

SUMMARY
Mechanical ventilation (MV) is used as therapy to support critically ill patients, however the mechanisms by which MV induces lung injury and inflammation remain unclear. EGFR mediated signaling plays a key role in various physiologic and pathologic processes, including those modulated by mechanical and shear forces, in various cell types. We hypothesized that EGFR-activated signaling plays a key role in ventilator induced lung injury and inflammation (VILI). To test this hypothesis, we assessed lung vascular and alveolar permeability, as well as inflammation, which are cardinal features of VILI, in mice treated with the EGFR inhibitor, AG1478. Inhibition of EGFR activity greatly diminished MV-induced lung alveolar permeability and neutrophil accumulation in the bronchoalveolar lavage (BAL) fluid, as compared to vehicle-treated controls. Similarly, AG1478 inhibition diminished lung vascular leak (as assessed by Evans blue extravasation), but did not affect interstitial neutrophil accumulation. Inhibition of the EGFR pathway also blocked expression of genes induced by MV. However, intratracheal instillation of EGF alone failed to induce lung injury. Collectively, our findings suggest that EGFR-activated signaling is necessary but not sufficient to produce ALI in mice.
INTRODUCTION
Mechanical ventilation (MV) is a cornerstone and only known effective therapy to support patients with the acute lung injury (ALI) and its more severe form, acute respiratory distress syndrome (ARDS). Although the outcome of ALI/ARDS patients subjected to MV has improved over the past few years, substantial mortality remains associated with this syndrome (~30–40%)1. Several studies have shown that mechanical and shear forces generated by injurious high tidal volumes related to MV can cause lung alveolar and vascular permeability as well as lung inflammatory responses. These events may further exacerbate a generalized inflammatory response and injury resulting in multi-system organ dysfunction, ultimately leading to death1. In vitro studies have demonstrated that MV can cause endothelial and epithelial cell deformation and physical disruption of plasma membrane integrity leading to alveolar protein leakage and activation of pro-inflammatory and pro-oxidant pathways (see reviews, 2, 3). An imbalance between prooxidants and antioxidants, and dysregulation of various cytokines and chemokines expression, play fundamental roles in lung injury and inflammation 3, 4; however, the exact mechanisms by which mechanical forces contribute to the pathogenesis of VILI remain unclear.
EGFR mediated signaling has been implicated in various cellular physiological and pathologic processes. EGFR-activated signaling regulates the downstream effector pathways, such as MAP kinase and PI3-K/Akt signaling, that activate various transcription factors in response to stimuli 5. The activation of EGFR-mediated signaling and subsequent MAP kinase activation by cyclic strain/stretch associated with MV have been demonstrated in various cell types, including lung epithelial cells, suggesting that EGFR acts as a mechanotransducer 6–8. To test our hypothesis that EGFR is a critical regulator of VILI in vivo, in the present study we compared lung injury and inflammatory responses to injurious MV in mice treated with pharmacological inhibition of EGFR. Our results support a role for EGFR in MV-induced lung injury in vivo, although receptor activation alone does not appear to be sufficient in driving this process.
METHODS
Mechanical ventilation (MV)
All experimental animal protocols were performed in accordance with guidelines approved by the animal care use committee at the Johns Hopkins University Bloomberg School of Public Health. The CD-1 strain of mice (females, 7–8 weeks old, ~30 gms) were used unless otherwise indicated. We have recently shown that MV induces lung injury and inflammation in this strain of mice9. Exposure to MV was performed as previously described 9. Briefly, mice were subjected to MV using model Inspira asv 55–7058, (Harvard Apparatus, MA) with injurious tidal volumes (30 ml/Kg body weight) for 2 hours. Sham-operated, anesthetized but breathing spontaneously (SpV), mice were used as a control group. At least three mice (n=3) were used in each experimental group unless otherwise indicated.
