

Abstract
Diverse species of pathogenic Gram-negative bacteria use secretion systems to export a variety of protein toxins and virulence factors that help establish and maintain infection. Disruption of such secretion systems is a potentially effective therapeutic strategy. We developed a high-throughput screen and identified a tris-aryl substituted 2-imino-5-arylidenethiazolidin-4-one, compound 1, as an inhibitor of the Type III secretion system. Expansion of this chemotype enabled us to define the essential pharmacophore for Type III secretion inhibition by this structural class. A synthetic diversity set helped us identify N-3 as the most permissive locus, and led to the design of a panel of novel N-3-dipeptide-modified congeners with improved activity and physiochemical properties. We now report on the synthesis of these compounds, including a novel solid phase approach to the rapid generation of the dipeptide-thiazolidinone hybrids, and their in vitro characterization as inhibitors of Type III secretion in Salmonella enterica serovar Typhimurium.
Introduction
Diverse species of pathogenic Gram-negative bacteria use Type II (T2SS) and Type III (T3SS) secretion systems to deliver virulence factors to host cells to establish and maintain bacterial infections. Some of the component proteins of these secretory apparati, such as the outer membrane secretin YscC/InvG, the inner membrane ring protein YscJ/PrgK, and the ATPase YscN/InvC/SpaI, demonstrate primary amino acid sequence conservation.1 Others demonstrate structural conservation.2 A potential advantage of targeting secretion is that it may result in less selection for resistant mutants, because secretion systems are not required for bacterial growth.3 Small molecules that inhibit secretion systems might be indicated for the prevention and/or treatment of infection from a wide variety of Gram-negative bacterial species and be applicable to diverse plant and animal diseases.4,5 The concept of secretion inhibition as a potentially effective broad-range therapeutic strategy is supported by the literature reports of in vitro activity against Yersina, Chlamydia,6, 7 and Salmonella8 by the salicylhydrazide class of T3SS inhibitors. The potential to disable the virulence processes of diverse Gram-negative pathogens9 prompted us to embark on a discovery project for novel T3SS inhibitors.
A high-throughput screen (HTS) was developed in our laboratory and used to evaluate 92,000 small molecules for inhibition of the Salmonella enterica serovar Typhimurium T3SS. This screen produced 89 initial hits, from which 25 were selected for confirmatory and secondary assays. In selecting compounds to advance to the next screens, we placed a priority on compounds likely to be functioning via our target mechanism of action. We rejected frank cytotoxic or cytostatic compounds, or transcriptional inhibitors, and favored those compounds that exhibited synthetic tractability and suitability for chemotype expansion. The results from our secondary assays led us to focus on the tris-aryl substituted 2-imino-5-arylidenethiazolidin-4-one 1 as our candidate hit. We assayed for secretion of SipA, a T3SS-secreted protein, and using both commercially available and our own synthetic compounds, generated a panel of analogs with which we were able to define the essential pharmacophore of this class of secretion inhibitors.10
Six distinct structural classes of T3SS inhibitors have been described previously. Two classes, the glycolipid caminosides reported by Linington et al11, 12 and the pseudotripeptide guadinomines recently reported by Iwatsuki et al,13 are natural products. Synthetic inhibitor classes include the N-substituted (2-acylamino-4-aryl)-2-pyridones,14 2,5-disubstituted oxazole-3-carboxamides,15 substituted (2-alkylamino)-1,3,5-triazines,16 and N-acylhydrazones of salicylaldehydes.17, 18 Mechanisms of action and molecular targets have not been determined for these compound classes, although currently the acylhydrazones are believed to act by inhibiting transcription of one or more T3SS proteins.6, 7 In contrast, our initial studies with the 2-imino thiazolidinones suggest that this class may act directly to disrupt a protein-protein interaction such as the interactions of the secretin protein involved in the assembly and/or stability of the secretion apparatus.10
The thiazolidinone nucleus appears in drug discovery programs in diverse therapeutic areas. Substituted thiazolidinones are under investigation as anticancer antagonists of the αvβ3 integrins,19 anti-inflammatory agents,20, 21 β3 adrenergic agonists,22,23 antischistosomal agents,24 general antifungal/antimicrobial agents,25–27 and inhibitors of the YycG histidine kinase of S. epidermidis.28 Many of these thiazolidinones are highly potent and target-specific, suggesting that the substituents impart the high degree of selectivity to the common core from which they are presented. This phenomenon in medicinal chemistry29 may be considered the small-molecule correlate of the observation that diverse proteins often share a similar fold without any primary sequence similarity.
Our initial panel of thiazolidinones was useful for characterizing the target and for supporting the hypothesis that very specific molecular interactions were disrupting a protein-protein interaction, but these compounds were poorly soluble and active only in high micromolar concentrations. In order to enhance solubility and activity, and to gain insight into the ligand-protein interface, we embarked on a synthetic program to first define, and then exploit, the permissive or conserved functional groups on the 2-imino thiazolidinone core. We synthesized a variety of compounds, with particular emphasis on ω-polar groups (Scheme 1), and, subsequently, the dipeptides (Schemes 2 and 3) fused to the thiazolidinone at N-3. We now present the preparation of these more hydrophilic 3-substituted 2-imino-5-arylidenethiazolidinones and their T3SS inhibitory activity,
Scheme 1.

Scheme 2.

Scheme 3.

Chemistry
Synthetic methods for thiazolidinones are well described in the literature. Our synthesis of the aminomethylphenyl and aminohexyl analogs, shown in Scheme 1, is an adaptation of the method of Klika et al.30
The regiochemical outcome of the cyclization to 2-iminothiazolidinone can be controlled by the choice of reagents and reaction conditions, but unless sufficient differences exist between the nucleophilic strengths of the two nitrogens the more thermodynamically favored 5-arylidene product will be obtained after the Knovenagel reaction.20, 21 Under the conditions in Scheme 1 we obtained a single compound from the cyclization of the alkyl/aryl thioureas but a mixture of regiomers for all aryl/aryl thioureas. We separated 5 and 7 by preparative reverse phase HPLC at the protected penultimate stage (optimum for this procedure) before cleaving the Boc group to, respectively, 6 and 8. The 1D and 2D 1H NMR spectra of 6 and 8 clearly indicated that the two were regiomers, and the mass spectra gave the same parent ions but a different fragmentation pattern for each compound. Absolute assignment of structure was obtained by crystallography of 8 (data not shown). The general method of Scheme 1 was used to generate the compounds in Table 1.
Table 1.
Synthetic diversity set generated to evaluate modifications of R1, R2, and R3. The IC50 values are calculated from the percent inhibition of SipA secretion, as determined by Western Blot, using a minimum of 3 concentrations of inhibitor. Compounds 36–40 are regiomeric pairs in which the major isomer has R2 as p-methoxyphenyl and R3 as phenyl, and the minor isomer has R3 as p-methoxyphenyl and R2 as phenyl.
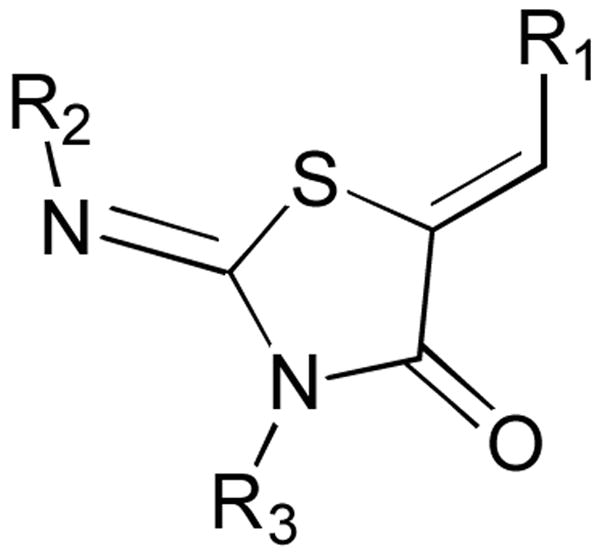 | |||||||
|---|---|---|---|---|---|---|---|
| R1 | R2 | R3 | IC50 (μM) | R1 | R2 | R3 | IC50 (μM) |
1 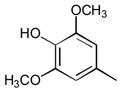
|
Ph | Ph | 83 |
31 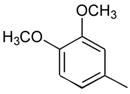
|
Ph | Ph | 278 |
7 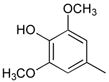
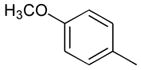
|
Ph |
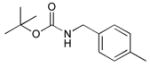
|
45 |
32 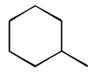
|
Ph |
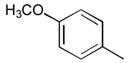
|
278 |
8 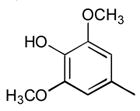
|
Ph |
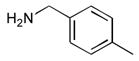
|
52 |
33 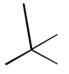
|
Ph |
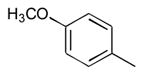
|
160 |
12 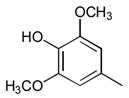
|
Ph |
|
54 |
34 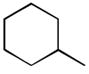
|
Ph | Ph | 225 |
23 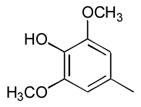
|
Ph |
|
36 |
6 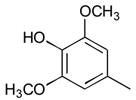
|
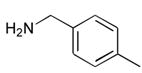
|
Ph | 101 |
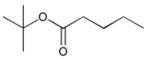
24 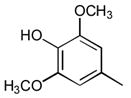
|
Ph |
|
4 |
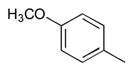
35 
|
|
Ph | 137 |
25 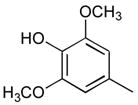
|
Ph |
|
66 |
36a 
|
|
Ph | 124 |
26 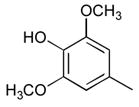
|
Ph |
|
51 |
37a 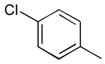
|
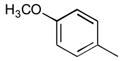
|
Ph | 198 |
27 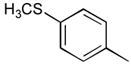
|
Ph | Ph | 26 |
38a 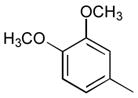
|
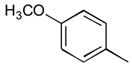
|
Ph | 80 |
28 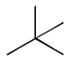
|
Ph | Ph | 244 |
39a 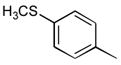
|
|
Ph | 52 |
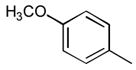
29 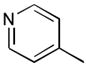
|
Ph | Ph | 376 |
40a 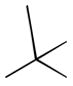
|
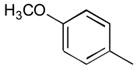
|
Ph | Šb |
30 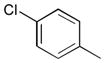
|
Ph | Ph | 322 | ||||
R2 and R3 were regiomeric mixtures of p-methoxyphenyl and phenyl, with the major regiomer indicated in the column.
