

CONSPECTUS
 Self-assembly of small molecules into one-dimensional nanostructures offers many potential applications in electronically and biologically active materials. The recent advances discussed in this Account demonstrate how researchers can use the fundamental principles of supramolecular chemistry to craft the size, shape, and internal structure of nanoscale objects. In each system described here, we used atomic force microscopy (AFM) and transmission electron microscopy (TEM) to study the assembly morphology. Circular dichroism, nuclear magnetic resonance, infrared, and optical spectroscopy provided additional information about the self-assembly behavior in solution at the molecular level.
Self-assembly of small molecules into one-dimensional nanostructures offers many potential applications in electronically and biologically active materials. The recent advances discussed in this Account demonstrate how researchers can use the fundamental principles of supramolecular chemistry to craft the size, shape, and internal structure of nanoscale objects. In each system described here, we used atomic force microscopy (AFM) and transmission electron microscopy (TEM) to study the assembly morphology. Circular dichroism, nuclear magnetic resonance, infrared, and optical spectroscopy provided additional information about the self-assembly behavior in solution at the molecular level.
Dendron rod–coil molecules self-assemble into flat or helical ribbons. They can incorporate electronically conductive groups and can be mineralized with inorganic semiconductors. To understand the relative importance of each segment in forming the supramolecular structure, we synthetically modified the dendron, rod, and coil portions. The self-assembly depended on the generation number of the dendron, the number of hydrogen-bonding functions, and the length of the rod and coil segments. We formed chiral helices using a dendron–rod–coil molecule prepared from an enantiomerically enriched coil.
Because helical nanostructures are important targets for use in biomaterials, nonlinear optics, and stereoselective catalysis, researchers would like to precisely control their shape and size. Tripeptide-containing peptide lipid molecules assemble into straight or twisted nanofibers in organic solvents. As seen by AFM, the sterics of bulky end groups can tune the helical pitch of these peptide lipid nanofibers in organic solvents. Furthermore, we demonstrated the potential for pitch control using trans-to-cis photoisomerization of a terminal azobenzene group. Other molecules called peptide amphiphiles (PAs) are known to assemble in water into cylindrical nanostructures that appear as nanofiber bundles. Surprisingly, TEM of a PA substituted by a nitrobenzyl group revealed assembly into quadruple helical fibers with a braided morphology. Upon photocleavage of this the nitrobenzyl group, the helices transform into single cylindrical nanofibers.
Finally, inspired by the tobacco mosaic virus, we used a dumbbell-shaped, oligo(phenylene ethynylene) template to control the length of a PA nanofiber self-assembly (<10 nm). AFM showed complete disappearance of long nanofibers in the presence of this rigid-rod template. Results from quick-freeze/deep-etch TEM and dynamic light scattering demonstrated the templating behavior in aqueous solution. This strategy could provide a general method to control size the length of non-spherical supramolecular nanostructures.
Introduction
Self-assembly spontaneously creates structures or patterns with a significant order parameter out of disordered components. The definition of spontaneity within the chemical field of self-assembly is that components which create the ordered structure achieve a specific outcome with minimal human or machine intervention. Although it is often useful for achieving highly ordered, thermodynamically stable systems, reversibility is not a strict requirement for self-assembly of the disorder-to-order transformation. In fact, irreversible self-assembly under non-equilibrium conditions is no less interesting or useful as long as the resulting systems are sufficiently ordered. Our criterion of “no human intervention” is a matter of degree; some processes are “templated” in a structural sense, and others must be “directed” by applied external forces. Self-assembling monolayers of thiols on gold surfaces are exemplary of templated self-assembly,1,2 whereas directed self-assembly could involve the application of external magnetic,3 electric,4 or flow fields5 to induce or nucleate self-assembly. We are interested in programming through molecular structure the self-assembly of nanostructures and their hierarchical organization across scales to create function. Our previous work has covered targets such as two-dimensional structures,6 zero-dimensional noncentrosymmetric nanostructures,7 and one-dimensional nanoscale objects.8,9 Our vision for hierarchical self-assembly across scales is exemplified by very recent work on three-dimensional macroscopic sacs and membranes through interactions among large and small molecules.10 In the context of molecular programming for self-assembly, the objective for the supramolecular chemist is to create nano- or macroscale structures composed of small molecules with precise control over size, shape, and spatial resolution or periodicity. A variety of biological systems provide an amazing level of control over these features in the assembly of complex, functional architectures like actin filaments,11 bacteriophages,11 and the ribosome.12,13 Early examples of designed self-assembly in systems such as helicates14 and melamine–cyanurate spheres15 focused on structure not function. More recent examples have gone further to demonstrate function in self-assembling organic structures, for example, piezoelectricity in our noncentrosymmetric clusters,16 and a significant increase in conductivity in hexabenzocoronene nanotubes17 and dendron–rod–coil ribbons.18 Meijer and co-workers have demonstrated both energy transfer19 and photoinduced electron transfer20 in supramolecular oligo-(phenylene vinylene) assemblies. Additionally, functional groups with known biological functionality can be appended to supramolecular structures to afford biomaterials with specific targets. We have found this to be particularly useful for the controlled growth and development of neuronal and vascular tissues.21-24 These bioactive groups appear to be presented in a highly favorable orientation and density for biological recognition. One-dimensional (1D) assemblies, such as nanotubes and nanoribbons,25 possess a single dimension that is much longer than the others. As with the examples above, 1D nanoscale structures exhibit a number of useful properties, including potential for alignment, conductivity, and biological interactions. As these 1D structures entangle, they behave like linear polymers by entrapping and slowing diffusion of solvent molecules. Materials of this type are observed macroscopically as self-supporting gels.26,27 Furthermore, helical structures are of particular interest because of their use in biomaterials, nonlinear optics,28 and stereoselective catalysis29 and, therefore, represent important targets for precise control of shape and length in supramolecular nanostructures. A number of synthetic systems, such as foldamers,30,31 have been developed that mimic the predictable tendency of proteins to fold into helical secondary or tertiary structures. However, the length of foldamers is limited by the significant synthetic effort required for their preparation. Self-assembly offers an alternative strategy to access helical structures with larger dimensions by effectively designing the periodic twisting of large arrays of molecules.32 One could view this process as “supramolecular folding”.