Assessment of lung injury and inflammation
To measure lung alveolar permeability and inflammation, at the end of experiments animals were given a lethal dose of the anesthetic agent and lungs were instilled with 1.5 ml (0.75 ml each time/twice) of sterile PBS and cells in the bronchoalveolar (BAL) fluid were counted using a hemocytometer. Differential cell counts were assessed with Diff-Quik stain set. BAL protein concentration was measured with a BCA Protein Assay kit (Pierce chemical co, IL). To measure lung vascular leak, mice were given a intraperitoneal injection of Evans blue dye (EBD) (Sigma, cat # E2129; 12 μl/g body weight) one hour prior to termination of MV. A blood sample was drawn from the inferior vena cava, centrifuged at 14000 rpm and serum was stored at −80°C. Lungs were perfused with a heparin solution (Sigma, cat # H3393), harvested en bloc, dabbed dry and stored in liquid nitrogen. Lungs from all experimental groups were placed individually into 1 ml formamide solution and incubated at 65°C for 48 h. The content of EBD present in the formamide solution and serum samples was read at 620 nm (with triplicate samples). Lungs were dried at 65°C for 48 h and weighed. EBD prepared in formamide solution was used to generate standard curve. The amount of EBD was calculated and expressed as mg/ml of lung solution using the following formula: (Lung EBD/Plasma EBD)/lung dry weight.
Supplementation of EGFR inhibitor
AG1478, which is known to specifically block EGFR activation, was obtained from EMD Biosciences. To assess the effects of EGFR inhibition on MV-induced pulmonary permeability and inflammation, mice received a single dose of the AG1478 (50 mg/kg body weight or ~1.5 mg/mouse) or (10 mg/kg or ~0.3 mg/mouse) by intraperitoneal injection 30 min prior to exposure to MV. Stock of this compound was prepared at concentration 200 mg/ml in DMSO and diluted freshly in PBS to minimize the DMSO concentration in mice. A similar volume of PBS containing the same concentration of DMSO was used as vehicle control.
Immunohistochemistry (IHC)
Lungs were inflated to 25 cm of water pressure with 10% buffered formalin containing 0.5% low-melting agarose, and embedded in paraffin; 5-μm sections were cut and stained with H&E. Tissue sections were incubated with a monoclonal antibody that specifically stains neutrophils (AbD Serotec, NC) followed by HRP-streptavidin conjugated secondary antibody, and the number of neutrophils in the lung sections were counted on images acquired with a Nikon E 800 Microscope, as described previously 9. The number of neutrophils in the lung (15 fields/lung section) was normalized per field, for each animal, and expressed as mean ± SE (n=4 per group) as detailed previously 9.
Real time RT-PCR analyses
Total RNA (2 μg) isolated from the left lobe of each mouse was reverse transcribed using superscript™ first strand cDNA synthesis system (Invitrogen Corp., CA), and real time RT-PCR reactions were performed in triplicate using TaqMan® gene expression assays for Egr1, Atf3, Areg, Cxcl2, and β-actin purchased from Applied Biosystems (CA). The CT values of individual sample for each gene were normalized to that of β-actin levels, and the relative value for the control groups was set as one. Three animals were used for each experimental group.
Western blot analysis
Lung tissues were homogenized in cold cell lysis buffer composed of 50 mM TrisHCl, pH 7.4, 150 mM NaCl, 1% Nonidet-P40, 5 mM EDTA, 50 mM NaF, 1 mM Na3VO5, 1 mM PMSF, and 1X protease inhibitor cocktail. The homogenates were then centrifuged at 12,000 rpm for 15 min at 4°C. The supernatants were collected and protein concentration was determined by the BCA method (Pierce Biochemicals, IL). Up to 100 μg of total protein was used for immunoblot analysis, which was performed using phospho-specific (Tyrosine-1068) and native-form of EGFR antibodies (Cell Signaling Technology, MA).
Statistical analysis
All data involving animal experimentation were collected in a double-blind fashion. Values are shown as means ± SE, with n = 3–5 for each experimental condition as indicated in the legends. Analysis of variance (ANOVA) was used to compare means of multiple groups. For paired data, Students’ t-test was used. Significance in all cases was defined as P ≤ 0.05.
