

Abstract
Protein Phosphatase 1 (PP1) is an essential and ubiquitous serine/threonine protein phosphatase that is regulated by more than 100 known inhibitor and targeting proteins. It is currently unclear how protein inhibitors distinctly and specifically regulate PP1 to enable rapid responses to cellular alterations. We demonstrate that two PP1 inhibitors, I-2 and DARPP-32, belong to the class of intrinsically unstructured proteins (IUPs). We show that both inhibitors have distinct preferences for transient local and long range structure. These preferences are likely their structural signature for their interaction with PP1. Furthermore, we show that upon phosphorylation of Thr34 in DARPP-32, which turns DARPP-32 into a potent inhibitor of PP1, neither local nor long range structure of DARPP-32 is altered. Therefore, our data suggests a role for these transient 3-dimensional topologies in binding mechanisms that enable extensive contacts with PP1's invariant surfaces. Together, these interactions enable potent and selective inhibition of PP1.
Keywords: Protein Phosphatase 1, Inhibitor-2, DARPP-32, serine/threonine signaling, NMR spectroscopy
Two distinct types of enzymes that are capable of transient covalent modifications of proteins are kinases and phosphatases. However, while the human genome encodes for an almost equal number of tyrosine (tyr) kinases and phosphatases (90:107), there is a dramatic disparity between the number of serine/threonine (ser/thr) kinases and phosphatases (428:∼40) (1). This suggests that ser/thr phosphatases might be more promiscuous than tyr phosphatases yet are presumably under strict temporal and spatial control. Protein phosphatase 1 (PP1) (2) is an essential ser/thr phosphatase found in all tissues with roles in cellular processes as diverse as cell cycle progression, protein synthesis, muscle contraction, carbohydrate metabolism, transcription and neuronal signaling. Due to its diverse roles, PP1 must be active towards a broad range of substrates while maintaining specificity. Its activity is under exceedingly tight and specific control: it is regulated by more than 100 inhibitor and targeting proteins, with predictions of up to 200 (3-5).
Two types of PP1 inhibitors are currently known: protein-based inhibitors and natural small molecular toxin inhibitors, such as microcystin-LR or tautomycin (6, 7). Most small molecular toxin inhibitors are potent against PP1, PP2A and to some degree to PP2B (the two related members of the PPP family of ser/thr protein phosphatases). This is not the case for protein based inhibitors, which are specific for a single PPP family member and are the focus of our work. Inhibitor proteins themselves come in two categories: pseudo-substrate inhibitors, which require phosphorylation to become potent inhibitors, and inhibitors that are potent without phosphorylation. Both types of inhibitors specifically bind PP1 and block its activity. A PP1 interaction motif [(R/K)x1x2x3(F/W)] has been identified as a requirement for PP1 binding (5). However, the presence of this motif alone is not adequate to confer binding to PP1. Therefore, it has been suggests by numerous PP1 research groups that regions outside of this interaction site are essential for PP1 regulation and that these regions provide the necessary specificity in the PP1:regulator interaction. Crystal structures of PP1 in complex with molecular toxins such as microcystin-LR (8-10), a peptide from the glycogen regulating subunit (11) and the MYPT1 regulatory subunit (12) have been reported. These structures show that the active site of PP1, which is directly targeted by PP1 inhibitors, is more than 20 Å away from the common PP1 interaction site. Despite differences in the PP1 isoforms crystallized, crystallization conditions used and crystal packing contacts observed PP1's overall structure is highly invariable. The only reported exception is an interaction of seven C-terminal PP1 residues with the ankyrin repeats in the PP1:MYPT1 regulatory subunit complex, which become structured upon interaction with PP1δ (12). Therefore, it has been established that inhibitor proteins contact distal sites in PP1 simultaneously.
Here, we studied the 3-dimensional structures of two biologically important PP1 inhibitors. First, DARPP-32 (dopamine- and cyclic AMP-regulated phosphoprotein of molecular weight 32 kDa) is a PP1 inhibitor enriched in the striatum that requires phosphorylation at Thr34 to become active as a pseudo-substrate inhibitor (13, 14). The phosphorylation state of DARPP-32 Thr34 is controlled by protein kinase A (PKA) and calcineurin, which are themselves individually up-regulated by various neurotransmitter pathways in striatal neurons. Thus, DARPP-32 is a key component in the integration of multiple signaling pathways in the brain (14). Second, I-2 (inhibitor-2) is a PP1 inhibitor that does not require phosphorylation to achieve inhibition (4, 15-18). I-2 is ubiquitously expressed, though its level and cellular localization fluctuate during cell cycle, suggesting a role in cell division. In vitro, I-2 forms an inactivate complex with PP1, which can be reactivated by phosphorylation of Thr72 on I-2 (GSK-3) without the release of I-2 (19). Although this reactivated complex has not yet been demonstrated in vivo, phospho-threonine I-2 has been found in vivo and a GSK-3:PP1:I-2 complex has been shown in cells over-expressing these three proteins (16, 20). The sequential backbone assignment of similar DARPP-32 (21) and I-2 (22) constructs, as well as the equally important a PP1:I-2 crystal structure (23), were recently published.
In the present study we characterized the unbound states of I-29-164 and DARPP-321-118 at atomic resolution. Additionally, as DARPP-32 must be phosphorylated at Thr34 for PP1 inhibition, we characterized the unbound form of Thr34 phosphorylated DARPP-321-118 (pDARPP-321-118). We leverage our data by direct comparison with literature reported biochemical data for the interactions of these regulators with PP1. Thus, studying these PP1 inhibitors at atomic resolution enables a detailed comparison of their unstructured states and, more importantly, enables us to propose likely scenarios for their binding interactions with PP1. We show that their inherent characteristics, namely high flexibility and discernible structural preferences in regions biochemically identified as important for their interactions with PP1, are likely important determinants for the specificity of their different interactions with PP1. Lastly, our data also shows that Thr34 phosphorylation of DARPP-321-118 does not affect local transient structure and dynamics, or long range contacts present in its unbound state.
Material and Methods
Inhibitor Expression and Purification
Constructs representing the biochemically identified PP1 binding domains of human I-29-164 and rat DARPP-321-118 C72S (21, 22) were subcloned into a pET28a-modified vector which encodes a Thio6His6 expression/purification tag and a TEV (tobacco etch virus protease) cleavage site (24). The C72S mutation of DARPP-321-118 was created because wild-type DARPP-321-118 readily formed a dimer in solution, as shown by analytical size exclusion chromatography (Superdex 200 10/300 GL, GE Healthcare) (data not shown). The plasmids were transformed into Escherichia coli strain BL21-CodonPlus (DE3)-RIL (Stratagene). The expression of uniformly 13C- and/or 15N-labeled protein was carried out by growing freshly transformed cells in M9 minimal media containing 4 g/L [13C]-d-glucose and/or 1 g/L 15NH4Cl (CIL) as the sole carbon and nitrogen sources, respectively, after induction with 1 mM IPTG.
All cells were lysed by high-pressure homogenization (Avestin C-3 Emulsiflex). For the first purification step the soluble proteins were loaded onto a HisTrap HP column (GE Healthcare) and were eluted with an imidazole gradient. Fractions containing the proteins were pooled, dialyzed into fresh buffer and incubated with TEV NIa (S219V) protease fused to an in-frame His6-tag until cleavage was complete. The samples were then loaded onto a Ni-NTA column (Invitrogen) from which they eluted in the flow through. For the final purification step, the proteins were loaded onto either a Superdex 75 26/60 or 16/60 size exclusion column (GE Healthcare) equilibrated with 20 mM NaPO4 (pH 6.5), 50 mM NaCl (I-29-164) or 50 mM NaPO4 (pH 5.5), 50 mM NaCl (DARPP-321-118). Fractions containing the pure proteins, as confirmed by SDS-PAGE, were pooled and concentrated to 0.75 mM. 0.25 mM phenylmethylsulfonyl fluoride (PMSF) was added as a protease inhibitor and 10% D2O was added for NMR experiments.
The activity of the purified proteins was tested by their ability to form complexes with recombinantly expressed PP1, as seen with size exclusion chromatography (Superdex 75 26/60, GE Healthcare; supporting information). Expression and Purification of PKA is also described in the supporting information.
Phosphorylation of DARPP-321-118
Purified DARPP-321-118 was exchanged into a pH 8.4 buffer for phosphorylation. The final reaction conditions were 20 mM Tris, pH 8.4, 50 mM NaCl, 200 μM ATP, 10 mM MnCl2, 1 mM EDTA, 8 μg/mL PKA. Stoichiometric phosphorylation of DARPP-321-118 at Thr34 occurred in 90 minutes at 30° C and was confirmed by mass spectrometry. Following phosphorylation, pDARPP-321-118 was loaded onto a Superdex 75 26/60 size exclusion column (GE Healthcare) equilibrated with 50 mM NaPO4 (pH 5.5), 50 mM NaCl. Fractions containing the pure protein, as confirmed by SDS-PAGE, were pooled and concentrated to 0.55 mM (13C,15N-sample) or 0.75 mM (15N-sample). 0.25 mM phenylmethylsulfonyl fluoride (PMSF) was added as a protease inhibitor and 10% D2O was added for NMR experiments.
