

Abstract
Convergent total syntheses of the potent cytotoxins (+)-tedanolide (1) and (+)-13-deoxytedanolide (2) are described. The carbon framework of these compounds was assembled via a stereoselective aldol reaction that unifies the C(1)–C(12) ketone fragment 5 with a C(13)–C(23) aldehyde fragment 6 (for 13-deoxytedanolide) or 52 (for tedanolide). Multiple obstacles were encountered en route to (+)-1 and (+)-2 that required very careful selection and orchestration of the stereochemistry and functionality of key intermediates. Chief among these issues was the remarkable stability and lack of reactivity of hemiketals 33b and 34 that prevented the tedanolide synthesis from being completed from aldol 4. Key to the successful completion of the tedanolide synthesis was the observation that the 13-deoxy hemiketal 36 could be oxidized to C(11,15)-diketone 38 en route to 13-deoxytedanolide. This led to the decision to pursue the tedanolide synthesis via C(15)-(S)-epimers, since this stereochemical change would destabilize the hemiketal that plagued the attempted synthesis of tedanolide via C(15)-(R) intermediates. However, use of C(15)-(S) configured intermediates required that the side chain epoxide be introduced very late in the synthesis, owing to the ease with which the C(15)-(S)-OH cyclized onto the epoxide of intermediate 50.
Introduction
The tedanolides are a group of 18-membered macrolactones that display remarkable biological activity. (+)-Tedanolide (1), reported by Schmitz and co-workers in 1984 after isolation from the Caribbean fire sponge Tedania ignis, is highly cytotoxic against human nasopharynx carcinoma (ED50 = 250 pg/mL) and lymphocytic leukemia (ED50 = 16 pg/mL) cell lines and causes cell accumulation in the S phase at concentrations as low as 10 ng/mL.1 In addition, tedanolide has been shown to increase the lifespan of mice implanted with lymphocytic leukemia by 23% at 1.5 μg/kg of body weight.2 The closely related macrolide (+)-13-deoxytedanolide (2) was isolated by Fusetani and co-workers in 1991 from the Japanese sponge Mycale adhaerens and demonstrates potent cytotoxicity against P388 murine leukemia cells (IC50 = 94 pg/mL).3 In an elegant study designed to probe the mechanism of 13-deoxytedanolide activity, Fusetani and co-workers demonstrated that the macrolide binds to the eukaryotic 60S ribosomal subunit4a and identified the C(11)–C(23) region of the natural product as the pharmacophore.4b,5 A third member of this macrolide family, (+)-tedanolide C (3), was reported by Ireland and co-workers in 2006 from the extract of Papua New Guinea marine sponge Ircinia sp.6 The reported structure of tedanolide C has different stereochemistry and a different methylation and oxygenation pattern than the two older congeners. Nevertheless, tedanolide C exhibits strong activity (IC50 = 57 ng/mL) against the HTC-116 colorectal cancer cell line.
The impressive biological profiles and the synthetic challenges presented by the tedanolides have encouraged our laboratory7,8 and others to pursue their synthesis.9–17 The complex architecture of tedanolide (1) is underscored by a heavily oxygenated and methylated carbon skeleton containing 13 stereocenters and two polysubstituted olefins—one of which is incorporated into a β,γ-unsaturated ketone with the potential to migrate into conjugation with the carbonyl. In addition, tedanolide possesses a sensitive α-hydroxy trisubstituted epoxide and four β-hydroxy ketone units that are potentially susceptible to retro aldol decomposition. Total syntheses of tedanolide were recently completed by Kalesse9a,b (2006) and Smith10a (2007); a few years earlier, Smith10c,d (2003) and our laboratory7 (2005) reported total syntheses of 13-deoxytedanolide.
Results and Discussions
In planning our synthetic approach to tedanolide and 13-deoxytedanolide, we initially envisioned that 4 would serve as a common intermediate that would permit entry into both macrolides (Scheme 1). We planned to assemble 4 from the convergent aldol coupling of methyl ketone 5 and aldehyde 6, which would establish the C(13)-alcohol stereochemistry of tedanolide via Felkin addition of 5 to 6. We chose the (R)-configuration for the C(15)-position of 6 after previous studies from our group demonstrated that the lithium enolates of methyl ketone models of 5 add to 2,3-anti aldehydes (e.g., 6) with greater Felkin selectivity than additions to 2,3-syn aldehydes (e.g., 39, vide infra).18 We envisaged that aldol 4 could be converted to tedanolide via a sequence involving C(15)-oxidation to the requisite carbonyl and macrolactonization. After considerable experimentation,8b our decision to mask the C(1)-acid in 5 as an allyl ester and the C(16)-hydroxymethyl group in 6 as an allyl carbonate (Alloc) was motivated by our desire to remove both protection groups simultaneously prior to macrolactonization. We envisioned that aldol adduct 4 could also be converted to 13-deoxytedanolide by using a similar sequence that also included deoxygenation of the C(13)-alcohol.
Scheme 1.

Retrosynthetic analysis of the tedanolides
We developed a convergent route that assembles the C(1)–C(12) ketone 5 via the aldol reaction of ethyl ketone 7 and aldehyde 8. The synthesis of ethyl ketone 7 began with asymmetric hydrogenation19 of β-keto ester 9 followed by alkylation of the β-hydroxy ester under Frater conditions20 to access the 2,3-anti ester 10 (Scheme 2). The hydroxyl group of 10 was protected as a DMPM ether in 11 through a sequence involving ester reduction with LiAlH4, conversion of the 1,3-diol to the 3,4-dimethoxybenzylidene acetal, and regioselective acetal reductive opening with DIBAL. The primary alcohol of 11 was oxidized under Swern conditions,21 and the resulting aldehyde was treated with 3 equiv of γ-siloxy allylstannane 1222a and BF3-OEt2 in CH2Cl2 at −78 °C22b to afford the 3,4-syn-4,5-syn-homoallylic alcohol with >95:5 dr. Methylation of the alcohol (MeOTf, 2,6-di-tert-butyl-4-methylpyridine)23 and oxidative cleavage of the olefin provided aldehyde 14, which in turn was converted to allyl ester 15 via chlorite oxidation to the carboxylic acid24 and esterification with allyl alcohol under Mitsunobu conditions.25
Scheme 2.

