

Abstract
Obesity is a serious international health problem that increases the risk of several common diseases. The genetic factors predisposing to obesity are poorly understood. A genome-wide search for type 2 diabetes–susceptibility genes identified a common variant in the FTO (fat mass and obesity associated) gene that predisposes to diabetes through an effect on body mass index (BMI). An additive association of the variant with BMI was replicated in 13 cohorts with 38,759 participants. The 16% of adults who are homozygous for the risk allele weighed about 3 kilograms more and had 1.67-fold increased odds of obesity when compared with those not inheriting a risk allele. This association was observed from age 7 years upward and reflects a specific increase in fat mass.
Obesity is a major cause of morbidity and mortality, associated with an increased risk of type 2 diabetes mellitus, heart disease, metabolic syndrome, hypertension, stroke, and certain forms of cancer. It is typically measured clinically with the surrogate measure of body mass index (BMI), calculated as weight divided by height squared. Individuals with a BMI ≥ 25 kg/m2 are classified as overweight, and those with a BMI ≥ 30 kg/m2 are considered obese. The prevalence of obesity is increasing worldwide, probably as the result of changed lifestyle. In 2003–2004, 66% of the U.S. population had a BMI ≥ 25 kg/m2, and 32% were obese (1).
Twin and adoption studies have demonstrated that genetic factors play an important role in influencing which individuals within a population are most likely to develop obesity in response to a particular environment (2). However, despite considerable efforts, there are, as yet, no examples of common genetic variants for which there is widely replicated evidence of association with obesity in the general population. Monogenic forms of obesity at present account for ∼7% of children with severe, young-onset obesity (3), but as this severity of obesity is only seen in <0.01% of the population, these mutations are rare in the general population. Recent attempts to identify gene variants predisposing to common, polygenic obesity have proven controversial. Initial reports of promising associations between common variants in the GAD2 (4–7), ENPP1 (5, 8, 9) and INSIG2 (9–12) genes and altered BMI have not been widely replicated.
Obesity is a major risk factor for type 2 diabetes, and variants that influence the development of obesity may also predispose to type 2 diabetes. As part of the Wellcome Trust Case Control Consortium (WTCCC), we recently completed a genome-wide association study comparing 1924 U.K. type 2 diabetes patients and 2938 U.K. population controls for 490,032 autosomal single-nucleotide polymorphisms (SNPs) (Wellcome Trust Case Control Consortium). SNPs in the FTO (fat mass and obesity associated) gene region on chromosome 16 were strongly associated with type 2 diabetes (e.g., rs9939609, OR = 1.27; 95% CI = 1.16 to 1.37; P = 5 × 10−8). This association was replicated by analyzing SNP rs9939609 in a further 3757 type 2 diabetes cases and 5346 controls (OR = 1.15; 95% CI = 1.09 to 1.23; P = 9 × 10−6). Analysis of BMI as a continuous trait was possible in the initial diabetes cases and in all replication samples but not in the initial control samples. The diabetes-risk alleles at FTO were strongly associated with increased BMI (Table 1). In the replication samples, the association between FTO SNPs and type 2 diabetes was abolished by adjustment for BMI (OR = 1.03; 95% CI = 0.96 to 1.10; P = 0.44), which suggests that the association of these SNPs with T2D risk is mediated through BMI. The major signal for association with BMI coincides perfectly with that for type 2 diabetes, and rs9939609 represents a cluster of 10 SNPs in the first intron of FTO that are associated with both traits (Fig. 1). All BMI-associated SNPs (P ranging from 1 × 10−4 to 1 × 10−5) are highly correlated with each other (r2 from 0.52 to 1.0). SNP rs9939609 was used in all further studies, because among the cluster of most highly associated SNPs it had the highest genotyping success rate (100%). The HapMap (haplotype map of the human genome) population frequencies of the rs9939609 A allele are 0.45 in the CEPH (Centre d'Etude du Polymorphisme Humain) Europeans, 0.52 in Yorubans, and 0.14 in Chinese and Japanese.
Table 1.
