

Abstract
Mammalian α-defensins all have a conserved triple-stranded β-sheet structure that is constrained by an invariant tridisulfide array, and the peptides exert bactericidal effects by permeabilizing the target cell envelope. Curiously, the disordered, disulfide-null variant of mouse α-defensin cryptdin-4 (Crp4), termed (6C/A)-Crp4, has equal or greater bactericidal activity than the native peptide, providing rationale for comparing the mechanisms by which the peptides interact with and disrupt phospholipid vesicles of defined composition. For both live E. coli ML35 cells and model membranes, disordered (6C/A)-Crp4 induced leakage similar to Crp4 but had less overall membrane-permeabilizing activity. Crp4 induction of fluorophore leakage from electronegative liposomes was strongly dependent on vesicle lipid charge and composition, and the incorporation of cardiolipin into liposomes of low electronegative charge to mimic bacterial membrane composition conferred sensitivity to Crp4- and (6C/A)-Crp4-mediated vesicle lysis. Membrane perturbation studies using biomimetic lipid/polydiacetylene vesicles showed that Crp4 had more pronounced bilayer surface interactions than (6C/A)-Crp4 in low rather than high negatively charged liposomes, correlating directly with measurements of induced leakage. Fluorescence resonance energy transfer experiments provided evidence that Crp4 translocates across highly charged or cardiolipin-containing membranes, in a process coupled with membrane permeabilization, but (6C/A)-Crp4 did not translocate across lipid bilayers and consistently displayed membrane surface association. Thus, despite the greater in vitro bactericidal activity of (6C/A)-Crp4, native, β-sheet containing Crp4 induces membrane permeabilization more effectively than disulfide-null Crp4 by translocating and forming transient membrane defects. (6C/A)-Crp4, on the other hand, appears to induce greater membrane disintegration.
Paneth cells, which reside at the base of the crypts of Lieberkϋhn in mammalian small intestine, synthesize and release α-defensins, termed cryptdins (Crps) in mice. Crps account for ∼70% of the bactericidal peptide activity in Paneth cell secretions and evidence implicates them as key components of mouse innate enteric immunity (1,2). Cryptdin-4 (Crp4) is the most bactericidal of the known mouse Crps with potent activity against both Gram positive and negative bacteria (3,4). Its bactericidal activity is mediated via membrane disruption, but the molecular mechanisms of membrane permeabilization have yet to be determined in detail. Crp4 is a 32 amino acid amphiphilic peptide, with a charge of +8.5 at pH 7.4, and it is monomeric in solution. Its tertiary structure consists of a triple-stranded antiparallel β-sheet that is constrained by three disulfide bonds whose pairings are invariant in the α-defensin family, connected by a series of tight turns and a β-hairpin (5,6). Functional analyses of the Crp4 disulfide array revealed that disulfide bonds confer protection from degradation during proteolytic conversion of the inactive proform to functional peptide. Disrupting disulfide pairings by site-directed mutagenesis, altered Crp4 from a triple-stranded, β-sheet molecule, changing it to a disordered, linear variant. Interestingly, the bactericidal potency of disulfide-null Crp4 in which the six Cys residue positions were substituted with Ala (6C/A)-Crp4), exceeds that of native Crp4 against Gram-positive and Gram-negative pathogens (7).
To understand Crp4 membrane disruption mechanisms in greater detail, we employed complementary assays to investigate the vesicle permeabilization, membrane localization and translocation properties of both Crp4 and (6C/A)-Crp4 in phospholipid bilayers, and the dependence of those interactions on lipid charge and composition. Vesicle permeabilization abilities of Crp4 and (6C/A)-Crp4 were assessed by measuring the efflux of an encapsulated water soluble fluorescent dye (carboxyfluorescein) from liposomes (8-11). A sensitive colorimetric technique that employs biomimetic lipid/polydiacrylate (PDA) vesicles for probing the relative penetration of peptides into lipid bilayers was used to gain insight into the membrane localization of the peptides (12,13). Implementation of this assay for studying membrane processes is based upon the unique biochromatic properties of the mixed assemblies (13,14). Previous studies have confirmed that peptides which localize superficially at the lipid/water interface induce higher colorimetric responses than those which incorporate into the hydrophobic core of the lipid component of the mixed vesicles (13-15). Also, the presence of the PDA matrix within phospholipid/PDA vesicles does not interfere with peptide–lipid interactions in these systems, and nonspecific interactions of membrane peptides with the PDA moieties in the mixed assemblies are minimal (16). Finally, fluorescence resonance energy transfer (FRET) measurements were employed to investigate the translocation of Crp4, i.e., its movement from the outer to the inner membrane leaflets, by measuring the amount of peptide that remains on the outer leaflet after incubation with liposomes and therefore quantifying the fraction of peptide that has translocated (17). The results showed that Crp4 and (6C/A)-Crp4 peptide membrane interactions were dependent on vesicle lipid composition. Specifically, increasing the content of negatively charged phospholipids or incorporating cardiolipin into vesicles enhanced vesicle sensitivity to Crp4 peptide-mediated membrane disruption dramatically. Also, despite having lower overall bactericidal activity, the native β-sheet-containing Crp4 molecule induced greater vesicle leakage, by penetrating into and translocating the bilayer more extensively than (6C/A)-Crp4. These findings show that even though the disulfide array is not essential for bactericidal activity, it is a major determinant of Crp4 mode of action.
MATERIALS AND METHODS
Materials
Native Crp4, G1W-Crp4, and disulfide-null (6C/A)-Crp4 α-defensins were produced by recombinant methods as described (7,15), and (6C/A)G1W-Crp4 was purchased from CPC Scientific, Inc. (San Jose, CA). Melittin from bee venom and the lipids L-α-phosphatidylethanolamine (PE) from Escherichia. coli, L-α-phosphatidylcholine (PC) from egg yolk, L-α-phosphatidylglycerol sodium salt (PG), palmitoyloleoylphosphatidylglycerol (POPG), dioleoylphosphatidylethanolamine (DOPE), cardiolipin (CL), cholesterol and α-Chymotrypsin from bovine pancreas were obtained from Sigma. E.coli TLE (Total Lipid Extract) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(5-dimethylamino-1-naphthalenesulfonyl) (DNS-PE) were purchased from Avanti Polar Lipids, Inc. (Alabaster, AL). 5-(and-6)-carboxyfluorescein (CF) was purchased from Molecular Probes (Invitrogen, Carlsbad, CA). Chemicals for phosphorus analysis, ONPG conversion assays, and potassium efflux measurememts were purchased from Sigma. The diacetylenic monomer, 10,12-tricosadiynoic acid, was purchased from GFS Chemicals (Powell, OH), washed in chloroform, and filtered through a 0.45 μm filter prior to use. Other chemicals were reagent grade and used without further purification.
The lipids used in this study were selected to mimic bacterial cell membranes, which are negatively charged due to the presence of phosphatidylglycerol and cardiolipin and often rich in phosphatidylethanolamine.
Peptide-induced permeabilization of E. coli ML35
E. coli strain ML35 (lacZc, lacY-) does not take up the lactose analogue 2-nitrophenyl β-D-pyranoside (ONPG) unless permeabilized by membrane disruptive agents, including defensins (18). Upon membrane disruption, ONPG diffuses into the bacterial cell cytoplasm, is hydrolyzed by β-galactosidase, and converted to ONP which can be measured by absorbance at 405 nm (18). Log-phase E. coli ML35 cells were washed and resuspended in PIPES buffer (10 mM PIPES, pH 7.4, supplemented with 0.1 % (v/v) trypticase soy broth). In triplicate, bacteria (5 × 106 cells/ml) were exposed to peptides in the presence of 2.5 mM ONPG for 2 h at 37 °C. The kinetics of ONPG hydrolysis and conversion to ONP was measured by absorbance at 405 nm using a Spectra-Max plate spectrophotometer (Molecular Devices, Sunnyvale, CA).