Preparation of allyl ester 15
The conversion of allyl ester 15 to the ethyl ketone 7 was complicated by the competitive formation of lactone 16 when 15 was treated with DDQ26 followed by oxidation of the resulting secondary alcohol via the Parikh-Doering method (eq 1).21 Lactone 16 is produced during isolation of the C(5)-alcohol after DDQ-mediated benzyl ether cleavage. This side reaction became more prominent when the debenzylation-oxidation sequence was performed on gram scale. The alcohol intermediate proved very sensitive to acid,27 and great care was taken to minimize the formation of lactone 16 by removing the mildly acidic dihydroquinone (DDQ byproduct) with aqueous sodium bicarbonate extractions.
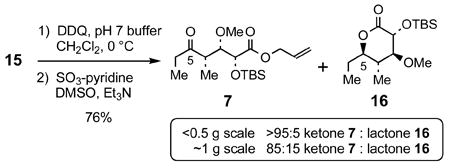 |
(1) |
Aldehyde 8 (to be coupled with ethyl ketone 7 en route to C(1)–C(12) ketone 5) was synthesized from the readily available anti-β-hydroxy-α-methylbutyrate 1728 as shown in Scheme 3. Ester 17 was converted to aldehyde 18 via ester reduction with LiAlH4, dimethoxybenzylidene acetal formation, regioselective acetal reductive opening, and Swern oxidation of the resulting primary alcohol. Wittig olefination of 18 with the stabilized ylide Ph3P=C(Me)CO2Et provided the α,β-unsaturated ester 19 with excellent selectivity (>95:5 E/Z). Reduction of 19 to the allylic alcohol followed by Parikh-Doering oxidation21 of the primary alcohol furnished aldehyde 8 in 70% yield over three steps.
Scheme 3.

Synthesis of unsaturated aldehyde 8
The stereoselective aldol coupling of ethyl ketone 7 and aldehyde 8 was effected by using TiCl4 and i-Pr2NEt and provided 20 in 73% yield with 10:1 diastereoselectivity (Scheme 4).29 The selectivity of this transformation stems from a transition state TS in which the C-O bond of the titanium enolate eclipses the α′-C-H bond, allowing the aldehyde to approach from the less-hindered face of the enolate.29 Extensive optimization was required to minimize the formation of 21 via intramolecular cyclization of the titanium enolate onto the allyl ester. This competitive pathway was suppressed by treating ketone 7 with TiCl4 and i-Pr2NEt for a maximum of 8 min; longer exposure times led to substantially lower yields (<30%) of 20 and correspondingly increased amounts of 21. The synthesis of C(1)–C(12) ketone 5 was completed in three steps with the silylation of the C(7)-alcohol of 20, DDQ cleavage of the dimethoxybenzyl ether,26 and TPAP oxidation30 of the liberated secondary alcohol.
Scheme 4.

Completion of the synthesis of C(1)–C(12) ketone 5
The synthesis of C(13)–C(23) aldehyde 6 is summarized in Scheme 5. Wittig olefination of the known aldehyde 2231 with the stabilized ylide Ph3P=C(Me)CO2Et furnished the α,β-unsaturated ester in excellent selectivity (97:3 E/Z), and DIBAL reduction provided the allylic alcohol. Subsequent Sharpless epoxidation32 of the allylic alcohol (>18:1 dr) and Parikh-Doering oxidation of the hydroxyl group afforded epoxyaldehyde 23. Evans aldol reaction33a of 23 with the chiral crotonate imide 24,33b silylation of the newly-formed alcohol and reduction of the acyl oxazolidinone with LiBH4 in aqueous THF34 provided homoallylic alcohol 25a. The primary alcohol of 25a was temporarily protected as a 2-bromoethyl carbonate (BEC); unfortunately, the Alloc group we desired at this position in 6 could not be directly installed due to its incompatibility with subsequent olefin oxidative cleavage steps. Standard oxidative cleavage of the vinyl group of BEC-protected 25b furnished an aldehyde that was converted to 27 via asymmetric crotylboration with (S, S)-26.35
Scheme 5.

Synthesis of C(13)–C(23) aldehyde 6
The conversion of 27 to the targeted aldehyde 6 required the installation of the C(21)–C(22) (Z)-olefin and conversion of the terminal C(13)-alkene to the aldehyde. Our initial efforts to chemoselectively functionalize the C(13)-alkene in the presence of the C(21)–C(22) olefin were unsuccessful; as a result, it was necessary to functionalize the terminal alkene before introducing the C(21)–C(22) olefin. This was accomplished by silylation of the secondary alcohol of 27, dihydroxylation of the terminal alkene, and protection of the resulting diol as two α-methoxyacetate esters in 28. The C(21)–C(22) (Z)-olefin was installed upon DDQ oxidative cleavage of the PMB ether of 28, Dess-Martin oxidization36 of the primary alcohol, and Wittig olefination of the resulting aldehyde. Treatment of 29 with allyl alcohol and MeMgBr, which generates the magnesium alkoxide in situ, converted the bromoethyl carbonate (BEC) to the allyl carbonate (Alloc) and also cleaved the two α-methoxyacetate esters. Treatment of the liberated diol with Pb(OAc)4 then furnished the targeted C(13)–C(23) aldehyde 6.
Completion of the synthesis of the tedanolide backbone was accomplished by converting methyl ketone 5 to its lithium enolate with LHMDS followed by the addition of aldehyde 6 (Scheme 6). This aldol reaction provided adduct 4 as a single diastereomer37 in 59% yield, along with recovered ketone 5 (27%) and aldehyde 6 (11%). Surprisingly, our efforts to protect the newly-formed C(13)-alcohol as a silyl ether (TMS or TES) or acetate ester (Ac or AcOMe) were unsuccessful, and we carried the unprotected aldol 4 forward. Cleavage of the allyl ester and allyl carbonate units by treatment with Pd(PPh3)4 and n-Bu3SnH38 liberated the seco-acid for macrolactonization;39 however, efforts to obtain macrolactone 30 were complicated by the formation of 14-membered macrolactone 31 via competitive cyclization onto the unprotected C(13)-alcohol. We did not observe equilibration of 30 and 31 upon treatment of these macrolactones with titanium isopropoxide.
Scheme 6.

