptake was approximately linear with time in all cases. The highest uptake was observed with 623-Tamra complexed with Lipofectamine 2000, followed by RPAO (either 623 or mis-matched), followed by CPA-623 and free 623-Tamra.
Figure 7. Cellular accumulation.
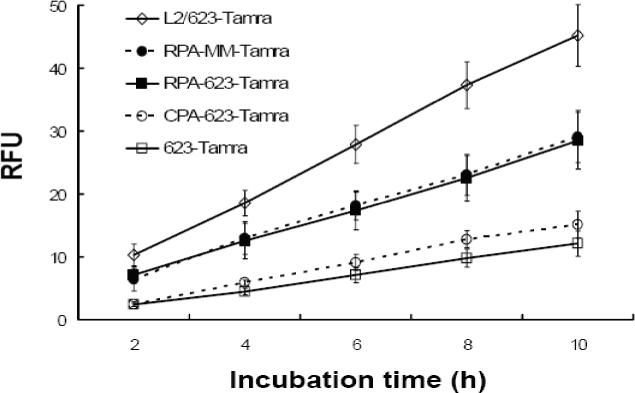
Cells were treated with 100 nM of either free 623-Tamra, 623-Tamra complexed with Lipofectamine 2000 (1.5 ug/ml) (L2/623), RPA-MM-Tamra, RPA-623-Tamra or CPA-623-Tamra. After each incubation period, cells were washed and lysates were analyzed using a Nanodrop fluorimeter for uptake measurements. Results are means and standard errors of three determinations.
We also examined the subcellular distribution of the 623-Tamra labled oligonucelotide after delivery by the conjugates or via complexation with Lipofectamine. As seen in Figure 8, live cells treated with either RPA-623-Tamra or a Lipofectamine/623-Tamra complex displayed substantial intracellular Tamra fluorescence at 24 h, including material present in cytoplasmic vesicles as well as in the nucleus. By contrast cells treated with free 623-Tamra or with CPA-623-Tamra exhibited less intracellular fluorescence with no evidence of nuclear accumulation. These observations suggest that the RGD-PEG-albumin conjugate can provide effective delivery of the pendant oligonucleotides to the cell nucleus.
Figure 8. Confocal microscopy analysis of 623 uptake.

Cells were treated with 100 nM of either (A) free 623-Tamra, (B) 623-Tamra complexed with Lipofectamine 2000 (1.5 ug/ml), (C) CPA-623-Tamra or (D) RPA-623-Tamra as described in experimental procedures. After 24 h, cells were washed with DMEM containing 10% FBS and placed in DMEM supplemented with 1% FBS without phenol red for analysis on an Olympus confocal fluorescence microscope as described in experimental procedures. Arrows indicate Tamra fluorophore accumulated in nuclei.
To further understand the cellular uptake and trafficking of the conjugates, we co-incubated them with well-known markers for different endocytotic pathways and then compared their subcellular distributions. In these studies we used transferrin as a marker for clathrin-coated vesicle mediated uptake, and dextran as a marker for smooth vesicle endocytosis (30). These molecules were labeled with Alexa 488, a green fluorophore, while the RPA-623-Tamra conjugate displays a red fluorescence. As seen in Figure 9, at early time points (2 h) there was little overlap of the RPA-623-Tamra fluorescence with that of transferrin, but there was substantial overlap with the dextran fluorescence. At later time points, there was fluorescence overlap in both cases, although it was most pronounced for dextran. Nuclear accumulation of Tamra fluorescence was observed at the later time points, similar to that seen in Fig 8. These observations suggest that the RGD-PEG-albumin-oligonucleotide conjugate (RPAO) is initially taken up via smooth vesicle endocytosis but later the material enters endomembrane compartments that are accessible via both the coated vesicle and smooth vesicle pathways. Inhibitor studies were consistent with this interpretation. Thus, as seen in Figure 10, cellular accumulation of the RPA-623-Tamra was inhibited by non-toxic concentrations of beta-cyclodextrin and by cytochalasin D (as well as by excess RGD peptide). This indicates that the RPA-623-Tamra is taken up by an actin-dependent pathway that involves smooth vesicles rich in lipid-raft components (30).
Figure 9. Co-localization of 623 with endosomal pathway markers.