Potassium Efflux from E. coli ML35
E. coli ML35 cells were grown to mid-exponential growth phase in 5 ml of trypticase soy broth at 37°C, deposited and washed twice with PIPES buffer (10 mM PIPES, pH 7.4), and resuspended in PIPES buffer at 2.5 × 108 CFU/ml, i.e., 6.25 × 107 CFU in 250 μl final volume at 37°C. Potassium efflux was measured using MI-442 potassium selective and MI-409F reference microelectrodes (Microelectrodes, Bedford, NH) fitted to an Orion SensorLink PCM-700 pH/ISE meter inserted into microfuge tubes containing 5 × 106 CFU/ml in 250 μl (19). Potassium efflux was monitored continuously prior to and immediately after addition of peptide samples and corresponding control solutions. The tip of the reference electrode was fitted with a salt bridge consisting of 2.0 M NaCl in molecular biology grade agarose to suppress electrode drift (20). In each experiment, extracellular potassium was measured at 10 sec intervals, and after 90 sec of monitoring potassium, peptides were added in 2 μl of resuspension buffer to a final concentration of 25 μg/ml. Measurements of potassium were taken for an additional 3-4 min.
Liposome Preparation
Unilamellar liposomes were prepared for carboxyfluorescein leakage assays by the extrusion method. The vesicles were made of PC/Chol (10:1), PE/PG (7:3), PE/PG (3:7), PE/PG/CL (7:1:1) molar ratios and E. coli total lipid extract (100%). Briefly, stock solutions of lipids in chloroform were mixed in appropriate quantities (∼2 mg total lipid), and the solvent was evaporated under N2 for 30 min. Dry lipid films were hydrated for 1 h in 1 mL carboxyfluorescein buffer (10 mM HEPES pH 7.4, 100 mM carboxyfluorescein, pH was adjusted to 7.4 with NaOH) for 1 hour. After hydration, the lipid mixture was subjected to five freeze-thaw cycles by freezing in liquid nitrogen and thawing at 50°C. Small unilamellar liposomes, were generated by 21 extrusions through polycarbonate filters (pore size 50 nm) mounted in a LiposoFast Basic mini-extruder (Avestin, Ottawa, Canada) at room temperature. Untrapped carboxyfluorescein was removed by gel permeation chromatography using a 1.5 × 10 cm Sepharose 6B column and 10 mM HEPES, 150 mM NaCl (pH 7.4) as eluent buffer. Phospholipid concentrations were determined by phosphate analysis (21), and the results are expressed in terms of the concentration of inorganic phosphorus. Liposomes were stored at 4°C and used within 48 h. For the lipid/PDA assay, mixed vesicles consisting of POPG/DOPE/PDA (3:7:15), POPG/DOPE/PDA (7:3:15) and CL/POPG/DOPE/PDA (1:1:7:14) relative molar ratios were prepared by dissolving all lipid constituents in chloroform/ethanol (1/1) and drying together in vacuo to constant weight. The lipid films were suspended in deionized water by probe sonication at 70°C for 3 minutes. The vesicle suspension was cooled to room temperature, incubated overnight at 4°C, and polymerized by irradiation at 254 nm for 30 sec, resulting in solutions having an intense blue appearance.
Carboxyfluorescein leakage assay
Liposome lysis assays were performed in the wells of polyvinyl chloride microtiter plates (Costar). Carboxyfluorescein-loaded liposomes were diluted in 10 mM HEPES, 150 mM NaCl (pH 7.4) to a final concentration of 200 μM phospholipid, based on phosphorus analysis. Various concentrations of peptides (100 μL total volume) or buffer (blank) were loaded in the wells of the microtiter plate, followed by automatic injection of 100 μL of liposomes. The peptide-induced leakage of carboxyfluorescein was monitored for 90 min at room temperature by measuring the increase in fluorescence intensity (decrease of self-quenching) on a Synergy HT fluorescence microplate reader (Bio-Tek) with excitation and emission wavelengths at 485/20 nm and 528/20 nm, respectively. One hundred percent efflux of carboxyfluorescein was determined after repeated cycles of freezing and thawing of the vesicles. The peptide-induced leakage was calculated by the following equation:
where F is the fluorescence intensity induced by the peptide, F0 is the fluorescence of intact vesicles and Ft represents the intensity at 100% leakage.
Lipid/PDA binding assay
Peptides, 0.3 to 6 μM, were added to 60 μl vesicle suspensions consisting of 0.5 mM total lipid in 25 mM Tris-base (pH 8). After peptide addition, solutions were diluted to 1 mL and spectra were acquired at 28°C on a Jasco V-550 spectrophotometer (Jasco Corp., Tokyo) using a 1 cm optical path cell. To quantify the extent of blue-to-red color transitions within the vesicle suspensions, the colorimetric response (% CR), was defined and calculated as follows (22):
where PB = Ablue/(Ablue + Ared), and A is the absorbance at 640 nm, the “blue” component of the spectrum, or at 500 nm, the “red” component (“blue” and “red” refer to the visual appearance of the material, not actual absorbance). PB0is the blue/red ratio of the control sample before induction of a color change, and PBIis the value obtained for the vesicle solution after the colorimetric transition has occurred. More reddish appearance of the vesicle suspensions indicates higher CR values.
Fluorescence Resonance Energy Transfer (FRET)
Symmetrically labeled liposomes were prepared by hydrating lipid films composed of either E. coli TLE/DNS-PE (10:0.5), PE/PG/DNS-PE (7:3:0.5), PE/PG/DNS-PE (3:7:0.5) or PE/PG/CL/DNS-PE (7:1:1:0.5) molar ratio with 10 mM HEPES, 150 mM NaCl (pH 7.4). Following extrusion through 50 nm polycarbonate filters, 50 μL liposomes (final concentration 100 μM) were injected into 100 μL (final concentration 5.9 μM) Crp4 solutions. Chymotrypsin (50 μL, final concentration 125 μg/mL) was added to peptide solutions that had been incubated with dansyl-labeled liposomes for various times.
Fluorescence Resonance Energy Transfer (FRET) from the Trp residue of the peptides to the dansyl chromophore in the membrane was monitored by observing the fluorescence intensity of the dansyl group at 528 nm upon excitation of Trp at 280 nm. Peptide binding to the liposomes was evident by an increase in dansyl fluorescence due to FRET from the tryptophan residue of the peptide to dansyl labeled lipids. A subsequent decrease in fluorescence upon addition of chymotrypsin implied digestion of the externally bound peptide by the enzyme.
RESULTS
Permeabilization of E. coli ML35 by Crp4 and (6C/A)-Crp4
α—Defensins generally induce bacterial cell death by permeabilization of the target cell membrane (15,23-24) with eventual disruption of chemiosmotic gradients (18,16). To test whether the differential bactericidal activities of Crp4 and (6C/A)-Crp4 (7) results from differing modes of peptide-induced bacterial cell permeabilization (18), Crp4 and (6C/A)-Crp4 were compared in real-time permeabilization assays and for their ability to induce K+ efflux from live E. coli ML35 cells (Experimental Procedures). Membrane permeabilization of β-galactosidase-constitutive E. coli ML35 cells enables diffusion of the lactose analogue ONPG into the cells, where β-gal converts colorless ONPG to ONP, which absorbs at 405 nm (18). In this assay system, 3 μM Crp4 induced rapid ONPG conversion (Figure 1A), and (6C/A)-Crp4 induced both more rapid and extensive E. coli cell permeabilization, consistent with its superior bactericidal activity (7). As expected from its lack of bactericidal activity (7,23,25), proCrp4 induced minimal E. coli ML35 permeabilization in this assay. Both peptides induced K+ efflux, with (6C/A)-Crp4 causing slightly more rapid but much less overall potassium release, a sensitive index of cell death, than Crp4. Thus, even though (6C/A)-Crp4 lacks structure in solution (7), its bactericidal effects are the result of membrane disruptive mechanisms. Therefore, to investigate the mechanisms of (6C/A)-Crp4 bactericidal activity, studies were performed to distinguish the membrane disruptive effects of the two bactericidal peptides.
FIGURE 1.