Aldol coupling and macrolactonization
In an attempt to circumvent the competitive formation of 31, we tried to alter the conformation of the seco-acid by installing the C(15)-ketone prior to macrolactonization. Selective cleavage of the C(15)-TES ether of 4 with aqueous AcOH in THF led to secondary alcohol 32a that cyclized onto the C(11)-carbonyl to form hemiketal 33a (Scheme 7). Silylation of the C(13)-alcohol of 33a with TESCl provided the hemiketal 33b which proved to be remarkably stable and unreactive toward oxidants. Extensive efforts to intercept the ring-opened isomer 32b with oxidants (Dess-Martin periodinane,36 SO3-pyridine in DMSO,21 H2CrO6 (Jones oxidation),40 PDC,41 TPAP/NMO30) en route to the C(15)-ketone failed. We postulated that converting these intermediates to the macrolactone 34 might shift the hemiketal/hydroxy ketone equilibrium to the hydroxy ketone isomer 35 and permit C(15)-oxidation. Therefore, conversion of 33b to the seco-acid and then macrocyclization as described for 4 provided macrocyclic hemiketal 34 (Scheme 8). However, the hydroxy ketone tautomer 35 could not be intercepted with oxidants to afford the targeted C(11,15)-dione, and under various oxidation conditions the hemiketal 34 was recovered intact. As a result, we were unable to elaborate 34/35 to tedanolide even though these intermediates are only two synthetic steps from the natural product (i.e., C(15)-oxidation and global desilylation).
Scheme 7.

Hemiketal formation of C(11, 15)-hydroxy ketone
Scheme 8.

Hemiketal stability in the macrolactone
Although it was not possible to convert hemiketal 33b or 34 to tedanolide, we were able to use 33a for the synthesis of 13-deoxytedanolide (Scheme 9). By analogy to the principles of the Thorpe-Ingold effect,42,43 which stipulates that increased numbers of substituents (typically in a geminal arrangement) between two reactive termini lead to an increased rate of ring formation, we speculated that removal of the C(13)-alcohol would decrease the stability of the hemiketal relative to the hydroxy ketone tautomer and enable oxidation of C(15)-OH to the C(11,15)-dione. Excision of C(13)-OH of 33a was accomplished via conversion of the alcohol to the pentafluorophenylthiocarbonate44 and subsequent treatment with Et3B and n-Bu3SnH.45 Although the resulting hemiketal 36 did not appear to be in equilibrium with hydroxy ketone isomer 37 according to 1H NMR analysis, we were gratified that treatment of 36 with Dess-Martin periodinane furnished the triketone 38 in 71% yield. Ketone 38 was converted to 13-deoxytedanolide (2) in three steps via seco-acid formation and Yonemitsu-modified Yamaguchi lactonization,39 followed by removal of the three TBS ethers with Et3N-buffered Et3N-3HF (thereby generating Et3N-2HF in situ).46 Synthetic 13-deoxytedanolide was identical in all respects (1H NMR, 13C NMR, IR, optical rotation, HRMS) to an authentic sample.
Scheme 9.

Synthesis of (+)-13-deoxytedanolide (2)
Having completed the synthesis of the 13-deoxy congener, we continued our efforts to synthesize tedanolide which had been thwarted by the stability of hemiketal intermediates 33b and 34. We hypothesized that inversion of the C(15)-(R)-alcohol stereochemistry in our first generation intermediates to the C(15)-(S)-configuration would destabilize the corresponding hemiketal B via increased 1,3-diaxial interactions and permit the requisite C(15)-oxidation (Scheme 10).47,48 Thus, we targeted aldehyde 39 containing the (S)-configuration at the C(15)-position.
Scheme 10.

Proposed effect of C(15)-configuration on hemiketal stability
We were unable to install the C(15)-(S)-OH of 39 via crotylboration (Scheme 11) of 40 (the immediate precursor to 27, Scheme 6), which was surprising since a similar crotylboration reaction set the C(15)-(R)-OH of aldehyde 6 (vide supra). The reactions of (R, R)-tartrate crotylboronate35 or (−)-Ipc2-(Z)-crotylboronane49 with aldehyde 40 did not provide 41, but rather gave either the undesired C(15)-(R)-OH diastereomer (i.e., 27) or unidentified products lacking the diagnostic olefin protons in the 1H NMR spectrum. Alternatively, we pursued this key stereocenter using an Evans aldol approach.33a Reaction of aldehyde 40 with chiral imide 42 provided the aldol adduct 43 with excellent yield and selectivity. Treatment of the alcohol with TESCl afforded the silyl ether. However, efforts to cleave the acyl oxazolidinone unit of 43 led to competitive reduction of the 2-bromoethyl carbonate, and so we looked to replace the carbonate protection group with a silyl ether as shown in Scheme 12. Aldol reaction of chiral imide 42 with aldehyde 45 and subsequent reduction of the acyl oxazolidinone provided 1,3-diol 47. The primary alcohol of 47 was temporarily protected as a methoxyacetate ester before silylation of the secondary alcohol. Subsequent cleavage of the methoxyacetate ester followed by oxidation of the resulting alcohol completed the synthesis of aldehyde 49 with the desired (S)-configuration at the C(15)-position.
Scheme 11.

Unsuccessful efforts toward aldehyde 39
Scheme 12.