RPA-623-Tamra (100 nM) prepared by conjugating a HSA surface thiol group to a cysteine instead of Alexa 488, was co-incubated with (A) Transferrin-Alexa 488 (200 nM) for 2 h, (B) Transferrin-Alexa 488 (100 nM) for 24 h, (C) Dextran-Alexa-488 (2 uM) for 2 h, (D) Dextran-Alexa-488 (2 uM) for 24 h. Live cells were observed for 1) Alexa-488, 2) Tamra, 3) Merged images of Alexa-488 and Tamra and 4) differential interference contrast (DIC) image using an Olympus confocal fluorescence microscope as described in experimental procedures. Arrows represent colocalization of Alexa 488 and Tamra fluorophores.
Figure 10. Cellular uptake inhibition.
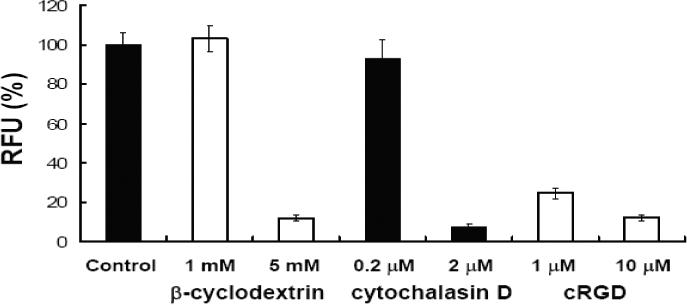
Cells were treated with either β-cyclodextrin, cytochalasin D or cyclo RGDfV (cRGD) for 30 min prior to treatment with either free 623-Tamra (100 nM) control or RPA-623-Tamra (100 nM). After 4h, cells were washed and lysates were analyzed using a Nanodrop fluorimeter for uptake measurements. Results (RFU) are expressed as percentages relative to RFU of control (no inhibition) and are means and standard errors of three determinations.
Lack of Cytotoxicity of the Conjugates
We examined the cytotoxicity of the RPA-623 conjugate using both short-term and long-term assays. A375MLuc705 cells were treated with RPA-623, free 623 or 623 complexed with Lipofectamine. For the short-term assay cell number was determined immediately after the incubation period. The long term assay involved colony formation in soft agar over a 14-day period. As seen in Fig 11, there was little acute or long-term toxicity of RPA-623 even when used at concentrations needed to obtain a strong pharmacological effect.
Figure 11. Toxicity studies.
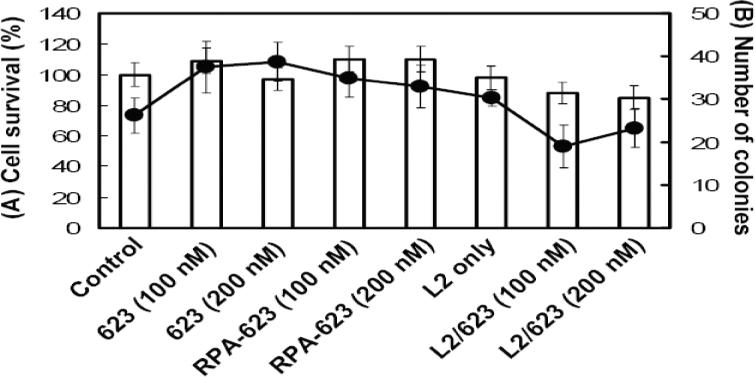
Cells were treated with free 623, 623 complexed with Lipofectamine 2000 (1.5 ug/ml) (L2/623) or RPA-623 under the same conditions as used for luciferase induction experiments. After 48 h, cells were trypsinized and viable cells were counted for short term studies (A, shown as bars). Alternatively cells were replated in 6 well plates containing a mixture of 1% low gelling temperature agarose and complete DMEM medium with 10% FBS for long term toxicity studies (B, shown as circles and a line). After 14 days, surviving colonies larger than 25 cells were counted. Survival is expressed as colonies per 100 cells plated.
DISCUSSION
Albumin has a long history as a biodegradable, non-toxic carrier for conventional low molecular weight drugs (28, 29). Here we have employed human serum albumin as a targetable carrier for antisense oligonucleotides. The splice-switching oligonucleotide (SSO) termed 623 was conjugated to albumin via reversible S-S linkages. RGD-PEG chains were also conjugated in order to provide stabilization and the ability to target a specific cell surface receptor, the αvβ3 integrin. This approach has provided a highly efficient carrier system that could be exploited in a variety of ways. The overall strategy is very flexible and one could easily visualize using many other high-affinity ligands in addition to RGD peptides, thus offering the possibility of ‘targeting’ to many types of cell surface receptors. The ‘payload’ of the conjugates is quite high, with an average of 10 oligonucleotides of approximately 7000 MW conjugated per albumin; this means that the molecular mass of the payload is similar to the total mass of the albumin. PEGlyation seems necessary, since in preliminary studies (not shown) we found that conjugation of the oligonucleotides to albumin in the absence of PEG led to non-specific binding to cells. Interestingly the PEG moieties also seemed to provide some protection against oligonucleotide degradation mediated by an endonuclease.