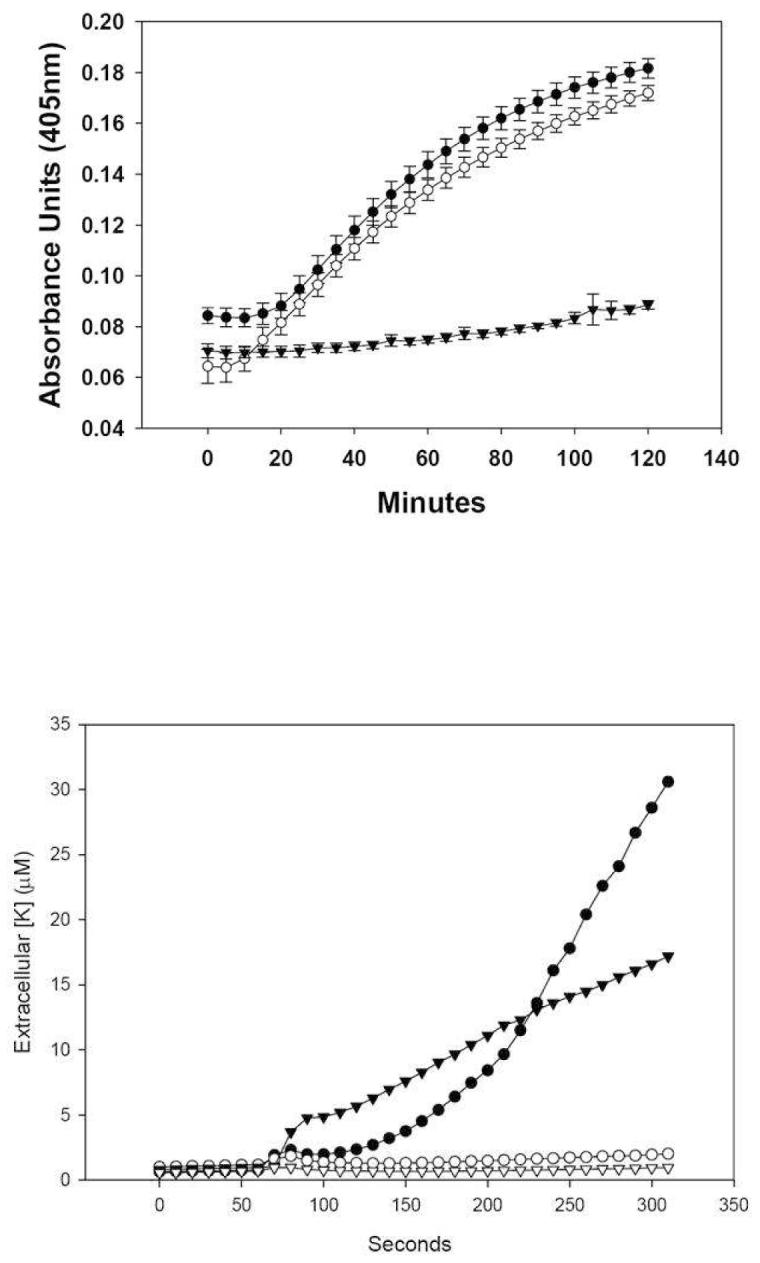
Panel A: Permeabilization of E. coli ML35 by Crp4 and (6C/A)-Crp4. E. coli ML35 growing in log-phase were exposed to 3 μM peptide concentrations in the presence of ONPG at 37 °C. ONPG hydrolysis was measured to determine the amount of permeabilization caused by the experimental peptides. Symbols: Crp4 (-●-), (6C/A)-Crp4 (-○-), and proCrp4, (-▼-). Panel B: Potassium Efflux from E. coli ML35. E. coli ML35 cells were resuspended at 2.5 × 108 CFU/ml in a final volume of 250 μl. After a 90 sec equilibration period, 2 μl of peptide solution were added to a final concentration of 25 μg peptide per ml (6.66 μM Crp4, 6.65 μM (6C/A)-Crp4, and 3.04 μM proCrp4) K+ efflux was monitored as described (Materials and Methods). Although the proCrp4 concentration was approximately half that of the Crp4 peptides, 6 μM proCrp4 does not induce E. coli cell permeabilization (25) or potassium efflux (not shown) Symbols: Crp4 (-●-), (6C/A)-Crp4 (-▼-), proCrp4 (-○-), no peptide addition (-▽-).
Native Crp4 and (6C/A)-Crp4 permeabilize lipid vesicles
The interaction of native Crp4 and disulfide-null (6C/A)-Crp4 with unilamellar liposomes of different lipid compositions and charge densities was studied by carboxyfluorescein (CF) leakage assays. Due to its strong lytic activity and linear structure, melittin, a widely studied bee venom peptide, was used as a positive, linear control (26). CF dye leakage from vesicles is a frequent test for peptide-induced membrane disruptive behavior in vesicle systems in which the fluorescent marker is self-quenched at the high concentrations captured within the liposomes, with increased fluorescence as a measure of dye release (11). In these experiments, CF release was monitored by fluorescence spectroscopy for up to 90 min after addition of 0.4 to 5.9 μM (1.25-20 μg/mL) Crp4 and (6C/A)-Crp4. A time-course experiment at the highest peptide concentrations noted above showed that Crp4 induced leakage was dependent on surface charge. Specifically, incubation of liposomes having a 70% negative surface charge, i.e., a PE/PG ratio of 3:7, with Crp4 induced immediate CF efflux followed by slower dye release with time (Figure 2A). In contrast, liposomes with higher zwitterionic phospholipid content, i.e., a PE/PG ratio of 7:3 (30% negative surface charge), were resistant to membrane permeabilization by either Crp4 or (6C/A)-Crp4 (Figure 2B). Similar findings were obtained with zwitterionic lipid liposomes consisting of PC and Chol (data not shown). For CF-loaded vesicles prepared using total lipids extracted from E. coli cells, Crp4 and (6C/A)-Crp4 induced extensive dye leakage despite the low ∼35% electronegative surface charge of the E. coli liposomes (Figure 2C). In contrast and as expected, melittin lysed all vesicle preparations effectively, causing 100% efflux of CF at 2 μM peptide (Figure 2).
FIGURE 2.

Time course of dye leakage from (A) PE/PG (3:7, 70% charge), (B) PE/PG (7:3, 30% charge), (C) E. coli (∼35% charge) and (D) PE/PG/CL (7:1:1, 30% charge) liposomes induced in the presence of (▲) melittin (2 μM), (◆) (6C/A)-Crp4 (5.9 μM) and (○) native Crp4 (5.9 μM).. The leakage is measured over the entire 90 min course of the assay (usually a plateau was reached within the first 15 min) with peptides and liposomes mixed at t = 2 min. An increase in fluorescence corresponds to the release of dye and is indicative of liposome leakage or lysis. A result average of three independent experiments is shown for each liposome preparation.
Therefore, it became apparent that unlike the comparably charged PE/PG (7:3) vesicles, E. coli cell lipid liposomes were permeable to the Crp4 peptides. Accordingly, we investigated whether a particular lipid component of E. coli liposomes was responsible for the increased susceptibility to permeabilization by Crp4. The lipid content of the E. coli phospholipid extract is approximately 56% PE, 15% PG, 10% CL and 18% other components (www.avantilipids.com). Because of its two electronegative phosphates, we tested the hypothesis that cardiolipin (CL) is the key E. coli lipid constituent that confers sensitivity to Crp4-induced lysis. Indeed, unilamellar liposomes consisting of PE/PG/CL (7:1:1 molar ratio) with a 30% net negative charge were permeabilized by both Crp4 and the (6C/A)-Crp4 (Figure 2D), consistent with reports of CL incorporation inducing vesicle sensitivity to rabbit neutrophil α-defensins (27). Crp4- and (6C/A)-Crp4-induced leakage increased markedly in the presence of CL compared to the responses of CL-lacking vesicles bearing similar overall charge (30%). It should be noted that both native Crp4-mediated lysis and (6C/A)-Crp4-mediated lysis consistently were 50% greater when LUVs were prepared with E. coli whole cell lipid extracts rather than of defined phospholipid composition as is evident at the plateau of the release kinetics after 25 min of peptide exposure, suggesting that additional whole-cell extract components affect Crp4-membrane interactions.
Native Crp4 induces leakage more effectively than (6C/A)-Crp4
To investigate the concentration dependence of peptide-induced membrane disruption, the fractional dye leakage attained at the plateau region of the dye release kinetics was monitored for negatively charged liposomes. The percent leakage as a function of peptide concentration was averaged over three independent experiments (Figure 3). By this measure, a molar ratio of lipid to Crp4 of 17 to 1 was required to trigger substantial dye release from the 70% charged PE/PG (3:7) liposomes, whereas the 30% charged PE/PG (7:3) liposomes remained intact over all peptide concentrations tested. Even though Crp4 and (6C/A)-Crp4 both carry 8.5 units of electropositive charge at pH 7.4, they induce differing extents of leakage and for all lipid vesicle compositions investigated, disordered (6C/A)-Crp4 only had approximately 50% the membrane permeabilizing activity of native Crp4. In contrast, melittin induced CF release independent of vesicle lipid charge or composition and was proportionate to peptide concentration (Figure 3C). Furthermore, partial substitution of PG with CL, with no change of the total lipid charge, increased the vesicle permeabilization activity of both Crp4 and (6C/A)-Crp4 peptides dramatically (Figure 3). Thus, cellular phospholipid composition, e.g., cardiolipin, determines membrane interactions of certain peptides, e.g., Crp4 but not melittin, as well as peptide selectivity for certain target cells.