RESULTS
Inhibition of EGFR activity attenuates MV-induced lung injury and inflammation
To determine the relevance of EGFR signaling in VILI, mice were subjected to MV supplemented with two different doses (10 mg or 50 mg/kg body weight) of the EGFR inhibitor AG1478. DMSO was used as vehicle control. Similarly, sham-operated and anesthetized (hereafter referred to as SpV) mice were treated with vehicle or AG1478 and were used as respective control groups. Lung alveolar protein leakage and leukocyte infiltration are hallmarks of ALI/ARDS. Thus, the effects of AG1478 on MV-induced lung alveolar permeability and neutrophilaccumulation in BAL fluid were assessed (Fig. 1). Consistent with previous results, we found a significant increase in total protein accumulation in vehicle-treated mice following MV as compared to the SpV control group. However, protein leakage in the BAL fluid of AG1478-treated mice subjected to MV was significantly lower as compared to the respective DMSO-treated mice (Fig. 1A). As anticipated, we found that inhibition of EGFR activation was more effective at higher (50 mg/ml) compared to lower (10 mg/ml) dose. Differential cell count analysis revealed decreased numbers of neutrophils in the BAL fluid of mice supplemented with AG1478 compared to DMSO-treated animals (Fig. 1C) following MV. However, EGFR inhibition at high dose significantly increased the number of macrophages in animals subjected to SpV or MV (Fig. 1D). These results suggest that the EGFR pathway modulates MV-induced lung injury and its associated inflammatory response.
Figure 1. Effect of EGFR inhibition on MV induced lung injury and inflammatory responses.
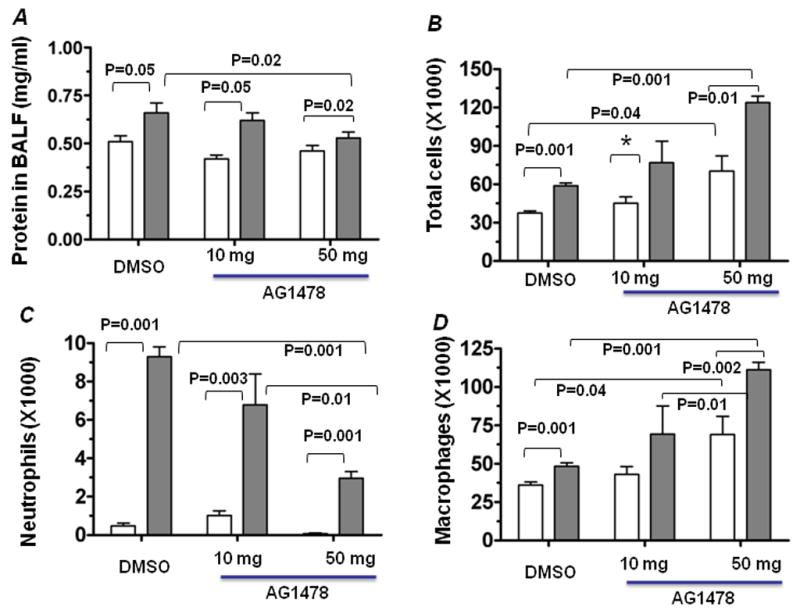

Mice were pretreated for 30 min with AG1478 (10 mg or 50/mg per kg body weight) or DMSO and then randomly assigned to SpV (open bars) or MV (filled bars) groups. Mice were subjected to MV or breathed spontaneously (SpV) for 2 h, and BAL fluid was obtained and protein was quantified as detailed in Methods. (A) The effect of AG1478 on MV induced lung alveolar permeability. The number of cells (B), neutrophils (C), and total macrophages (D) in the BAL fluid collected from mice was counted by differential staining with Diff Quick’s. Values represent means ± SE (n = 3 for SpV group and n=5 for MV group) and the respective P values are displayed. (E) EGFR activation by MV. Mice were exposed to SpV or MV for 30 min supplemented with either DMSO or AG1478. The right lobe from each animal was separately homogenized in MAP kinase lysis buffer and immunoblotted using antibodies specific for the phosphorylated form of EGFR. The native form of EGFR was used as loading control. Lanes 2 and 3 represent lung samples from two different mice treated with DMSO and then subjected to MV, whereas lanes 5 and 6 represent that of AG1478-treated MV group. Lanes 1 and 2 represent lung samples isolated from SpV mice pre-treated with DMSO and AG1478, respectively.
To verify the status EGFR activation in our experimental conditions, mice were pretreated with DMSO (vehicle) or AG178 for 30 min, and then subjected to MV or SpV for 30 min. Lung cell lysates were probed with antibodies specific for the phosphorylated form of EGFR by Western blot analysis (1E). As expected, MV increased the levels of EGFR phosphorylation as compared to SpV control, which was blocked by AG1478 treatment. These results support EGFR activation by MV and the effectiveness of this mode of pharmacological inhibition.