Generation of cysteine mutants and spin labeling
The QuikChange Site-Directed Mutagenesis Kit (Stratagene) was used to create the C72S single mutant and the G5C/C72S, A36C/C72S, S51C/C72S and S94C/C72S double mutants of DARPP-321-118, as well as the G12C/C85S, D47C/C85S, S121C/C85S and N152C/C85S double mutants of I-29-164, following the manufacturer's protocol. The presence of the mutations was confirmed by sequencing analysis.
(1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanethiosulfonate (MTSL) (Toronto Research Chemicals) was covalently linked to single cys residues by incubating the proteins with MTSL overnight. [1H,15N] HSQCs were measured on the proteins in a buffer with DTT before MTSL incubation; the sample was then quickly exchanged into the same buffer without DTT using a HiPrep 26/10 desalting column (GE Healthcare) immediately before incubation with MTSL. To remove free MTSL, the proteins were purified by size exclusion chromatography (Superdex 75 26/60 or 16/60, equilibrated with their typical NMR buffers). Spin label incorporation for most samples was confirmed by mass spectrometry (Voyager Pro DE MALDI-TOF mass spectrometer, Applied Biosystems) and dynamic light scattering (see below); 2D [1H,15N] HSQC spectra of the samples before and after spin label incorporation ensured that the label did not perturb the overall structure of the proteins (supporting information). Results were within experimental error, indicating no change in overall structure of the proteins upon addition of MTSL.
NMR measurements
Most NMR measurements were performed at 298 K on a Bruker AvanceII 500 MHz spectrometer. In addition, measurements of R2 relaxation rates were repeated at 800 MHz at the Brandeis University Nuclear Magnetic Resonance Facility. Both spectrometers are equipped with TCI HCN z-gradient cryoprobes. Proton chemical shifts were referenced directly to internal 3-(trimethylsilyl)-1-propanesulfonic acid, sodium salt (DSS) and 13C and 15N chemical shifts were referenced indirectly using the absolute frequency ratios. The NMR spectra were processed with Topspin 1.3 (Bruker) and analyzed with the CARA software package (www.nmr.ch).
15N longitudinal (R1) and transverse (R2) relaxation rates and the [1H,15N] heteronuclear NOE (hetNOE) were measured as described previously (25-27). The relaxation spectra were processed using NMRPipe version 97.027.12.56 (http://spin.niddk.nih.gov/NMRPipe) (28) and peak intensities were analyzed with NMRView version 5.2.2.01 (http://www.onemoonscientific.com/nmrview/download.html) (29). The exponential decay function I = Ae(-x/B) (where I = peak intensity, A = Amplitude, t = relaxation delay and R = R1 or R2) was fit to the peak intensities using the jitter function in NMRView to determine the R1 and R2 relaxation rates. HetNOEs were calculated by dividing the intensity of the peaks in the spectra without presaturation by the intensity of the peaks in the presaturated spectra.
Chemical Shift Assignment
The following spectra were used to achieve sequence-specific backbone and side chain assignments of all aliphatic residues (30): two-dimensional (2D) 1H-15N HSQC, 3D HNCACB, 3D CBCA(CO)NH, 3D CC(CO)NH, 3D HNCO, 3D HN(CA)CO, 3D HNCA (DARPP-321-118 only), 3D HBHA(CO)NH, 3D 15N-resolved 1H-1H NOESY (mixing time of 175 ms) and 3D 15N-resolved 1H-1H TOCSY (mixing time of 60 ms). Chemical shift assignments of I-29-164, DARPP-321-118 and pDARPP-321-118 were deposited in the BMRB as entries 15179, 15176 and 15865 respectively.
Chemical shift indexing and secondary structure propensity
The Secondary Structure Propensity program (SSP) (31) was used to calculate both the chemical shift indices (CSI) and secondary structure propensities (ssp) of I-29-164, DARPP-321-118 and pDARPP-321-118 using the ((Cαexp –Cβexp) – (Cαdatabase –Cβdatabase)) method (32). The RefDB (33), Wang and Jardetzky (34) and Wishart (35) random coil databases were used in calculations. Data from residues before prolines was excluded; in addition, ssp scores were calculated with a seven-residue moving-average window size.
Analysis of relaxation results
ProtScale, on the ExPASy bioinformatics server (http://us.expasy.org), was used to calculate the hydrophobicity profiles using the Abraham & Leo scale (36), the bulkiness profiles and the average area buried on transfer from standard state to folded protein (AABUF) profiles (37). In each case, a seven-residue moving-average window size was used and the weight of the window edges was set to 100% compared to the center; values were scaled for ease of visualization. The segmental motion model for the prediction of random coil R2 relaxation rates was calculated as described (38, 39), using a chain persistence length of 7 and an R2int constant of 0.225. Additional relaxation analysis information is provided in the supporting information
Hydrodynamic radius - dynamic light scattering measurements
DLS measurements using a Viscotek model 802 dynamic light scattering instrument (Viscotek Corporation, Houston, TX) were performed to measure the hydrodynamic radius (Rh) of I-29-164 and DARPP-321-118 (25°C). Numerous empirical derived power factors have been established for folded and denatured proteins (40, 41). By direct comparison of the estimated radius for folded and unfolded proteins, protein compaction factors, , were calculated.
Results
I-29-164 and DARPP-321-118 have a sequence bias, with frequent Gln, Ser, Pro, Glu, Lys and Gly residues (disorder inducing amino acids) and few hydrophobic residues (Figure 1 A, B), suggesting they belong to the family of intrinsically unstructured proteins (IUPs). This is in agreement with the report of Huang et al. (22) and Lin et al. (21), which first described the sequence specific backbone assignment of I-21-172 and DARPP-321-118, respectively.
Figure 1.

Sequence bias and 2D [1H,15N] HSQC spectra demonstrate that I-29-164 and DARPP-321-118 are intrinsically unstructured. Sequences of (A) I-29-164 and (B) DARPP-321-118 are colored according to Dyson and Wright to highlight the amino acid residue preferences of IUPs (43). Green: small residues, uncharged hydrophilic residues and proline residues (Asn, Gln, Ser, Thr, Gly, Ala, Pro); yellow: hydrophobic residues (Val, Leu, Ile, Phe, Tyr); red: acidic residues (Asp, Glu); blue: basic residues (Lys, Arg, His); purple: low-frequency residues (Cys, Trp). Black boxes indicate sequences important for PP1-binding: residues 10-13 (IKGI), 42-46 (KSQKW) and 144-147 (KLHY) in I-29-164; 7-11 (KKIQF) in DARPP-321-118. Black circles show important phosphorylation sites: Thr72 (GSK3) in I-29-164; Thr34 (PKA) and Ser102 (CK2) in DARPP-321-118. 2D [1H,15N] HSQC spectra are shown for (C) I-29-164 and (D) DARPP-321-118. Blue bar in D indicates the lack of dispersion in the 1H dimension of unstructured proteins, compared to the considerably wider dispersion of peaks in the 1H dimension of structured proteins. Blue circles in D demonstrate the amino acid specific clustering of peaks in unstructured proteins. Red asterisks indicate side chain NH2 groups of Asn and Gln.
NMR spectroscopy is the only atomic resolution technique able to investigate the structural and dynamical characteristics of IUPs (42). An IUP “structure” has been described as an ensemble of inter-converting conformers, often with preferences for certain secondary structure elements and long-range interactions, collectively referred to as transient structure. Increasing evidence has shown that this transient structure is correlated with function (43). 2D [1H,15N] HSQC spectra of I-29-164 and DARPP-321-118 are shown in Figure 1 C, D; the limited peak dispersion in the 1H dimension indicates that they are in fact unstructured. While it has been shown that I-21-172 and DARPP-321-118 are intrinsically unstructured (21, 22), a detailed analysis of their unstructured states, including extensive study of local and, most importantly, long range transient structure, has not been reported. Furthermore, while previous studies have indicated that the overall conformation of DARPP-321-118 is not affected by Thr34 phosphorylation (44), this has not been studied in molecular detail. Figure 2 shows the overlay of the DARPP-321-118 and pDARPP-321-118 2D [1H,15N] HSQCs and also a chemical shift difference analysis. This clearly shows that chemical shift changes between the two spectra are limited to the immediate vicinity of Thr34. Finally, though I-29-164 and DARPP-321-118 are unstructured, they are active, as shown by their ability to bind PP1 (supporting information).
Figure 2.