FIGURE 3.
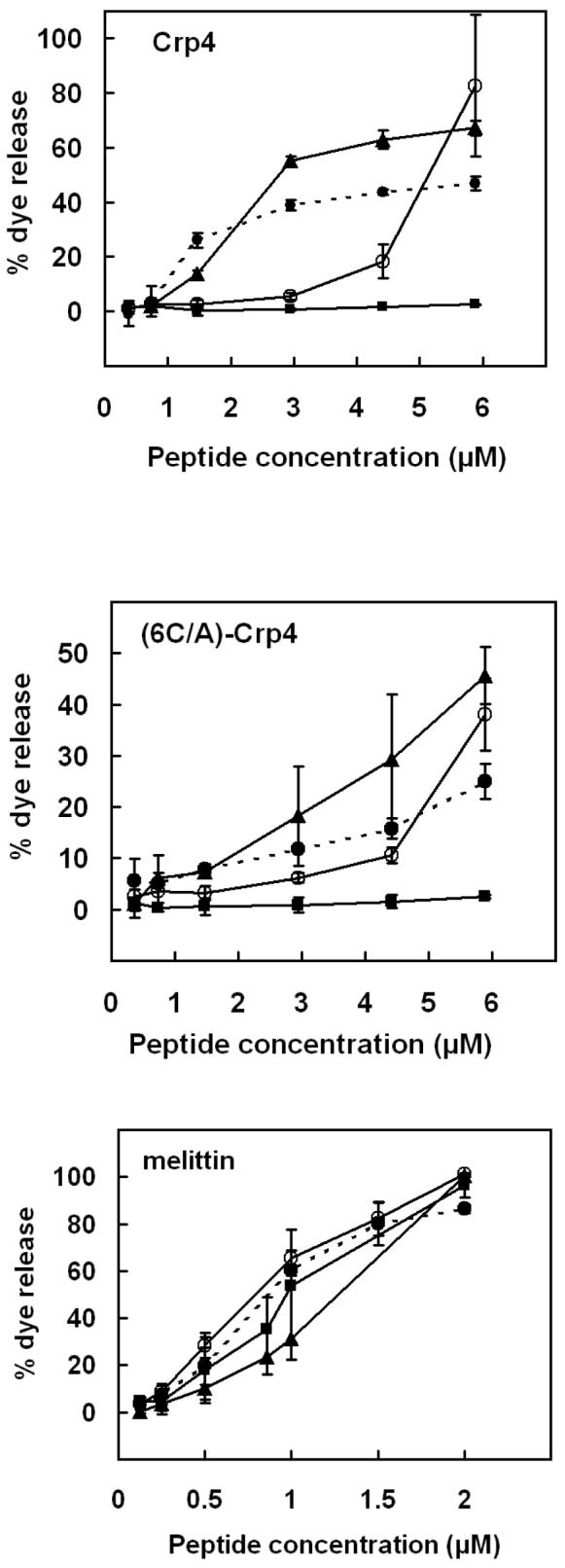
CF leakage from (○) PE/PG (3:7, 70% charge), (■) PE/PG (7:3, 30% charge), (●) PE/PG/CL (7:1:1, 30% charge) and (▲) E. coli (∼35% charge) liposomes as a function of concentration of native Crp4 (top panel), (6C/A)-Crp4 (middle panel), and melittin (bottom panel). Liposomes containing carboxyfluoerescein as a fluorescent marker were incubated with various concentrations of peptides for 90 min and the percentage of dye released was determined by measuring the fluorescence as described in “Materials and methods”. Averages of three experiments and standard deviations are shown.
Lipid/PDA assays
Membrane perturbation activities of the peptides were analyzed by measuring colorimetric transitions in the lipid/PDA vesicle system (22). Previous studies have shown that peptides in general (14) and cryptdins in particular (16) interact selectively with phospholipid domains of the mixed phospholipid/PDA assemblies and chromatic transitions of 100% PDA constructs are minimal. Therefore, percentages of surface negative charge of the PDA-containing vesicles were calculated considering solely the phospholipid content. In POPG/DOPE/PDA (7:3:15) assemblies consisting of 70% electronegative phospholipids, disulfide-null (6C/A)-Crp4 induced the greatest chromatic shift among the peptides tested, suggesting that it occupies interfacial binding sites on the lipid domains of the mixed vesicles. On the other hand, Crp4 and melittin caused lower % CR values, evidence of their deeper insertion into the acyl chain region of the lipid bilayer (Figure 4A). Incorporation of cardiolipin into vesicles containing 30% negative charge (CL/POPG/DOPE/PDA, 1:1:7:14) resulted in stronger association of both Crp4 and (6C/A)-Crp4 with lipid headgroups than melittin, as demonstrated by the higher %CR induced by cryptdins (Figure 4B) (14).
FIGURE 4.
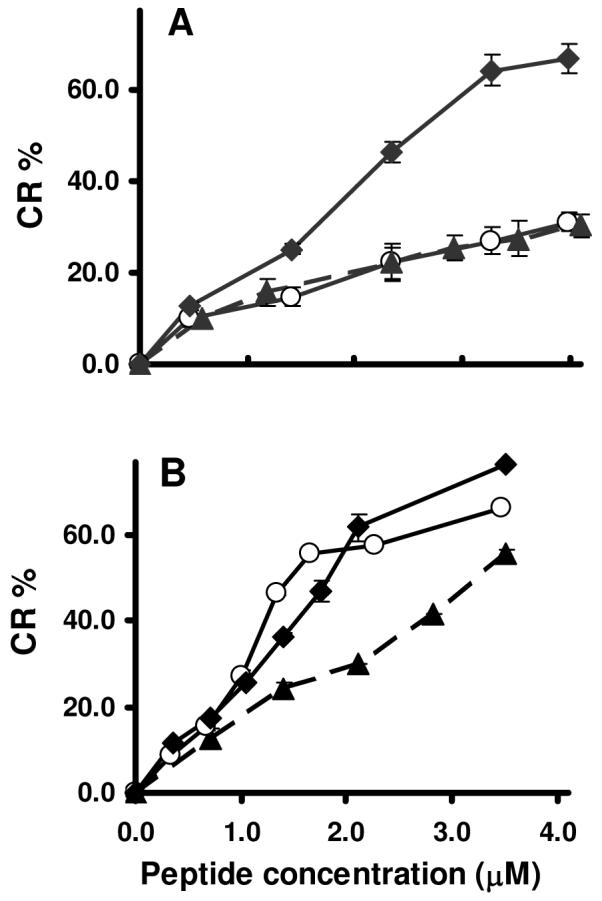
%CR transitions induced by Crp4, (6C/A)-Crp4 and melittin in vesicles of different lipid composition. Vesicle solutions containing (A) POPG/DOPE/PDA (7:3:15, 70% charge) and (B) CL/POPG/DOPE/PDA (1:1:7:14, 30% charge) were exposed to Crp4 (-○-), (6C/A)-Crp4 (-◆-) or melittin (--▲--). The net charge of the lipid assemblies pertains exclusively to the phospholipid components of the phospholipid/PDA mixed vesicles. Higher %CR indicates more pronounced interfacial peptide binding.
The presence of CL in lipid assemblies comprised of CL/POPG/DOPE/PDA (1:1:7:14, 30% charge) decreased the colorimetric response to Crp4 by 10% relative to the %CR of POPG/DOPE/PDA (3:7:15, 30% charge), showing more substantial penetration of Crp4 into the hydrocarbon chain region of the vesicles (Figure 5). In contrast however, the disulfide mutant (6C/A)-Crp4 reacted more superficially at the bilayer-water interface of CL/POPG/DOPE/PDA system, demonstrated by an increased %CR for these vesicles (Figure 5). These collective findings are consistent with the hypothesis that Crp4 and (6C/A)-Crp4 differ with respect to their binding and localization in these model membranes. Specifically, membrane insertion of the natural peptide, unlike its disulfide-null analog, is facilitated by lipid charge and cardiolipin content.
FIGURE 5.