Significant (≥50%) inhibition only at 1 mM
Short peptides are an expeditious way to survey functional groups, stereochemistry, and molecular shape. Significantly for this work, by appending dipeptides to the non-peptidic thiazolidinone scaffold, the hydrophilicities of the ensuing hybrid molecules can be varied across several orders of magnitude. In order to investigate the SAR of the dipeptide component we prepared a diverse set of analogs presenting the dipeptide from R3 and holding the vinylidene (R1) and 2-imino (R2) groups constant. Our selection of amino acids emphasized arginine, tryptophan, and tyrosine in accordance with residues that are statistically overrepresented in protein-protein interactions.31 Other residues were selected for their neutrality (alanine), charge (ornithine, glutamic acid), steric demand (valine), or to probe hydrogen–bonding (thiazolidinylalanine, pyridylalanine) or hydrophobic contacts (biphenylalanine, tetrahydroisoquinoline carboxylate). Suitably protected dipeptides were assembled and incorporated into the original synthetic route. With arginine in the terminal position, a protected ornithine was used to provide a common precursor to both the primary amine 20a and, upon guanylation,32 22a. The solution synthesis of dipeptidyl thiazolidinones is illustrated by the preparation of 22a (Scheme 2).
The linear directionality of the Scheme 2 route, from peptide C-terminus to complete N3-peptidyl thiazolidinone, prompted the investigation of a solid phase route for the rapid generation of dipeptide libraries. A Tentagel Rink resin was loaded with the C-terminal residue, and the Scheme 1 and Scheme 2 methods were adapted to solid phase by increasing the stoichiometry of the reagents (Scheme 3). In the solid phase route, arginine could be added directly as the pentamethylsulfonylbenzofuran (Pbf) protected derivative. Yields and purities were comparable to those obtained in solution chemistry. The chiral integrity was generally preserved for the dipeptides, with the exception of the ValTyr (42), AlaTrp (44), and OrnDht (49) analogs. The D-alanine dipeptides 20b and 22b were obtained from D-alanine, the D-biphenylalanine dipeptide 55b was obtained from D-biphenylalanine.
Results and Discussion
Compounds were evaluated as T3SS inhibitors by measuring secretion of the Salmonella enterica serovar Typhimurium effector protein SipA (Tables 1 and 2 and Figure 2).10 Replacement of the syringyl ring (28–38, 40), with the exception of p-thiomethylphenyl (27, 39) or 3,4-dimethoxy (38) was generally a poorly tolerated modification. The 2-imino-substituted analog 6 was also somewhat weaker in inhibitory activity than 1. Amido/imino regiomeric pairs that had proven difficult to separate (36–40) were each tested as a mixture with the intention of resolving them only if the inhibitory activity warranted additional effort. Within this data set, none of these compounds qualified. In contrast, multiple analogs with different N-3 groups were active (Table 1), particularly the N-3 ω-carboxy- (23) and carbalkoxy- (24) analogs that retained a significant degree of SipA inhibition at concentrations of 1μM or less. We used N-3 as the position of choice for a series of dipeptides with the intention of identifying additional beneficial functional groups.
Table 2.
Dipeptides presented from N-3. The IC50 values are calculated from the percent inhibition of SipA secretion, as determined by Western Blot, using a minimum of 3 concentrations of inhibitor. The carbons are all S (retention of the L-amino acid stereochemistry) unless otherwise designated. For the discrete epimers 44a/44b and 49a/49b the absolute configurations were not determined. Dihydrotryptophan analogs 49a/49b and 50, derived from commercially supplied racemic material, are correspondingly racemic at the γ carbon.
 | |||||
|---|---|---|---|---|---|
| R | X | IC50 (μM) | R | X | IC50 (mu;M) |
| 41 CH3 | AlaCONH2 | 62 |
49a 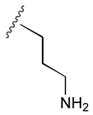
|
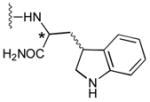
|
52 |
| 42 CH(CH3)2 | TyrCONH2 | 52 |
49b 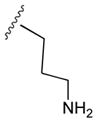
|
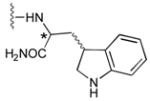
|
56 |
| 43 CH3 | TyrCONH2 | 112 |
50 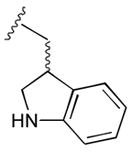
|
ArgCONH2 | 50 |
| 44a CH3 (isomer A) | TrpCONH2 | 30 |
51 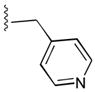
|
ArgCONH2 | 109 |
| 44b CH3 (isomer B) | TrpCONH2 | 29 | |||
| 20a CH3 (S) | OrnCONH2 | 16 |
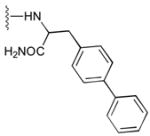
52 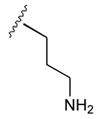
|
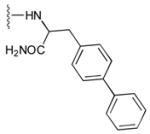
|
13 |
| 20b CH3 (R) | OrnCONH2 | 9 |
53 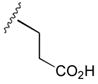
|
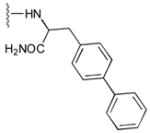
|
50 |
| 22a CH3 (S) | ArgCONH2 | 77 |
54 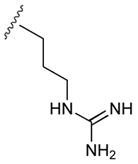
|
|
50 |
| 22b CH3 (R) | ArgCONH2 | 38 | |||
45 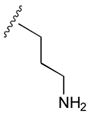
|
AlaCONH2 | 32 |
55a 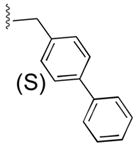
|
ArgCONH2 | 3 |
46 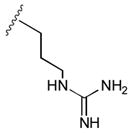
|
AlaCONH2 | 41 |
55b 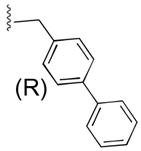
|
ArgCONH2 | 8 |
47 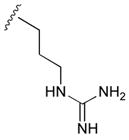
|
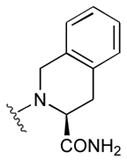
|
115 | |||
48 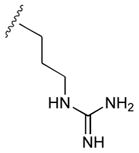
|
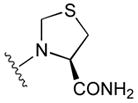
|
65 |
56 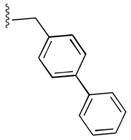
|
OrnCONH2 | 55 |
Figure 2. Inhibition of the secretion of the virulence protein SipA into the culture medium by S. typhimurium.

Representative Western Blots of SipA secreted into culture medium from S. typhimurium grown in the presence of compound 1 (above) and 52 (below) at the concentrations (in μM) indicated above each blot. The results for 55a were comparable to those shown for 52.10 Secretion in the absence of compound but the presence of 5% DMSO is shown (‘0’) at the far left.
The inhibitory potencies of the dipeptides are shown in Table 2. Many of the dipeptides, while having improved solubility,33 retained but did not exceed a level of potency comparable to that of 1. We selected 1, 7, 8, 52, and 55a as representative compounds in the 5-substituted-2-arylimino-thiazolidinone class to test for growth inhibitory activity against S. typhimurium, and no effect was observed at concentrations up to 1 mM (data not shown). These data support the conclusion that effects of these compounds are mediated by direct inhibition of the T3SS secretory process and not a result of inhibition of bacterial growth.
The more inhibitory dipeptides, 55a and 55b, and to a lesser extent 20a and 20b, have a common motif of a neutral residue (alanine or biphenylalanine) in the proximal position coupled to a cationic residue (ornithine or arginine) in the terminal position. It is interesting that other dipeptides that ostensibly share this motif are much weaker: Arginine is in the terminal position of analogs 22a, 22b, 50 and 51, and ornithine is in the terminal position of 56, but the inhibitory activity of all of these remains comparable to that of 1. Appending a peptide does not necessarily improve the inhibitory activity of the 3-substituent: glutamyl dipeptide 53, that might be regarded as a congruent hybrid of two modestly active analogs, acid 25 (66 μM) and biphenyl peptide 52 (13 μM), showed no significant improvement over acid 25 and was weaker than the corresponding t-butyl ester 24. Taken together, these data suggest that an interactive relationship between the two residues is likely to be more important than the independent contributions of the individual sidechain functional groups. The two residues of the epimeric active dipeptides might be acting in concert to fold the ligand into the correct pose for the binding epitope, and there may be more than a single solution to the problem of peptide-induced, or peptide-stabilized, ligand folding. The higher activities of 52 vs. 56, and of 55a/b vs. 54, indicate that the dipeptide’s effect, however subtle and complex, is the result of specific contacts rather than some general phenomenon such as increased access to the target.
Protein-protein interactions are extended, often over 1000–3000 Å2, but are neither random nor unstructured. The interfaces are frequently coiled coils of helices or other highly defined secondary structures.34 The residues making the most significant contributions to this interface may be buried inside the shape they create, limiting access to external ligands.35 This model describes quaternary structures in the T3SS, including the 3500 Å2 surface of the EscJ trimeric interface in which 35% of that interface is buried.36 Strategies to design ligands for protein-protein interfaces have been described. These strategies emphasize the predominance of hydrophobic interactions and aromatic residues in the ‘hotspots’ that drive the particular protein-protein interaction.34, 37 In this work we have identified an epimeric pair of compounds, 55a and 55b, that increase the T3SS inhibition of our initial HTS hit more than 10-fold. These dipeptides are characterized by a combination of a cationic group and a functional group having the potential for hydrophobic interactions. Our current effort to determine the optimum consensus pharmacophore takes into account the accumulated data from the Table 1 and Table 2 compounds. From the perspective of future drug evolution, it is encouraging to observe that the more active compounds such as 55a are an order of magnitude more hydrophilic than 1, suggesting that potent inhibitors of T3SS can be developed that will have acceptable pharmacokinetic properties.