Functional Nanoribbons from Dendron–Rod–Coil Molecules
Inspired by our previous work on molecules termed rod–coils that combined rigid and flexible segments,7,33 we designed a related modular structure termed the dendron–rod–coil (DRC) molecule.9 These molecules incorporate a dendritic block34 at the end of the rod segment. DRC 1 formed a self-supporting gel in dichloromethane at concentrations as low as 0.2 wt % (Figure 1).9 Transmission electron microscopy (TEM) and atomic force microscopy (AFM) indicate the formation of high aspect ratio supramolecular ribbons that are ∼10 nm across and ∼2 nm thick. Other microscopic changes, such as 1H NMR line broadening, are also observed under assembly conditions.
FIGURE 1.

1D assemblies of DRC molecules: (a) TEM microscopy of the nanofiber structure; (b) molecular graphics representation of the nanofiber ribbon. Adapted with permssion from ref 9. Copyright 2001 American Chemical Society.
To understand the relative importance of the each segment in forming the supramolecular structure, we systematically modified each of the dendron, rod, and coil portions through synthesis (Figure 2).35
FIGURE 2.

Chemical structure of DRC 1 and structural modifications, such as the number of hydrogen-bonding groups on the dendron (blue) and changes to the length of the rod (red).
Dendron
The DRC terminated with a G1 dendron (1) forms ribbon assemblies with head-to-head hydrogen bonds between the dendron segments of adjacent molecules. Higher-generation dendrons (G2–G4) afforded only isolated aggregates by TEM. Presumably, these bulkier dendrons inhibit self-assembly. The number of available hydrogen-bonding groups on the dendron also affected the assembly. Monomers with no hydroxyls give isotropic solutions; those with two hydroxyls did not form the ribbon assemblies and instead precipitate at room temperature. Compounds displaying four or six hydroxyls per monomer yielded purple gels in nonpolar solvents. Small-angle X-ray scattering (SAXS) confirmed that the crystalline order of the assembly increased with the number of hydrogen-bonding groups. This rigid portion likely offsets some of the entropic penalty of bringing so many molecules together. To more precisely understand the specific intermolecular interactions between the dendron units, we obtained an X-ray crystal structure of a partial dendron–rod (2).9 In the crystalline state, the dendrons formed cyclic tetramers held together by four hydrogen bonds and eight additional hydrogen bonds linked adjacent tetramers into a stacked array (Figure 3).
FIGURE 3.

In the crystalline state, the dendron–rod 2 crystallizes as a stack of cyclic tetramers. Hydrogen bonds between adjacent tetramers are indicated by dashed lines. Reproduced with permission from ref 9. Copyright 2001 American Chemical Society.
Rod
A single biphenyl ester in the rod portion gives a viscous, isotropic solution. A rod portion with two biphenyl esters affords a weak gel. As the rod portion is further extended to three or four biphenyl ester units, the resulting purple bire-fringent gels form self-supporting gels. Solid-state SAXS studies indicate that the longer rods result in bilayer structures with sharper peaks and larger d spacings (110 Å) compared with the single biphenyl ester (62 Å). Thus, the mechanical properties and crystallinity are affected similarly by the rod length. As discussed below, the oligophenylene unit can also be replaced with other rigid aromatic groups to afford greater functionality.
Coil
The coil segment is essential to solubilize the DRC molecules. In addition to the polyisoprene coil of compound 2, the assembly also forms with a shorter, branched 2-octyldodecyl coil.9 A shorter, linear tail (e.g., n-dodecyl) results in only insoluble material.9
Despite these structural requirements, we have found that the assembly is still quite robust to other mutations, including the introduction of functional components. For example, the rigid oligo(biphenyl ester) rod was replaced with oligothiophenes and oligo(phenylene vinylene)s.18 The conductivity of I2-doped films of the oligothiophene 3 (Figure 4) increases from 8.0 × 10−8 S/cm when unassembled to 7.9 × 10−5 S/cm in the assembled state. In contrast, the biphenyl ester rods remain completely insulating.
FIGURE 4.

Chemical structure of oligothiophene-containing DRC 3.
Oligo(biphenyl ester)s (R)-4 and (S)-4 were synthesized with enantiomerically enriched coils and were found to self-assemble into twisted ribbons with opposite handedness as shown by AFM (Figure 5).36 The circular dichroism (CD) spectrum of the enantiopure DRC in acetonitrile showed π–π* transitions by the achiral biphenyls, indicating chiral induction by the coil and exciton coupling between adjacent biphenyls. Even a slight enantiomeric excess shows a strong CD response, suggesting a “majority rules” behavior in which the overall chirality of the system is determined by the component in excess.37,38 No signal is observed in THF (nonassembling solvent) or in dichloromethane (a solvent that induces the formation of only flat ribbons). This supramolecular chirality could be exploited as a scaffold for chiral catalysis.
FIGURE 5.

(a) Molecular structure of chiral DRC 4, (b) AFM images of (R)-4 (left) and (S)-4 (right) drop cast on mica from acetonitrile solutions–the horizontal and vertical scale bars are identical for both images, and the white lines indicate handedness, and (c) the CD signal at 300 nm is plotted as a function of mole percent (S)-4. Adapted with permission from ref 36. Copyright 2005 American Chemical Society.