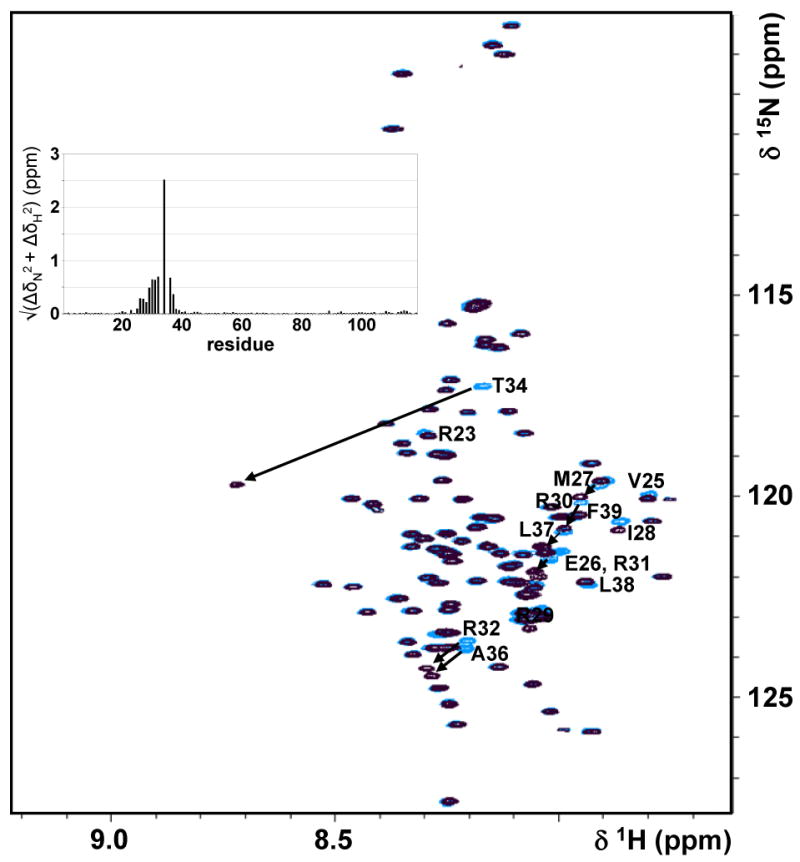
Phosphorylation of Thr34 on DARPP-321-118 does not alter the conformation of DARPP-321-118. Overlay of 2D [1H,15N] HSQC spectra of DARPP-321-118 (blue) with pDARPP-321-118 (black); changes in the 1HN and 15N chemical shifts between the DARPP-321-118 and pDARPP-321-118 2D [1H,15N] HSQC are shown in a insert graph. Peak shifts in the pDARPP-321-118 spectrum are minor, with the exception of that of Thr34, and are restricted to residues within the vicinity of the phosphorylation site. Residues with peak shifts are indicated with residue numbers and arrows.
I-29-164 and DARPP-321-118 have preferred secondary structure
To understand the basis of IUP:PP1 recognition it is important to elucidate any transient structural preferences of the unbound IUPs, including local structural propensities and long distance interactions, as these might be the critical signatures of their interactions with PP1. NMR chemical shifts are highly sensitive to φ/ψ angle propensities. Therefore, subtracting random coil chemical shifts from experimental chemical shifts (Δδ), known as the chemical shift index (CSI) (32, 35, 45), is a powerful tool to detect local regions of transient secondary structure in IUPs. It is known that different nuclei show differential sensitivity towards certain φ/ψ combinations, i.e. Cα chemical shifts are far more sensitive to α-helical behavior and Cβ are more sensitive to β-sheet behavior. Furthermore, ΔCα is positive in a helix and negative in extended structures, while ΔCβ is the opposite (negative and positive, respectively). Therefore, throughout this work we used ((Cαexp –Cβexp) – (Cαdatabase –Cβdatabase)) values (32) to ensure that no single secondary structure propensity is either over- or under-emphasized. Positive deviations from random coil values represent preferred α-helical conformations, whereas negative deviations from random coil values represent preferred β-strand or extended structure regions. The ΔCα - ΔCβ CSI values for I-29-164, DARPP-321-118 and pDARPP-321-118, calculated using the RefDB random coil chemical shift database, are shown in Figure 3. Areas of significantly populated transient α-helices are detected in both I-29-164 and DARPP-321-118; no significant change in transient secondary structure is seen upon Thr34 phosphorylation of DARPP-321-118 (Figures 2 and 3C).
Figure 3.

CSI scores of (A) I-29-164, (B) DARPP-321-118 and (C) pDARPP-321-118 indicate the presence of transient structural preferences in I-29-164 and DARPP-321-118; preferences are not significantly affected by Thr34 phosphorylation of DARPP-321-118. The (ΔCα – ΔCβ) CSI is reported, calculated using the RefDB (33) random coil database. Positive deviations from random coil values indicate transient α-helices. Cartoon representations above each indicate the presence of transient α-helices, as determined by ssp scores (Figure 4), with grey cylinders.
In fully formed secondary structure, certain amino acids types give stronger CSIs than others. A secondary structure propensity (ssp) score (31) normalizes this by calculating the ratio of the experimental CSI for each residue to the CSI for that specific amino acid type in fully formed secondary structures. This calculation provides an estimate of the percent of time each transient secondary structure element is populated, where an ssp score of 1 indicates an α-helix populated 100% of the time and an ssp score of -1 indicates extended structure populated 100% of the time. We consider areas of transient structure significant if 5 or more sequential residues have ssp scores greater than 20%. The ssp scores for I-29-164, DARPP-321-118 and pDARPP-321-118 are shown in Figure 4A, B and C (top), respectively; transient α-helices are indicated by grey cylinders above the graphs. Three structural hotspots can be detected in I-29-164: residues 36-42, 96-106 and 127-154, with residues populating the α-helical conformation space ∼30, 48 and 67% of the time, respectively. Importantly, residues 132-138 have >98% α-helical character based on this analysis. Indeed, this strong helical character is supported by experimentally identified HN-HN NOE cross peaks between residues 132-138 in the 3D 15N-resolved 1H-1H NOESY spectrum. In DARPP-321-118 one transient helix is detected: residues 22-29, populated ∼36% of the time, are located immediately N-terminal to the highly conserved Thr34 PKA phosphorylation site whose phosphorylation is required for PP1 inhibition. Phosphorylation of Thr34 slightly decreases the average percent of time this transient helix is populated from 36% to 33%. Additionally, one region of transient extended structure is detected in DARPP-321-118 between residues 62-72, populated on average ∼24% of the time. pDARPP-321-118 does not exhibit ssp scores indicative of significantly populated transient structure in this region.
Figure 4.

Ssp scores and 15N[1H]-NOE (hetNOE) data indicate regions of transient structure and reduced backbone motions in I-29-164, DARPP-321-118 and pDARPP-321-118. The region of backbone restriction is increased upon Thr34 phosphorylation of DARPP-321-118. (A) I-29-164 ssp (top) and 15N[1H]-NOE (bottom), (B) DARPP-321-118 ssp (top) and 15N[1H]-NOE (bottom), (C) pDARPP-321-118 ssp (top) and 15N[1H]-NOE (bottom). Areas of transient structure are considered significant if five residues or more have a ssp score > 0.2, indicating a transient α-helix, or < -0.2, indicating a region with extended structure. Cartoon representations above the ssp scores indicate the presence of transient α-helices with grey cylinders. Transient α-helices correspond well with regions of restricted backbone motions (positive 15N[1H]-NOEs) and an additional region of restricted motion is found in the C-terminus of DARPP-321-118. Phosphorylation of DARPP-32 Thr34 extends the region of restricted backbone motion and makes the single region of transient extended structure insignificant. Regions of structurally insignificant ssp scores (-0.2 to 0.2) are shown in grey boxes. Asterisks indicate known phosphorylation sites important for PP1 interaction/inhibition: Thr72 (GSK3) in I-29-164; Thr34 (PKA) and Ser102 (CK2) in DARPP-321-118.
Flexibility highlights potential important functional regions in DARPP-32 and I-2
To further examine these IUPs beyond local structure and to gain insight into the molecular foundation for their preferred transient structures, we carried out auto-correlated 15N relaxation experiments, including measurement of 15N[1H]-NOE and 15N longitudinal (R1) and transverse (R2) relaxation rates. The data from these experiments reports on the flexibility of the backbone along the protein chain. Larger 15N[1H]-NOEs indicate restricted backbone motions, while small or even negative 15N[1H]-NOEs indicate increased high-frequency backbone mobility. In structured proteins, 15N[1H]-NOE values are positive and follow a flattened bell-shaped curve with the termini exhibiting slightly more flexibility than the remainder of the chain. In contrast, most 15N[1H]-NOE values are negative in unfolded proteins, indicating a high degree of backbone mobility. Figure 4 A, B and C (bottom) shows the 15N[1H]-NOE for I-29-164, DARPP-321-118 and pDARPP-321-118, respectively. Rigid regions (positive 15N[1H]-NOE) of I-29-164 and DARPP-321-118 correlate remarkably with the transient α-helices, confirming the presence of preferred structure in these regions(15, 46). In I-29-164, positive 15N[1H]-NOEs extend beyond the first transient helix (residues 36-42) to include residues up to Tyr55, indicating restricted motion well beyond the transient helix. Phosphorylation of Thr34 on DARPP-321-118 increases the rigidity of this region, as seen by the extension of positive 15N[1H]-NOEs beyond the transient helix to residue 37 (positive 15N[1H]-NOEs end at residue 32 in DARPP-321-118). In addition, residues 103-114 of DARPP-321-118 and pDARPP-321-118 show reduced flexibility that is not mirrored by significant ssp values. Interestingly, Ser102 is a known CK2 phosphorylation site, whose phosphorylation has been reported to enhance phosphorylation of Thr34. A group of negatively charged amino acids trails the Ser102 phosphorylation site which may contribute to this increased backbone rigidity. Furthermore, although they do not meet our strict criteria for transient structure, residues 109-111 have an average ssp score of 0.21, which may contribute to the localized rigidity.