Conclusion
Virulence targets represent an emerging concept in antibacterial therapy3, 38, 39 for which there are examples of compounds that inhibit virulence functions and have demonstrated efficacy in mouse40,41, 42 and other8 in vivo models. The challenge for the further development of anti-virulence therapeutics will require the development of compounds with adequate pharmacokinetic and activity profiles to promote incentive for further development. A key issue when considering a given virulence target is whether drugs successfully directed against it will have sufficiently broad spectrum efficacy to be clinically useful. This work suggests that dipeptide derivatives of the thiazolidinone scaffold may provide a critical step toward the validation of this strategy and the development of novel therapeutics.
Experimental
Chemistry
General
All reactions were run under an atmosphere of dry nitrogen. Reagents and solvents were obtained in the highest available purity and used without further purification unless indicated. 1H NMR spectra were obtained on a 300 MHz (Bruker AV300 or AV301) or 500 MHz (Bruker AV500 or Varian) instrument. 13C NMR spectra were obtained on a 500 MHz Bruker AV500. Identity of the compounds was confirmed by mass spectrometry. The compound solution was infused into the electrospray ionization source operating in positive ion mode. Low resolution spectra were obtained on the Esquire LC ion trap mass spectrometer (Bruker Daltonics, Billerica, MA). Accurate mass measurements were performed on the APEX Qe 47 Fourier transform ion cyclotron resonance mass spectrometer (Bruker Daltonics, Billerica, MA). LC-MS measurements to determine logP values were obtained on a Waters Quattro Micro mass spectrometer interfaced with a Waters Alliance 2795 liquid chromatography instrument. Normal phase silica gel purifications were done using a Biotage SP4 instrument using the cartridges supplied by Biotage. RP-HPLC was done on a Varian instrument equipped with a diode array ultraviolet detector. For preparative reverse phase chromatography a 10 × 250 mm C18 5μ column at a flow rate of 4.6 mL/min was used; for analytical reverse phase chromatography a 4.6 × 250 mm C18 5μ column at a flow rate of 1 mL/min was used. Ultraviolet detection was at 215 and either 254 or 360 nm. Unless otherwise specified, buffer A was 0.05% TFA in H2O, buffer B was 0.05% TFA in acetonitrile. Thin layer chromatography was done using 0.2 mm polygram SIL G/UV plates (Alltech, Deerfield, Ill) or Si250F (J. T Baker, Phillipsburg, NJ) plates, developed using mobile phases of varying compositions of ethyl acetate/hexane, MeOH/CH2Cl2, or MeOH/CHCl3, and visualized by UV light supplemented by vanillin, ninhydrin, and other solution stains where appropriate.
Scheme 1
General method A for generation of thioureas is illustrated for the preparation of t-Butyl 4-(3-phenylthioureido)benzylcarbamate 2. To a stirred solution of phenylisothiocyanate (244 μL, 2 mmol) in 50 mL CH2Cl2 was added (Boc) 4-aminomethylaniline (244 mg, 2 mmol), and the solution stirred for 5 days until TLC (95:5 CH2Cl2/MeOH) showed the reaction to be complete. The solvent was removed in vacuo and the white solid collected and washed with hexane/diethyl ether to give 2. Yield: 6.14 mg, 1.72 mmol. 1H NMR (300 MHz, CDCl3, δ): 1.47 (s, 9H), 4.32 (d, J = 5.9 Hz, 2H), 4.91 (br, 1H), 7.28–7.86 (m, 9H). MS m/z 358 [M + H]+, 380 [M + Na]+, 324 [M − t-Bu + H]+, 346 [M − t-Bu + Na]+.
t-Butyl 6-(3-phenylthioureido)hexylcarbamate 9 was prepared by General method A to give 9 (593 mg, 1.70 mmol) that was used without further purification. 1H NMR (300 MHz, CDCl3, δ): 1.50 (s, 9H), 1.34–1.58 (m, 8H), 3.10 (dd, J = 6.3, 6.3 Hz, 2H), 3.65 (dd, J = 6.3, 6.3 Hz, 2H), 7.21–7.57 (m, 5H). MS m/z 374 [M + Na]+, 274 [M − Boc + Na]+.
General method B for generation of 2-iminothiazolidinones is illustrated for the preparation of (Z)-t-Butyl-4-(4-oxo-2-phenylthizolidin-3-ylideneamino)benzylcarbamate 3 and (Z)-t-Butyl4-(4-oxo-3-phenylthizolidin-2-ylideneamino)benzylcarbamate 4. To a stirred solution of 2 (435 mg, 1.22 mmol) in 40 mL CH2Cl2 were added successively DIEA (424 μL, 2.44 mmol) and methyl bromoacetate (115 μL, 1.22 mmol). The reaction mixture was stirred overnight at ambient temperature, concentrated in vacuo, and the residue purified via silica gel chromatography using a gradient from 2 to 20% MeOH in CH2Cl2 to give 3 and 4 (484 mg, 1.2 mmol) as a mixture of regiomers. 1H NMR (300 MHz, CDCl3, δ): 1.48 (s, 9H), 3.99 (s, 2H), 4.29 (br d, J = 5.1 Hz, 1H), 4.37 (br d, J = 5.7 Hz, 1H), 4.90 (br, 1H), 6.89–7.57 (m, 9H). MS m/z 398 [M + H]+, 420 [M + Na]+, 342 [M − t-Bu + H]+, 364 [M − t-Bu + Na]+.
(Z)–t-Butyl 6-(4-oxo-2-phenylimino)thiazolidin-3-yl)hexylcarbamate 10 was prepared by General method B on a 1.14 mmol scale from 9 to give 471 mg, 1.1 mmol as a colorless oil. 1H NMR (300 MHz, CDCl3, δ): 1.51 (s, 9H), 1.38–1.77 (m, 8H), 3.11 (dd, J = 6.3, 6.3 Hz, 2H), 3.81 (s, 2H), 3.86 (dd, J = 7.2, 7.2 Hz, 2H), 4.53 (br, 1H), 6.95–7.39 (m, 5H). 13C NMR (500 MHz, CDCl3, δ): 26.4, 26.5, 27.1, 285, 29.9, 32.7, 40.5, 43.07, 79.0, 121.0, 124.6, 133.3, 148.26, 154.4, 155.9, 171.8. MS m/z 392 [M + H]+, 414 [M + Na]+, 336 [M − t-Bu + H]+, 292 [M - Boc + H]+.
General method C to introduce the 5-arylidene or 5-vinylidene substituent is illustrated by the synthesis of t-Butyl4-((Z)-((Z)-5-(4-hydroxy-3,5-dimethoxybenzylidine)-4-oxo-3-phenylthiazolidin-2-ylidene)amino)benzylcarbamate 5 and t-Butyl 4-((Z)-((Z)-5-(4-hydroxy-3,5-dimethoxybenzylidne)-4-oxo-2-phenylthiazolidin-3-ylidene)amino)benzylcarbamate 7 A solution of the mixture of 3 and 4 (484 mg, 1.22 mmol), syringaldehyde (222 mg, 1.22 mmol), and piperidine (163 μL, 1.64 mmol) in 15 mL of EtOH was refluxed for 19 h. The reaction mixture was cooled to ambient temperature and treated with hexane to precipitate the product, which was collected, washed with additional hexane, and dried to give 565 mg of a mixture of 5 and 7. Preparative reverse phase HPLC using a gradient of 65 to 85% B in A over 15 min enabled the separation of the two pure compounds as well as an overlap band that was held in reserve for further purification. 5 (48.5 mg, 0.09 mmol) 1H NMR (300 MHz, DMSO-d6, δ): 1.41 (s, 9H), 3.77 (s, 6H), 4.06–4.14 (m, 2H), 6.90 (s, 2H), 6.95–7.59 (m, 9H), 7.75 (s, 1H), 9.32 (br, 1H). MS m/z 562 [M + H]+, 584 [M + Na]+ 7 (54.2 mg, 0.10 mmol) 1H NMR (300 MHz, DMSO-d6, δ): 1.43 (s, 9H), 3.72 (s, 6H), 4.15–4.23 m, 2H), 6.89 (s, 2H), 7.00–7.64 (m, 9H), 7.76 (s, 1H), 9.31 (br, 1H). MS m/z 562 [M + H]+, 584 [M + Na]+.
t-Butyl-6-((2Z,5Z)-6-(4-hydroxy-3,5-dimethoxybenzlidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)hexylcarbamate 11 was prepared by General method C to give 548 mg, 0.95 mmol, as an orange solid. 1H NMR (300 MHz, CDCl3, δ): 1.42 (s, 9H), 1.46–1.54 (m, 6H), 1.71–1.81 (m, 2H), 3.03 (dd, J = 6.5, 6.6 Hz, 2H), 3.86 (s, 6H), 3.96 (dd, J = 4.2, 4.2 Hz, 2H), 5.28 (br, 1H), 6.81 (s, 2H), 7.07 (d, J = 7.5 Hz, 2H), 7.23 (t, J = 7.4 Hz, 1H), 7.43 (t, J = 7.9 Hz, 2H), 7.66 (s, 1H). 13C NMR (500 MHz, CDCl3, δ): 23.6, 24.4, 26.4, 26.5, 27.4, 28.5, 29.5, 42.9, 45.3, 55.7, 56.0, 81.4, 108.0, 121.0, 121.3, 124.5, 129.2, 129.2, 132.6, 148.5, 149.4, 151.0, 167.3. MS m/z 556 [M + H]+, 578 [M + Na]+, 500 [M − t-Bu + H]+.