The RGD-PEG-albumin-oligonucleotide conjugate (RPA-623-Tamra) was taken up by cells in an RGD-dependent fashion and was initially delivered to smooth-walled endosomes rich in lipid rafts and then trafficked to additional endomembrane compartments. Thus the initial path of entry of the RGD-conjugates seems similar to that of integrins themselves, which utilize smooth walled vesicles, including caveolae, for internalization (31, 32). Eventually, 623 oligonucleotide was delivered to the nucleus where it exerted a strong effect in terms of correcting splicing of the aberrant luciferase reporter gene. This effect was attained when the conjugate was used at oligonucleotide concentrations in the 25−200 nanomolar range.
It is interesting to note that the degree of nuclear delivery and the subsequent effect on luciferase were very similar using the RPA conjugate and utilizing a Lipofectamine 2000 complex. However, given the long history of therapeutic use of albumin and of various PEGylated proteins (33), it seems likely that delivery using the albumin conjugates will be far less toxic in vivo than delivery obtained with cationic lipid complexes (34). It will also be of interest to see if the current albumin carrier technology can be adapted to the delivery of duplex siRNAs, as well as single stranded antisense compounds.
The RPA-623 complex is polydisperse with an average hydrodynamic radius of about 6 nm. This is substantially smaller than the size of most nanoparticles currently being used to carry therapeutic agents (35); however, it is large by molecular standards. Thus egress of the RPA-623 conjugates from the bloodstream in many normal tissues will be limited to some degree. It seems likely that RPA-623 will be able to exit capillaries via the so called “large pore” system that allows egress of proteins as large as pentavalent IgM, but will have restricted access to the more abundant small pore system that largely excludes molecules >4.5 nm (18). In some tumors, as well as inflamed tissues, gaps in the endothelium allow egress of larger molecules, possible including the RPA conjugates (36). In contrast, an unconjugated oligonucleotide (5−15,000 daltons) is likely to have a much broader tissue distribution. In the current case it may be possible to devise albumin-oligonucleotide-PEG conjugates that utilize smaller or fewer PEG molecules, thus reducing hydrodynamic radius and allowing more extensive tissue distribution; this issue remains to be explored. In any case, further development of this intermediated-sized delivery system for oligonucleotides will entail substantial in vivo experimentation. Here we have simply explored the basic aspects of the system at the cellular level.
ACKNOWLEGEMENTS
This work was supported by NIH grant P01GM059299 to RLJ. We thank Betsy Clarke for expert editorial assistance. Authors also thank Dr. Ashutosh Tripathy of the UNC Macromolecular Interactions Core Facility for his helpful advice on light scattering experiments.
Footnotes
SUPPORTING INFORMATION. There is no supplemental information with this manuscript.
References
- 1.Chan JH, Lim S, Wong WS. Antisense oligonucleotides: from design to therapeutic application. Clin Exp Pharmacol Physiol. 2006;33:533–40. doi: 10.1111/j.1440-1681.2006.04403.x. [DOI] [PubMed] [Google Scholar]
- 2.de Fougerolles A, Vornlocher HP, Maraganore J, Lieberman J. Interfering with disease: a progress report on siRNA-based therapeutics. Nat Rev Drug Discov. 2007;6:443–53. doi: 10.1038/nrd2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Crooke ST. Progress in antisense technology. Annu Rev Med. 2004;55:61–95. doi: 10.1146/annurev.med.55.091902.104408. [DOI] [PubMed] [Google Scholar]