Synthesis of C(13)–C(23) aldehyde 49
The aldol reaction of the lithium enolate generated from C(1)–C(12) ketone 5 and C(13)–C(23) aldehyde 49 led to the Felkin adduct 50 as the major product37 with 78:22 diastereoselectivity (Scheme 13). The diminished diastereoselectivity (compared to the aldol coupling of 5 and 2,3-anti aldehyde 6) was expected due to the mismatched 1,3-induction imparted by the 2,3-syn aldehyde 49.18,50 Specifically, the expected Felkin chair-like transition state (TS-1) for the aldol reaction of 5 with 2,3-syn aldehyde 49 is destabilized relative to the Felkin chair-like transition state (TS-2) for the 2,3-anti aldehyde 6 due to the stereoelectronic repulsion of interacting dipoles of the aldehyde carbonyl and C(15)-hydroxy TES ether.50 In an attempt to move the synthesis forward, aldol product 50 was treated with aqueous AcOH in THF to cleave the TES ether. Unfortunately, these desilylation conditions led to the 5-exo-cyclization of the liberated C(15)-alcohol onto the epoxide51 to form tetrahydrofuran 51. The analogous cyclization was not observed during manipulation of aldol 4 deriving from the 2, 3-anti-aldehyde 6 (Scheme 7). We hoped that cleavage of the C(15)-TES ether under buffered or basic conditions (Et3N-3HF, TBAF, TAS-F) might avoid the epoxide-opening, but these efforts led to competitive desilylation of the various TBS ethers. Nevertheless, even by using these neutral or weakly basic conditions, evidence for cyclization of C(15)-OH onto the epoxide was observed (e.g., 51 and desilylated versions of 51 were detected). Thus, the change in stereochemistry of C(15)-OH significantly affected the reactivity of intermediate 50 (Scheme 13). As a result, we were unable to elaborate these intermediates to tedanolide.
Scheme 13.

Aldol coupling of ketone 5 and aldehyde 49
At this point, we targeted aldehyde 52 as a third-generation coupling partner for ketone 5 en route to tedanolide (eq 2). The C(13)–C(23) fragment 52 maintains the (S)-configuration at the C(15)-position but lacks the epoxide. This revision to our approach to tedanolide would now require a late-stage, C(17)-OH-directed epoxidation of the C(18)–C(19) olefin,12c,52 a strategy utilized by Smith10c,d in the synthesis of 13-deoxytedanolide and by Kalesse9a,b and Smith10a in their syntheses of tedanolide. Aldehyde 52 was designed with protecting groups for the C(15)- and C(17)-alcohols that could be selectively removed prior to C(15)-alcohol oxidation and C(17)-directed epoxidation, respectively, at appropriate junctions of the synthesis following the aldol coupling with 5.
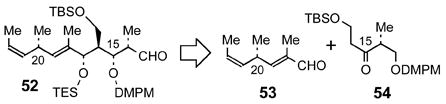 |
(2) |
We sought to construct the C(13)–C(23) framework of aldehyde 52 via the anti-aldol reaction of aldehyde 53 and ketone 54 (eq 2) using conditions developed by Paterson (enol borinate formation with c-Hex2BCl, Et3N in Et2O at 0 °C followed by aldehyde addition).53 This convergent assembly would set the C(16)- and C(17)-stereocenters in a single operation through a transition state believed to minimize electronic repulsion between the oxygen lone pairs of the enol borinate and the benzyl ether.53b We were encouraged by literature precedent showing that β-siloxy ethyl ketones participate in the anti-aldol reaction under Paterson’s conditions without β-elimination of the siloxy substituent.12c Furthermore, after the completion of this work,8a Kalesse and co-workers reported the successful anti-aldol coupling of aldehyde 53 with the C(13)-OPMB analog of ketone 54 under similar conditions.9a
Fragments 53 and 54 were synthesized by using easily scalable routes (Scheme 14). The synthesis of 539a,d commenced with the Wittig olefination of trityl-protected aldehyde 55 followed by cleavage of the trityl ether under acidic conditions. Swern oxidation of alcohol 56 afforded β,γ-unsaturated aldehyde 57. The stabilized ylide Ph3P=C(Me)CO2Et was added directly to the Swern reaction mixture to generate α,β-unsaturated ester 58 in a one-pot oxidation-olefination sequence. Reduction of the ester and subsequent MnO2 oxidation of the allylic alcohol then provided aldehyde 53. Ketone 54 was prepared from the DMPM-protected 6054 in four synthetic steps. Addition of the lithium enolate of tert-butyl acetate to 60 resulted in a β-hydroxy ester that was reduced to a 1,3-diol with LiAlH4. Selective silylation of the primary alcohol and oxidation of the secondary alcohol furnished the requisite ketone 54.
Scheme 14.

Synthesis of aldehyde 53 and ketone 54
Our early efforts to unify aldehyde 53 and ketone 54 using Paterson’s anti-aldol conditions led to 61 as the major of two diastereomers with 80:20 selectivity (Scheme 15). Literature precedent9a,12c suggested that 62 would be the minor product; however, Mosher ester analysis55 revealed that the C(17)-alcohol of the major and minor aldols both have identical (S)-configurations. Independent conversion of the two aldol products to their corresponding benzylidene acetals (of type 65) and acetonides (of type 66) revealed through spectroscopic analysis56 that both aldol adducts have identical stereochemistry within the C(14)–C(17) positions; that is, both aldol diastereomers are anti-aldol products with (S)-configurations at C(17)-OH.
Scheme 15.