Testing the molecular foundations for preferred conformation and restricted dynamics
To gain insight into the molecular foundation for these IUPs' preferred transient structures, auto-correlated R1 and R2 relaxation data was recorded in addition to the 15N[1H]-NOE data. The interpretation of relaxation rates in IUPs is different from that in structured proteins because internal motions in structured proteins occur on a different time scale than the global motion of the protein; thus, and consistent with previous studies, we interpreted the data with spectral density mapping at three frequencies (J(0), J(0.87ωH), J(ωN) (supporting information). The main contributions to J(0) come from both ps-ns (15N[1H]-NOE) and slower ns-ms (R2, among others) backbone motions, and it has been shown that the latter may be responsible for the deviations of R2 rates from predicted random coil behavior in IUPs (47). Consequently, we focus here on the qualitative analysis of R2 values since they are especially sensitive to local structure variations. The measurement of the R2 relaxation rates at two magnetic fields (500 and 800 MHz) ensured that conformational exchange can likely be excluded as the reason for the deviations of R2 rates in our IUPs. The R2 relaxation rates of I-29-164, DARPP-321-118 and pDARPP-321-118 are shown in Figure 5. With the exception of the region near the Thr34 phosphorylation site, shown by 15N[1H]-NOEs to be more rigid upon phosphorylation, the R2 relaxation rates of DARPP-321-118 and pDARPP-321-118 agree well. Figure 5 also includes predicted R2 relaxation rates for random coil proteins of the respective lengths of I-29-164 and DARPP-321-118, calculated using the segmental motion model. This model was previously shown to accurately predict the R2 relaxation rates of denatured proteins (39), however, it does not predict the R2 relaxation rates of I-29-164 or DARPP-321-118 within reasonable error.
Figure 5.

Experimental R2 relaxation rates of I-29-164 and DARPP-321-118 cannot be described using a model for random coil polymers. Experimental R2 relaxation rates of (A) I-29-164, (B) DARPP-321-118 and (C) pDARPP-321-118 are shown as black squares with error bars; rates calculated using the segmental motion model for random coil polymers of the same length are shown as solid black lines. Cartoon representations above each indicate the presence of transient α-helices with grey cylinders, based on ssp scores (Figure 4).
Therefore, different residue specific properties such as hydrophobicity, bulkiness, and average area buried upon folding (AABUF) were used to determine whether inherent characteristics of the protein sequence provide the basis for the experimentally detected deviations from random coil behavior in I-29-164 and DARPP-321-118 (supporting information). These parameters have been recommended previously to predict these deviations in denatured and unstructured proteins (48). In our case, it can be readily seen that for low populated transient local structure states, such as helix22-29 in DARPP-321-118 or helix36-42 in I-29-164, AABUF profiles correlate well with the experimental R2 profiles. Therefore, the dynamics of these low-populated states are dictated by the individual residue specific characteristics of side chain size and hydrophobicity. However, highly populated secondary structure elements, such as helix127-154 in I-29-164, can only be correlated in profile, not in magnitude, indicating that cooperativity between residues in a highly populated transient helix, while not directly measureable using CSI data, most likely dominates over individual amino acid properties.
DARPP-321-118 and I-29-164 show a preferred transient 3-dimensional structure
IUPs have more extended 3-dimensional topologies than folded proteins, however long-range contacts may still be present, and preferred 3-dimensional topologies may play an essential role in the distinct interactions between PP1 and its regulatory proteins. NOE spectroscopy, the most common and reliable source for structure information of folded proteins, cannot be used for IUPs due to their high intrinsic flexibilities. Therefore, dynamic light scattering was used to rapidly asses the hydrodynamic radii of these proteins in order to determine their overall compactness (40). These measurements demonstrate that the inhibitor proteins are ∼30% more compact than extended polymer chains of the same length (supporting information). Therefore, long-range interactions likely occur in our IUPs, and might contribute to their distinct topologies and PP1-binding selectivities.
To gain thorough molecular insight into the preferred 3-dimensional topologies of these two IUPs we used the paramagnetic spin label MTSL (1-oxyl-2,2,5,5-tetramethyl-3-pyrroline-3-methyl) methanethiosulfonate) which covalently attaches to free sulfhydryl groups (49). Spin labels cause substantial line broadening of the resonances of spins within ∼20Å of the labeling site through paramagnetic relaxation enhancement (50-52). Along the protein chain, the ratio of the peak intensity, i.e. in a 2D [1H,15N] HSQC spectrum, of the paramagnetic sample to that of the diamagnetic sample is correlated with the spatial distance of each residue from the spin label (53). However, an IUP is a highly flexible conformational ensemble; therefore the ratio is an average ratio for the distance of the spin label from that residue in the ensemble. A ratio of 1 indicates no relaxation enhancement, or that the particular residue is not within 20 Å of the spin label; a ratio of 0 indicates that the residue is in very close proximity to the spin label in many structures in the ensemble. Because of the innate high backbone flexibility of our IUPs, we did not extract quantitative distances from these measurements, but instead we used them to obtain qualitative descriptions of the preferred 3-dimensional topologies of our IUPs.
I-29-164, DARPP-321-118, and pDARPP-321-118 were spin labeled at their wild-type cysteine residues (C85, and C72, respectively). Additionally, four single cysteine mutants of I-29-164 and DARPP-321-118 were spin labeled based on the results of the wild-type spin labeling experiments to further test their overall topologies (raw data in supporting information). The paramagnetic spin label data was translated into contact heat maps and topological models of our IUPs. It is important to point out that these are not computational models of our structures, merely models that were created to allow for better understanding of our available short and long-range data. In I-29-164 (Figures 6 A, B), the most prominent observation is a topological core formed around its single tryptophan residue (Trp46), which includes residues ∼35-60, ∼90-110 and ∼150-155. Furthermore, the region just N-terminal to helix127-154 spends little time in contact with the rest of I-29-164. Thus, the first half of helix127-154 protrudes from the core of the molecule. DARPP-321-118's topology (Figure 6 C, D) is distinct from that of I-29-164. Residues 80-118 spend a significant amount of time as a compact core. The N-terminus, which includes the PP1 binding motif, spends very little time in contact with this core, which may allow it to seek out PP1 as a potential first interaction site. Phosphorylation of Thr34 does not affect the profile of long range contacts on DARPP-321-118 (supporting information), as indicated by the similarity in the raw MTSL data between the two forms of DARPP-321-118.
Figure 6.

Long-range contact (heat) maps indicate the presence of long-range interactions in (A) I-29-164 and (C) DARPP-321-118. Blue circles designate sites of MTSL spin labeling. Red indicates the highest amount of interaction between the spin label and the residue (paramagnetic:diamagnetic peak intensity ratio 0-0.2), orange and yellow indicate a medium amount of interaction (paramagnetic:diamagnetic peak intensity ratios 0.2-0.4 and 0.4-0.6, respectively) and white indicates very little interaction (paramagnetic:diamagnetic peak intensity ratio >0.6). Residues without data, including prolines and peaks with severe chemical shift overlap, are colored in gray. Cartoon representations across the top of each indicate presence of secondary structure preferences based on ssp evaluation (Figure 4). (B) and (D) show topographical representations of unbound I-29-164 and DARPP-321-118, respectively, and include both local and long-range data. Important phosphorylation sites are highlighted (purple circles, described in Figures 1 and 4), as are PP1 binding motifs (green boxes, described in Figure 1). Cylinders represent transient α-helices as determined by ssp scores and blue circles are sites of paramagnetic MTSL spin labels, which were used to determine the transient tertiary structure of these proteins.
Discussion
In the current study we have used a variety of approaches to identify the individual structural preferences of two essential protein inhibitors of PP1: I-29-164 and DARPP-321-118. Additionally, we studied Thr34 phosphorylated DARPP-321-118 to determine whether phosphorylation alters the structural preferences of DARPP-321-118. Many reported IUPs exhibit transient secondary structure preferences which are important to their function. Indeed, these preferences can often be correlated with fully formed secondary structures following a “folding-upon-binding” transition which occurs when the IUP binds its target molecule (54-56).
Both tertiary and local structures are present in the unbound states of I-29-164 and DARPP-321-118. As discussed in further detail below, the results we obtained with I-29-164, DARPP-321-118, and pDARPP-321-118 are consistent with previously reported biochemical studies of these proteins in their unbound forms. Taken together this data allows us to describe potential, distinctive molecular models for the interactions of these two proteins with PP1. The transient structural preferences we report for I-29-164 and DARPP-321-118 are likely to play important roles in their recognition and selectivity for PP1.
Inhibitor-2
Several structure/function studies of I-2 have identified a number of “domains” that are involved in its interaction with PP1 (15, 17, 19, 57, 58). A recent study has also presented a crystal structure of the PP1:I-2 complex (23). Together, these studies have establish that three short regions of I-2 interact directly with PP1: 1) residues 42-54 that contain the KSQKW42-46 sequence which interacts with the common (R/K)x1x2x3(F/W)-motif docking site. 2) residues 11-16, which form a unique N-terminal domain, KGILKN11-16, that binds to a site in PP1 adjacent to the KSQKW42-46 docking site (17, 57). 3) I-2 contains a C-terminal region between amino acids 128-167 that is largely helical in structure and that covers the acidic and hydrophobic grooves as well as the active site of PP1, making a majority of contacts near the active site (23).
Our studies of unbound I-29-164 identified three preferred helical regions (residues 36-42, 96-106 and 127-154). In the crystal structure of the PP1:I-2 complex, only ∼25% of all I-2 residues are visible in the experimental electron density map: residues 11-16, 42-54, and 128-167. Residues which compose the first two transient helices in unbound I-29-164 (residues 36-42 and 96-106) have no corresponding experimental electron density map in the crystal structure of the PP1:I-2 complex. Thus, direct comparison between unbound and bound I-2 is not possible for these regions.