EGFR inhibition augments MV-induced lung vascular neutrophil infiltration
Lung vascular leukocyte infiltration also contributes to the development of ALI. To assess whether inhibition of EGFR also attenuates MV-induced vascular neutrophil infiltration, we determined neutrophil accumulation in lung tissue sections by immunohistochemical analysis, as previously described 9. Tissue sections (Fig. 2A) were stained with anti-neutrophil antibody (Fig. 2B) and the number of neutrophils present in the alveolar septae was quantified (Fig. 2C) as detailed in “Methods”. MV caused a significant increase (in neutrophil accumulation in lungs of DMSO-treated mice as compared to the SpV group. Surprisingly, AG1478–treated mice displayed greater levels of neutrophil accumulation as compared to vehicle-treated mice in response to MV.
Figure 2. Effect of AG1478 on MV-induced pulmonary neutrophil infiltration.

(A) H& E staining of lung tissues (magnification 400×). Arrows indicate the position of positive antibody staining for the presence of neutrophils. (B) Assessment of MV-induced neutrophil infiltration in lung parenchyma of mice supplemented with vehicle or AG1478 was performed using anti-neutrophil antibody as detailed in “Methods”. (C) Quantification of neutrophils present in the alveolar space. Digital images of tissue sections stained with anti-neutrophil antibody were obtained and neutrophils were quantified as detailed elsewhere 9. Data represent means ± SE of an average of at least 15 high-power fields for each experimental group (n =4). P values are shown for respective experimental groups.
EGFR inhibition attenuates MV-induced lung vascular leak
The above results suggest that EGFR activated signaling may specifically participate in migration of neutrophils from the interstitial space to the alveolar compartment or may play a prominent role in regulating MV-induced epithelial (rather than vascular) barrier function. To test this hypothesis, we examined the effect of EGFR inhibition on MV-induced vascular leak. Evans blue dye extravasation assays revealed decreased levels of MV-induced lung vascular leak in mice supplemented with the EGFR inhibitor compared with mice supplemented with DMSO (Fig. 3). These results suggest that EGFR regulates MV-induced lung vascular permeability.
Figure 3. Effects of exogenous A1478 on MV-induced vascular leak.

Mice were treated for 30 min with AG1478 (50 mg/kg b.w) or DMSO prior to MV exposure, and randomly assigned to MV or SpV. Mice were injected with Evans blue dye intraperitoneally 1 h prior to the end of MV exposure. Lungs were flushed and vascular leakage was assessed by the extravasation of Evans blue into lung parenchyma as detailed in Methods. Data are presented as means ± SE. The following number of mice was used in each experimental group include: N=5 for DMSO-SpV; N=9 for DMSO-MV; N=5 for AG1478-SpV, and N=7 for AG1478-MV. P values for respective groups are indicated.
EGFR modulates MV-induced gene expression in the lung
To determine if inhibition of EGFR activity ultimately leads to alteration in MV-induced gene expression, we analyzed the expression of selective genes that are induced by MV by quantitative RT-PCR (Fig. 4). We chose 4 candidate genes encoding for the transcription factors Egr1 and Atf3, for a pro-inflammatory chemokine Cxcl2, and an EGF-like EGFR ligand, amphiregulin (Areg). These genes are greatly induced by MV and are known to be involved in lung injury and inflammation 10, 11. As shown in Fig. 4, there was a marked increase in the lung expression of Atf3, Areg, and Cxcl2 in response to MV in DMSO-treated mice; however, this induction was significantly inhibited in AG1478-treated counterpart animals. Although MV significantly increased the expression of Egr1 gene, this was not significantly affected by treatment with AG1478 (bar 4). On the contrary, EGFR inhibition significantly elevated the expression levels of Egr1 under basal conditions, when compared to similarly treated SpV control (bar 3). These results suggest that EGFR regulates the expression levels of genes induced by MV in the lung.
Figure 4. Effects of EGFR inhibition on MV-induced gene expression.