Aldol coupling of 53 and 54
We speculated that 63 might be the correct structure of the minor aldol product, and that it might arise from epimerization of the bisallylic C(20)-stereocenter during the synthesis of aldehyde 53. This hypothesis was supported by our observation that only one diastereomer is formed from the aldol coupling of ketones 54 or 67 with methacrolein under identical conditions (eq 3). Furthermore, Mosher ester analysis of the primary alcohol 59 (precursor to 53, Scheme 14) revealed an 80:20 ratio of diastereomers at the C(20)-bisallylic stereocenter. We concluded that C(20)-epimerization was occurring during the oxidation-olefination sequence used in the conversion of alcohol 56 (>95:5 er as determined by Mosher ester analysis) to α,β-unsaturated ester 58. After vigorously investigating different oxidation and olefination conditions,8a we found that treatment of 56 with Dess-Martin periodinane at 0 °C, followed by ylide addition and olefination at 0 °C provided ester 58 (and subsequently aldehyde 53) with 94:6 enantiomeric ratio. When aldehyde 53 (of 94:6 er) was used in the anti-aldol reaction with 54, we obtained a 94:6 mixture of aldol diastereomers 61 and 63 (eq 4). It is remarkable that the aldol selectivity perfectly matched the enantiomeric purity of aldehyde 53. These experiments were also crucial to our ability to bring sufficient material forward en route to tedanolide.
 |
(3) |
 |
(4) |
The synthesis of C(13)–C(23) aldehyde 52 from 61 was completed in five steps (Scheme 16). Reduction of the β-hydroxy ketone 61 to the syn-1,3-diol with DIBAL and selective silylation57 of the allylic alcohol provided TES ether 70. The dimethoxybenzyl ether of 70 was transferred to the secondary alcohol via benzylidene acetal formation followed by regioselective acetal reductive opening with DIBAL. Finally, oxidation of primary alcohol 71 using the Dess-Martin periodinane provided aldehyde 52 in excellent yield.
Scheme 16.

Completion of the synthesis of aldehyde 52
Addition of the lithium enolate of ketone 5 to aldehyde 52 provided Felkin adduct 72 (as determined by NMR analysis37 and Mosher ester analysis55) in 77% yield with 75:25 selectivity (Scheme 17). The diastereomeric ratio for this transformation is similar to that observed previously for the aldol reaction of 2,3-syn aldehyde 49 (vide supra), and variations to the stoichiometry of aldehyde 52 (0.8–1.4 equiv) relative to ketone did not affect the yield (based on limiting reagent) or selectivity. At this point, we looked to protect the C(13)-alcohol of 72 before further elaborating the intermediate to the natural product. Treatment of 72 with TBSOTf and 2,6-lutidine in CH2Cl2 led to the product 73 as a mixture of TES and TBS ethers at the C(17)-position. It seemed unlikely that this substitution is due to the direct silylation of the C(17)-OTES oxygen, as the TES:TBS ratio at this position was unchanged when a mixture of product 73-TES and 73-TBS was resubjected to the silylation reaction conditions. However, attempts to suppress the scrambling of the silyl ether protecting group at this position were unsuccessful.
Scheme 17.

Aldol coupling of ketone 5 and aldehyde 52
Instead, protection of the C(13)-OH of 72 as a SEM ether proceeded without complication in quantitative yield (Scheme 18). At this point, we planned to convert the C(15)-dimethoxybenzyl ether to the C(15)-ketone via cleavage of the DMPM group and subsequent oxidation. Treatment of SEM-protected 72 with DDQ liberated the C(15)-(S)-OH of 74; however, hemiketal 75 was observed as the exclusive product by spectroscopic analysis. Furthermore, efforts to intercept the hydroxy ketone isomer 74 with oxidants (e.g., DMP, SO3-pyridine in DMSO) were unsuccessful, and we were unable to obtain the desired C(15)-ketone 76.
Scheme 18.

Attempted C(15)-oxidation of hemiketal 75
Our next course of action was to form the macrolactone prior to C(15)-oxidation (Scheme 19). We postulated that conformational constraints imposed by the macrocycle might destabilize the hemiketal, or even disfavor hemiketal formation from the hydroxy ketone. Therefore, the C(13)-alcohol of aldol adduct 72 was protected as a SEM ether, and the seco-acid was liberated via stepwise cleavage of the primary TBS ether (along with the C(17)-TES ether58) to provide 77, followed by removal of the allyl ester. Macrolactonization of this seco-acid was effected at room temperature using the Yonemitsu modification of the Yamaguchi protocol.39 The cyclization was completely selective for the primary alcohol (no reaction at the secondary C(17)-OH was observed)59 and the macrolactone was obtained in 32% overall yield from 72 (4 steps). Finally, the C(17)-OH was protected as a SEM ether to provide 78.
Scheme 19.