The longest of the three preferred helical regions in unbound I-29-164 is residues 127-154. Previously the region containing this helix was biochemically shown to play a critical role in the PP1:I-2 interaction: a peptide of residues 135-151 antagonizes the inhibitory potency of I-2 as well as DARPP-32, indicating that this region of I-2 shares a binding site on PP1 with DARPP-32 (57). The crystal structure shows that in the PP1:I-2 complex residues 128-167 bind PP1 in an extended α-helical conformation disrupted only at residues 147-151 (23). Residues LHYNE145-149 have a dual role, blocking access to PP1's active site and potentially pushing one or both metals out of the active site. Our data shows a transient helix in this region of unbound I-29-164, complimenting the crystal structure of I-2 bound to PP1. Taken together, this data indicates that the identified transient helix performs an important role in mediating PP1 binding and inactivation. Importantly, no folding-upon-binding transition is identified for this helix.
While full inhibition of PP1 by I-2 requires the extended helical region at the C-terminus of the protein (residues 128-167), it is clear that the N-terminal region is also involved in the interaction with PP1. Truncation mutagenesis has found that partial fragments of I-2 as short as residues 1-114 (IC50 ∼10 nM) (57), or 1-99 (IC50 ∼3 nM)11, have strong inhibitory activity comparable to that of full-length I-2 (IC50 ∼1-2 nM). Thus N-terminal fragments of I-2 act as potent inhibitors without any occlusion of the active site of PP1. It is possible that although the structure of PP1 is not believed to be grossly affected upon binding targeting or inhibitor proteins (8, 12, 23), the N-terminus of I-2 may have an allosteric effect on the active site of PP1. One possibility, is that binding of the N-terminal region of I-2 may contribute to the loss of one of the catalytic metals from PP1, a feature observed in the crystal structure of the PP1:I-2 complex (23). Residues LHYNE145-149 in the C-terminal helix may then bind to the active site and stabilize the inactive enzyme.
Using paramagnetic spin labels it was possible to identify the preferred 3-dimensional structure of I-29-164. It has been proposed that transient local and long range interactions in IUPs expose primary interaction sites to enable a more effective means of binding to target molecules (59). It is our hypothesis that the 3-dimensional transient structures of the IUPs studied here provide a molecular signature for their PP1 interactions. A major feature revealed by this analysis of I-29-164 is the preference observed for a topological core formed around its single tryptophan residue (Trp46), which includes residues ∼35-60, ∼90-110 and ∼150-155. The crystal structure of the PP1:I-2 complex shows that Trp46 is part of a degenerate common PP1 interaction motif, KSQKW42-46. The KSQKW42-46 region is located near, but not within, the first transient helix in unbound I-29-164, allowing this motif to bind the common PP1 interaction site in the required extended confirmation.
A second significant 3-dimensional feature in I-29-164 is that the N-terminus of the long and significantly populated helix127-154 protrudes from I-29-164's core. Because of the strong local bias towards secondary structure that is found in unbound and PP1-bound I-2, and the long-range structural features found for unbound I-29-164, a second potential PP1 interaction scenario could be that helix127-154 acts as a primary contact site in I-29-164 which initiates PP1 interaction. This would allow for stabilization of helix127-154 and folding of I-2 around the PP1 scaffold. While it is the more popular belief that PP1 regulatory proteins interact first via their common PP1 binding motifs, the existence of a core structure in I-29-164 may allow for a different mode of binding to PP1 where helix127-154 instead plays a prominent role as the initial contact site for I-29-164.
Lastly, phosphorylation of Thr72, a GSK-3β phosphorylation site that is important for the formation of the re-activated ATP-Mg-dependent PP1 phosphatase complex, has been experimentally shown to not alter the overall structure of unbound I-29-164 (60). Our data is consistent with these results since Thr72 is part of a longer loop, including residues ∼75-90, that shows modest interaction with the I-29-164 core. Residues in this region of I-29-164 are missing from the crystal structure of the PP1:I-2 complex, however, phosphorylation of Thr72 on I-2 when bound to PP1 increases the fluorescence anisotropy of I-2, suggesting a conformational change upon phosphorylation of bound I-2 (61). Flexibility in this region of I-2 even after binding PP1 is likely critical to reactivation of the PP1:I-2 complex.
DARPP-321-118
In contrast to I-29-164, only a single transient helix (residues 22-29) was identified in the pseudo-substrate inhibitor DARPP-321-118. This region directly connects the two biochemically identified “subdomains” of DARPP-32 that interact with PP1: the common PP1 binding motif (KKIQF7-11) (62) and the region surrounding Thr34, the essential phosphorylation site whose phosphorylation is needed for strong PP1 inhibition (15, 62, 63). While it is possible that secondary structure elements are necessary for some phosphorylation events, structures of PKA in a complex with a pseudosubstrate peptide indicates that the substrate interacts with the active site in an extended manner (64-67). Furthermore, the Nairn laboratory has previously found that several hydrophobic residues within and near this helix are important for inhibition of PP1. To a lesser extent, the number of residues separating the two “subdomains” of DARPP-32 are also important: mutations and deletions in this region resulted in an increased IC50 for PP1 inhibition by pDARPP-32 (15). Therefore, we suggest that the transient helix likely plays an important role in the binding interaction between pDARPP-32 and PP1, acting as a geometric lead to place the pseudo-substrate pThr34 at PP1's active site.
PP1 contains three characteristic surface grooves (acidic, hydrophobic and C-terminal) which emanate from the active site and form the known binding sites for the molecular toxin PP1 inhibitors (6). Additionally, these are potential sites of interaction with PP1 substrates. The stretch of residues between helix22-29 and Thr34 is highly positively charged with numerous arginine residues. This is likely a PP1 interaction signature of DARPP-32, as these residues complement the PP1 acidic groove, which lies between the common interaction site and the active site, in an ideal manner. It has been previously reported that not all areas of this groove are DARPP-32 accessible (15); it is thus possible that the transient helix guides DARPP-32 into a certain binding configuration. Thus, in both inhibitors transient secondary structure likely plays a role in their recognition and binding modes: while helix127-154 of I-2 plays a central role in PP1 inhibition by blocking the PP1 active site, helix22-29 in DARPP-32 likely guides pThr34 into PP1's active site.
A single region of transient extended structure in DARPP-321-118 (residues 62-72) was also identified. One possibility is that transient structure in this region is important for the interaction with PP1 and that it could form a β-sheet with a complementary strand from PP1. However, this is unlikely as pDARPP-321-118 does not exhibit transient extended structure in this region. Another possibility is that because it is N-terminal to the Thr75 phosphorylation site (Cdk5) which, when phosphorylated, activates DARPP-32 as an inhibitor of PKA (68), the transient structure in this region may play a role in phosphorylation of Thr75 or inhibition of PKA.
Based on the results from the use of paramagnetic spin labels, DARRP-321-118 shows a distinctive 3-dimensional topology. The C-terminal residues of DARPP-321-118 form a compact core. Residues near the Thr34 phosphorylation site show some interaction with the core, and, significantly, also with Ser102. Phosphorylation of Ser102 is known to increase the rate of phosphorylation of Thr34 (69), thus our data presents a topological rationale for this enhancement. Lastly, we show that a region of restricted backbone motion can be detected N-terminal of the Ser102 phosphorylation site; this may play a role in the phosphorylation of Ser102. A recent study also indicates that Ser102 is involved in regulating the nuclear export of DARPP-32, which contains a potent nuclear exporting sequence that includes the important residues Leu108 and Leu114. As Stipanovich et al. (70) have shown, when DARPP-32 is phosphorylated at Ser102 the nuclear exporting sequence is active and DARPP-32 is pumped out of the nucleus. However activation of PP2A can dephosphorylate Ser102, prohibiting nuclear export and thus at steady state gives the appearance of DARPP-32 translocating to the nucleus.
Another important topological feature of DARPP-321-118 is that the common PP1 interaction motif (KKIQF7-11) protrudes from the C-terminal core. Mutation of key residues in the motif results in a large decrease in inhibitory potency (15). Furthermore, a peptide containing this motif is able to antagonize inhibition of PP1 by pDARPP-32 (62), and deletion of the motif significantly decreases the binding affinity of pDARPP-32 for PP1, as seen by surface plasmon resonance (46). The freely accessible KKIQF7-11 motif may therefore act as an initial docking site for binding to PP1. Docking may stabilize helix22-29, which could then wrap around the acidic groove of PP1, and allow placement of the pseudo-substrate pThr34 into PP1's active site. Lastly, residues 40-65 form a loosely defined extended loop, as a spin label on residue 51 is only in proximity to its neighboring residues. Very little is so far known about the function of this loop. Ser45 is a defined substrate for CK2 in vitro (69) and is likely phosphorylated in vivo (71); additionally, it has been proposed that this site might play a role in increasing the efficacy of phosphorylation by PKA (69, 71).