General method D for deprotection of acid-labile protecting groups is illustrated by the preparation of (2Z,5Z)-2-(4-(aminomethyl)phenylimino)-5-(4-hydroxy-3,5-dimethoxybenzyidene)-3-phenylthiazolidin-4-one 6. Ice cold TFA (0.5 mL) was added to neat 5 (4 mg, 0.007 mmol) cooled in an ice bath. The reaction was complete by HPLC in 15 min, and the reaction mixture was concentrated in vacuo to a sticky brown oil. Trituration with diethyl ether gave a solid that was purified on reverse phase HPLC (65 to 85 % B in A over 12 min) to give 6 (3.0 mg, 0.004 mmol) as the TFA salt. 1H NMR (500 MHz, CD3CN, δ): 3.83 (s, 6H), 4.25 (s, 2H), 6.85 (s, 2H), 7.02 (d, J = 8.5 Hz, 2H), 7.22 (t, J = 8.2 Hz, 1H), 7.42 (t, J = 8.5 Hz, 2H), 7.60 (d, J = 8.4 Hz, 2H), 7.67 (d, J = 8.4 Hz, 2H), 7.74 (s, 1H). MS m/z 462 [M + H]+. HRMS (m/z): [M + H]+ calcd for C25H23N3O4S, 462.1488; found 462.1491.
(2Z,5Z)-3-(4-(Aminomethyl)phenylimino)-5-(4-hydroxy-3,5-dimethoxybenzyidene)-2-phenylthiazolidin-4-one 8 was obtained from 7 (6.9 mg, 0.01 mmol) by General method D, and purified on reverse phase HPLC (65 to 85 % B in A over 12 min) to give 8 (5.5 mg, 0.009 mmol) as the TFA salt. 1H NMR (500 MHz, CD3CN, δ): 3.84 (s, 6H), 4.14 (s, 2H), 6.86 (s, 2H), 7.05 (d, J = 8.3 Hz, 2H), 7.46–4.62 (m, 7H) 7.75 (s, 1H). MS m/z 462 [M + H]+. HRMS (m/z): [M + H]+ calcd for C25H23N3O4S, 462.1488; found 462.1486.
(2Z, 5Z)-3-(6-Aminohexyl)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-2-(phenylimino)thiazolidin4-one 12 was obtained from 11 on a 0.95 mmol scale to give, after two silica gel chromatographies, the first using a gradient from 0 to 10 % MeOH in CHCl3 and the second from 1 to 50% MeOH in CHCl3, 456 mg (0.80 mmol). 1H NMR (300 MHz, CDCl3, δ): 1.42–1.45 (m, 4H), 1.64–1.66 (m, 2H), 1.77–1.79 (m, 2H), 2.65 (br, 2H), 2.89 (t, J = 7.5 Hz, 2H), 3.84 (s, 6H), 3.97 (t, J = 7.5 Hz, 2H), 6.71 (s, 2H), 6.99 (d, J = 8.4 Hz, 2H), 7.14–7.19 (m, 1H), 7.28–7.38 (m, 2H), 7.63 (s, 1H). 13C NMR (500 MHz, DMF-d7, δ): 30.2, 30.5, 31.3, 43.6, 47.0, 53.0, 60.3, 112.5, 120.6, 122.1, 123.0, 125.5, 129.0, 133,5, 133.6, 135.5, 152.6, 152.8, 154.4, 162.9, 163.1, 163.4, 166.1, 166.4, 166.6, 170.5. MS rs m/z 456 [M + H]+. HRMS (m/z): [M + H]+ calcd for C24H30N3O4S, 456.1957; found 456.1946.
Scheme 2
General method E for amidation is illustrated by the preparation of (S)-benzyl 1-amino-5-t-butoxycarbonylamino-1-oxopentan-2-ylcarbamate 13. To a suspension of Z-Orn(Boc)-OH (733 mg, 2.0 mmol) in 3.5 mL THF was added N-methyl morpholine (0.48 mL, 4.4 mmol). The reaction mixture was cooled in an ice bath, and isobutyl chloroformate (0.31 mL, 2.4 mmol) was added. After 1 h of stirring on ice, concentrated NH4OH (0.60 mL, 10 mmol) was added, and the reaction mixture stirred an additional 30 min. The reaction mixture was partitioned between 4:1 CHCl3/isopropanol and saturated aqueous sodium carbonate. The organic phase was separated, washed with saturated brine, dried over Na2SO4, and concentrated in vacuo. The crude product was purified on silica gel using a gradient from 0 to 25% MeOH in CH2Cl2 to give 13 670.2 mg (1.84 mmol).1H NMR (500 MHz, CD3OD, δ): 1.46 (s, 9H), 1.51–1.72 (m, 3H), 1.72–1.88 (m, 1H), 2.99–3.16 (m, 2H), 4.07–4.18 (m, 1H), 5.05–5.22 (m, 2H), 7.26–7.46 (m, 5H). 13C NMR (500 MHz, CD3OD, δ): 30.08, 31.4, 33.3, 43.4, 58.5, 70.3, 82.55, 131.5, 131.6, 132.1, 140.8, 161.1, 180.2. MS m/z 366 [M + H]+, 388 [M + Na]+.
General method F for hydrogenolysis, coupling, and subsequent deprotection to the free α-amino dipeptide amide is illustrated by the synthesis of t-Butyl (S)-5-amino-4-((S)-2-aminopropanamido)-5-oxopentylcarbamate 16. N-Protected 13 (664 mg, 1.82 mmol) was hydrogenated over 10% palladium on carbon (650 mg) in 50 mL 4:1 EtOH/DMF for 2.5 h. The catalyst was filtered off and the filtrate concentrated in vacuo to give 14 (420 mg, 1.82 mmol) that was used without further purification. (1H NMR (300 MHz, CD3OD, δ): 1.45 (s, 9H), 1.51–1.65 (m, 3H), 1.65–1.84 (m, 1H), 3.07 (t, J = 6.2 Hz, 2H), 3.09–3.24 (m, 1H); 13C NMR (500 MHz, CD3OD, δ): 29.8, 31.4, 36.3, 43.6, 58.0, 82.5, 158.6, 180.3; MS m/z 232 [M + H]+, 254 [M + Na]+). Tetrahydrofuran (5 mL) was added to a dry mixture of Cbz L-alanine (406 mg, 1.82 mmol) and carbonyldiimidazole (353 mg, 2.18 mmol), and the reaction mixture allowed to stir for 1.5 h. To this was added amine 14, along with DIEA (350 μL, 2.00 mmol), and the reaction mixture stirred for 3 days until determined complete by TLC (95:5 CH2Cl2/MeOH). The volatiles were removed in vacuo, and a 4% aqueous solution of NaHCO3 (10 mL) was added to precipitate the product. The crude solid was collected and purified on silica gel using a gradient from 0 to 10% MeOH in CH2Cl2 to give α-Cbz-δ-Boc dipeptide 15 (226 mg, 0.52 mmol). 1H NMR (300 MHz, CD3OD, δ): 1.36 (d, J = 7.2 Hz, 3H), 1.44 (s, 9H), 1.48–1.74 (m, 3H), 1.76–1.94 (m, 1H), 2.97–3.12 (m, 2H), 4.15 (q, J = 7.1 Hz, 1H), 4.27–4.41 (m, 1H), 5.13 (s, 2H), 7.26–7.43 (m, 5H); m/z 437 [M + H]+, 459 [M + Na]+. Hydrogenolysis of the Cbz group over 10% palladium on carbon (225 mg) in 38 mL 14:1 EtOH/DMF for 2 h gave α-amino δ-Boc dipeptide amide 16 (147.8 mg, 0.49 mmol). 1H NMR (300 MHz, CD3OD, δ): 1.30 (d, J = 6.9 Hz, 3H), 1.45 (s, 9H), 1.50–1.75 (m, 3H), 1.77–1.89 (m, 1H), 3.08 (td, J = 6.6, 1.7 Hz, 2H), 3.48 (q, J = 6.9 Hz, 1H), 4.30–4.49 (m, 1H). MS m/z 303 [M + H]+.
General method G for the formation of N-phenyl-N′-amidodipeptidyl thioureas is illustrated by the preparation of t-Butyl(S)-5-amino-5-oxo-4-((S)-2-(3-phenylthioureido)propanamido)pentylcarbamate 17. To a solution of 16 (144 mg, 0.48 mmol) in 1.5 mL MeOH was added phenylisothiocyanate (180 μL, 0.95 mmol) and the reaction mixture stirred overnight, during which time the product precipitated. The volatiles were removed in vacuo, hexane was added, and the suspension cooled at 4 °C for 1 h. The crude thiourea was collected and washed with diethyl ether to give 17 (204 mg, 0.47 mmol). 1H NMR (500 MHz, CD3OD, δ): 1.45 (s, 9H), 1.46 (d, J = 7.4 Hz, 3H), 1.51–1.65 (m, 2H), 1.65–1.76 (m, 1H), 1.85–1.94 (m, 1H), 2.98–3.17 (m, 2H), 4.32–4.44 (m, 1H), 4.97 (q, J = 7.0 Hz, 1H), 7.22 (t, J = 7.3 Hz, 1H), 7.39 (t, J = 7.9 Hz, 2H), 7.45 (d, J = 7.9 Hz, 2H). MS m/z 438 [M + H]+, 460 [M + Na]+.
t-Butyl (S)-5-amino-5-oxo-4-((S)-2-((Z)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentylcarbamate 18 was prepared by General Method B in THF on a 1.77 mmol scale to give, after silica gel chromatography using a gradient from 0 to 10% MeOH in CH2Cl2, 603.8 mg (1.27 mmol) product. 1H NMR (300 MHz, CD3OD, δ): 1.44 (s, 9H), 1.49–1.78 (m, 3H), 1.66 (d, J = 7.0 Hz, 3H), 1.82–2.01 (m, 1H), 2.50–3.10 (m, 2H), 3.97 (s, 2H), 4.33–4.49 (m, 1H), 5.25 (q, J = 7.0 Hz, 1H), 6.98 (d, J = 7.9 Hz, 2H), 7.14 (t, J = 7.4 Hz, 1H), 7.35 (t, J = 7.8 Hz, 2H); 13C NMR (500 MHz, CDCl3, δ): 17.7, 31.0, 32.5, 32.8, 36.8, 43.3, 56.5, 56.9, 83.5, 125.1, 129.0, 133.4, 151.3, 157.5, 160.8, 173.0, 175.6, 178.2; m/z 478 [M + H]+, 500 [M + Na]+.
t-Butyl(S)-5-amino-4-((S)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)-5-oxopentylcarbamate 19 was prepared by General method C from the corresponding thiazolidinone on a 0.44 mmol scale. The crude solid was purified via silica gel chromatography using a gradient from 0 to 10% MeOH in CH2Cl2 to give 224 mg (0.35 mmol) product. 1H NMR (300 MHz, CD3OD, δ): 1.41 (s, 9H), 1.47–1.68 (m, 3H), 1.72 (d, J = 7.0 Hz, 3H), 1.81–2.02 (m, 1H), 3.03 (t, J = 6.5 Hz, 2H), 3.83 (s, 6H), 4.35–4.45 (m, 1H), 5.43 (q, J = 7.0 Hz, 1H), 6.81 (s, 2H), 7.07 (d, J = 7.4 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.71 (s, 1H). 13C NMR (500 MHz, CDCl3, δ): 18.0, 30.82, 32.4, 32.9, 43.3, 56.5, 57.0, 60.4, 83.4, 111.4, 122.3, 125.2, 125.3, 129.1, 129.2, 133.4, 136.3, 141.2, 151.39, 153.3, 160.7, 170.4, 173.2, 178.2. MS m/z 642 [M + H]+, 664 [M + Na]+.