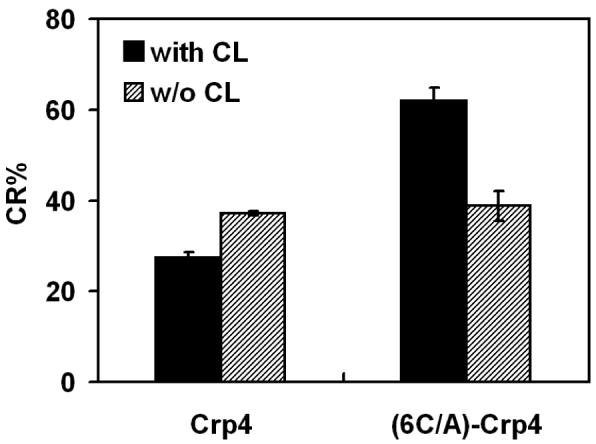
Distinctive sensitivity of Crp4 and (6C/A)-Crp4 membrane interactions to lipid composition. The degree of blue-to-red transitions (%CR) induced by Crp4 and (6C/A)-Crp4 was measured in mixed vesicles composed of: POPG/DOPE/PDA (3:7:15) and CL/POPG/DOPE/PDA (1:1:7:14). Both types of vesicles possess 30% of surface negative charge (accounting only to the phospholipid components of the mixed vesicles).
Detection of peptide translocation
Fluorescence Resonance Energy Transfer (FRET) experiments demonstrated that the mechanisms of Crp4 and (6C/A)-Crp4 lytic action are markedly different: With time, Crp4 becomes less exposed to the outer membrane surface of liposomes, but the disordered (6C/A)-Crp4 variant does not display such behavior. FRET is particularly applicable for measurement of untranslocated peptides remaining in the outer monolayers of liposomes (17), because peptides that remain at the vesicle surface can be removed by proteolysis. We used Crp4 peptides that possess a tryptophan residue and are vulnerable to proteolytic digestion. G1W-Crp4 and (6C/A-G1W)-Crp4, variants with N-terminal Trp for Gly substitutions, have the same bactericidal peptide activities as Gly terminated peptides and thereby were judged to be valid, biologically-relevant fluorescent probes (16). Hence, peptide-membrane associations were studied by monitoring FRET from the N-terminal Trp residue to the dansyl chromophore incorporated into the lipid vesicles.
The exposure of dansyl liposomes (100 μM) to Crp4 (5.9 μM) increased dansyl fluorescence at 528 nm significantly following selective excitation of the peptide Trp residue at 280 nm, thus indicating FRET due to binding of the peptides to the membrane (Figure 6). G1W-Crp4 bound rapidly to labeled PE/PG/CL liposomes, being complete within 10 s. At various incubation intervals of peptides with liposomes, chymotrypsin was added to the mixtures so that hydrolysis of peptides on the membrane surface would be expected to desorb them from the membrane, relieving FRET which would be evident as a decrease in dansyl fluorescence. As controls, G1W-Crp4 and (6C/A-G1W)-Crp4 also were exposed to chymotrypsin before their addition to liposomes. Disulfide-null (6C/A-G1W)-Crp4 peptide was completely digested by chymotrypsin, and subsequent addition of liposomes to the digested fragments had no effect on dansyl fluorescence (Figures 6C and 6D, dotted line). However, pre-incubation of G1W-Crp4 with chymotrypsin decreased but did not abolish dansyl fluorescence (Figures 6A and 6B, trace 1). Fluorescence observed in this case probably arises from a small hydrophobic Trp containing fragment of the peptide that is able to bind on the membrane and result in FRET, and was used as a baseline value.
FIGURE 6.
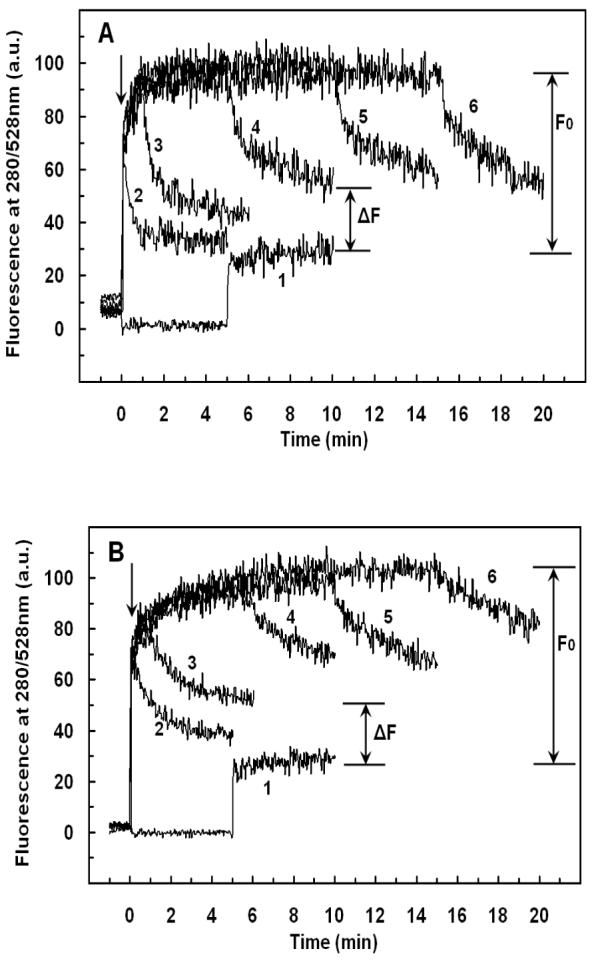
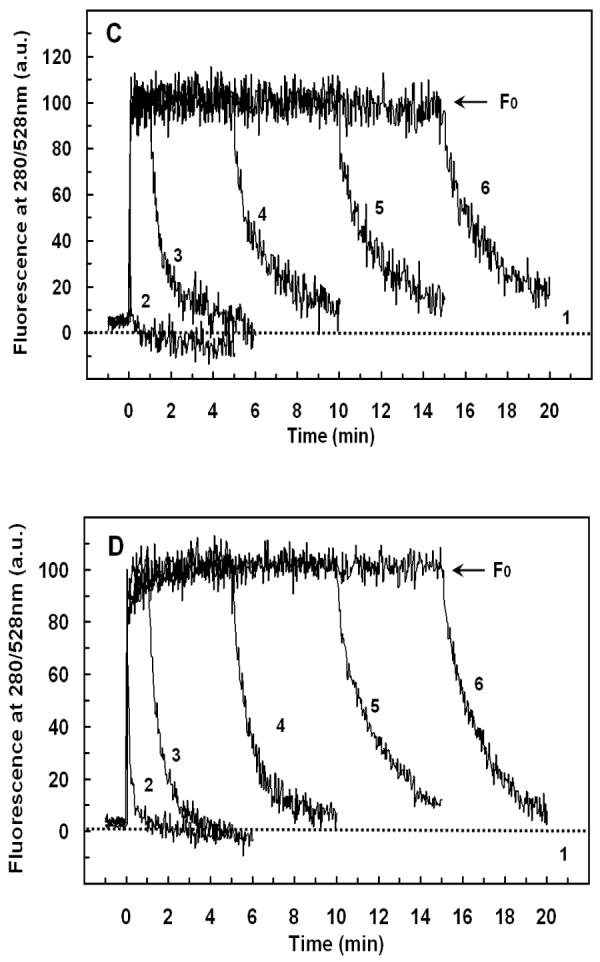
Detection of peptide translocation by selective digestion of G1W-Crp4 or (6C/A)G1W-Crp4 on the outer leaflet. Dansyl-labeled vesicles were injected into the peptide solution at the time indicated by the arrow and the fluorescence intensity of the dansyl group was recorded at 528nm (excitation at 280nm). Binding of the peptide increased fluorescence due to FRET. Addition of chymotrypsin (0.125 μg/mL) at various incubation intervals (0, 1, 5, 10 or 15 min, traces 2-6 respectively) resulted in digestion of the peptide molecules from the outer leaflet in a time-dependent manner, indicating that prolonged incubation protected the peptide from enzyme attack. Pre-incubation of the peptides with the enzyme for 5 min (trace 1 in A, B and dotted line in C, D) before addition of liposomes was used as a control. (A) G1W-Crp4 and PE/PG/CL liposomes with 30% negative charge (B) G1W-Crp4 and PE/PG liposomes with 70% negative charge (C) (6C/A)G1W-Crp4 and PE/PG/CL liposomes with 30% negative charge (D) (6C/A)G1W-Crp4 and PE/PG liposomes with 70% negative charge. The final peptide and lipid concentrations were 5.9 μM and 100 μM, respectively. Each trace is the average of three to four experiments.