Total RNA was isolated from the left lobe of each mice treated with the AG1478 inhibitor or vehicle and subjected to SpV (n=3) or MV (n=3) for 2 h. cDNA was prepared separately for each RNA sample and real-time RT-PCR was performed using Taqman assays specific for mouse Areg, Atf3, Cxcl2, and Egr1 genes. GAPDH was used as an internal control. Data are presented as means ± SE. P values for respective each experimental groups are shown.
Administration of EGF into the lung does not induce injury
Our results suggest so far that EGFR regulates VILI in vivo. We therefore examined whether stimulation of EGFR activation alone is sufficient to mimic lung injury and inflammation induced by MV exposure. Therefore, we assessed alveolar lung injury and leukocyte accumulation in the BAL fluid after EGF instillation into the lung. We used two different doses (20 ng and 100 ng) of EGF, which are known to stimulate EGFR-activated signaling. Administration of EGF at a dose of 20 ng/mouse had no significant effect on protein (Fig. 5A) or neutrophil (Fig. 5C) accumulation in the BAL fluid as compared to saline instillation in the control group. Similarly, administration of EGF at a dose of 100 ng/mouse caused no significant effect on protein levels (Fig. 5A). However, there was a trend towards an increase in neutrophil numbers in BAL fluid (N=5) with 100 ng dose of EGF (Fig. 5C), which mechanistically supports the inhibitory effect of AG1478 on BAL fluid neutrophils demonstrated in Figure 1C. These results suggest that activation of EGFR alone may not be sufficient to produce acute lung injury in mice.
Figure 5. Effects of exogenous EGF on lung injury and inflammatory response.

Mice were briefly anesthetized and EGF was instilled intratracheally at two different doses of 20 ng or 100 ng (n=4/each group). Saline (50 μl) instillation was used as vehicle control group (n=5). After 2 h of EGF or vehicle administration, BAL fluid was collected for cell count and protein estimation. Total protein (A) and cells (B), and neutrophils (C) and macrophages (D) in the BAL fluid were quantified as detailed in Fig. 1.
DISCUSSION
Protein leakage and neutrophil accumulation, cardinal features of ALI and ARDS patients, can be reproduced in experimental mouse models of injurious MV and ALI 1. The present study suggests a role for EGFR in MV-induced lung injury. There was a significant decrease in MV-induced lung alveolar and vascular permeability as well as alveolar neutrophil accumulation in mice supplemented with the inhibitor AG1478 suggesting a role for EGFR-activated signaling in VILI. However, we also observed that administration of EGF ligand into the lung did not produce lung injury and inflammation, suggesting that EGFR-activated signaling is necessary but may not sufficient to produce ALI in mice.
Capillary barrier dysfunction plays key roles in accumulation of neutrophils and protein edema in the lung vasculature12. The accumulation of neutrophils can further lead to enhanced levels of inflammatory cytokines, which play fundamental roles in the development and/or perpetuation of lung injury, including VILI 12. Assessment of lung vascular permeability using Evans blue dye extravasation revealed that EGFR regulates MV-induced lung vascular leak. However, quantitative immunohistochemical analysis revealed that EGFR inhibition had no significant inhibitory effect on MV-induced neutrophil accumulation in the interstitial space. In contrast, we detected elevated levels of neutrophil accumulation in the vasculature. It is likely that an increased level of neutrophils in the vasculature may be attributed to the effects of the EGFR inhibitor on neutrophil migration into the alveolar space. Although further studies are needed to validate this notion, we would also like to point out that discordance between the influx of neutrophils and lung injury has been well documented in various experimental models of ALI/ARDS. For example, Martin et al demonstrated recruitment of neutrophils in alveolar space without a change in protein permeability 13. Moreover, we have recently demonstrated significant neutrophil accumulation, without change in BAL protein levels, in response to low tidal volume MV 9. We noted that intratracheal instillation of LPS at doses ranging from 100 to 500 ng/mouse greatly induces leukocyte accumulation but does not alter protein levels in the BAL fluid (data not shown).