(S)-5-Amino-2-((S)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentanamide 20a was obtained from 19 (102 mg, 0.16 mmol) by General method D, and purified on reverse phase HPLC (10 to 95% B in A over 25 min) to give 20a (5.1 mg, 0.008 mmol) as the TFA salt. 1H NMR (300 MHz, CD3OD, δ): 1.75 (d, J = 7.0 Hz, 3H), 1.79–1.92 (m, 3H), 1.96–2.18 (m, 1H), 2.85–3.05 (m, 2H), 3.83 (s, 6H), 4.44–4.57 (m, 1H), 5.46 (q, J = 7.0 Hz, 1H), 6.80 (s, 2H), 7.07 (d, J = 7.3 Hz, 2H), 7.22 (t, J = 7.4 Hz, 1H), 7.42 (t, J = 7.8 Hz, 2H), 7.71 (s, 1H). 13C NMR (500 MHz, CD3OD, δ): 16.7, 27.7, 31.9, 42.7, 56.4, 56.6, 59.5, 111.7, 121.5, 125.0, 128.4, 128.9, 133.1, 136.0, 142.5, 151.8, 152.2, 154.0, 170.6, 174.4, 178.7. MS m/z 542 [M + H]+, 564 [M + Na]+. HRMS (m/z): [M + H]+ calcd for C26H32N5O6S, 542.2073; found 542.2070; [M + Na]+ calcd for C26H31N5O6NaS, 564.1893; found 564.1890.
(S)-5-Amino-2-((R)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentanamide 20b. Compound 20b was prepared by General methods C and D on a 0.58 mmol scale and purified via silica gel chromatography using a gradient from 1 to 10% MeOH in CH2Cl2. Fractions containing protected penultimate intermediate (1H NMR (300 MHz, CDCl3, δ): 1.35 (s, 9H), 1.50–1.74 (m, 2H), 1.76 (d, J = 7.0 Hz, 3H), 1.97–2.15 (m, 1H), 3.00–3.39 (m, 2H), 3.85 (s, 3H), 3.98 (s, 3H), 4.70–5.10 (m, 1H), 5.34 (br s, 1H), 5.44 (q, J = 6.9 Hz, 1H), 6.64 (s, 2H), 6.93 (br s, 1H), 7.00 (d, J = 7.5 Hz, 2H), 7.19 (t, J = 7.0 Hz, 2H), 7.37 (t, J = 7.7 Hz, 2H), 7.65 (s, 1H). MS m/z 642 [M + H]+, 664 [M + Na]+) were treated with neat TFA for 45 min to give, after silica gel chromatography using a gradient from 1 to 10% MeOH in CH2Cl2, 20b (224 mg, 0.34 mmol) as the TFA salt. 1H NMR (300 MHz, CD3OD, δ): 1.74 (d, J = 7.0 Hz, 3H), 1.77–1.91 (m, 3H), 1.93–2.13 (m, 1H), 2.93 (t, J = 6.9 Hz, 2H), 3.79 (s, 6H), 4.44–4.62 (m, 1H), 5.45 (q, J = 7.0 Hz, 1H), 6.73 (s, 2H), 7.06 (d, J = 7.4 Hz, 2H), 7.20 (t, J = 7.4 Hz, 1H), 7.40 (t, J = 7. 8 Hz, 2H), 7.68 (s, 1H). 13C NMR (500 MHz, CD3OD, δ): 16.8, 27.7, 32.1, 42.8, 56.4, 56.6, 59.4, 111.7, 121.5, 124.9, 128.4, 128.8, 133.1, 136.1, 142.3, 151.8, 152.1, 153.9, 170.8, 174.3, 178.0. MS m/z 542 [M + H]+. HRMS (m/z): [M + H]+ calcd for C26H32N5O6S, 542.2068; found 542.2064; [M + Na]+ calcd for C26H31N5O6NaS, 564.1887; found 564.1882.
(S)-5-Guanidino-2-((S)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentanamide (21a). General method H for the formation of bis-Boc-guanidine and subsequent deprotection to the free guanidine is illustrated by the synthesis of 22a. 1,3-bis-(Boc)-2-methyl-2-thiopseudourea (80 mg, 0.28 mmol), triethylamine (114 μL, 0.82 mmol), and HgCl2 (90 mg, 0.33 mmol) were added to a solution of 20a (150 mg, 0.23 mmol) in 2.8 mL DMF at 0 °C. The reaction mixture stirred for one hour at 0 °C then at room temperature overnight. The reaction mixture was diluted with ethyl acetate, filtered over celite, and the filtrate concentrated in vacuo. The crude solid was purified via silica gel chromatography using a gradient from 0 to 50% MeOH in CH2Cl2 to give 21a (80.7 mg, 0.10 mmol). 1H NMR (300MHz, (CD3)2CO, δ): 1.45 (s, 9H), 1.50 (s, 9H), 1.60–1.72 (m, 3H), 1.75 (d, J = 7.1 Hz, 3H), 1.83–1.96 (m, 1H), 3.24–3.51 (m, 2H), 3.83 (s, 6H), 4.48–4.62 (m, 1H), 5.40 (q, J = 7.0 Hz, 1H), 6.47 (br s, 1H), 6.86 (s, 2H), 7.01 (br s, 1H), 7.08 (d, J = 7.3 Hz, 2H), 7.19 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.67 (s, 1H), 7.69–7.77 (m, 1H), 8.32 (br s, 1H). 13C NMR (500 MHz, (CD3)2CO, δ): 17.3 29.7, 31.3, 31.6, 31.7, 44.1, 56.8, 59.9, 60.0, 82.2, 86.8, 112.1, 122.4, 125.3, 128.6, 128.8, 133.3, 135.4, 142.7, 152.0, 152.2, 153.3, 160.2, 167.7, 169.6, 169.9, 172.3, 177.4. MS m/z 784 [M + H]+, 806 [M + Na]+. 21a was taken up in 50:50 CH2Cl2/TFA (3 mL) and stirred at room temperature for one hour, then concentrated in vacuo. The crude solid was purified by preparative reverse phase HPLC using a gradient from 10 to 50% B in A over 30 min to give 22a (2 mg, 0.003 mmol) as the TFA salt. 1H NMR (300 MHz, CD3OD, δ): 1.57–1.73 (m, 3H), 1.74 (d, J = 7.0 Hz, 3H), 1.93–2.12 (m, 1H), 3.07–3.28 (m, 2H), 3.82 (s, 6H), 4.43–4.55 (m, 1H), 5.45 (q, J = 6.9 Hz, 1H), 6.78 (s, 2H), 7.06 (d, J = 7.6 Hz, 2H), 7.21 (t, J = 7.4 Hz, 1H), 7.41 (t, J = 7.7 Hz, 2H), 7.68 (s, 1H). 13C NMR (500 MHz, CD3OD, δ): 16.8, 29.0, 32.4, 44.5, 56.7, 57.0, 59.4, 111.6, 121.5, 125.0, 128.4, 128.9, 133.1, 136.0, 142.2, 151.7, 152.1, 153.7, 161.2, 170.5, 174.5, 179.2. MS m/z 584 [M + H]+. HRMS (m/z): [M + H]+ calcd for C27H34N7O6S, 584.2286; found 584.2306.
(S)-5-Guanidino-2-((R)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentanamide 22b was prepared by General method H via the free amine 20b (200 mg, 0.37 mmol), purified via silica gel chromatography using a gradient from 1 to 20% MeOH in CH2Cl2 to give the bis-Boc guanidine 21b (226 mg, 0.29 mmol) 1H NMR (300 MHz, CD3OD, δ): 1.47 (s, 9H), 1.52 (s, 9H), 1.61–1.85 (m, 3H), 1.76 (d, J = 6.9 Hz, 3H), 1.88–2.07 (m, 1H), 3.22–3.46 (br, solvent envelope over CH2), 3.82 (s, 6H), 4.45–4.56 (m, 1H), 5.37–5.55 (m, 1H), 6.79 (s, 2H), 7.04 (d, J = 7.7 Hz, 2H), 7.19 (t, J = 7.2 Hz, 1H), 7.39 (t, J = 7.1 Hz, 2H), 7.71 (s, 1H); 13C NMR (500 MHz, CD3OD, δ): 16.9, 29.5, 30.9, 30.9, 31.2, 43.9, 56.6, 57.2, 59.5, 83.0, 87.0, 110.6, 111.7, 121.8, 124.9, 128.5, 128.7, 133.0, 135.9, 151.9, 152.2, 156.6, 156.7, 160.3, 167.2, 170.7, 174.2, 179.3; MS m/z 785 [M + H]+; 807 [M + Na]+; to the free guanidine, purified on reverse phase HPLC (10 to 75 % B in A over 30 min), 22b (2.0 mg, 0.003 mmol) as the TFA salt. 1H NMR (300 MHz, CD3OD, δ): 1.57–1.73 (m, 3H), 1.75 (d, J = 7.0 Hz, 3H), 1.90–2.10 (m, 1H), 3.10 (t, J = 6.3 Hz, 2H), 3.83 (s, 6H), 4.42–4.58 (m, 1H), 5.45 (q, J = 7.0 Hz, 1H), 6.80 (s, 2H), 7.06 (d, J = 7.4 Hz, 2H), 7.21 (t, J = 7.5 Hz, 1H), 7.41 (t, J = 7.8 Hz, 2H), 7.72 (s, 1H). 13C NMR (500 MHz, CD3OD, δ): 16.8, 29.0, 32.4, 44.5, 56.6, 56.8, 59.5, 111.7, 119.5, 121.9, 124.9, 128.8, 133.1, 136.0, 151.8, 152.3, 153.9, 161.2, 165.7, 166.0, 174.4, 179.1. MS m/z 584 [M + H]+. HRMS (m/z): [M + H]+ calcd for C27H34N7O6S, 584.2286; found 584.2279; [M + Na]+ calcd for C27H33N7O6NaS, 606.2105; found 606.2115.