The ribbon formed from DRC 1 was used as a template for the mineralization of nanohelical 1D cadmium sulfide semi-conductors.39 By TEM, the mineralized helices showed widths of 35–100 nm, pitches of 60 nm, and lengths of several micrometers. The unmineralized assembly of the same compound gave nanohelices with a 20 nm pitch. These nanoribbons were also found to disperse ZnO, TiO2, and CdS semiconductor nanoparticles quite well compared with simple polymers like poly(vinyl alcohol). The hybrid assemblies with ZnO nanocrystals were aligned parallel to the poling direction of an applied electric field (dc, 1500 V/cm).40 The poled orientation was stable even after annealing to 100 °C for 24 h. The resulting material showed strongly anisotropic emission and ultraviolet lasing with a lower threshold than that for pure ZnO nanocrystals placed directly between two glass slides or for unpoled films.
Controlling Pitch in Supramolecular Helices Using Mechanical Torque
Controlling the architecture of helical structures remains a great challenge.41,42 Nolte et al. have shown that disk-shaped crown ether–phthalocyanines can assemble into one-dimensional coiled-coil aggregates.43 In that system, the helicity of the nanostructure can be switched on or off in response to potassium ions, but the degree of helicity is fixed by the geometric structure of the molecule. Recently, we showed that it is possible to tune the pitch of helical self-assemblies by inducing mechanical forces on the nanoscale though molecular design.32
Peptide–lipid (PL) compounds 5–15 represent a class of molecules composed of a simple surfactant molecule with a short peptide sequence in the middle (Table 1).44 The amino acids have the propensity to form intermolecular β-sheets, causing the formation of nanofibers in nonpolar solvents such as benzene and hexane.44 During our recent work on functionalizing these peptide–lipid molecules to template various materials,45 we found that attaching a bulky group to the end of one of the alkyl tails results in helical structure.32
TABLE 1.
 | |||
|---|---|---|---|
| Compounda | R | Pitch (nm)b | Van der Waals Volume (Å3) |
| 5 |  |
∞ | 58.1 |
| 6 |  |
22 ± 2 | 92.7 |
| 7 |  |
22 ± 2 | 110.0 |
| 8 |  |
∞ | 113.4 |
| 9 |  |
160 ± 30 | 130.7 |
| 10 |  |
40 ± 3 | 148.0 |
| 11 |  |
22 ± 2 | 165.3 |
| 12 |  |
22 ± 2 | 182.6 |
| 13 |  |
∞ | 104.4 |
| 14 |  |
90 ± 10 | 218.2 |
| 15 |  |
90 ± 10 | 235.5 |
| 16 | 74 ± 6 | 292.3 | |
All compounds were synthesized using l-amino acids.
The pitch values are averages obtained from AFM height images corresponding to tens of nanostructures.
At a concentration of 1 wt %, PLs 5 and 6 each dissolve in chlorocyclohexane at ∼80 °C and form a translucent self-supporting gel upon cooling to ambient temperature. These gels were diluted, dried on silicon substrates, and imaged by AFM. The micrographs revealed straight cylindrical fibers for compound 5, whereas the aggregates formed from 6 show left-handed helices with regular pitch of 22 nm (Figure 6). This dramatic change in morphology resulted from a rather subtle change in molecular structure, suggesting that the bulkier substituent at the terminus of the alkyl segment was responsible for the twisting of the cylindrical assemblies of 6.
FIGURE 6.

AFM images of the aggregates formed by compounds (a) 5 and (b) 6 in chlorocyclohexane. The height profile shows that these helices have a pitch of 22 ± 2 nm. Scale bars = 100 nm. Adapted with permission from ref 32. Copyright 2007 Wiley-VCH.
In a nonpolar solvent, the amphiphilic molecules assemble as a result of both solvophobicity of the charged tetraalkylammonium head groups and the formation of intermolecular hydrogen bonds between adjacent peptide segments.44 Infrared spectroscopy confirms parallel β-sheet formation. The cylindrical assemblies formed by assembly of 6 are probably “superhelical” (Figure 7). The β-sheets define the primary helices within the one-dimensional nanofiber. Torsional strain caused by steric repulsion among the bulky substituents may overwind the β-sheets. The axis of the β-sheets likely relaxes into a superhelix to accommodate the bulky end groups without disrupting the hydrogen-bonded network (Figure 7, left). The observed left-handed sense of the nanostructures formed by compound 6 is consistent with a superhelix formed by overwinding a left-handed primary helix (Figure 7, right).46 However, we are unable to directly observe the primary helix with the resolution available by AFM.
FIGURE 7.

Steric interactions between the bulky end groups on the periphery of the nanostructure (left, green spheres) cause a torque, which is relieved (right) as the nanofiber forms a superhelix. Adapted with permission from ref 32. Copyright 2007 Wiley-VCH.
To explore the scope of this torque-induced approach, we synthesized three series of compounds, 5–7, 8–12, and 13–15, covering a wide range of steric bulkiness (Table 1).32 All of these compounds formed self-supporting gels in chlorocyclohexane. Straight cylindrical fibers were considered as having infinite pitch. Within each of the series, the helical pitch decreases with increasing end group size. This trend is qualitatively consistent with the van der Waals theory of liquids and solids 47 in which molecular arrangements are primarily determined by the steric effects that result from short-ranged repulsive forces. Interestingly, the series 5–7 and 8–12 appear to have the same lower limit (∼22 nm), despite the structural differences of their end groups. This limiting value may result from a geometric requirement to maintain the β-sheets while allowing supercoiling of the nanofiber.
The helical pitch also appears to depend on the shape and packing of end groups, as seen when comparing compounds between series. For example, the phenyl groups in compound 8 are able to π–π stack and thereby reduce torsional strain in the assembly, resulting in an infinitely large pitch. Similarly, the nearly flat benzophenone 14 also displayed a large pitch (90 nm). The bulkier substituted aromatic end groups of 11 and 12 cause greater strain and reduce the observed pitch (22 nm).