Completion of the total synthesis of (+)-tedanolide (1)
Treatment of 78 with DDQ liberated the C(15)-alcohol. Spectroscopic analysis revealed that 79 exists as the open chain hydroxy ketone, and we did not detect the hemiketal isomer. We were gratified that treatment of 79 with Dess-Martin periodinane afforded the C(11,15)-dione 80 in 71% yield over two steps from DMPM ether 78. The synthesis of (+)-tedanolide (1) was then completed in three steps. Removal of the two SEM ethers was accomplished by using MgBr2 and EtSH in Et2O60 (as previously demonstrated by Smith10a). C(17)-Hydroxyl-directed epoxidation of the C(18)–C(19) olefin with m-CPBA12c gave a single epoxide diastereomer in 54% yield. Finally, global desilylation of the penultimate intermediate with Et3N-buffered Et3N-3HF46 provided synthetic (+)-tedanolide. The spectroscopic data (1H NMR, 13C NMR, IR, optical rotation, HRMS) from our synthetic tedanolide were in excellent agreement with data obtained for an authentic sample,1 as well as with data reported by Kalesse9a,b and Smith10a for synthetic tedanolide.
Summary
Convergent, stereocontrolled total syntheses of (+)-tedanolide and (+)-13-deoxytedanolide have been accomplished. A key transformation for the assembly of both natural products is the aldol coupling of the C(1)–C(12) methyl ketone 5 with a C(13)–C(23) aldehyde partner. Aldehyde 6 with 2,3-anti stereochemistry was initially employed, and its aldol reaction with the lithium enolate generated from 5 proceeded with >95:5 selectivity for the Felkin aldol 4. However, it proved impossible to elaborate 4 to tedanolide owing to the remarkable stability of hemiketals 33b and 34 and especially their lack of reactivity toward oxidants. Deoxygenation of hemiketal 33a provided the 13-deoxy hemiketal 36 which was successfully oxidized to the C(11,15)-diketone 38 by using the Dess-Martin periodinane reagent. The later intermediate was smoothly elaborated to (+)-13-deoxytedanolide.
Recognition that 13-deoxy hemiketal 36 could be oxidized by way of its hydroxy ketone tautomer 37 suggested that the remarkable stability and lack of reactivity of hemiketals 33b and 34 might be due to the equatorial conformation of ring substituents in 33b and 44 which stabilizes the hemiketal relative to the acyclic hydroxy ketone isomer. This then led to the decision to redesign the synthesis of tedanolide to proceed by way of the stereochemically inverted C(15)-(S)-alcohol intermediates, since hemiketal intermediates with C(15)-(S) series would be destabilized by having at least one large substituent in an axial position (Scheme 10). However, use of C(15)-(S)-configured intermediates also required that the side chain epoxide unit be introduced very late in the synthesis, owing to the ease with which C(15)-(S)-OH cyclized onto the epoxide in intermediate 50 (Scheme 13).
Therefore, 2,3-syn aldehyde 52 was employed in the aldol coupling with methyl ketone 5 which gave the Felkin aldol 72 with 75:25 selectivity. Elaboration of Felkin aldol 72 to macrolactone 78 set the stage for removal of the C(15)-DMPM ether and oxidation of the C(15)-OH to the required ketone in 80. Fortunately, the hemiketal that plagued the unsuccessful tedanolide syntheses via C(15)-(R)-configured intermediates 33b and 34 was not observed for the C(15)-(S)-hydroxy ketone 79 derived from 78. Intermediate 80 was smoothly elaborated to tedanolide via deprotection of C(17)-OH, which was used to direct the epoxidation of the adjacent C(18,19)-olefin. Final, global cleavage of all silyl ethers then completed the total synthesis of (+)-tedanolide.
Supplementary Material
Supporting Information Available: Experimental procedures and spectroscopic data and spectra for selected intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.
Figure 1.