2-((2Z,5Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)acetic acid 23 (329 mg, 0.70 mmol) was obtained by General method D, and purified by silica gel chromatography using a gradient from 0 to 10% MeOH in CH2Cl2 to give 23 (235.7 mg, 0.57 mmol). 1H NMR (300 MHz, DMF-d7, δ): 3.70 (br s, 1H), 4.02 (s, 6H), 4.85 (s, 2H), 7.11 (s, 2H), 7.25 (d, J = 7.4 Hz, 2H), 7.39 (t, J = 7.4 Hz, 1H), 7.62 (t, J = 7.8 Hz, 2H), 7.95 (s, 1H), 9.62 (br s, 1H). 13C NMR (500 MHz, DMF-d7, δ): 53.1, 60.3, 112.4, 112.6, 122.0, 125.5, 128.3, 129.1, 133.6, 133.7, 126.0, 143.5, 152.2, 152.8, 154.1, 170.2. MS m/z 415 [M + H]+, 437 [M + Na]+. HRMS (m/z): [M + Na]+ calcd for C20H18N2O6NaS, 437.0783; found 437.0787.
4-((2Z,5Z)-5-(4-Hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)butanoic acid 25 (190 mg, 0.38 mmol) was obtained by General method D, and purified by silica gel chromatography using a gradient from 0 to 10% MeOH in CHCl3 to give 25 (89 mg, 0.20 mmol). 1H NMR (300 MHz, CDCl3, δ): 1.86–2.14 (m, 4H), 2.62 (br s, 1H), 4.00 (s, 6H), 4.07–4.22 (m, 2H), 6.83 (s, 2H), 7.34 (d, J = 6.9 Hz, 2H), 7.47–7.64 (m, 3H), 7.84 (s, 1H). 13C NMR (500 MHz, CDCl3, δ): 13.8, 30.3, 60.6, 68.9, 111.6, 119.8, 128.5, 131.8, 133.9, 134.0, 134.8, 136.5, 137.7, 139.2, 142.0, 151.6, 176.5. MS m/z 443 [M + H]+, 465 [M + Na]+. HRMS (m/z): [M + Na]+ calcd for C22H22N2O6NaS, 465.1096; found 465.1106.
(S)-5-Amino-N-((S)-1-amino-3-(biphenyl-4-yl)-1-oxopropan-2-yl)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)pentanamide 52. Compound 52 was obtained by General method D on a 0.052 mmol scale and purified on reverse phase HPLC (10 to 75 % B in A over 30 min) to give 52 (6.0 mg, 0.007 mmol) as the TFA salt. 1H NMR (300 MHz, CD3OD, δ): 1.60–1.87 (m, 2H), 2.35 (q, J = 7.6 Hz, 2H), 2.94–3.13 (m, 3H), 3.16–3.28 (m, 1H), 3.77 (s, 6H), 4.56–4.71 (m, 1H), 5.35 (t, J = 7. 5 Hz, 1H), 6.72 (s, 2H), 6.91 (d, J = 7.5 Hz, 2H), 7.17 (t, J = 7.4 Hz, 1H), 7.21–7.37 (m, 8H), 7.40 (d, J = 8.1 Hz, 2H), 7.67 (d, J = 7.1 Hz, 1H), 7.69 (s, 1H). 13C NMR (500 MHz, CD3OD, δ): 28.1, 28.2, 40.7, 43.0, 58.6, 59.4, 59.9, 111.6, 120.9, 124.9, 128.2, 128.9, 130.3, 130.8, 130.8, 132.3, 133.0, 133.2, 136.5, 139.9, 142.50, 143.5, 144.4 151.3, 152.2, 153.5, 170.4, 172.9, 178.8. MS m/z 694 [M + H]+. HRMS (m/z): [M + H]+ calcd for C38H40N5O6S, 694.2694; found 694.2687; [M + Na]+ calcd for C38H39N5O6NaS, 716.2523; found 716.2513.
Scheme 3
General method I for the solid phase synthesis is illustrated by the preparation of 41. TentaGel S Ram (Advanced ChemTech, 500 mg, 0.25 mmol/g, 0.125 mmol) was swollen in 15 mL CH2Cl2 for 30 min, then deprotected by two 10 min cycles of 20% piperidine/DMF (10 mL) with two DMF washes in between. The resin was washed successively with 10 mL each CH2Cl2 (3x), DMF (3x), MeOH (2x), and CH2Cl2 (3x). A solution of Fmoc L-alanine (117 mg, 0.375 mmol) in 5 mL CH2Cl2 was treated successively with hydroxyazabenzotriazole (HOAt, 51 mg, 0.375 mmol), diisopropylcarbodiimide (DIC, 58 mL, 0.375 mmol), and diisopropylethylamine (109 mL, 0.525 mmol), and the reaction mixture stirred for 5 minutes, then added to the deprotected, washed resin, rinsing with an additional 10 mL CH2Cl2. The resin was shaken overnight, drained, and subjected to the above standard washing protocol. A sample of the beads gave a negative Kaiser test. After deprotection as above, the sample of beads gave a positive Kaiser test, and the second Fmoc L-alanine was added by the identical procedure. Overnight shaking gave a resin showing, after washing, a negative Kaiser test. The resin was deprotected, washed, checked, and treated with 30 μL (0.25 mmol) phenylisothiocyanate in 10 mL CH2Cl2. Kaiser test showed the reaction to be incomplete after overnight shaking, and two additional 150 μL (1.25 mmol) phenylisothiocyanate in 10 mL were added over two days to complete the reaction. After washing, a sample of beads were removed, cleaved with 95:5 TFA/CH2Cl2 for 90 min, and the filtrate analyzed by mass spectrometry to show predominant peaks for the N-amidoAlaAla-N′-phenylthiourea at m/z 317 [M + Na]+, 295 [M + H]+. The resin was treated with methyl bromoacetate (115 μL, 1.25 mmol) and DIEA (436 μL, 2.5 mmol) in 10 mL CH2Cl2, washed, and an aliquot cleaved and examined by mass spectrometry as above to show a predominant [M + H]+ molecular ion of 335. To the resin was added a solution of syringaldehyde (229 mg, 1.25 mmol) and piperidine (173 μL, 1.75 mmol) in 10 mL EtOH, and the suspension refluxed for 9 h. After draining and washing, a cleaved aliquot showed the presence of unreacted starting material, and the charge and reflux were repeated. The drained, washed resin was then cleaved by a 90 min treatment with 95:5:5 TFA/H2O/triisopropylsilane (10 mL). After filtering the resin, and twowashings with 10 mL MeOH each, the filtrate was concentrated in vacuo to give 39 mg of a crude product that was purified on RP-HPLC (20 to 95 % B in A over 18 min) to provide 9.84 mg (0.02 mmol) 41 that was identical to the 41 obtained by solution chemistry under Scheme 2. 1H NMR (500 MHz, CD3CN, δ): 1.35 (d, J = 5.8 Hz, 3H), 1.67 (d, J = 7.1 Hz, 1H), 3.83 (s, 6H), 4.35 (dq, J = 7.2, 7.2 Hz, 1H), 5.32 (q, J = 7.2 Hz, 1H), 5.7 (br, 1H), 7.01 (s, 2H), 7.05–7.46 (m, 5H), 7.70 (s, 1H). MS m/z 499 [M + H]+, 521 [M + Na]+ 411 [M − Ala + H]+. HRMS (m/z): [M + H]+ calcd for C24H26N4NaO6S, 521.1465; found 521.1467.
An earlier-eluting minor peak was identified as the D-Ala-L-AlaNH2 diastereomer m/z 499 [M + H]+, 521 [M + Na]+, 411 [M - Ala + H]+, 428 [M - C(CH3)CONH2 + H]+.
(S)-2-((S)-3-(Biphenyl-4-yl)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)-5-guanidinopentanamide 55a
1H NMR (500 MHz, CD3CN, δ): 1.65–1.70 (m, 2H), 3.12–3.20 (m, 2H), 3.61 (s, 2H), 3.80 (s, 6H), 4.29–4.33 (m, 1H), 5.52 (dd, J = 6.4, 6.3 Hz, 1H), 5.86 (br, 1H), 6.75 (s, 2H), 6.85 (d, J = 7.8 Hz, 2H), 7.19–7.62 (m, 13H), 7.87 (br, 1H). MS m/z 736 [M + H]+. HRMS (m/z): [M + H]+ calcd for C39H42N7O6S, 736.2912; found 736.2888.
(S)-N-((S)-1-Amino-3-(biphenyl-4-yl)-1-oxopropan-2-yl)-5-guanidino-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)pentanamide 54
1H NMR (500 MHz, CD3CN, δ): 1.46–1.50 (m, 2H), 2.91–3.10 (m, 2H), 3.12–3.20 (m, 2H), 3.65 (s, 2H), 4.30–4.45 (m, 1H), 5.10 (t, J = 7.6 Hz, 1H), 5.82 (br, 1H), 6.62 (s, 2H), 6.85 (d, J = 7.8 Hz, 2H), 6.81–7.56 (m, 13H). MS m/z 736 [M + H]+. HRMS (m/z): [M + H]+ calcd for C39H42N7O6S, 736.2912; found 736.2884.