pDARPP-321-118
Phosphorylation of Thr34 on DARPP-32 increases its inhibitory potency for PP1 ∼1000-fold, similar to the increased inhibitory potency of the myosin phosphatase holoenzyme (MYPT1:PP1 complex) inhibitor CPI-17 upon its phosphorylation at Thr38 (72-74). While DARPP-32 is an unstructured general PP1 inhibitor, CPI-17 is a structured inhibitor specific to the myosin phosphatase holoenzyme, which consists of PP1, the PP1 targeting protein MYPT1, and the M21 accessory subunit. Phosphorylation of CPI-17 induces a conformational change which arranges pThr38 on the surface of the inhibitor, poised for interaction with PP1 (75). Thus, the phosphorylation induced conformational change of this structured PP1 inhibitor is critical to its inhibitory role. Fluorescence anisotropy and quenching studies of DARPP-32 have indicated that the overall conformation of DARPP-32 is not altered by phosphorylation of Thr34 (44). Our data confirms these results by providing molecular level insight into the structural differences between DARPP-321-118 and pDARPP-321-118. The length of the single transient helix, helix22-29, is not altered by Thr34 phosphorylation, though the percent of time this helix is populated drops slightly from 36% to 33%. No changes in the 1H,1H-NOE pattern is detectable upon phosphorylation of Thr34 (supporting information). Importantly, more residues in this region experience restricted fast timescale backbone motion, indicated by positive 15N[1H]-NOEs, upon Thr34 phosphorylation. This restriction may be due to the strong negative charge of the phosphate group at Thr34. After binding of the common PP1 interaction motif to PP1, this additional backbone restriction may aid in the stabilization of helix22-29 and the placement of Thr34 in the active site of PP1. In addition, the single, significantly populated region of extended structure in DARPP-321-118, residues 62-72 indicated by ssp scores < -0.2, lost its significance upon phosphorylation of Thr34. This region is highly flexible in both forms of DARPP-321-118, as indicated by negative 15N[1H]-NOEs for all residues. Interestingly, fluorescence anisotropy experiments have previously reported that the region near Cys72 on DARPP-32 is slightly constrained in motion, though this is not altered by phosphorylation of Thr34 (44). Our data does not reflect this.
This study shows an important difference between inhibitor protein interactions with apo-PP1 and those with the myosin phosphatase holoenzyme. DARPP-321-118 does not undergo extensive structural changes upon phosphorylation, in contrast to CPI-17. This indicates that the presence of the strong negative charge of the phosphate group most likely contributes significantly to the binding and inhibition of PP1 by pDARPP-321-118.
Summary
In summary, we have determined an experimental 3-dimensional model for two PP1 inhibitor proteins: DARPP-32 and I-2. The high intrinsic flexibility of these PP1 inhibitor proteins provides a likely rationale for how PP1 can maintain an invariant structure while being selectively inhibited. While interaction between PP1 and its regulatory proteins could be accomplished by two rigid and invariant binding partners, our data highlights the advantages of interactions between a rigid (PP1, as shown in the PP1:I2 crystal structure) and a flexible (inhibitor protein) binding partner. First, the high flexibility of the inhibitor allows for multiple interaction points between the inhibitor and PP1, and also for a large interaction surface with PP1 without the need of excessive protein size. Second, the high intrinsic flexibility might be a way to tune the affinity of this interaction. Most PP1 regulatory proteins bind tightly (low nM Kd). However, due to the large interaction surfaces and the likely occurring loss in degrees of freedom in regulatory proteins upon binding, the entropic cost of this interaction carries an adequate energetic penalty to allow for rapid responses to changing signals of the cell or other regulatory proteins of PP1. Furthermore, while the PP1 binding motif in PP1 regulatory proteins is necessary for binding, their intrinsic structural preferences likely guide their binding interactions with PP1. Lastly, the intrinsic flexibility of these PP1 inhibitors also allow for the simultaneous binding of particular pairs of PP1 targeting and inhibitory proteins to PP1 (as it has been proposed for I-2 and spinophilin (18)), and therefore provides additional means for rapid modulation of PP1 by cellular signals.
Supplementary Material
Further information about used methods, including protein expression and purification, NMR analysis, NMR raw data and DARPP-321-118 and I-29-164 behavior in crowded environment is summarized in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
W.P. is the Manning Assistant Professor for Medical Science at Brown University. We thank Dr. Tun-Li Shen for help with the MS measurements and Dr. Rebecca Page for careful reading of the manuscript.
Funding This material is based upon work supported under a National Science Foundation Graduate Research Fellowship to B.D. The project described was supported by Grant Number R01NS056128 from the National Institute Of Neurological Disorders And Stroke to W.P and by P50 MH074866 from the National Institute of Mental Health to A.C.N. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute Of Neurological Disorders And Stroke or the National Institutes of Health. Some NMR data was recorded on the Bruker AVANCE II 800 MHz at Brandies University (NIH S10-RR017269). The CD spectrometer is part of the RI-EPSCoR Proteomics Facility which is supported by the National Science Foundation under Grant No. 0554548 and by NIH NCRR grant (1S10RR020923). The DLS is supported by a Seed-Fund grant from the Office of the Vice President of Research at Brown University.
Abbreviations
- PP1
Protein Phosphatase 1
- NMR
Nuclear Magnetic Resonance
- IUP
intrinsically unstructured proteins
- DARPP-32
dopamine- and cyclic AMP-regulated phosphoprotein of molecular weight 32 kDa
- I-2
Inhibitor-2
References
- 1.Moorhead GB, Trinkle-Mulcahy L, Ulke-Lemee A. Emerging roles of nuclear protein phosphatases. Nat Rev Mol Cell Biol. 2007;8:234–244. doi: 10.1038/nrm2126. [DOI] [PubMed] [Google Scholar]
- 2.Cohen PT. Protein phosphatase 1--targeted in many directions. J Cell Sci. 2002;115:241–256. doi: 10.1242/jcs.115.2.241. [DOI] [PubMed] [Google Scholar]
- 3.Bollen M. Combinatorial control of protein phosphatase-1. Trends Biochem Sci. 2001;26:426–431. doi: 10.1016/s0968-0004(01)01836-9. [DOI] [PubMed] [Google Scholar]
- 4.Watanabe T, Huang HB, Horiuchi A, da Cruze Silva EF, Hsieh-Wilson L, Allen PB, Shenolikar S, Greengard P, Nairn AC. Protein phosphatase 1 regulation by inhibitors and targeting subunits. Proc Natl Acad Sci U S A. 2001;98:3080–3085. doi: 10.1073/pnas.051003898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meiselbach H, Sticht H, Enz R. Structural analysis of the protein phosphatase 1 docking motif: molecular description of binding specificities identifies interacting proteins. Chem Biol. 2006;13:49–59. doi: 10.1016/j.chembiol.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 6.Holmes CF, Maynes JT, Perreault KR, Dawson JF, James MN. Molecular enzymology underlying regulation of protein phosphatase-1 by natural toxins. Current medicinal chemistry. 2002;9:1981–1989. doi: 10.2174/0929867023368827. [DOI] [PubMed] [Google Scholar]
- 7.Honkanen RE, Golden T. Regulators of serine/threonine protein phosphatases at the dawn of a clinical era? Current medicinal chemistry. 2002;9:2055–2075. doi: 10.2174/0929867023368836. [DOI] [PubMed] [Google Scholar]
- 8.Goldberg J, Huang HB, Kwon YG, Greengard P, Nairn AC, Kuriyan J. Three-dimensional structure of the catalytic subunit of protein serine/threonine phosphatase-1. Nature. 1995;376:745–753. doi: 10.1038/376745a0. [DOI] [PubMed] [Google Scholar]
- 9.Kita A, Matsunaga S, Takai A, Kataiwa H, Wakimoto T, Fusetani N, Isobe M, Miki K. Crystal structure of the complex between calyculin A and the catalytic subunit of protein phosphatase 1. Structure (Camb) 2002;10:715–724. doi: 10.1016/s0969-2126(02)00764-5. [DOI] [PubMed] [Google Scholar]
- 10.Maynes JT, Bateman KS, Cherney MM, Das AK, Luu HA, Holmes CF, James MN. Crystal structure of the tumor-promoter okadaic acid bound to protein phosphatase-1. J Biol Chem. 2001;276:44078–44082. doi: 10.1074/jbc.M107656200. [DOI] [PubMed] [Google Scholar]