The sensitized fluorescence intensity recorded in the (G1W-Crp4)-liposome mixtures decreased slowly when chymotrypsin was added, as surface bound peptide molecules were digested (Figures 6A & 6B). However, the decreased fluorescence intensity recorded exceeded the FRET measured when (G1W)-Crp4 was pre-digested with enzyme, showing that the enzyme did not have access to the total amount of peptide in the mixture i.e. the peptide became less exposed to the outer surface with time. Since prolonged incubation protected the peptide from enzymatic proteolysis, it is possible that some (G1W)-Crp4 molecules have translocated across the lipid bilayer and into the inner leaflet. In contrast to the behavior of (G1W)-Crp4, exposure of the disordered (6C/A-G1W)-Crp4 peptide to chymotrypsin abolished FRET, evidence that this peptide did not translocate across the lipid membranes of PE/PG (3:7) or PE/PG/CL liposomes (Figures 6C & 6D). Unlike its behavior with 70% electronegative PE/PG and 30% electronegative PE/PG/CL vesicles, G1W-Crp4 did not show evidence of translocation across 30% electronegative PE/PG liposomes that lacked cardiolipin (data not shown). Thus, for PE/PG (7:3) vesicles, 30% electronegative charge alone is insufficient to enable (G1W)-Crp4 translocation to the inner leaflet.
Relationship between peptide translocation and membrane permeabilization
To determine the relation between Crp4 translocation and membrane leakage, we first plotted the fractional leakage of CF from vesicles interacting with 5.9 μM peptide in a time course of membrane permeabilization (Figures 7A & 7B, solid curve). Then, the percent translocation values for liposomes of both compositions were estimated as 100 × ΔF/F0 from Figure 6, where ΔF/F0 corresponds to the fraction of translocated peptide. A strong association exists between leakage and translocation (Figures 7A & 7B), suggesting that (G1W)-Crp4 can be found in the inner membrane leaflet after peptide-induced membrane defects cease. In our experiments, approximately 40% of (G1W)-Crp4 peptides translocated through PE/PG/CL and 60% through PE/PG (3:7) lipid bilayers within 10 min. After 10 min of exposure, leakage becomes attenuated, possibly due to a reduction of externally bound peptides that can accumulate and result in membrane defects. Although pore formation-peptide translocation mechanisms have been proposed for certain α-helical peptides including magainin (17) and melittin (28) and the β-sheet AMP tachyplesin I (29), evidence shows that Crp4 does not form stable pores in model membranes (15,30,31). In figure 7 also, an attempt was made to relate vesicle leakage induced by the disulfide-null (6C/A-G1W)-Crp4 peptide to its possible translocation activity, which as mentioned earlier appears to be negligible. Indeed, the low % translocation that was calculated from Figure 6 (10-20%), along with the relatively high variability between experiments on different days, points to the fact that this peptide does not inflict a major impact on the membranes that were investigated, neither in the form of leakage nor translocation.
FIGURE 7.
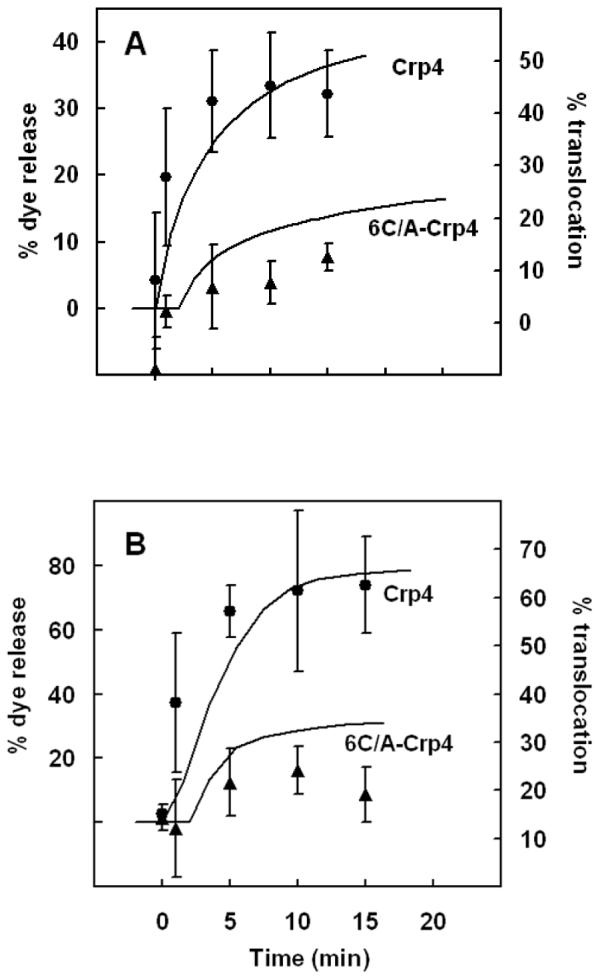
Relationship between Crp4 translocation and membrane leakage. The time courses of carboxyfluorescein leakage and peptide translocation are shown by solid curves and symbols, respectively: (A) trace and ▲, PE/PG/CL liposomes with 30% negative charge; (B) trace and ●, PE/PG liposomes with 70% negative charge. Percent translocation was estimated as 100× (ΔF/F0) from Figs. 7A & 7B. The peptide and lipid concentrations were 5.9 μM and 100 μM respectively.
DISCUSSION
The bactericidal activity of most AMPs correlates directly with the ability to destabilize and disrupt target cell membranes, although individual peptides differ in their modes of action (32,33). Here, we investigated the membrane disruption mechanisms of two α-defensins, the mouse Crp4 and its linear (6C/A)-Crp4 analog. Our results indicate that the two peptides utilize distinctly different mechanisms of membrane interaction and disruption. First, Crp4 showed stronger membrane permeabilizing activities than (6C/A)-Crp4 regardless of vesicle lipid composition. Second, both peptides exhibited different membrane perturbation profiles in membranes of defined lipid composition. For example, the existence of 70% electronegative membrane charge or incorporation of cardiolipin into 30% negatively-charged vesicles enhances insertion of the parent Crp4 peptide into the lipid bilayer, but facilitates retention of (6C/A)-Crp4 at the lipid headgroup region. Third, evidence indicates that Crp4, unlike (6C/A)-Crp4, can translocate across lipid bilayers.
Action mechanism of Crp4
Crp4 triggered the release of the low molecular weight analyte CF (MW=326) from the liposomes in a time-dependent mode. The gradual increase of leakage signal and the subsequent attenuation after approximately 15 min of peptide-liposome incubation, suggest that the formation of transient membrane defects is a plausible mechanism of action. This leakage mode is in agreement with previous reports on membrane permeabilization of POPC/POPG liposomes by Crp4 (34). The maximum leakage values observed for Crp4 were 80% for PE/PG (3:7), 65% for E. coli TLE and 45% for PE/PG/CL liposomes. On the other hand, Crp4 did not induce dye release from less electronegative PE/PG (7:3) liposomes. Thus, increasing lipid charge by addition of PG enhances peptide-induced leakage as previously observed, showing a correlation between vesicle surface potential and peptide-mediated vesicle leakage (15,34). The interaction of Crp4 with 70% charged liposomes assessed with the colorimetric lipid/PDA vesicle assay revealed that the peptide permeates into the hydrophobic lipid region. These findings corroborate CF leakage experiments (Figure 2A) and show that dye release from 70% charged vesicles induced by Crp4 and melittin is mediated by peptide insertion into the hydrophobic core of the bilayer, although only Crp4 is sensitive to vesicle phospholipid composition. The membrane localization of Crp4 in this vesicle system also correlates with its capacity to translocate through the lipid bilayer.