To demonstrate the potential utility for actuation or sensing, we constructed a system in which the pitch could be controlled reversibly.32 Irradiating a chlorocyclohexane suspension of nanofibers made of peptide–lipid 16 induces the trans-to-cis isomerization of the azobenzene group. Because the cisisomer is less planar than the trans-isomer, isomerization should increase the sterically induced torque and reduce the superhelical pitch. Indeed, the light-induced isomerization led to a smaller pitch of the supramolecular helices by AFM (Figure 8). This photoinduced pitch reduction varies over the sample, suggesting an inhomogeneous degree of isomerization between different nanostructures. However, the pitch is constant within a particular nanostructure, indicating that the torque-induced actuation is the result of a relaxation process that acts uniformly over the entire assembly.
FIGURE 8.
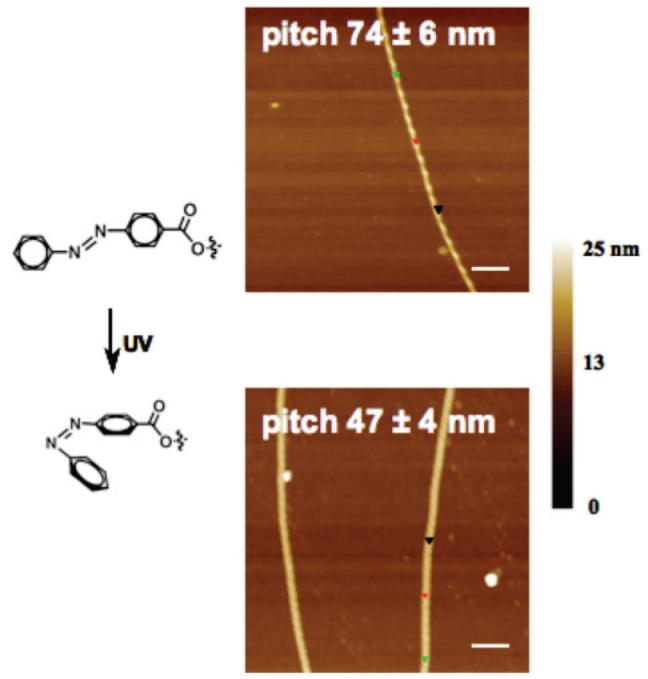
Reversible control of superhelical pitch by trans-cis photoisomerization of peptide lipid 16. Adapted with permission from ref 32. Copyright 2007 Wiley-VCH.
Photoinduced Dissociation of Quadruple Helix into Single Fibers
We have also explored the torque-mediated strategy with the self-assembling, water-soluble peptide amphiphiles (PAs) previously developed in our laboratory.8 These molecules self-assemble into nanofibers as a result of hydrophobic collapse of the alkyl chains and β-sheet formation among the peptide segments. PAs 17 and 18 were synthesized containing a palmitoyl tail and the oligopeptide segment GV3A3E3.48 The nitrogen of the N-terminal amide of 17 was further appended with a 2-nitrobenzyl group that can be cleaved by irradiation at 350 nm to afford 18. Hartgerink et al. reported that hydrogen bonds on the amino acids close to the core play a crucial role in directing the self-assembly into cylindrical structure.49 Even a small N-methyl group on the amide considerably affected the secondary structure. Because of the bulkiness of the photocleavable group and a lack of the hydrogen bond on the amide closest to the core, we expected that 17 and 18 would differ in their supramolecular architectures.
TEM of 17 revealed helical, high aspect ratio architectures with a nearly uniform width and helical pitch of 33 ± 2 and 92 ± 4 nm, respectively (Figure 9a).48 As seen in Figure 9c, the frayed terminus of one of the supramolecular structures reveals it is actually a quadruple helix: it is composed of two smaller helices (blue arrows) and each of these is further composed of two individual fibrils (red arrows). After irradiation, the helical structures disappeared completely, and only cylindrical fibrils with a diameter of 11 nm were observed (Figure 9b). We hypothesized that increased steric bulk induces torsional strain that is released as the fibers coil into the observed structures. This system suggests future strategies to create functional, photoresponsive materials that may be useful in sensing or actuation.
FIGURE 9.

TEM micrographs of PA 17 (pH 11, 7.4 × 10−4 M, annealed at 80 °C and slowly cooled to 25 °C) (a) before and (b) after photoirradiation (350 nm, 250 W, 5 min) to give 18 and (c) TEM showing that the unirradiated helices (yellow arrow) were composed of braids of four individual fibrils (red arrows). Adapted with permission from ref 48. Copyright 2008 American Chemical Society.
Templated Length Control
In systems that form supramolecular objects on the nano- and micrometer scale, it is desirable to precisely control nanostructure size in all dimensions. While precise structural control is well-known for closed, spherical structures (e.g., micelles), the control of open structures is much more challenging. In supramolecular polymers, this can be controlled to a limited extent by the monomer concentration or the use of end caps.50,51 Kelly et al. demonstrated a molecular vernier approach to control aggregate size using hydrogen bonding (Figure 10a).52 The overall stability of such a system is limited by the weakest link, in this case a single pair of hydrogen bonds. Hunter and Tomas later designed a triple-stranded molecular vernier that was stable enough for characterization by size exclusion chromatography.53 However, the vernier approach requires precise spacings of the complementary binding groups and is only suitable for relatively small aggregates. The general problem of length control of self-assembled systems with nanoscale dimensions requires a more versatile approach.
FIGURE 10.

Strategies for controlling length of self-assemblies: (a) vernier approach requires two complementary but different recognition groups (indicated as a ball and socket); (b) a small amount of a rigid-rod template (blue) can limit the length of a 1D nanostructure (green).