Our study also demonstrates that administration of EGF, a potent activator of EGFR, failed to mimic lung injury and inflammation produced by MV (Fig.5). Even at a high dose, EGF had no significant effect on alveolar permeability and neutrophil infiltration as compared to MV-exposed mice. These results imply that EGFR-activated signaling is necessary but may not be sufficient to mediate MV-induced lung injury. Thus, it is possible that other pathways, in addition to EGFR-activated signaling, act in concert to mediate MV-induced lung injury and inflammation. In support of this notion, we have recently shown a requirement of both cytoskeletal remodeling and EGFR-activated signaling for cyclic stretch induced gene expression in lung epithelial and endothelial cells 14. Macrophages are known to be required for the proper resolution of neutrophils. Macrophage activation can have pro- and anti-inflammatory effects in the regulation of lung injury 15. Thus, it is unclear whether increased levels of macrophages observed in response to EGFR inhibition play a role in attenuation of neutrophil accumulation in the alveolar space, or whether they elicit pro- and anti-inflammatory responses in our acute model of VILI.
Expression profiling analysis performed by us and others have revealed that exposure to injurious MV induces the expression of several genes that regulate injury, repair, and inflammation (see reviews, 10, 11). Analysis of some selected genes demonstrated that inhibition of EGFR activity diminishes the expression levels of genes induced by MV suggesting that EGFR-activated signaling regulates VILI by modulating gene expression involved in injury, repair, and inflammation (Fig. 4). For example, we have shown elevated levels of Cxcl2 (MIP2α) expression by MV in the lungs of vehicle-treated mice but not in AG1478-supplemented mice, suggesting that activation of EGFR-signaling increases the induction of this chemokine which regulates neutrophil infiltration. Similarly, we also found that EGFR regulates AREG expression induced by MV. AREG has been shown to transactivate EGFR in response to compressive stress in lung epithelial cells 10, 16, and MV induces elevated levels of AREG expression in the lungs of rodents 17, 18. Moreover, we have recently shown that the AREG-EGFR pathway is critical for regulating Akt and ERK1/2 signaling and subsequent downstream target gene expression induced by cyclic stretch in lung epithelial cells 14. Because AREG is an EGF-related molecule which binds and activates EGFR, it is unclear whether EGFR and AREG interact, in either an autocrine or paracrine manner, and regulate VILI in our experimental setting. Inhibition of EGFR activity significantly affected the expression levels of ATF3, a stress response transcription factor 19. The mechanisms by which EGFR signaling causes enhanced levels of Cxcl2, AREG, and ATF3 expression in response to MV are unclear and warrant further investigation.
In summary, our data suggest that EGFR regulates MV-induced lung injury. Inhibition of EGFR markedly attenuates both lung alveolar and vascular permeability. These changes were accompanied by diminished levels of MV-induced gene expression that are known to regulate lung injury and inflammatory responses. This is the first study to address the role of EGFR in VILI using a pharmacologic approach, which has been extensively used to investigate the functions of EGFR in vivo by various laboratories, despite the fact that this inhibitor exhibits some non-specific effects against other kinases. Thus, further studies using genetic models and siRNA approaches are warranted to determine whether a dysfunctional EGFR signaling may play a role in enhancing susceptibility to the pathogenesis of VILI and whether targeting EGFR may be of potential therapeutic value in this syndrome.
Acknowledgments
This work was supported by NIH grants SCCOR P50 HL073994 (to SPR and PH), HL66109 (to SPR) and HL049441 (PH). We thank the Pathology Core of the ALI SCCOR for assisting in immunohistochemical and histopathological analysis.