- 4.Kim DH, Rossi JJ. Strategies for silencing human disease using RNA interference. Nat Rev Genet. 2007;8:173–84. doi: 10.1038/nrg2006. [DOI] [PubMed] [Google Scholar]
- 5.Mescalchin A, Detzer A, Wecke M, Overhoff M, Wunsche W, Sczakiel G. Cellular uptake and intracellular release are major obstacles to the therapeutic application of siRNA: novel options by phosphorothioate-stimulated delivery. Expert Opin Biol Ther. 2007;7:1531–8. doi: 10.1517/14712598.7.10.1531. [DOI] [PubMed] [Google Scholar]
- 6.Russ V, Wagner E. Cell and tissue targeting of nucleic acids for cancer gene therapy. Pharm Res. 2007;24:1047–57. doi: 10.1007/s11095-006-9233-9. [DOI] [PubMed] [Google Scholar]
- 7.Derfus AM, Chen AA, Min DH, Ruoslahti E, Bhatia SN. Targeted quantum dot conjugates for siRNA delivery. Bioconjug Chem. 2007;18:1391–6. doi: 10.1021/bc060367e. [DOI] [PubMed] [Google Scholar]
- 8.Fattal E, Couvreur P, Dubernet C. “Smart” delivery of antisense oligonucleotides by anionic pH-sensitive liposomes. Adv Drug Deliv Rev. 2004;56:931–46. doi: 10.1016/j.addr.2003.10.037. [DOI] [PubMed] [Google Scholar]
- 9.Gilmore IR, Fox SP, Hollins AJ, Akhtar S. Delivery strategies for siRNA-mediated gene silencing. Curr Drug Deliv. 2006;3:147–5. doi: 10.2174/156720106776359159. [DOI] [PubMed] [Google Scholar]
- 10.Li W, Szoka FC., Jr. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res. 2007;24:438–49. doi: 10.1007/s11095-006-9180-5. [DOI] [PubMed] [Google Scholar]
- 11.Sanguino A, Lopez-Berestein G, Sood AK. Strategies for in vivo siRNA delivery in cancer. Mini Rev Med Chem. 2008;8:248–55. doi: 10.2174/138955708783744074. [DOI] [PubMed] [Google Scholar]
- 12.Vinogradov SV, Batrakova EV, Kabanov AV. Nanogels for oligonucleotide delivery to the brain. Bioconjug Chem. 2004;15:50–60. doi: 10.1021/bc034164r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Abes S, Moulton H, Turner J, Clair P, Richard JP, Iversen P, Gait MJ, Lebleu B. Peptide-based delivery of nucleic acids: design, mechanism of uptake and applications to splice-correcting oligonucleotides. Biochem Soc Trans. 2007;35:53–5. doi: 10.1042/BST0350053. [DOI] [PubMed] [Google Scholar]
- 14.Juliano RL. Peptide-oligonucleotide conjugates for the delivery of antisense and siRNA. Curr Opin Mol Ther. 2005;7:132–6. [PubMed] [Google Scholar]
- 15.Alam MR, Dixit V, Kang H, Li ZB, Chen X, Trejo J, Fisher M, Juliano RL. Intracellular delivery of an anionic antisense oligonucleotide via receptor-mediated endocytosis. Nucleic Acids Res. 2008;36:2764–76. doi: 10.1093/nar/gkn115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cesarone G, Edupuganti OP, Chen CP, Wickstrom E. Insulin receptor substrate 1 knockdown in human MCF7 ER+ breast cancer cells by nuclease-resistant IRS1 siRNA conjugated to a disulfide-bridged D-peptide analogue of insulin-like growth factor 1. Bioconjug Chem. 2007;18:1831–40. doi: 10.1021/bc070135v. [DOI] [PubMed] [Google Scholar]
- 17.Jain RK. Transport of molecules, particles, and cells in solid tumors. Annu Rev Biomed Eng. 1999;1:241–63. doi: 10.1146/annurev.bioeng.1.1.241. [DOI] [PubMed] [Google Scholar]
- 18.Rippe B, Rosengren BI, Carlsson O, Venturoli D. Transendothelial transport: the vesicle controversy. J Vasc Res. 2002;39:375–90. doi: 10.1159/000064521. [DOI] [PubMed] [Google Scholar]
- 19.Scherphof GL. In vivo behavior of liposomes; interaction with the mononuclear phagocyte system and implications for drug targeting. In: Juliano RL, editor. Targeted Drug Delivery. Springer Verlag; Berlin: 1991. [Google Scholar]
- 20.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 21.van Vlerken LE, Vyas TK, Amiji MM. Poly(ethylene glycol)-modified nanocarriers for tumor-targeted and intracellular delivery. Pharm Res. 2007;24:1405–14. doi: 10.1007/s11095-007-9284-6. [DOI] [PubMed] [Google Scholar]