The tobacco mosaic virus (TMV) assembly represents an excellent example effective control over size of self-assembled structures with a template. Thousands of coat proteins self-assemble into a 300-nm-long cylinder around a single molecule of RNA. The overall length of the assembly is determined by the length of the enclosed RNA template.54 Without the template, the TMV coat proteins assemble into cylinders with the same diameter as the untemplated capsids but variable length. The RNA template prevents the unlimited self-assembly of the capsid proteins through specific molecular interactions. Inspired by this strategy, we designed a rigid template to control the length of synthetic self-assembled PA nanofibers (e.g., 19, Figure 11).8 The length of these nanofibers was previously impossible to control.
FIGURE 11.

Chemical structures of (a) peptide amphiphile monomer 19 and (b) PEG-terminated oligo(phenylene ethynylene) template 20 (R = 2-octyldodecyl).
We designed and synthesized a dumbbell-shaped template molecule 20 (Figure 11) that contains an oligo(p-phenylene ethynylene) core.55 The hydrophobic rigid rod defines a precise length that can be encapsulated within the hydrophobic core of the PA nanofibers. In contrast to the TMV system, we could not rely on specific molecular interactions to truncate the assembly, so bulky poly(ethylene glycol) (PEG) groups were chosen as end caps to block fiber growth. We hypothesized that PA 19 and template 20 would coaggregate through hydrophobic collapse.
An aqueous mixture of 19 and 20 (200:1 molar ratio) revealed small aggregates by AFM, rather than the nanofibers observed for 19 alone (Figure 12a,b). These mixed aggregates have heights of 5.5 ± 0.7 nm compared with 5.3 ± 0.6 nm for the untemplated fibers. In samples with a higher molar fraction of PA 19 (500:1 or 1000:1), both small, micelle-like nanostructures and longer nanofibers are observed, indicating that there is insufficient template to fully suppress the one-dimensional PA assembly. To better understand the structure of the assembly without the effects of drying, we prepared additional TEM samples according to a quick-freeze/deep-etch (QFDE) protocol. Slightly elongated nanostructures were observed to be ∼9.2 nm long and ∼6.4 nm across (Figure 12c).
FIGURE 12.

AFM height images of (a) peptide amphiphile 1 alone (heights = 5.3 ± 0.6 nm) or (b) 200:1 molar ratio mixture of 19 and 20 showed no fibers and (c) QFDE TEM of 200:1 molar ratio mixture of 19 to 20 revealing the aspect ratio of the small aggregates. Reproduced with permission from ref 55. Copyright 2008 American Chemical Society.
Dynamic light scattering (DLS) measurements of PA 19 showed structures with a hydrodynamic radius >1 μm, whereas solutions of template 20 showed a hydrodynamic radius of ∼17.4 nm. The mixed sample consisting of 200:1 molar ratio gave a mean hydrodynamic radius of ~17.9 nm. We expected little change in hydrodynamic radius of the assembly compared with the dumbbell, since the long axis of the assembly is defined by the length of the rigid template. CD spectroscopy confirmed the presence of β-sheets in solution of PA 19 with and without the template, showing that the intermolecular basis of PA self-assembly remains unchanged. While it remains difficult to spectroscopically determine the molecular interactions between 19 and 20, microscopy and light scattering data provide compelling evidence that template 20 can limit the length of self-assembling nanofibers.
Conclusion/Outlook
The past couple of decades experienced great growth in the simultaneous development of supramolecular chemistry and nanochemistry. Nanoscience emerged mostly from physics and materials science with the notion that physical properties of matter at the nanoscale would undergo unique transitions and was mostly envisioned for inorganic substances. Supramolecular chemistry, popularized by Lehn, is a very different axis directed to the notion that chemists could control noncovalent bonds in all structures with the same precision achieved by synthetic organic chemistry. The molecular stories discussed in this manuscript demonstrate the possibility of using supramolecular chemistry principles to craft the size, shape, and internal structure of nanoscale objects. They dealt specifically with organic molecules and their self-assembly into soft, as opposed to hard, nanostructures such as carbon nanotubes and metallic nanoparticles. Soft nanostructures, not the original focus of nanoscience, remain the least explored systems in terms of physical properties and value as a toolbox to assemble novel materials. One of the reasons is that synthetic organic chemists continue to be focused on methodologies to construct the complex molecules that cater to pharmacological targets inspired by natural products. Over the next two decades, the field could turn with new generations of chemists focusing on organic supramolecular chemistry to create soft functional nanostructures, including the exploration of their physical properties and use as components of materials and macroscopic devices. This last objective will require a “systems chemistry”56 approach in which molecular design and synthesis of specific nanostructures are integrated with new ideas on how to approach design in multicomponent systems. This is the approach that will build bottom-up, self-assembling structures with macroscopic dimension and function such as photovoltaic, catalytic, and biomedical systems.
As for the one-dimensional nanostructures discussed here, the potential is great given their inherent ability to serve as conduits of charge and to align elements in their surroundings such as molecules, macromolecules, and even cells. One-dimensional objects are ubiquitous in biology and serve critical functions such as the fibrils in the structural and signaling machinery of extracellular matrices, the cellular cytoskeleton, axons, dendrites, and many others. One-dimensional nanostructures, as demonstrated by the systems we have studied, readily form three-dimensional networks giving rise to gel matter from solutions. Gel-like matter with high content of one-dimensional soft nanostructures is the substrate to create functionality in mammalian biology (organs, tissues), and there must be an equivalent phase space of synthetic structures that could be accessed and designed for novel functions. The field needs to go toward mapping strategies to use molecular interactions to achieve dimensional and shape precision in organic nanostructures, and most importantly, to control structure across the scales. For example, how does one dial in the bundling of 10–100 one-dimensional nanostructures the way triple-helical bundles of collagen make fibrils for a wide variety of functions with a single type of nanounit? Even though the technological targets may require top-down approaches, some of which have been popularized by chemists, the goal of chemical research should be to create the structures bottom-up from nano- to macroscales.
Acknowledgments
The authors are grateful for support of this work by grants from Department of Energy (Grant DE-FG02-00ER45810), the National Institutes of Health (Grants BRP, 5 R01 EB003806-05, NIDCR 5 R01 DE015920-04, and NCI/CCNE 5 U54 CA119341-03), and the National Science Foundation (Grant DMR-0605427).