The tedanolide natural products
Acknowledgments
Support from the National Institutes of Health (GM 038436) and a Bristol-Myers Squibb Graduate Fellowship to L.D.J. is gratefully acknowledged. We thank Gregory C. Lane for preliminary work8c involving the synthesis of alcohol 25a from aldehyde 22 (Scheme 5). We also thank Professor Schmitz for supplying an authentic sample of (+)-tedanolide, and Professors Fusetani and Matsunaga for supplying an authentic sample of (+)-13-deoxytedanolide.
References
- 1.Schmitz FJ, Gunasekera SP, Yalamanchili G, Hossain MB, van der Helm D. J Am Chem Soc. 1984;106:7251. [Google Scholar]
- 2.Schmitz, F. J.; Gunasekera, S. P.; Hossain, M. B.; van der Helm, D.; Yalamanchili, G. (1988) U.S. Patent Application 87-7347.
- 3.Fusetani N, Sugawara T, Matsunaga S. J Org Chem. 1991;56:4971. [Google Scholar]
- 4.(a) Nishimura S, Matsunaga S, Yoshida M, Hirota H, Yokoyama S, Fusetani N. Bioorg Med Chem. 2005;13:449. doi: 10.1016/j.bmc.2004.10.012. [DOI] [PubMed] [Google Scholar]; (b) Nishimura S, Matsunaga S, Yoshida S, Nakao Y, Hirota H, Fusetani N. Bioorg Med Chem. 2005;13:455. doi: 10.1016/j.bmc.2004.10.014. [DOI] [PubMed] [Google Scholar]
- 5.For structure-activity relationship studies of the structurally-related myriaporones 3 and 4: Hines J, Roy M, Cheng H, Agapakis CM, Taylor R, Crews CM. Mol BioSyst. 2006;2:371. doi: 10.1039/b602936a.
- 6.Chevallier C, Bugni TS, Feng X, Harper MK, Orendt AM, Ireland CM. J Org Chem. 2006;71:2510. doi: 10.1021/jo052285+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Total synthesis of (+)-13-deoxytedanolide: Julian LD, Newcom JS, Roush WR. J Am Chem Soc. 2005;127:6186. doi: 10.1021/ja050729d.
- 8.Previous efforts toward tedanolide: Dunetz JR, Roush WR. Org Lett. 2008;10:2059. doi: 10.1021/ol800546g.Roush WR, Newcom JS. Org Lett. 2002;4:4739. doi: 10.1021/ol0272343.Roush WR, Lane GC. Org Lett. 1999;1:95. doi: 10.1021/ol990572s.
- 9.(a) Ehrlich G, Hassfeld J, Eggert U, Kalesse M. Chem Eur J. 2008;14:2232. doi: 10.1002/chem.200701529. [DOI] [PubMed] [Google Scholar]; (b) Ehrlich G, Hassfeld J, Eggert U, Kalesse M. J Am Chem Soc. 2006;128:14038. doi: 10.1021/ja0659572. [DOI] [PubMed] [Google Scholar]; (c) Hassfeld J, Eggert U, Kalesse M. Synthesis. 2005:1183. [Google Scholar]; (d) Ehrlich G, Kalesse M. Synlett. 2005:655. [Google Scholar]; (e) Hassfeld J, Kalesse M. Synlett. 2002:2007. [Google Scholar]
- 10.(a) Smith AB, III, Lee D. J Am Chem Soc. 2007;129:10957. doi: 10.1021/ja073329u. [DOI] [PubMed] [Google Scholar]; (b) Cho CG, Kim WS, Smith AB., III Org Lett. 2005;7:3569. doi: 10.1021/ol051376q. [DOI] [PubMed] [Google Scholar]; (c) Smith AB, III, Adams CM, Lodise Barbosa SA, Degnan AP. Proc Natl Acad Sci USA. 2004;101:12042. doi: 10.1073/pnas.0402084101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Smith AB, III, Adams CM, Lodise Barbosa SA, Degnan AP. J Am Chem Soc. 2003;125:350. doi: 10.1021/ja0289649. [DOI] [PubMed] [Google Scholar]; (e) Smith AB, III, Lodise SA. Org Lett. 1999;1:1249. doi: 10.1021/ol9909233. [DOI] [PubMed] [Google Scholar]
- 11.(a) Jung ME, Zhang T-h. Org Lett. 2008;10:137. doi: 10.1021/ol702729u. [DOI] [PubMed] [Google Scholar]; (b) Jung ME, Yoo D. Tetrahedron Lett. 2008;49:816. doi: 10.1016/j.tetlet.2008.10.039. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Jung ME, Yoo D. Org Lett. 2007;9:3543. doi: 10.1021/ol0714038. [DOI] [PubMed] [Google Scholar]; (d) Jung ME, Lee CP. Org Lett. 2001;3:333. doi: 10.1021/ol000329p. [DOI] [PubMed] [Google Scholar]; (e) Jung ME, Lee CP. Tetrahedron Lett. 2000;41:9719. [Google Scholar]; (f) Jung ME, Marquez R. Org Lett. 2000;2:1669. doi: 10.1021/ol005675l. [DOI] [PubMed] [Google Scholar]; (g) Jung ME, Marquez R. Tetrahedron Lett. 1999;40:3129. [Google Scholar]
- 12.(a) Wong CM, Loh TP. Tetrahedron Lett. 2006;47:4485. [Google Scholar]; (b) Loh TP, Feng LC. Tetrahedron Lett. 2001;42:6001. [Google Scholar]; (c) Loh TP, Feng LC. Tetrahedron Lett. 2001;42:3223. [Google Scholar]
- 13.(a) Matsui K, Zheng B-Z, Kusaka S-i, Kuroda M, Yoshimoto K, Yamada H, Yonemitsu O. Eur J Org Chem. 2001:3615. [Google Scholar]; (b) Zheng B-Z, Yamauchi M, Dei H, Kusaka S-i, Matsui K, Yonemitsu O. Tetrahedron Lett. 2000;41:6441. [Google Scholar]; (c) Matsushima T, Nakajima N, Zheng BZ, Yonemitsu O. Chem Pharm Bull. 2000;48:855. doi: 10.1248/cpb.48.855. [DOI] [PubMed] [Google Scholar]; (d) Zheng B-Z, Maeda H, Mori M, Kusaka S-i, Yonemitsu O, Matsushima T, Nakajima N, Uenishi J-i. Chem Pharm Bull. 1999;47:1288. doi: 10.1248/cpb.47.1288. [DOI] [PubMed] [Google Scholar]; (e) Matsushima T, Mori M, Zheng B-Z, Maeda H, Nakajima N, Uenishi J-i, Yonemitsu O. Chem Pharm Bull. 1999;47:308. doi: 10.1248/cpb.47.308. [DOI] [PubMed] [Google Scholar]; (f) Matsushima T, Zheng B-Z, Maeda H, Nakajima N, Uenishi J-i, Yonemitsu O. Synlett. 1999:780. doi: 10.1248/cpb.47.308. [DOI] [PubMed] [Google Scholar]; (g) Matsushima T, Mori M, Nakajima N, Maeda H, Uenishi J-i, Yonemitsu O. Chem Pharm Bull. 1998;46:1335. doi: 10.1248/cpb.47.308. [DOI] [PubMed] [Google Scholar]; (h) Matsushima T, Horita K, Nakajima N, Yonemitsu O. Tetrahedron Lett. 1996;37:385. [Google Scholar]
- 14.Liu JF, Abiko A, Pei Z, Buske DC, Masamune S. Tetrahedron Lett. 1998;39:1873. [Google Scholar]
- 15.Iwata Y, Tanino K, Miyashita M. Org Lett. 2005;7:2341. doi: 10.1021/ol050569a. [DOI] [PubMed] [Google Scholar]
- 16.(a) Taylor RE, Hearn BR, Ciavarri JP. Org Lett. 2002;4:2953. doi: 10.1021/ol026356s. [DOI] [PubMed] [Google Scholar]; (b) Taylor RE, Ciavarri JP, Hearn BR. Tetrahedron Lett. 1998;39:9361. [Google Scholar]
- 17.(a) Nyavanandi VK, Nadipalli P, Nanduri S, Naidu A, Iqbal J. Tetrahedron Lett. 2007;48:6905. [Google Scholar]; (b) Nyavanandi VK, Nanduri S, Dev RV, Naidu A, Iqbal J. Tetrahedron Lett. 2006;47:6667. [Google Scholar]