(S)-5-Amino-2-((S)-3-(biphenyl-4-yl)-2-((2Z,5Z)-5-(4-hydroxy-3,5-dimethoxybenzylidene)-4-oxo-2-(phenylimino)thiazolidin-3-yl)propanamido)pentanamide 56
1H NMR (500 MHz, CD3CN, δ): 1.77–1.80 (m, 2H), 1.97–1.99 (m, 2H), 3.06–3.10 (m, 2H), 3.60–3.64 (m, 2H), 3.78 (s, 6H), 4.42–4.55 (m, 1H), 5.64 (dd, J = 10.3, 5.4 Hz, 1H), 6.08 (br, 1H), 6.74 (s, 2H), 6.82 (d, J = 7.6 Hz, 2H), 7.19–7.62 (m, 16H). MS m/z 694 [M + H]+. HRMS (m/z): [M + H]+ calcd for C38H40N5O6S, 694.2694; found 694.2681.
Sip A secretion assay
Salmonella typhimurium secretion assays: Bacteria were grown for 18 h in LB broth and bacterial cells were pelleted by centrifugation. Culture supernates were filtered through a 0.2 μm polyethersulfone syringe filter. 10% TCA was added to culture supernates to precipitate proteins and incubated on ice for 30 min and spun at 16K rpm for 20 min. Pellets were washed with acetone and resuspended in SDS-PAGE sample buffer. Proteins were separated by SDS-PAGE and Western Blotted with polyclonal anti-SipA antibodies.43
MIC determinations
To 30 μL aliquots of a 2 mL overnight culture of S. typhimurium (ATCC 14028) 0.1, 1.0, 2.0, 3.0, 6.0, and 15.0 μL of each test compound in DMSO was added. The treated cultures were incubated overnight and growth determined by reading the OD600. Each sample was run in triplicate. DMSO alone was used as a control and gave partial growth inhibition at 20% v/v in LB (comparable to 2 mM compound) and complete growth inhibition at 50% v/v (comparable to 5 mM compound).
Supplementary Material
Experimental data for the intermediates and final compounds not given in the main body of the text, and a Table of HPLC retention times and conditions, can be found in the Supporting Information. This material is free of charge via the Internet at http://pubs.acs.org.
Figure 1.
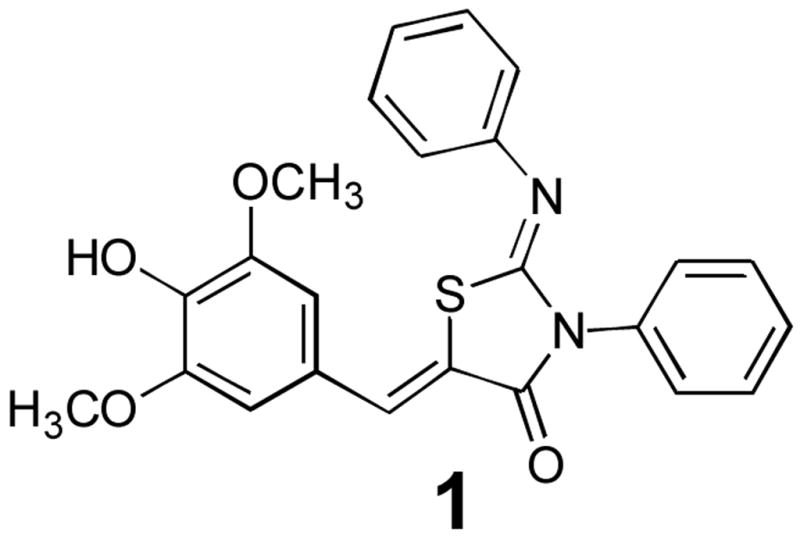
The lead structure emerging from our high-throughput screen for T3SS inhibitors
Acknowledgments
The authors would like to thank Ross Lawrence (Department of Medicinal Chemistry) for the assistance with the high resolution mass spectroscopy data, and Martin Sadilek (Department of Chemistry) for mass spectroscopy, particularly in the determinations of the logP values. The crystal structure of 8 was obtained by Werner Kaminsky (Department of Chemistry). V. Koronakis (Department of Pathology, University of Cambridge) generously provided the SipA antibodies. We are grateful to Wei Deng (Department of Biochemistry) for helpful discussions regarding the SAR data. The support of NIAID (U54 A105714) is acknowledged. The investigators are members of the Northwest Regional Center of Excellence for Biodefense and Emerging Infectious Diseases Research.
Abbreviations
- Ala
alanine
- Arg
arginine
- ATPase
adenosine triphosphate hydrolase
- ATCC
American Type Culture Collection
- Boc
tert-butoxycarbonyl
- Cbz
benzyloxycarbonyl
- Dht
dihydrotryptophan
- DIC
diisopropylcarbodiimide
- DIEA
diisopropylethylamine
- DMSO
dimethylsulfoxide
- Fmoc
fluorenylmethyloxycarbonyl
- HOAt
hydroxyazabenzotriazole
- HTS
high-throughput screen
- LB
Luria-Bertoni broth
- O.D
optical density
- Orn
ornithine
- Pbf
pentamethylsulfonylbenzofuran
- RP-HPLC
reverse phase high performance liquid chromatography
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel electrophoresis
- T2SS
Type II Secretion System
- T3SS
Type III Secretion System
- TCA
trichloroacetic acid
- TFA
trifluoroacetic acid
- TIS
triisopropylsilane
- TLC
thin layer chromatography
- Tyr
tyrosine
- Trp
tryptophan
- Val
valine
- Z
benzyloxycarbonyl
References and notes
- 1.Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4:811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 2.Rosqvist R, Hakansson S, Forsberg A, Wolf-Watz H. Functional conservation of the secretion and translocation machinery for virulence proteins of yersiniae, salmonellae and shigellae. Embo J. 1995;14:4187–4195. doi: 10.1002/j.1460-2075.1995.tb00092.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith PA, Romesberg FE. Combating bacteria and drug resistance by inhibiting mechanisms of persistence and adaptation. Nat Chem Biol. 2007;3:549–556. doi: 10.1038/nchembio.2007.27. [DOI] [PubMed] [Google Scholar]
- 4.Baron C, Coombes B. Targeting bacterial secretion systems: benefits of disarmament in the microcosm. Infect Disord Drug Targets. 2007;7:19–27. doi: 10.2174/187152607780090685. [DOI] [PubMed] [Google Scholar]
- 5.Preston GM. Metropolitan microbes: type III secretion in multihost symbionts. Cell Host & Microbe. 2007;2:291–294. doi: 10.1016/j.chom.2007.10.004. [DOI] [PubMed] [Google Scholar]
- 6.Bailey L, Gylfe A, Sundin C, Muschiol S, Elofsson M, Nordstrom P, Henriques-Normark B, Lugert R, Waldenstrom A, Wolf-Watz H, Bergstrom S. Small molecule inhibitors of type III secretion in Yersinia block the Chlamydia pneumoniae infection cycle. FEBS Lett. 2007;581:587–595. doi: 10.1016/j.febslet.2007.01.013. [DOI] [PubMed] [Google Scholar]
- 7.Negrea A, Bjur E, Ygberg SE, Elofsson M, Wolf-Watz H, Rhen M. Salicylidene acylhydrazides that affect type III protein secretion in Salmonella enterica serovar typhimurium. Antimicrob Agents Chemother. 2007;51:2867–2876. doi: 10.1128/AAC.00223-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hudson DL, Layton AN, Field TR, Bowen AJ, Wolf-Watz H, Elofsson M, Stevens MP, Galyov EE. Inhibition of type III secretion in Salmonella enterica serovar Typhimurium by small-molecule inhibitors. Antimicrob Agents Chemother. 2007;51:2631–2635. doi: 10.1128/AAC.01492-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Troisfontaines P, Cornelis GR. Type III secretion: more systems than you think. Physiology (Bethesda) 2005;20:326–339. doi: 10.1152/physiol.00011.2005. [DOI] [PubMed] [Google Scholar]
- 10.Felise HB, Nguyen HV, Pfuetzner RA, Barry KC, Jackson SR, Blanc MP, Brotnstein PA, Kline T, Miller SI. An Inhibitor of Bacterial Virulence Protein Secretion. Cell Host & Microbe. 2008 doi: 10.1016/j.chom.2008.08.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Linington RG, Robertson M, Gauthier A, Finlay BB, van Soest R, Andersen RJ, Caminoside A. An antimicrobial glycolipid isolated from the marine sponge Caminus sphaeroconia. Org Lett. 2002;4:4089–4092. doi: 10.1021/ol0268337. [DOI] [PubMed] [Google Scholar]
- 12.Linington RG, Robertson M, Gauthier A, Finlay BB, MacMillan JB, Molinski TF, van Soest R, Andersen RJ. Caminosides B-D, antimicrobial glycolipids isolated from the marine sponge Caminus sphaeroconia. J Nat Prod. 2006;69:173–177. doi: 10.1021/np050192h. [DOI] [PubMed] [Google Scholar]
- 13.Iwatsuki M, Uchida R, Yoshijima H, Ui H, Shiomi K, Kim YP, Hirose T, Abe A, Tomoda H, Omura S. Guadinomines, Type III Secretion System Inhibitors, Produced by Streptomyces, sp. K01-0509. J Antibiot. 2008;61:230–236. doi: 10.1038/ja.2008.33. [DOI] [PubMed] [Google Scholar]
- 14.Li, X. Pyridone Compounds As Inhibitors of Bacterial Type II Protein Secretion Systems. US 7,987,622 B2 2006.
- 15.Li, X. Inhibitors of Bacterial Type III Secretion Systems. US patent 0272784 A1 2005.
- 16.Li, X., Kang, F.-A., Macielag, M. J. Triazine Compounds as Inhibitors of Bacterial Type III Protein Secretion Systems. PCT/US2005/015808 WO2005/111017 2005.