Biographies
Liam C. Palmer was born in Halifax, Nova Scotia, in 1977 and grew up in Columbia, South Carolina. He received his B.S. in Chemistry from the University of South Carolina in 1999. He completed his Ph.D. on molecular encapsulation under the supervision of Prof. Julius Rebek, Jr., at The Scripps Research Institute in 2005. He is now working as a Research Assistant Professor in the group of Prof. Samuel Stupp at Northwestern University. His current research focuses on strategies to control the self-assembly of amphiphilic small molecules.
Samuel I. Stupp earned his B.S. in Chemistry from the University of California at Los Angeles and his Ph.D. in materials science and engineering from Northwestern University in 1977. He was a member of the faculty at Northwestern until 1980 and then spent 18 years at the University of Illinois at Urbana–Champaign, where he was appointed in 1996 Swanlund Professor of Materials Science and Engineering, Chemistry, and Bioengineering. In 1999, he returned to Northwestern as a Board of Trustees professor of Materials Science, Chemistry, and Medicine and later was appointed Director of Northwestern's Institute for BioNano-technology in Medicine. His research is focused on self-assembly of materials with special interest in regenerative medicine, cancer therapies, and solar energy technology.
REFERENCES
- 1.Bain CD, Whitesides GM. Molecular-Level Control over Surface Order in Self-Assembled Monolayer Films of Thiols on Gold. Science. 1988;240(4848):62–63. doi: 10.1126/science.240.4848.62. [DOI] [PubMed] [Google Scholar]
- 2.Nuzzo RG, Fusco FA, Allara DL. Spontaneously Organized Molecular Assemblies. 3. Preparation and Properties of Solution Adsorbed Monolayers of Organic Disulfides on Gold Surfaces. J. Am. Chem. Soc. 1987;109(8):2358–2368. [Google Scholar]
- 3.Tanase M, Silevitch DM, Hultgren A, Bauer LA, Searson PC, Meyer GJ, Reich DH. Magnetic Trapping and Self-Assembly of Multicomponent Nanowires. J. Appl. Phys. 2002;91:8549–8551. [Google Scholar]
- 4.Smith PA, Nordquist CD, Jackson TN, Mayer TS, Martin BR, Mbindyo J, Mallouk TE. Electric-Field Assisted Assembly and Alignment of Metallic Nanowires. Appl. Phys. Lett. 2000;77:1399–1401. [Google Scholar]
- 5.Huang Y, Duan X, Wei Q, Lieber CM. Directed Assembly of One-Dimensional Nanostructures into Functional Networks. Science. 2001;291(5504):630–633. doi: 10.1126/science.291.5504.630. [DOI] [PubMed] [Google Scholar]
- 6.Stupp SI, Son S, Lin HC, Li LS. Synthesis of 2-Dimensional Polymers. Science. 1993;259(5091):59–63. doi: 10.1126/science.259.5091.59. [DOI] [PubMed] [Google Scholar]
- 7.Stupp SI, LeBonheur V, Walker K, Li LS, Huggins KE, Keser M, Amstutz A. Supramolecular Materials: Self-Organized Nanostructures. Science. 1997;276(5311):384–389. doi: 10.1126/science.276.5311.384. [DOI] [PubMed] [Google Scholar]
- 8.Hartgerink JD, Beniash E, Stupp SI. Self-Assembly and Mineralization of Peptide-Amphiphile Nanofibers. Science. 2001;294(5547):1684–1688. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 9.Zubarev ER, Pralle MU, Sone ED, Stupp SI. Self-Assembly of Dendron Rodcoil Molecules into Nanoribbons. J. Am. Chem. Soc. 2001;123(17):4105–4106. doi: 10.1021/ja015653+. [DOI] [PubMed] [Google Scholar]
- 10.Capito RM, Azevedo HS, Velichko YS, Stupp SI. Self-Assembly of Large and Small Molecules into Hierarchically Ordered Sacs and Membranes. Science. 2008;319(5871):1812–1816. doi: 10.1126/science.1154586. [DOI] [PubMed] [Google Scholar]
- 11.Berger B, Shor PW. On the Structure of the Scaffolding Core of Bacteriophage T4 and Its Role in Head Length Determination. J. Struct. Biol. 1998;121(3):285–294. doi: 10.1006/jsbi.1997.3942. [DOI] [PubMed] [Google Scholar]
- 12.Wimberly BT, Brodersen DE, Clemons WM, Morgan-Warren RJ, Carter AP, Vonrhein C, Hartsch T, Ramakrishnan V. Structure of the 30S Ribosomal Subunit. Nature. 2000;407(6802):327–339. doi: 10.1038/35030006. [DOI] [PubMed] [Google Scholar]
- 13.Ban N, Nissen P, Hansen J, Moore PB, Steitz TA. The Complete Atomic Structure of the Large Ribosomal Subunit at 2.4 Angstrom Resolution. Science. 2000;289(5481):905–920. doi: 10.1126/science.289.5481.905. [DOI] [PubMed] [Google Scholar]
- 14.Hasenknopf B, Lehn JM, Boumediene N, Dupont-Gervais A, Van Dorsselaer A, Kneisel B, Fenske D. Self-Assembly of Tetra- and Hexanuclear Circular Helicates. J. Am. Chem. Soc. 1997;119:10956–10962. [Google Scholar]