- 11.Egloff MP, Johnson DF, Moorhead G, Cohen PT, Cohen P, Barford D. Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. Embo J. 1997;16:1876–1887. doi: 10.1093/emboj/16.8.1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Terrak M, Kerff F, Langsetmo K, Tao T, Dominguez R. Structural basis of protein phosphatase 1 regulation. Nature. 2004;429:780–784. doi: 10.1038/nature02582. [DOI] [PubMed] [Google Scholar]
- 13.Svenningsson P, Nishi A, Fisone G, Girault JA, Nairn AC, Greengard P. DARPP-32: an integrator of neurotransmission. Annu Rev Pharmacol Toxicol. 2004;44:269–296. doi: 10.1146/annurev.pharmtox.44.101802.121415. [DOI] [PubMed] [Google Scholar]
- 14.Greengard P, Allen PB, Nairn AC. Beyond the dopamine receptor: the DARPP-32/protein phosphatase-1 cascade. Neuron. 1999;23:435–447. doi: 10.1016/s0896-6273(00)80798-9. [DOI] [PubMed] [Google Scholar]
- 15.Huang HB, Horiuchi A, Watanabe T, Shih SR, Tsay HJ, Li HC, Greengard P, Nairn AC. Characterization of the inhibition of protein phosphatase-1 by DARPP-32 and inhibitor-2. J Biol Chem. 1999;274:7870–7878. doi: 10.1074/jbc.274.12.7870. [DOI] [PubMed] [Google Scholar]
- 16.DePaoli-Roach AA, Lee FT. Phosphoprotein phosphatase inhibitor-2 is phosphorylated at both serine and threonine residues in mouse diaphragm. FEBS Lett. 1985;183:423–429. doi: 10.1016/0014-5793(85)80824-3. [DOI] [PubMed] [Google Scholar]
- 17.Connor JH, Frederick D, Huang H, Yang J, Helps NR, Cohen PT, Nairn AC, DePaoli-Roach A, Tatchell K, Shenolikar S. Cellular mechanisms regulating protein phosphatase-1. A key functional interaction between inhibitor-2 and the type 1 protein phosphatase catalytic subunit. J Biol Chem. 2000;275:18670–18675. doi: 10.1074/jbc.M909312199. [DOI] [PubMed] [Google Scholar]
- 18.Terry-Lorenzo RT, Elliot E, Weiser DC, Prickett TD, Brautigan DL, Shenolikar S. Neurabins recruit protein phosphatase-1 and inhibitor-2 to the actin cytoskeleton. J Biol Chem. 2002;277:46535–46543. doi: 10.1074/jbc.M206960200. [DOI] [PubMed] [Google Scholar]
- 19.Park IK, DePaoli-Roach AA. Domains of phosphatase inhibitor-2 involved in the control of the ATP-Mg-dependent protein phosphatase. J Biol Chem. 1994;269:28919–28928. [PubMed] [Google Scholar]
- 20.Sakashita G, Shima H, Komatsu M, Urano T, Kikuchi A, Kikuchi K. Regulation of type 1 protein phosphatase/inhibitor-2 complex by glycogen synthase kinase-3beta in intact cells. J Biochem. 2003;133:165–171. doi: 10.1093/jb/mvg020. [DOI] [PubMed] [Google Scholar]
- 21.Lin TH, Huang YC, Chin ML, Chen YC, Jeng HH, Lin FM, Shiao MS, Horiuchi A, Greengard P, Nairn AC, Huang HB. 1H, 15N, and 13C resonance assignments of DARPP-32 (dopamine and cAMP-regulated phosphoprotein, Mr. 32,000)--a protein inhibitor of protein phosphatase-1. Journal of biomolecular NMR. 2004;28:413–414. doi: 10.1023/B:JNMR.0000015358.20495.b8. [DOI] [PubMed] [Google Scholar]
- 22.Huang HB, Chen YC, Tsai LH, Wang H, Lin FM, Horiuchi A, Greengard P, Nairn AC, Shiao MS, Lin TH. Backbone 1H, 15N, and 13C resonance assignments of inhibitor-2 -- a protein inhibitor of protein phosphatase-1. Journal of biomolecular NMR. 2000;17:359–360. doi: 10.1023/a:1008355428294. [DOI] [PubMed] [Google Scholar]
- 23.Hurley TD, Yang J, Zhang L, Goodwin KD, Zou Q, Cortese M, Dunker AK, DePaoli-Roach AA. Structural basis for regulation of protein phosphatase 1 by inhibitor-2. J Biol Chem. 2007;282:28874–28883. doi: 10.1074/jbc.M703472200. [DOI] [PubMed] [Google Scholar]
- 24.Peti W, Page R. Strategies to maximize heterologous protein expression in Escherichia coli with minimal cost. Protein Expr Purif. 2007;51:1–10. doi: 10.1016/j.pep.2006.06.024. [DOI] [PubMed] [Google Scholar]
- 25.Kay LE, Torchia DA, Bax A. Backbone dynamics of proteins as studied by 15N inverse detected heteronuclear NMR spectroscopy: application to staphylococcal nuclease. Biochemistry. 1989;28:8972–8979. doi: 10.1021/bi00449a003. [DOI] [PubMed] [Google Scholar]
- 26.Akke M, Palmer AG. Monitoring macromolecular motions on microsecond to millisecond time scales by R1p - R1 constant relaxation time NMR spectroscopy. Journal of the American Chemical Society. 1996;118:2. [Google Scholar]
- 27.Palmer AG, Case DA. Molecular dynamics analysis of NMR relaxation in a zinc-finger peptide. Journal of the American Chemical Society. 1992;114:9. [Google Scholar]
- 28.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 29.Johnson BA. Using NMRView to visualize and analyze the NMR spectra of macromolecules. Methods Mol Biol. 2004;278:313–352. doi: 10.1385/1-59259-809-9:313. [DOI] [PubMed] [Google Scholar]
- 30.Sattler J, Schleucher J, Griesinger C. Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradients. Progress in NMR Spectroscopy. 1999;34:93–158. [Google Scholar]
- 31.Marsh JA, Singh VK, Jia Z, Forman-Kay JD. Sensitivity of secondary structure propensities to sequence differences between alpha- and gamma-synuclein: implications for fibrillation. Protein Sci. 2006;15:2795–2804. doi: 10.1110/ps.062465306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Spera S, Bax A. Empirical correlation between protein backbone conformation and Ca and Cb 13C nuclear magnetic resonance chemical shifts. Journal of the American Chemical Society. 1991;113:3. [Google Scholar]
- 33.Zhang H, Neal S, Wishart DS. RefDB: a database of uniformly referenced protein chemical shifts. Journal of biomolecular NMR. 2003;25:173–195. doi: 10.1023/a:1022836027055. [DOI] [PubMed] [Google Scholar]
- 34.Wang Y, Jardetzky O. Probability-based protein secondary structure identification using combined NMR chemical-shift data. Protein Sci. 2002;11:852–861. doi: 10.1110/ps.3180102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wishart DS, Bigam CG, Holm A, Hodges RS, Sykes BD. 1H, 13C and 15N random coil NMR chemical shifts of the common amino acids. I. Investigations of nearest-neighbor effects. Journal of biomolecular NMR. 1995;5:67–81. doi: 10.1007/BF00227471. [DOI] [PubMed] [Google Scholar]
- 36.Abraham DJ, Leo AJ. Proteins: Structure, function and genetics. Vol. 2. 1987. p. 23. [Google Scholar]
- 37.Rose GD, Geselowitz AR, Lesser GJ, Lee RH, Zehfus MH. Hydrophobicity of amino acid residues in globular proteins. Science (New York, NY) 1985;229:834–838. doi: 10.1126/science.4023714. [DOI] [PubMed] [Google Scholar]
- 38.Schwalbe H, Fiebig KM, Buck M, Jones JA, Grimshaw SB, Spencer A, Glaser SJ, Smith LJ, Dobson CM. Structural and dynamical properties of a denatured protein. Heteronuclear 3D NMR experiments and theoretical simulations of lysozyme in 8 M urea. Biochemistry. 1997;36:8977–8991. doi: 10.1021/bi970049q. [DOI] [PubMed] [Google Scholar]
- 39.Wirmer J, Peti W, Schwalbe H. Motional properties of unfolded ubiquitin: a model for a random coil protein. Journal of biomolecular NMR. 2006;35:175–186. doi: 10.1007/s10858-006-9026-9. [DOI] [PubMed] [Google Scholar]
- 40.Wilkins DK, Grimshaw SB, Receveur V, Dobson CM, Jones JA, Smith LJ. Hydrodynamic radii of native and denatured proteins measured by pulse field gradient NMR techniques. Biochemistry. 1999;38:16424–16431. doi: 10.1021/bi991765q. [DOI] [PubMed] [Google Scholar]
- 41.Kohn JE, Millett IS, Jacob J, Zagrovic B, Dillon TM, Cingel N, Dothager RS, Seifert S, Thiyagarajan P, Sosnick TR, Hasan MZ, Pande VS, Ruczinski I, Doniach S, Plaxco KW. Random-coil behavior and the dimensions of chemically unfolded proteins. Proc Natl Acad Sci U S A. 2004;101:12491–12496. doi: 10.1073/pnas.0403643101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eliezer D. Characterizing residual structure in disordered protein states using nuclear magnetic resonance. Methods Mol Biol. 2007;350:49–67. doi: 10.1385/1-59745-189-4:49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dyson HJ, Wright PE. Intrinsically unstructured proteins and their functions. Nat Rev Mol Cell Biol. 2005;6:197–208. doi: 10.1038/nrm1589. [DOI] [PubMed] [Google Scholar]
- 44.Neyroz P, Desdouits F, Benfenati F, Knutson JR, Greengard P, Girault JA. Study of the conformation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, by fluorescence spectroscopy. J Biol Chem. 1993;268:24022–24031. [PubMed] [Google Scholar]
- 45.Wishart DS, Sykes BD, Richards FM. The chemical shift index: a fast and simple method for the assignment of protein secondary structure through NMR spectroscopy. Biochemistry. 1992;31:1647–1651. doi: 10.1021/bi00121a010. [DOI] [PubMed] [Google Scholar]