Crp4 did not disrupt PE/PG (7:3) liposomes with 30% charged lipid headgroups, but the reason these vesicles are resistant is unclear. However, previous reports on Crp4 and POPC/POPG vesicles showed that vesicles leaked at more than 25 mol% POPG ratio, implying that there is a threshold of surface charge required for leakage to occur (34). Nevertheless, incorporation of cardiolipin into the 30% electronegative lipid bilayer promoted deeper insertion of Crp4 into the membrane and induced liposomal leakage. Such effect could be attributed to the ability of cardiolipin to increase fluidity of lipid bilayers in model membranes (35) and in bacteria (36). In addition, cardiolipin reportedly forms CL-containing lipid domains in E. coli cells (37), leading us to speculate that CL domains may serve as highly charged “gates” to facilitate Crp4 movement into and through lipid membranes. Our findings show that the presence of cardiolipin in the lipid matrix promotes permeation of amphipathic, triple ß-sheet Crp4 into the membrane without the necessity to alter the overall net negative charge of those lipid vesicles. In addition, in highly charged or cardiolipin containing liposomes, translocation of Crp4 was strongly coupled to leakage. These findings support the hypothesis that Crp4 inserts into the membrane via transient membrane defects and then translocates to the inner membrane leaflet as a consequence of closure and disintegration of these short-lived formations.
Mechanism of (6C/A)-Crp4 membrane disruption
The membrane permeabilization activities of (6C/A)-Crp4 were generally weaker than those of Crp4 and were mediated by a distinct mechanism. The maximum leakage induced by (6C/A)-Crp4 was 40%, 45% and 25% for highly charged PE/PG (3:7), E. coli TLE and PE/PG/CL liposomes, respectively. Increasing lipid electronegative charge or incorporating cardiolipin into low charge vesicles enhanced leakage activity as it did for Crp4. However, the distinctive membrane localization pattern of this peptide relative to Crp4 is indicative of a differing mechanism of action. Contrary to Crp4, (6C/A)-Crp4 consistently displayed bilayer surface association similar to the accumulation of varied linear peptides at the lipid/water interface, including magainin and tachyplesin (38), dermaseptin B (39), and NK-lysin (40). This phenomenon depends both on the nature of the peptide and the lipids in that membrane constituents that induce negative curvature strain on the lipid assemblies commonly tolerate superficial binding of large quantities of the peptides before the membrane breaks down (41). Cardiolipin has a conical three dimensional structure due to its multiple acyl chains and thus imposes a strong negative curvature strain that may facilitate accumulation of (6C/A)-Crp4 at the lipid headgroup region. Unavoidable destabilization of the membrane structure could explain the observed moderate 10-20% leakage of the fluorescent marker from the vesicles. Generally, peptides capable of accumulating at the lipid/water interface tend to lyse model membranes at much higher concentrations than molecules which form short-lived defects in lipid bilayers (41). Apparently, native Crp4 which was shown to incorporate deeper and translocate through cardiolipin containing vesicles, induced higher leakage than its disulfide-null analog at the same concentrations, and our FRET experiments do not support a model of (6C/A)-Crp4 translocation through either PE/PG/CL or PE/PG 70% liposomes.
Overall, these data support the view that membrane disruptive action of Crp4 is linked to peptide translocation, which is facilitated by the presence of the α-defensin disulfide array. In addition, the results presented here are consistent with previous findings which pointed to the crucial role that cardiolipin plays in the interactions of rabbit α-defensins with unilamellar vesicles (27). Accordingly, the greater bactericidal activity of (6C/A)-Crp4 relative to Crp4 may result from favorable interactions of the disordered variant with bacterial cell envelope components that are absent in reconstituted vesicles.
ACKNOWLEDGMENT
We would like to acknowledge Dr A. Tsortos for helpful discussions. We would also like to thank Xiaoqing Qu for excellent technical assistance.
Footnotes
This work was supported by a HFSP grant. A.J.O. acknowledges support of NIH grants AI059346 and DK044632.
- Crp4
- cryptdin-4
- FRET
- fluorescence resoncance energy transfer
- PDA
- polydiacetylene
- PE
- phosphatidylethanolamine
- PC
- phosphatidylcholine
- PG
- Phosphatidylglycerol
- POPG
- palmitoyloleoylphosphatidylglycerol
- DOPE
- dioleoylphosphatidylethanolamine
- %CR
- percentage colorimetric response
- HEPES
- N-(2-hydroxyethyl)-piperazine-N’-2-ethanesulfonic acid
REFERENCES
- 1.Ayabe T, Satchell DP, Wilson CL, Parks WC, Selsted ME, Ouellette AJ. Secretion of microbicidal alpha-defensins by intestinal Paneth cells in response to bacteria. Nat. Immunol. 2000;1:113–118. doi: 10.1038/77783. [DOI] [PubMed] [Google Scholar]
- 2.Salzman NH, Ghosh D, Huttner KM, Paterson Y, Bevins CL. Protection against enteric salmonellosis in transgenic mice expressing a human intestinal defensin. Nature. 2003;422:522–526. doi: 10.1038/nature01520. [DOI] [PubMed] [Google Scholar]
- 3.Ouellette AJ, Hsieh MM, Nosek MT, Cano-Gauci DF, Huttner KM, Buick RN, Selsted ME. Mouse Paneth cell defensins: primary structures and antibacterial activities of numerous cryptdin isoforms. Infect. Immun. 1994;62:5040–5047. doi: 10.1128/iai.62.11.5040-5047.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Selsted ME, Miller SI, Henschen AH, Ouellette AJ. Enteric defensins: antibiotic peptide components of intestinal host defense. J. Cell Biol. 1992;118:929–936. doi: 10.1083/jcb.118.4.929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jing W, Hunter HN, Tanabe H, Ouellette AJ, Vogel HJ. Solution structure of cryptdin-4, a mouse paneth cell alpha-defensin. Biochemistry. 2004;43:15759–15766. doi: 10.1021/bi048645p. [DOI] [PubMed] [Google Scholar]
- 6.Rosengren KJ, Daly NL, Fornander LM, Jonsson LM, Shirafuji Y, Qu X, Vogel HJ, Ouellette AJ, Craik DJ. Structural and functional characterization of the conserved salt bridge in mammalian paneth cell alpha-defensins: solution structures of mouse CRYPTDIN-4 and (E15D)-CRYPTDIN-4. J. Biol. Chem. 2006;281:28068–28078. doi: 10.1074/jbc.M604992200. [DOI] [PubMed] [Google Scholar]
- 7.Maemoto A, Qu X, Rosengren KJ, Tanabe H, Henschen-Edman A, Craik DJ, Ouellette AJ. Functional analysis of the alpha-defensin disulfide array in mouse cryptdin-4. J. Biol. Chem. 2004;279:44188–44196. doi: 10.1074/jbc.M406154200. [DOI] [PubMed] [Google Scholar]
- 8.Hancock RE, Rozek A. Role of membranes in the activities of antimicrobial cationic peptides. FEMS Microbiol. Lett. 2002;206:143–149. doi: 10.1111/j.1574-6968.2002.tb11000.x. [DOI] [PubMed] [Google Scholar]
- 9.Netz DJ, Mdo C. Bastos, Sahl HG. Mode of action of the antimicrobial peptide aureocin A53 from Staphylococcus aureus. Appl. Environ. Microbiol. 2002;68:5274–5280. doi: 10.1128/AEM.68.11.5274-5280.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yamaji A, Sekizawa Y, Emoto K, Sakuraba H, Inoue K, Kobayashi H, Umeda M. Lysenin, a novel sphingomyelin-specific binding protein. J. Biol. Chem. 1998;273:5300–5306. doi: 10.1074/jbc.273.9.5300. [DOI] [PubMed] [Google Scholar]