- 15.Seto CT, Mathias JP, Whitesides GM. Molecular Self-Assembly through Hydrogen Bonding: Aggregation of Five Molecules To Form a Discrete Supramolecular Structure. J. Am. Chem. Soc. 1993;115:1321–1329. [Google Scholar]
- 16.Pralle MU, Urayama K, Tew GN, Neher D, Wegner G, Stupp SI. Piezoelectricity in Polar Supramolecular Materials. Angew. Chem., Int. Ed. 2000;39(8):1486–1489. doi: 10.1002/(sici)1521-3773(20000417)39:8<1486::aid-anie1486>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]
- 17.Hill JP, Jin WS, Kosaka A, Fukushima T, Ichihara H, Shimomura T, Ito K, Hashizume T, Ishii N, Aida T. Self-Assembled Hexa-peri-hexabenzocoronene Graphitic Nanotube. Science. 2004;304(5676):1481–1483. doi: 10.1126/science.1097789. [DOI] [PubMed] [Google Scholar]
- 18.Messmore BW, Hulvat JF, Sone ED, Stupp SI. Synthesis, Self-Assembly, And Characterization of Supramolecular Polymers from Electroactive Dendron Rodcoil Molecules. J. Am. Chem. Soc. 2004;126(44):14452–14458. doi: 10.1021/ja049325w. [DOI] [PubMed] [Google Scholar]
- 19.Hoeben FJ, Herz LM, Daniel C, Jonkheijm P, Schenning AP, Silva C, Meskers SC, Beljonne D, Phillips RT, Friend RH, Meijer EW. Efficient Energy Transfer in Mixed Columnar Stacks of Hydrogen-Bonded Oligo(p-phenylene vinylene)s in Solution. Angew. Chem., Int. Ed. 2004;43(15):1976–1979. doi: 10.1002/anie.200353451. [DOI] [PubMed] [Google Scholar]
- 20.Schenning AP, V Herrikhuyzen J, Jonkheijm P, Chen Z, Würthner F, Meijer EW. Photoinduced Electron Transfer in Hydrogen-Bonded Oligo(p-phenylene vinylene)-perylene Bisimide Chiral Assemblies. J. Am. Chem. Soc. 2002;124(35):10252–10253. doi: 10.1021/ja020378s. [DOI] [PubMed] [Google Scholar]
- 21.Silva GA, Czeisler C, Niece KL, Beniash E, Harrington DA, Kessler JA, Stupp SI. Selective Differentiation of Neural Progenitor Cells by High-Epitope Density Nanofibers. Science. 2004;303(5662):1352–1355. doi: 10.1126/science.1093783. [DOI] [PubMed] [Google Scholar]
- 22.Rajangam K, Behanna HA, Hui MJ, Han XQ, Hulvat JF, Lomasney JW, Stupp SI. Heparin Binding Nanostructures To Promote Growth of Blood Vessels. Nano Lett. 2006;6(9):2086–2090. doi: 10.1021/nl0613555. [DOI] [PubMed] [Google Scholar]
- 23.Tysseling-Mattiace VM, Sahni V, Niece KL, Birch D, Czeisler C, Fehlings MG, Stupp SI, Kessler JA. Self-Assembling Nanofibers Inhibit Glial Scar Formation and Promote Axon Elongation after Spinal Cord Injury. J. Neurosci. 2008;28:3814–3823. doi: 10.1523/JNEUROSCI.0143-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Behanna HA, Donners JJJM, Gordon AC, Stupp SI. Coassembly of Amphiphiles with Opposite Peptide Polarities into Nanofibers. J. Am. Chem. Soc. 2005;127(4):1193–1200. doi: 10.1021/ja044863u. [DOI] [PubMed] [Google Scholar]
- 25.Hartgerink JD, Zubarev ER, Stupp SI. Supramolecular One-Dimensional Objects. Curr. Opin. Solid State Mater. Sci. 2001;5:355–361. [Google Scholar]
- 26.Estroff LA, Hamilton AD. Water Gelation by Small Organic Molecules. Chem. Rev. 2004;104(3):1201–1218. doi: 10.1021/cr0302049. [DOI] [PubMed] [Google Scholar]
- 27.Weiss RG, Terech P. Molecular Gels: Materials with Self-Assembled Fibrillar Networks. Dordrecht, The Netherlands; Springer: 2006. [Google Scholar]
- 28.Kauranen M, Verbiest T, Boutton C, Teerenstra MN, Clays K, Schouten AJ, Nolte RJM, Persoons A. Supramolecular 2nd-Order Nonlinearity of Polymers with Orientationally Correlated Chromophores. Science. 1995;270(5238):966–969. [Google Scholar]
- 29.Sato I, Kadowaki K, Urabe H, Jung JH, Ono Y, Shinkai S, Soai K. Highly Enantioselective Synthesis of Organic Compound Using Right- And Left-Handed Helical Silica. Tetrahedron Lett. 2003;44(4):721–724. [Google Scholar]
- 30.Hill DJ, Mio MJ, Prince RB, Hughes TS, Moore JS. A Field Guide to Foldamers. Chem. Rev. 2001;101(12):3893–4012. doi: 10.1021/cr990120t. [DOI] [PubMed] [Google Scholar]
- 31.Goodman CM, Choi S, Shandler S, DeGrado WF. Foldamers as Versatile Frameworks for the Design and Evolution of Function. Nat. Chem. Biol. 2007;3(5):252–262. doi: 10.1038/nchembio876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li LS, Jiang H, Messmore BW, Bull SR, Stupp SI. A Torsional Strain Mechanism to Tune Pitch in Supramolecular Helices. Angew. Chem., Int. Ed. 2007;46:5873–5876. doi: 10.1002/anie.200701328. [DOI] [PubMed] [Google Scholar]
- 33.Pralle MU, Whitaker CM, Braun PV, Stupp SI. Molecular Variables in the Self-Assembly of Supramolecular Nanostructures. Macromolecules. 2000;33(10):3550–3556. [Google Scholar]
- 34.Smith DK, Hirst AR, Love CS, Hardy JG, Brignell SV, Huang BQ. Self-Assembly Using Dendritic Building Blocks - Towards Controllable Nanomaterials. Prog. Polym. Sci. 2005;30:220–293. [Google Scholar]