- 46.Lin TH, Tsai PC, Liu HT, Chen YC, Wang LH, Hsieh FK, Huang HB. Characterization of the Protein Phosphatase 1-Binding Motifs of Inhibitor-2 and DARPP-32 by Surface Plasmon Resonance. J Biochem (Tokyo) 2005;138:697–700. doi: 10.1093/jb/mvi167. [DOI] [PubMed] [Google Scholar]
- 47.Klein-Seetharaman J, Oikawa M, Grimshaw SB, Wirmer J, Duchardt E, Ueda T, Imoto T, Smith LJ, Dobson CM, Schwalbe H. Long-range interactions within a nonnative protein. Science (New York, NY) 2002;295:1719–1722. doi: 10.1126/science.1067680. [DOI] [PubMed] [Google Scholar]
- 48.Schwarzinger S, Wright PE, Dyson HJ. Molecular hinges in protein folding: the urea-denatured state of apomyoglobin. Biochemistry. 2002;41:12681–12686. doi: 10.1021/bi020381o. [DOI] [PubMed] [Google Scholar]
- 49.Lietzow MA, Jamin M, Jane Dyson HJ, Wright PE. Mapping long-range contacts in a highly unfolded protein. J Mol Biol. 2002;322:655–662. doi: 10.1016/s0022-2836(02)00847-1. [DOI] [PubMed] [Google Scholar]
- 50.Schmidt PG, Kuntz ID. Distance measurements in spin-labeled lysozyme. Biochemistry. 1984;23:4261–4266. doi: 10.1021/bi00313a038. [DOI] [PubMed] [Google Scholar]
- 51.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. II. Distance restraints from paramagnetic relaxation and calculation of an ensemble of structures. J Mol Biol. 1997;268:170–184. doi: 10.1006/jmbi.1997.0953. [DOI] [PubMed] [Google Scholar]
- 52.Gillespie JR, Shortle D. Characterization of long-range structure in the denatured state of staphylococcal nuclease. I. Paramagnetic relaxation enhancement by nitroxide spin labels. J Mol Biol. 1997;268:158–169. doi: 10.1006/jmbi.1997.0954. [DOI] [PubMed] [Google Scholar]
- 53.Teilum K, Kragelund BB, Poulsen FM. Transient structure formation in unfolded acyl-coenzyme A-binding protein observed by site-directed spin labelling. J Mol Biol. 2002;324:349–357. doi: 10.1016/s0022-2836(02)01039-2. [DOI] [PubMed] [Google Scholar]
- 54.Dyson HJ, Wright PE. Coupling of folding and binding for unstructured proteins. Curr Opin Struct Biol. 2002;12:54–60. doi: 10.1016/s0959-440x(02)00289-0. [DOI] [PubMed] [Google Scholar]
- 55.Tompa P. The interplay between structure and function in intrinsically unstructured proteins. FEBS Lett. 2005;579:3346–3354. doi: 10.1016/j.febslet.2005.03.072. [DOI] [PubMed] [Google Scholar]
- 56.Radhakrishnan I, Perez-Alvarado GC, Dyson HJ, Wright PE. Conformational preferences in the Ser133-phosphorylated and non-phosphorylated forms of the kinase inducible transactivation domain of CREB. FEBS Lett. 1998;430:317–322. doi: 10.1016/s0014-5793(98)00680-2. [DOI] [PubMed] [Google Scholar]
- 57.Yang J, Hurley TD, DePaoli-Roach AA. Interaction of inhibitor-2 with the catalytic subunit of type 1 protein phosphatase. Identification of a sequence analogous to the consensus type 1 protein phosphatase-binding motif. J Biol Chem. 2000;275:22635–22644. doi: 10.1074/jbc.M003082200. [DOI] [PubMed] [Google Scholar]
- 58.Park IK, Roach P, Bondor J, Fox SP, DePaoli-Roach AA. Molecular mechanism of the synergistic phosphorylation of phosphatase inhibitor-2. Cloning, expression, and site-directed mutagenesis of inhibitor-2. J Biol Chem. 1994;269:944–954. [PubMed] [Google Scholar]
- 59.Csizmok V, Bokor M, Banki P, Klement E, Medzihradszky KF, Friedrich P, Tompa K, Tompa P. Primary contact sites in intrinsically unstructured proteins: the case of calpastatin and microtubule-associated protein 2. Biochemistry. 2005;44:3955–3964. doi: 10.1021/bi047817f. [DOI] [PubMed] [Google Scholar]
- 60.Lin TH, Chen YC, Chyan CL, Tsay LH, Tang TC, Jeng HH, Lin FM, Huang HB. Phosphorylation by glycogen synthase kinase of inhibitor-2 does not change its structure in free state. FEBS Lett. 2003;554:253–256. doi: 10.1016/s0014-5793(03)01097-4. [DOI] [PubMed] [Google Scholar]
- 61.Picking WD, Kudlicki W, Kramer G, Hardesty B, Vandenheede JR, Merlevede W, Park IK, DePaoli-Roach A. Fluorescence studies on the interaction of inhibitor 2 and okadaic acid with the catalytic subunit of type 1 phosphoprotein phosphatases. Biochemistry. 1991;30:10280–10287. doi: 10.1021/bi00106a028. [DOI] [PubMed] [Google Scholar]
- 62.Kwon YG, Huang HB, Desdouits F, Girault JA, Greengard P, Nairn AC. Characterization of the interaction between DARPP-32 and protein phosphatase 1 (PP-1): DARPP-32 peptides antagonize the interaction of PP-1 with binding proteins. Proc Natl Acad Sci U S A. 1997;94:3536–3541. doi: 10.1073/pnas.94.8.3536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hemmings HC, Jr, Nairn AC, Elliott JI, Greengard P. Synthetic peptide analogs of DARPP-32 (Mr 32,000 dopamine- and cAMP-regulated phosphoprotein), an inhibitor of protein phosphatase-1. Phosphorylation, dephosphorylation, and inhibitory activity. J Biol Chem. 1990;265:20369–20376. [PubMed] [Google Scholar]
- 64.Kim C, Vigil D, Anand G, Taylor SS. Structure and dynamics of PKA signaling proteins. Eur J Cell Biol. 2006;85:651–654. doi: 10.1016/j.ejcb.2006.02.004. [DOI] [PubMed] [Google Scholar]
- 65.Meinkoth JL, Alberts AS, Went W, Fantozzi D, Taylor SS, Hagiwara M, Montminy M, Feramisco JR. Signal transduction through the cAMP-dependent protein kinase. Mol Cell Biochem. 1993;127-128:179–186. doi: 10.1007/BF01076769. [DOI] [PubMed] [Google Scholar]
- 66.Taylor SS, Kim C, Vigil D, Haste NM, Yang J, Wu J, Anand GS. Dynamics of signaling by PKA. Biochim Biophys Acta. 2005;1754:25–37. doi: 10.1016/j.bbapap.2005.08.024. [DOI] [PubMed] [Google Scholar]
- 67.Taylor SS, Yang J, Wu J, Haste NM, Radzio-Andzelm E, Anand G. PKA: a portrait of protein kinase dynamics. Biochim Biophys Acta. 2004;1697:259–269. doi: 10.1016/j.bbapap.2003.11.029. [DOI] [PubMed] [Google Scholar]
- 68.Nairn AC, Svenningsson P, Nishi A, Fisone G, Girault JA, Greengard P. The role of DARPP-32 in the actions of drugs of abuse. Neuropharmacology. 2004;47 1:14–23. doi: 10.1016/j.neuropharm.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 69.Girault JA, Hemmings HC, Jr, Williams KR, Nairn AC, Greengard P. Phosphorylation of DARPP-32, a dopamine- and cAMP-regulated phosphoprotein, by casein kinase II. J Biol Chem. 1989;264:21748–21759. [PubMed] [Google Scholar]
- 70.Stipanovich A, Valjent E, Matamales M, Nishi A, Ahn JH, Maroteaux M, Bertran-Gonzalez J, Brami-Cherrier K, Enslen H, Corbille AG, Filhol O, Nairn AC, Greengard P, Herve D, Girault JA. A phosphatase cascade by which rewarding stimuli control nucleosomal response. Nature. 2008;453:879–884. doi: 10.1038/nature06994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jin M, Bateup H, Padovan JC, Greengard P, Nairn AC, Chait BT. Quantitative analysis of protein phosphorylation in mouse brain by hypothesis-driven multistage mass spectrometry. Anal Chem. 2005;77:7845–7851. doi: 10.1021/ac051519m. [DOI] [PubMed] [Google Scholar]
- 72.Desdouits F, Cheetham JJ, Huang HB, Kwon YG, da Cruz e Silva EF, Denefle P, Ehrlich ME, Nairn AC, Greengard P, Girault JA. Mechanism of inhibition of protein phosphatase 1 by DARPP-32: studies with recombinant DARPP-32 and synthetic peptides. Biochem Biophys Res Commun. 1995;206:652–658. doi: 10.1006/bbrc.1995.1092. [DOI] [PubMed] [Google Scholar]
- 73.Ohki S, Eto M, Kariya E, Hayano T, Hayashi Y, Yazawa M, Brautigan D, Kainosho M. Solution NMR structure of the myosin phosphatase inhibitor protein CPI-17 shows phosphorylation-induced conformational changes responsible for activation. J Mol Biol. 2001;314:839–849. doi: 10.1006/jmbi.2001.5200. [DOI] [PubMed] [Google Scholar]
- 74.Ohki S, Eto M, Shimizu M, Takada R, Brautigan DL, Kainosho M. Distinctive solution conformation of phosphatase inhibitor CPI-17 substituted with aspartate at the phosphorylation-site threonine residue. J Mol Biol. 2003;326:1539–1547. doi: 10.1016/s0022-2836(03)00048-2. [DOI] [PubMed] [Google Scholar]
- 75.Eto M, Kitazawa T, Matsuzawa F, Aikawa S, Kirkbride JA, Isozumi N, Nishimura Y, Brautigan DL, Ohki SY. Phosphorylation-induced conformational switching of CPI-17 produces a potent myosin phosphatase inhibitor. Structure. 2007;15:1591–1602. doi: 10.1016/j.str.2007.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Further information about used methods, including protein expression and purification, NMR analysis, NMR raw data and DARPP-321-118 and I-29-164 behavior in crowded environment is summarized in the supporting information. This material is available free of charge via the Internet at http://pubs.acs.org.