- 17.Nordfelth R, Kauppi AM, Norberg HA, Wolf-Watz H, Elofsson M. Small-molecule inhibitors specifically targeting type III secretion. Infect Immun. 2005;73:3104–3114. doi: 10.1128/IAI.73.5.3104-3114.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kauppi AM, Andersson CD, Norberg HA, Sundin C, Linusson A, Elofsson M. Inhibitors of type III secretion in Yersinia: Design, synthesis and multivariate QSAR of 2-arylsulfonylamino-benzanilides. Bioorg Med Chem. 2007;15:6994–7011. doi: 10.1016/j.bmc.2007.07.047. [DOI] [PubMed] [Google Scholar]
- 19.Dayam R, Aiello F, Deng J, Wu Y, Garofalo A, Chen X, Neamati N. Discovery of small molecule integrin alphavbeta3 antagonists as novel anticancer agents. J Med Chem. 2006;49:4526–4534. doi: 10.1021/jm051296s. [DOI] [PubMed] [Google Scholar]
- 20.Ottana R, Maccari R, Barreca ML, Bruno G, Rotondo A, Rossi A, Chiricosta G, Di Paola R, Sautebin L, Cuzzocrea S, Vigorita MG. 5-Arylidene-2-imino-4-thiazolidinones: design and synthesis of novel anti-inflammatory agents. Bioorg Med Chem. 2005;13:4243–4252. doi: 10.1016/j.bmc.2005.04.058. [DOI] [PubMed] [Google Scholar]
- 21.Ottana R, Maccari R, Ciurleo R, Vigorite MG, Panico AM, Cardile V, Garufi F, Ronsivalle S. Synthesis and in vitro evalutation of 5-Arylidene-3-hydroxyalkyl-2phenylimino-4-thiazolidinones with antigenerative activity on human chondrocyte cultures. Bioorg Med Chem. 2007;15:7618–7625. doi: 10.1016/j.bmc.2007.09.001. [DOI] [PubMed] [Google Scholar]
- 22.Hu B, Malamas M, Ellingboe J, Largis E, Han S, Mulvey R, Tillett J. New oxadiazolidinedione derivatives as potent and selective human beta3 agonists. Bioorg Med Chem Lett. 2001;11:981–984. doi: 10.1016/s0960-894x(01)00147-0. [DOI] [PubMed] [Google Scholar]
- 23.Hu B, Malamas M, Ellingboe J. Synthesis of Heterocycle Substituted 1-aryl-4-piperidones. Heterocycles. 2002;57:857–865. [Google Scholar]
- 24.Manhi FM, Manhoud MR. Studies on the Reactivity of Fused Thiazole Toward Nucleophilic Reagents: Synthesis of New Thiazolo-Derivatives of Potential Antischistomsomal Activity. Heteroatom Chemistry. 2007;16:121–131. [Google Scholar]
- 25.Khare RK, Srivastava MK, Singh H. Synthesis and fungicidal activity of some 5-methylene-2-[5′-aryl-1′,3′,4′-oxa(thia)-diazol-[2′-yl]amino-4-thiazolones. Indian Journal of Chemistry. 1995;34B:828–831. [Google Scholar]
- 26.Rajanarendar EA, Karunakar D. Synthesis of isoxazolylpryrazolo[3,4-d]thiazoles and isoxazolylthiazoles and their antibacterial and antifungal activity. Indian Journal of Chemistry. 2004;43B:168–173. [Google Scholar]
- 27.Merja BC, Joshi AM, Parikh KA, Parikh AR. Synthesis and biological evaluation of pyrido-[1,2-1]pyrimidie an disoxazoline derivatives. Indian Journal of Chemistry. 2004;43B:909–912. [Google Scholar]
- 28.Qin Z, Zhang J, Xu B, Chen L, Wu Y, Yang X, Shen X, Molin S, Danchin A, Jiang H, Qu D. Structure-based discovery of inhibitors of the YycG histidine kinase: new chemical leads to combat Staphylococcus epidermidis infections. BMC Microbiol. 2006;6:96. doi: 10.1186/1471-2180-6-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Evans BE, Rittle KE, Bock MG, DiPardo RM, Freidinger RM, Whitter WL, Lundell GF, Veber DF, Anderson PS, Chang RS, Lotti VJ, Cerino DJ, Chen TB, Kling PJ, Kunkel KA, Springer JP, Hirshfield J. Methods for drug discovery: development of potent, selective, orally effective cholecystokinin antagonists. J Med Chem. 1988;31:2235–2246. doi: 10.1021/jm00120a002. [DOI] [PubMed] [Google Scholar]
- 30.Klika KD, Valtamo P, Janovec L, Suchar G, Kristian P, Imrich J, Kivela H, Alfoldi J, Pihlaja K. Regioselective syntheses, structural characterization, and electron ionization mass spectrometric behavior of regioisomeric 2,3-disubstituted 2-imino-1,3-thiazolidin-4-ones. Rapid Commun Mass Spectrom. 2004:87–95. doi: 10.1002/rcm.1290. [DOI] [PubMed] [Google Scholar]
- 31.Yin H, Hamilton AD. Strategies for targeting protein-protein interactions with synthetic agents. Angew Chem Int Ed Engl. 2005;44:4130–4163. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 32.Tian X, Field TB, Switzer AG, Mazur AW, Ebetino FH, Wos JA, Berberich SM, Jayasinghe LR, Obringer CM, Dowty ME, Pinney BB, Farmer JA, Crossdoersen D, Sheldon RJ. Design, synthesis, and evaluation of proline and pyrrolidine based melanocortin receptor agonists. A conformationally restricted dipeptide mimic approach. J Med Chem. 2006;49:4745–4761. doi: 10.1021/jm060384p. [DOI] [PubMed] [Google Scholar]
- 33.Calculated logP values for the dipeptides, using the algorithm provided by ChemDraw v.9.01, gave a range of values from 0.98 (24, 48) to 4.47 (54, 55a), compared to a calculated logP value of 4.87 for 1. Experimentally derived values were somewhat lower although the ranking did not change. These values were determined for 55a (logP= 2.63) and 1 (logP=3.34 by monitoring the respective molecular ions on LC-MS and using calibration curves ranging from 0.1 to 10 μM (MeOH) to obtain the concentration of each compound partitioned into otcanol and water from the respective dry samples. The LC gradient was25 to 95% Bin A over 15 minutes, where A was 94:5:1 water:acetonitrile: acetic acid and B was 99:1 acetonitrile:acetic acid. The column was a Zorba SB-C18 (2.1 × 100 mm) maintained at 30 °C. All samples ere analyzed in positive ionization mode using the respective specific precursor/product ions 433.1/195.2 (dwell 0.1min, cone 45, coll energy 35V) for 1 and 736.3/563.2 (dwell 0.1 min, cone 25, coll energy 33V) for 55a.
- 34.Xu Y, Lu H, Kennedy JP, Yan X, McAllister LA, Yamamoto N, Moss JA, Boldt GE, Jiang S, Janda KD. Evaluation of “credit card” libraries for inhibition of HIV-1 gp41 fusogenic core formation. J Comb Chem. 2006;8:531–539. doi: 10.1021/cc0600167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jager M, Michalet X, Weiss S. Protein-protein interactions as a tool for site-specific labeling of proteins. Protein Sci. 2005;14:2059–2068. doi: 10.1110/ps.051384705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yip CK, Kimbrough TG, Felise HB, Vuckovic M, Thomas NA, Pfuetzner RA, Frey EA, Finlay BB, Miller SI, Strynadka NC. Structural characterization of the molecular platform for type III secretion system assembly. Nature. 2005;435:702–707. doi: 10.1038/nature03554. [DOI] [PubMed] [Google Scholar]
- 37.Gestwicki JE, Cairo CW, Strong LE, Oetjen KA, Kiessling LL. Influencing receptor-ligand binding mechanisms with multivalent ligand architecture. J Am Chem Soc. 2002;124:14922–14933. doi: 10.1021/ja027184x. [DOI] [PubMed] [Google Scholar]
- 38.Alksne LE, Projan SJ. Bacterial virulence as a target for antimicrobial chemotherapy. Curr Opin Biotechnol. 2000;11:625–636. doi: 10.1016/s0958-1669(00)00155-5. [DOI] [PubMed] [Google Scholar]
- 39.Marra A. Targeting virulence for antibacterial chemotherapy: identifying and characterising virulence factors for lead discovery. Drugs R D. 2006;7:1–16. doi: 10.2165/00126839-200607010-00001. [DOI] [PubMed] [Google Scholar]
- 40.Bowser TE, Bartlett VJ, Grier MC, Verma AK, Warchol T, Levy SB, Alekshun MN. Novel anti-infection agents: Small-molecule inhibitors of bacterial transcription factors. Bioorg Med Chem Lett. 2007;17:5652–5655. doi: 10.1016/j.bmcl.2007.07.072. [DOI] [PubMed] [Google Scholar]
- 41.Liu CI, Liu GY, Song Y, Yin F, Hensler ME, Jeng WY, Nizet V, Wang AH, Oldfield E. A cholesterol biosynthesis inhibitor blocks Staphylococcus aureus virulence. Science. 2008;319:1391–1394. doi: 10.1126/science.1153018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hilleringmann M, Pansegrau W, Doyle M, Kaufman S, MacKichan ML, Gianfaldoni C, Ruggiero P, Covacci A. Inhibitors of Helicobacter pylori ATPase Cagalpha block CagA transport and cag virulence. Microbiology. 2006;152:2919–2930. doi: 10.1099/mic.0.28984-0. [DOI] [PubMed] [Google Scholar]
- 43.Pegues DA, Hantman MJ, Behlau I, Miller SI. PhoP/PhoQ transcriptional repression of Salmonella typhimurium invasion genes: evidence for a role in protein secretion. Mol Microbiol. 1995;17:169–181. doi: 10.1111/j.1365-2958.1995.mmi_17010169.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Experimental data for the intermediates and final compounds not given in the main body of the text, and a Table of HPLC retention times and conditions, can be found in the Supporting Information. This material is free of charge via the Internet at http://pubs.acs.org.