- 22.Agrawal S, Zhang R. Pharmacokinetics of oligonucleotides. Ciba Found Symp. 1997;209:60–75. doi: 10.1002/9780470515396.ch6. discussion 75−8. [DOI] [PubMed] [Google Scholar]
- 23.Cossum PA, Sasmor H, Dellinger D, Truong L, Cummins L, Owens SR, Markham PM, Shea JP, Crooke S. Disposition of the 14C-labeled phosphorothioate oligonucleotide ISIS 2105 after intravenous administration to rats. J Pharmacol Exp Ther. 1993;267:1181–90. [PubMed] [Google Scholar]
- 24.DeLong RK, Nolting A, Fisher M, Chen Q, Wickstrom E, Kligshteyn M, Demirdji S, Caruthers M, Juliano RL. Comparative pharmacokinetics, tissue distribution, and tumor accumulation of phosphorothioate, phosphorodithioate, and methylphosphonate oligonucleotides in nude mice. Antisense Nucleic Acid Drug Dev. 1997;7:71–7. doi: 10.1089/oli.1.1997.7.71. [DOI] [PubMed] [Google Scholar]
- 25.Amantana A, Moulton HM, Cate ML, Reddy MT, Whitehead T, Hassinger JN, Youngblood DS, Iversen PL. Pharmacokinetics, biodistribution, stability and toxicity of a cell-penetrating peptide-morpholino oligomer conjugate. Bioconjug Chem. 2007;18:1325–31. doi: 10.1021/bc070060v. [DOI] [PubMed] [Google Scholar]
- 26.Stupack DG, Cheresh DA. Integrins and angiogenesis. Curr Top Dev Biol. 2004;64:207–38. doi: 10.1016/S0070-2153(04)64009-9. [DOI] [PubMed] [Google Scholar]
- 27.Sazani P, Kole R. Therapeutic potential of antisense oligonucleotides as modulators of alternative splicing. J Clin Invest. 2003;112:481–6. doi: 10.1172/JCI19547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Di Stefano G, Fiume L, Baglioni M, Busi C, Bolondi L, Farina C, Mori F, Chieco P, Pariali M, Kratz F, Molin L, Salmaso S, Caliceti P. Doxorubicin coupled to lactosaminated albumin: effect of heterogeneity in drug load on conjugate disposition and hepatocellular carcinoma uptake in rats. Eur J Pharm Sci. 2008;33:191–8. doi: 10.1016/j.ejps.2007.11.005. [DOI] [PubMed] [Google Scholar]
- 29.Temming K, Meyer DL, Zabinski R, Senter PD, Poelstra K, Molema G, Kok RJ. Improved efficacy of alphavbeta3-targeted albumin conjugates by conjugation of a novel auristatin derivative. Mol Pharm. 2007;4:686–94. doi: 10.1021/mp0700312. [DOI] [PubMed] [Google Scholar]
- 30.Kirkham M, Parton RG. Clathrin-independent endocytosis: new insights into caveolae and non-caveolar lipid raft carriers. Biochim Biophys Acta. 2005;1745:273–86. doi: 10.1016/j.bbamcr.2005.06.002. [DOI] [PubMed] [Google Scholar]
- 31.Caswell PT, Norman JC. Integrin trafficking and the control of cell migration. Traffic. 2006;7:14–21. doi: 10.1111/j.1600-0854.2005.00362.x. [DOI] [PubMed] [Google Scholar]
- 32.White DP, Caswell PT, Norman JC. alpha v beta3 and alpha5beta1 integrin recycling pathways dictate downstream Rho kinase signaling to regulate persistent cell migration. J Cell Biol. 2007;177:515–25. doi: 10.1083/jcb.200609004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ryan SM, Mantovani G, Wang X, Haddleton DM, Brayden DJ. Advances in PEGylation of important biotech molecules: delivery aspects. Expert Opin Drug Deliv. 2008;5:371–83. doi: 10.1517/17425247.5.4.371. [DOI] [PubMed] [Google Scholar]
- 34.Lv H, Zhang S, Wang B, Cui S, Yan J. Toxicity of cationic lipids and cationic polymers in gene delivery. J Control Release. 2006;114:100–9. doi: 10.1016/j.jconrel.2006.04.014. [DOI] [PubMed] [Google Scholar]
- 35.Juliano RL. Biological Barriers to Nanocarrier-Mediated Delivery of Therapeutic and Imaging Agents. In: Mirkin C, Niemeyer C, editors. Nanobiotechnology II. Wiley-VCH; Weinheim, Germany: 2007. pp. 263–278. [Google Scholar]
- 36.Greish K. Enhanced permeability and retention of macromolecular drugs in solid tumors: a royal gate for targeted anticancer nanomedicines. J Drug Target. 2007;15:457–64. doi: 10.1080/10611860701539584. [DOI] [PubMed] [Google Scholar]