- 11.Zhang L, Rozek A, Hancock RE. Interaction of cationic antimicrobial peptides with model membranes. J. Biol. Chem. 2001;276:35714–35722. doi: 10.1074/jbc.M104925200. [DOI] [PubMed] [Google Scholar]
- 12.Jelinek R, Kolusheva S. Polymerized lipid vesicles as colorimetric biosensors for biotechnological applications. Biotechnol. Adv. 2001;19:109–118. doi: 10.1016/s0734-9750(00)00064-1. [DOI] [PubMed] [Google Scholar]
- 13.Kolusheva S, Boyer L, Jelinek R. A colorimetric assay for rapid screening of antimicrobial peptides. Nat. Biotechnol. 2000;18:225–227. doi: 10.1038/72697. [DOI] [PubMed] [Google Scholar]
- 14.Kolusheva S, Shahal T, Jelinek R. Peptide-membrane interactions studied by a new phospholipid/polydiacetylene colorimetric vesicle assay. Biochemistry. 2000;39:15851–15859. doi: 10.1021/bi000570b. [DOI] [PubMed] [Google Scholar]
- 15.Satchell DP, Sheynis T, Kolusheva S, Cummings J, Vanderlick TK, Jelinek R, Selsted ME, Ouellette AJ. Quantitative interactions between cryptdin-4 amino terminal variants and membranes. Peptides. 2003;24:1795–1805. doi: 10.1016/j.peptides.2003.08.020. [DOI] [PubMed] [Google Scholar]
- 16.Satchell DP, Sheynis T, Shirafuji Y, Kolusheva S, Ouellette AJ, Jelinek R. Interactions of mouse Paneth cell alpha-defensins and alpha-defensin precursors with membranes: Prosegment inhibition of peptide association with biomimetic membranes. J. Biol. Chem. 2003;278:13838–13846. doi: 10.1074/jbc.M212115200. [DOI] [PubMed] [Google Scholar]
- 17.Matsuzaki K, Murase O, Fujii N, Miyajima K. Translocation of a channel-forming antimicrobial peptide, magainin 2, across lipid bilayers by forming a pore. Biochemistry. 1995;34:6521–6526. doi: 10.1021/bi00019a033. [DOI] [PubMed] [Google Scholar]
- 18.Lehrer RI, Barton A, Ganz T. Concurrent assessment of inner and outer membrane permeabilization and bacteriolysis in E. coli by multiple-wavelength spectrophotometry. J. Immunol. Methods. 1988;108(12):153–158. doi: 10.1016/0022-1759(88)90414-0. [DOI] [PubMed] [Google Scholar]
- 19.Tincu JA, Menzel LP, Azimov R, Sands J, Hong T, Waring AJ, Taylor SW, Lehrer RI. Plicatamide, an antimicrobial octapeptide from Styela plicata hemocytes. J Biol. Chem. 2003;278:13546–13553. doi: 10.1074/jbc.M211332200. [DOI] [PubMed] [Google Scholar]
- 20.Orlov DS, Nguyen T, Lehrer RI. Potassium release, a useful tool for studying antimicrobial peptides. J. Microbiol. Methods. 2002;49:325–328. doi: 10.1016/s0167-7012(01)00383-9. [DOI] [PubMed] [Google Scholar]
- 21.Bartlett GR. Phosphorus assay in column chromatography. J. Biol. Chem. 1959;234:466–468. [PubMed] [Google Scholar]
- 22.Jelinek R, Okada S, Norvez S, Charych D. Interfacial catalysis by phospholipases at conjugated lipid vesicles: colorimetric detection and NMR spectroscopy. Chem. Biol. 1998;5:619–629. doi: 10.1016/s1074-5521(98)90290-3. [DOI] [PubMed] [Google Scholar]
- 23.Shirafuji Y, Tanabe H, Satchell DP, Henschen-Edman A, Wilson CL, Ouellette AJ. Structural determinants of procryptdin recognition and cleavage by matrix metalloproteinase-7. J. Biol. Chem. 2003;278:7910–7919. doi: 10.1074/jbc.M210600200. [DOI] [PubMed] [Google Scholar]
- 24.White SH, Wimley WC, Selsted ME. Structure, function, and membrane integration of defensins. Curr. Opin. Struct. Biol. 1995;5:521–527. doi: 10.1016/0959-440x(95)80038-7. [DOI] [PubMed] [Google Scholar]
- 25.Weeks CS, Tanabe H, Cummings JE, Crampton SP, Sheynis T, Jelinek R, Vanderlick TK, Cocco MJ, Ouellette AJ. Matrix metalloproteinase-7 activation of mouse paneth cell pro-alpha-defensins: SER43 down arrow ILE44 proteolysis enables membrane-disruptive activity. J. Biol. Chem. 2006;281:28932–28942. doi: 10.1074/jbc.M602041200. [DOI] [PubMed] [Google Scholar]
- 26.Shai Y. Mode of action of membrane active antimicrobial peptides. Biopolymers. 2002;66:236–248. doi: 10.1002/bip.10260. [DOI] [PubMed] [Google Scholar]
- 27.Hristova K, Selsted ME, White SH. Critical role of lipid composition in membrane permeabilization by rabbit neutrophil defensins. J. Biol. Chem. 1997;272:24224–24233. doi: 10.1074/jbc.272.39.24224. [DOI] [PubMed] [Google Scholar]
- 28.Matsuzaki K, Yoneyama S, Miyajima K. Pore formation and translocation of melittin. Biophys. J. 1997;73:831–838. doi: 10.1016/S0006-3495(97)78115-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Matsuzaki K, Yoneyama S, Fujii N, Miyajima K, Yamada K, Kirino Y, Anzai K. Membrane permeabilization mechanisms of a cyclic antimicrobial peptide, tachyplesin I, and its linear analog. Biochemistry. 1997;36:9799–9806. doi: 10.1021/bi970588v. [DOI] [PubMed] [Google Scholar]
- 30.Cummings JE, Vanderlick TK. Kinetics of cryptdin-4 translocation coupled with peptide-induced vesicle leakage. Biochemistry. 2007;46:11882–11891. doi: 10.1021/bi701110m. [DOI] [PubMed] [Google Scholar]
- 31.Tanabe H, Qu X, Weeks CS, Cummings JE, Kolusheva S, Walsh KB, Jelinek R, Vanderlick TK, Selsted ME, Ouellette AJ. Structure-activity determinants in paneth cell alpha-defensins: loss-of-function in mouse cryptdin-4 by charge-reversal at arginine residue positions. J. Biol. Chem. 2004;279:11976–11983. doi: 10.1074/jbc.M310251200. [DOI] [PubMed] [Google Scholar]
- 32.Ganz T, Lehrer RI. Antibiotic peptides from higher eukaryotes: biology and applications. Mol. Med. Today. 1999;5:292–297. doi: 10.1016/s1357-4310(99)01490-2. [DOI] [PubMed] [Google Scholar]
- 33.Hancock RE, Diamond G. The role of cationic antimicrobial peptides in innate host defences. Trends Microbiol. 2000;8:402–410. doi: 10.1016/s0966-842x(00)01823-0. [DOI] [PubMed] [Google Scholar]
- 34.Cummings JE,SDP, Shirafuji Y, Ouellette AJ, Vanderlick TK. Electrostatically controlled interactions of mouse Paneth cell a-defensins with phospholipid membranes. Australian J. Chem. 2005;56:1031–1034. [Google Scholar]
- 35.Kolusheva S, Wachtel E, Jelinek R. Biomimetic lipid/polymer colorimetric membranes: molecular and cooperative properties. J. Lipid Res. 2003;44:65–71. doi: 10.1194/jlr.m200136-jlr200. [DOI] [PubMed] [Google Scholar]
- 36.Bernal P, Segura A, Ramos JL. Compensatory role of the cis-trans-isomerase and cardiolipin synthase in the membrane fluidity of Pseudomonas putida DOT-T1E. Environ. Microbiol. 2007;9:1658–1664. doi: 10.1111/j.1462-2920.2007.01283.x. [DOI] [PubMed] [Google Scholar]
- 37.Mileykovskaya E, Dowhan W. Visualization of phospholipid domains in Escherichia coli by using the cardiolipin-specific fluorescent dye 10-N-nonyl acridine orange. J. Bacteriol. 2000;182:1172–1175. doi: 10.1128/jb.182.4.1172-1175.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Matsuzaki K. Why and how are peptide-lipid interactions utilized for self-defense? Magainins and tachyplesins as archetypes. Biochim. Biophys. Acta. 1999;1462:1–10. doi: 10.1016/s0005-2736(99)00197-2. [DOI] [PubMed] [Google Scholar]
- 39.La Rocca P, Shai Y, Sansom MS. Peptide-bilayer interactions: simulations of dermaseptin B, an antimicrobial peptide. Biophys. Chem. 1999;76:145–159. doi: 10.1016/s0301-4622(98)00232-4. [DOI] [PubMed] [Google Scholar]
- 40.Schroder-Borm H, Willumeit R, Brandenburg K, Andra J. Molecular basis for membrane selectivity of NK-2, a potent peptide antibiotic derived from NK-lysin. Biochim Biophys. Acta. 2003;1612:164–171. doi: 10.1016/s0005-2736(03)00115-9. [DOI] [PubMed] [Google Scholar]
- 41.Matsuzaki K, Sugishita K, Ishibe N, Ueha M, Nakata S, Miyajima K, Epand RM. Relationship of membrane curvature to the formation of pores by magainin 2. Biochemistry. 1998;37:11856–11863. doi: 10.1021/bi980539y. [DOI] [PubMed] [Google Scholar]