- 18.Gustin DJ, VanNieuwenhze MS, Roush WR. Tetrahedron Lett. 1995;36:3443. [Google Scholar]
- 19.Taber DF, Silverberg LJ. Tetrahedron Lett. 1991;32:4227.For a review: Noyori R, Ohkuma T. Angew Chem Int Ed. 2001;40:40.
- 20.Fráter G, Müller U, Günther W. Tetrahedron. 1984;40:1269. [Google Scholar]
- 21.For a review: Mancuso AJ, Swern D. Synthesis. 1981:165.
- 22.(a) Keck GE, Abbott DE, Wiley MR. Tetrahedron Lett. 1987;28:139. [Google Scholar]; (b) Evans DA, Dart MJ, Duffy JL, Yang MG. J Am Chem Soc. 1996;118:4322. [Google Scholar]
- 23.Evans DA, Ratz AM, Huff BE, Sheppard GS. Tetrahedron Lett. 1994;35:7171. [Google Scholar]
- 24.Lindgren BO, Nilsson T. Acta Chem Scand. 1973;27:888.For a modification using 2-methyl-2-butene as chlorine scavenger: Kraus GA, Taschner MJ. J Org Chem. 1980;45:1175.
- 25.For a review: Mitsunobu O. Synthesis. 1981:1.
- 26.Oikawa Y, Tanaka T, Horita K, Yoshioka T, Yonemitsu O. Tetrahedron Lett. 1984;25:5393. [Google Scholar]
- 27.A sample of secondary alcohol in CDCl3 (treated with K2CO3) completely converted to lactone 16 within 20 minutes, as observed by 1H NMR analysis.
- 28.Available from methyl acetoacetate (see ref. 20) via a sequence of asymmetric hydrogenation and alkylation that parallels the conversion of 9 to 10 (Scheme 2).
- 29.Evans DA, Rieger DL, Bilodeau MT, Urpi F. J Am Chem Soc. 1991;113:1047. [Google Scholar]
- 30.Griffith WP, Ley SV, Whitcombe GP, White AD. J Chem Soc, Chem Commun. 1987:1625.For a review: Ley SV, Norman J, Griffith WP, Marsden SP. Synthesis. 1994:639.
- 31.Harried SS, Lee CP, Yang G, Lee TIH, Myles DC. J Org Chem. 2003;68:6646. doi: 10.1021/jo034521r. [DOI] [PubMed] [Google Scholar]
- 32.For a review: Katsuki T, Martin VS. Org React. 1996;48:1.
- 33.(a) Evans DA, Bartroli J, Shih TL. J Am Chem Soc. 1981;103:2127. [Google Scholar]; (b) Evans DA, Sjogren EB, Bartroli J, Dow RL. Tetrahedron Lett. 1986;27:4957. [Google Scholar]
- 34.Penning TD, Djuric SW, Haack RA, Kalish VJ, Miyashiro JM, Rowell BW, Yu SS. Synth Commun. 1990;20:307. [Google Scholar]
- 35.Roush WR, Palkowitz AD, Ando K. J Am Chem Soc. 1990;112:6348. [Google Scholar]
- 36.Dess DB, Martin JC. J Org Chem. 1983;48:4155. [Google Scholar]
- 37.The stereochemistry was assigned by using NMR methods: Roush WR, Bannister TD, Wendt MD, VanNieuwehnze MS, Gustin DJ, Dilley GJ, Lane GC, Scheidt KA, Smith WJ., III J Org Chem. 2002;67:4284. doi: 10.1021/jo0164148.
- 38.Dangles O, Guibé F, Balavoine G, Lavielle S, Marquet A. J Org Chem. 1987;52:4984. [Google Scholar]
- 39.For a macrolactonization review: Parenty A, Moreau X, Campagne J-M. Chem Rev. 2006;106:911. doi: 10.1021/cr0301402.
- 40.For a review: Luzzio FA. Org React. 1998;53:1.
- 41.Corey EJ, Schmidt G. Tetrahedron Lett. 1979;20:399. [Google Scholar]
- 42.Beesley RM, Ingold CK, Thorpe JF. J Chem Soc. 1915;107:1080. [Google Scholar]
- 43.Implicit to this assumption is the recognition that the equilibrium constant for cyclization of the hydroxy ketone to the hemiketal is determined by the ratio of the rates of formation and breakdown of the hemiketal intermediate. Thus, while the Thorpe-Ingold effect is kinetic in nature, it may also impact the equilibrium constant of reversible processes.
- 44.Barton DHR, Jaszberenyi JC. Tetrahedron Lett. 1989;30:2619. [Google Scholar]
- 45.Nozaki K, Oshima K, Utimoto K. Tetrahedron Lett. 1988;29:6125. [Google Scholar]
- 46.(a) Giudicelli MB, Picq D, Veyron B. Tetrahedron Lett. 1990;31:6527. [Google Scholar]; (b) McClinton MA. Aldrichimica Acta. 1995;28:31. [Google Scholar]
- 47.Julian LD. Studies toward the total synthesis of the tedanolides: Total synthesis of 13-deoxytedanolide. University of Michigan; 2005. Ph.D. thesis. [Google Scholar]
- 48.While this work was in progress, Kalesse reported experimental evidence for this hypothesis (refs 9a and 9b). Kalesse credits us for this analysis (ref 9b).
- 49.Brown HC, Jadhav PK. J Am Chem Soc. 1983;105:2092. [Google Scholar]
- 50.(a) Evans DA, Duffy JL, Dart MJ. Tetrahedron Lett. 1994;35:8537. [Google Scholar]; (b) Evans DA, Dart MJ, Duffy JL, Yang MG. J Am Chem Soc. 1996;118:4322. [Google Scholar]
- 51.Loh and co-workers induced a similar cyclization to analyze epoxide stereochemistry in their tedanolide fragment synthesis (ref 12c).
- 52.For a review of hydroxy directed epoxidations of allylic alcohols: Adam W, Wirth T. Acc Chem Res. 1999;32:703.
- 53.(a) Paterson I, Goodman JM, Isaka M. Tetrahedron Lett. 1989;30:7121. [Google Scholar]; (b) Vulpetti A, Bernardi A, Gennari C, Goodman JM, Paterson I. Tetrahedron. 1993;49:685. Mechanistic discussion: [Google Scholar]
- 54.Prepared in analogous fashion to the known PMB-protected aldehyde: Organ MG, Wang J. J Org Chem. 2003;68:5568. doi: 10.1021/jo034371k.
- 55.Dale JA, Mosher HS. J Am Chem Soc. 1973;95:512.For a useful explanation and examples of Mosher ester analysis: Ohtani I, Kusumi T, Kashman Y, Kakisawa H. J Am Chem Soc. 1991;113:4092.
- 56.Acetonide analysis: Rychnovsky SD, Rogers BN, Richardson TI. Acc Chem Res. 1998;31:9.
- 57.Hicks JD, Huh CW, Legg AD, Roush WR. Org Lett. 2007;9:5621. doi: 10.1021/ol702588h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.We were unable to remove the primary TBS ether without also cleaving the TES ether.
- 59.Similar selectivity was also observed by Smith and co-workers in their syntheses of tedanolide and 13-deoxytedanolide (refs 10a, c, d).
- 60.Kim S, Kee IS, Park YH, Park JH. Synlett. 1991:183. SYNOPSIS TOC (Word Style “SN_Synopsis_TOC”) [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information Available: Experimental procedures and spectroscopic data and spectra for selected intermediates. This material is available free of charge via the Internet at http://pubs.acs.org.