Abbreviations
- ALI
Acute lung injury
- ARDS
Adult respiratory distress syndrome
- VILI
Ventilator induced lung injury and inflammation
- MV
Mechanical ventilation
- BAL
Bronchoalveolar lavage
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Matthay MA, Zimmerman GA. Acute lung injury and the acute respiratory distress syndrome: four decades of inquiry into pathogenesis and rational management. Am J Respir Cell Mol Biol. 2005;33:319–327. doi: 10.1165/rcmb.F305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Waters CM. Reactive oxygen species in mechanotransduction. Am J Physiol Lung Cell Mol Physiol. 2004;287:L484–485. doi: 10.1152/ajplung.00161.2004. [DOI] [PubMed] [Google Scholar]
- 3.Reddy SP, Hassoun PM, Brower R. Redox imbalance and ventilator-induced lung injury. Antioxid Redox Signal. 2007;9:2003–2012. doi: 10.1089/ars.2007.1770. [DOI] [PubMed] [Google Scholar]
- 4.Belperio JA, Keane MP, Lynch JP, 3rd, Strieter RM. The role of cytokines during the pathogenesis of ventilator-associated and ventilator-induced lung injury. Semin Respir Crit Care Med. 2006;27:350–364. doi: 10.1055/s-2006-948289. [DOI] [PubMed] [Google Scholar]
- 5.Fischer OM, Hart S, Gschwind A, Ullrich A. EGFR signal transactivation in cancer cells. Biochem Soc Trans. 2003;31:1203–1208. doi: 10.1042/bst0311203. [DOI] [PubMed] [Google Scholar]
- 6.Tschumperlin DJ, Shively JD, Swartz MA, et al. Bronchial epithelial compression regulates MAP kinase signaling and HB-EGF-like growth factor expression. Am J Physiol Lung Cell Mol Physiol. 2002;282:L904–911. doi: 10.1152/ajplung.00270.2001. [DOI] [PubMed] [Google Scholar]
- 7.Tschumperlin DJ. EGFR autocrine signaling in a compliant interstitial space: mechanotransduction from the outside in. Cell Cycle. 2004;3:996–997. doi: 10.4161/cc.3.8.1062. [DOI] [PubMed] [Google Scholar]
- 8.Correa-Meyer E, Pesce L, Guerrero C, Sznajder JI. Cyclic stretch activates ERK1/2 via G proteins and EGFR in alveolar epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2002;282:L883–891. doi: 10.1152/ajplung.00203.2001. [DOI] [PubMed] [Google Scholar]
- 9.Papaiahgari S, Yerrapureddy A, Reddy SR, et al. Genetic and Pharmacologic Evidence Links Oxidative Stressto Ventilator-Induced Lung Injury in Mice. Am J Respir Crit Care Med. 2007;176:1222–1235. doi: 10.1164/rccm.200701-060OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma SF, Grigoryev DN, Taylor AD, et al. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;289:L468–477. doi: 10.1152/ajplung.00109.2005. [DOI] [PubMed] [Google Scholar]
- 11.Wurfel MM. Microarray-based analysis of ventilator-induced lung injury. Proc Am Thorac Soc. 2007;4:77–84. doi: 10.1513/pats.200608-149JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Halbertsma FJ, Vaneker M, Scheffer GJ, van der Hoeven JG. Cytokines and biotrauma in ventilator-induced lung injury: a critical review of the literature. Neth J Med. 2005;63:382–392. [PubMed] [Google Scholar]
- 13.Martin TR, Pistorese BP, Chi EY, Goodman RB, Matthay MA. Effects of leukotriene B4 in the human lung. Recruitment of neutrophils into the alveolar spaces without a change in protein permeability. J Clin Invest. 1989;84:1609–1619. doi: 10.1172/JCI114338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Papaiahgari S, Yerrapureddy A, Hassoun PM, et al. EGFR-Activated Signaling and Actin Remodeling Regulate Cyclic Stretch-induced Nrf2-ARE Activation. Am J Respir Cell Mol Biol. 2006;36:304–312. doi: 10.1165/rcmb.2006-0131OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Serhan CN, Chiang N, Van Dyke TE. Resolving inflammation: dual anti-inflammatory and pro-resolution lipid mediators. Nat Rev Immunol. 2008;8:349–361. doi: 10.1038/nri2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chu EK, Foley JS, Cheng J, et al. Bronchial epithelial compression regulates epidermal growth factor receptor family ligand expression in an autocrine manner. Am J Respir Cell Mol Biol. 2005;32:373–380. doi: 10.1165/rcmb.2004-0266OC. [DOI] [PubMed] [Google Scholar]
- 17.Ma SF, Grigoryev DN, Taylor AD, et al. Bioinformatic identification of novel early stress response genes in rodent models of lung injury. Am J Physiol Lung Cell Mol Physiol. 2005;289:L468–477. doi: 10.1152/ajplung.00109.2005. [DOI] [PubMed] [Google Scholar]
- 18.Grigoryev DN, Ma SF, Simon BA, et al. In vitro identification and in silico utilization of interspecies sequence similarities using GeneChip technology. BMC Genomics. 2005;6:62. doi: 10.1186/1471-2164-6-62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hai T, Wolfgang CD, Marsee DK, Allen AE, Sivaprasad U. ATF3 and stress responses. Gene Expr. 1999;7:321–335. [PMC free article] [PubMed] [Google Scholar]