- 35.Zubarev ER, Sone ED, Stupp SI. The Molecular Basis of Self-Assembly of Dendron-Rod-Coils into One-Dimensional Nanostructures. Chem.—Eur. J. 2006;12(28):7313–7327. doi: 10.1002/chem.200600619. [DOI] [PubMed] [Google Scholar]
- 36.Messmore BW, Sukerkar PA, Stupp SI. Mirror Image Nanostructures. J. Am. Chem. Soc. 2005;127(22):7992–7993. doi: 10.1021/ja051183y. [DOI] [PubMed] [Google Scholar]
- 37.van Gestel J, Palmans AR, Titulaer B, Vekemans JA, Meijer EW. “Majority-Rules” Operative in Chiral Columnar Stacks of C3-Symmetrical Molecules. J. Am. Chem. Soc. 2005;127(15):5490–5494. doi: 10.1021/ja0501666. [DOI] [PubMed] [Google Scholar]
- 38.Jin W, Fukushima T, Niki M, Kosaka A, Ishii N, Aida T. Self-Assembled Graphitic Nanotubes with One-Handed Helical Arrays of a Chiral Amphiphilic Molecular Graphene. Proc. Natl. Acad. Sci. U.S.A. 2005;102(31):10801–10806. doi: 10.1073/pnas.0500852102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sone ED, Zubarev ER, Stupp SI. Semiconductor Nanohelices Templated by Supramolecular Ribbons. Angew. Chem., Int. Ed. 2002;41(10):1706–1709. [PubMed] [Google Scholar]
- 40.Li LM, Beniash E, Zubarev ER, Xiang WH, Rabatic BM, Zhang GZ, Stupp SI. Assembling a Lasing Hybrid Material with Supramolecular Polymers and Nanocrystals. Nat. Mater. 2003;2(10):689–694. doi: 10.1038/nmat983. [DOI] [PubMed] [Google Scholar]
- 41.Rowan AE, Nolte RJM. Helical Molecular Programming. Angew. Chem., Int. Ed. Engl. 1998;37:63–68. [Google Scholar]
- 42.Brizard A, Oda R, Huc I. Chirality Effects in Self-Assembled Fibrillar Networks. Top. Curr. Chem. 2005;256:167–218. doi: 10.1007/b107174. [DOI] [PubMed] [Google Scholar]
- 43.Engelkamp H, Middelbeek S, Nolte RJM. Self-Assembly of Disk-Shaped Molecules to Coiled-Coil Aggregates with Tunable Helicity. Science. 1999;284(5415):785–788. doi: 10.1126/science.284.5415.785. [DOI] [PubMed] [Google Scholar]
- 44.Yamada N, Ariga K, Naito M, Matsubara K, Koyama E. Regulation of β-Sheet Structures within Amyloid-like β-Sheet Assemblage from Tripeptide Derivatives. J. Am. Chem. Soc. 1998;120:12192–12199. [Google Scholar]
- 45.Li LS, Stupp SI. One-Dimensional Assembly of Lipophilic Inorganic Nanoparticles Templated by Peptide-Based Nanofibers with Binding Functionalities. Angew. Chem., Int. Ed. 2005;44(12):1833–1836. doi: 10.1002/anie.200462142. [DOI] [PubMed] [Google Scholar]
- 46.Bauer WR, Crick FHC, White JH. Supercoiled DNA. Sci. Am. 1980;243(1):118–133. [PubMed] [Google Scholar]
- 47.Chandler D, Weeks JD, Andersen HC. Van der Waals Picture of Liquids, Solids, and Phase-Transformations. Science. 1983;220(4599):787–794. doi: 10.1126/science.220.4599.787. [DOI] [PubMed] [Google Scholar]
- 48.Muraoka T, Cui H, Stupp SI. Quadruple Helix Formation of a Photoresponsive Peptide Amphiphile and Its Light-Triggered Dissociation into Single Fibers. J. Am. Chem. Soc. 2008;130:2946–2947. doi: 10.1021/ja711213s. [DOI] [PubMed] [Google Scholar]
- 49.Paramonov SE, Jun HW, Hartgerink JD. Self-Assembly of Peptide-Amphiphile Nanofibers: The Roles of Hydrogen Bonding and Amphiphilic Packing. J. Am. Chem. Soc. 2006;128(22):7291–7298. doi: 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- 50.Lehn JM. Supramolecular Polymer Chemistry-Scope and Perspectives. Polym. Int. 2002;51:825–839. [Google Scholar]
- 51.Brunsveld L, Folmer BJ, Meijer EW, Sijbesma RP. Supramolecular Polymers. Chem. Rev. 2001;101(12):4071–4098. doi: 10.1021/cr990125q. [DOI] [PubMed] [Google Scholar]
- 52.Kelly TR, Xie RL, Weinreb CK, Bregant T. A Molecular Vernier. Tetrahedron Lett. 1998;39:3675–3678. [Google Scholar]
- 53.Hunter CA, Tomas S. Accurate Length Control of Supramolecular Oligomerization: Vernier Assemblies. J. Am. Chem. Soc. 2006;128(27):8975–8979. doi: 10.1021/ja061928f. [DOI] [PubMed] [Google Scholar]
- 54.Klug A. The Tobacco Mosaic Virus Particle: Structure and Assembly. Philos. Trans. R. Soc. London., Ser. B: Biol. Sci. 1999;354(1383):531–535. doi: 10.1098/rstb.1999.0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bull SR, Palmer LC, Fry NJ, Greenfield MA, Messmore BW, Meade TJ, Stupp SI. A Templating Approach for Monodisperse Self-Assembled Organic Nanostructures. J. Am. Chem. Soc. 2008;130(9):2742–2743. doi: 10.1021/ja710749q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ludlow RF, Otto S. Systems Chemistry. Chem. Soc. Rev. 2008;37(1):101–108. doi: 10.1039/b611921m. [DOI] [PubMed] [Google Scholar]