Association of BMI with rs9939609 genotypes, corrected for sex, in type 2 diabetes cases from genome-wide and replication studies, control participants from replication studies, and adult population-based studies. P values represent the change per A allele. BMI presented as geometric means and back-transformed 95% confidence intervals.
| Study | Age, years (mean, SD) | Males (%) | N | Mean BMI (95% CI) by genotype | P | ||
|---|---|---|---|---|---|---|---|
| TT | AT | AA | |||||
| Type 2 diabetes | |||||||
| UK cases (WTCCC) | 58.6 (10.3) | 58 | 1913 | 30.15 (29.69, 30.62) | 30.47 (30.12, 30.83) | 31.99 (31.39, 32.59) | 8 × 10−6 |
| UK T2D Cases | 59.2 (8.6) | 58 | 609 | 30.89 (30.12, 31.69) | 31.14 (30.51, 31.78) | 33.46 (32.38, 34.58) | 0.001 |
| UKT2D GCC Cases | 64.1 (9.6) | 57 | 2961 | 30.59 (30.24, 30.95) | 30.96 (30.67, 31.26) | 31.98 (31.48, 32.50) | 3 × 10−5 |
| Combined T2D (I2) | 3 × 10−11 (15.6%) | ||||||
| Nondiabetic controls | |||||||
| EFSOCH | 31.8 (5.6) | 51 | 1746 | 24.50 (24.21, 24.80) | 25.21 (24.95, 25.47) | 25.41 (24.92, 25.91) | 0.0002 |
| UKT2D GCC Controls | 58.8 (11.9) | 52 | 3428 | 26.25 (26.02, 26.48) | 26.34 (26.13, 26.54) | 27.07 (26.71, 27.44) | 0.001 |
| Population-based studies | |||||||
| Adult | |||||||
| ALSPAC (mothers) | 28.4 (4.7) | 0 | 6376 | 22.42 (22.28, 22.56) | 22.73 (22.61, 22.85) | 23.27 (23.03, 23.51) | 3 × 10−10 |
| NFBC1966 (age 31) | 31 | 48 | 4435 | 24.12 (23.94, 24.31) | 24.43 (24.26, 24.60) | 24.82 (24.53, 25.12) | 5 × 10−5 |
| Oxford Biobank | 40.6 (6.1) | 55 | 765 | 25.48 (25.02, 25.94) | 25.36 (24.95, 25.78) | 26.43 (25.70, 27.17) | 0.09 |
| Older adult | |||||||
| Caerphilly | 56.7 (4.5) | 100 | 1328 | 26.10 (25.80, 26.40) | 26.48 (26.20, 26.76) | 26.69 (26.11, 27.28) | 0.03 |
| EPIC-Norfolk | 59.7 (9.0) | 47 | 2425 | 25.87 (25.63, 26.11) | 26.20 (25.99, 26.42) | 26.61 (26.22, 27.01) | 0.001 |
| BWHHS | 68.8 (5.5) | 0 | 3244 | 26.77 (26.51, 27.02) | 27.33 (27.09, 27.56) | 27.58 (27.17, 28.00) | 0.0002 |
| InCHIANTI | 74.3 (6.9) | 45 | 851 | 26.99 (26.53, 27.47) | 26.99 (26.61, 27.37) | 27.84 (27.23, 28.46) | 0.06 |
| Combined population studies (I2) | 2 × 10−20 (0%) | ||||||
| Combined population and control studies (I2) | 1 × 10−25 (0%) | ||||||
| All studies (I2) | 3 × 10−35 (0%) | ||||||
Fig. 1.

Associations of SNPs in the FTO/KIA1005 region of chromosome 16 with (A) type 2 diabetes using 1924 cases and 2938 controls and (B) adult BMI in type 2 diabetic patients. (C) Linkage disequilibrium (r2) between associated SNP rs9939609 and all other SNPs in HapMap data in Caucasian European samples. (D) Gene positions.
We studied the association of FTO gene variation with BMI and the risk of being overweight and obese in an additional 19,424 white European adults from seven general population-based studies (mean age 28 to 74 years, mean BMI 22.7 to 27.2 kg/m2) and in 10,172 white European children from two studies (mean age 7 to 14 years, mean BMI 16.1 to 19.2 kg/m2) [table S1 and supporting online text (13)].
In all adult population-based studies, we found that the type 2 diabetes–associated A allele of rs9939609 (frequency 39%) was associated with increased BMI (Table 1) with a median per-allele change of ∼0.36 kg/m2 (range 0.34 to 0.46 kg/m2). In each study, carriers of two A alleles had a higher BMI than heterozygote individuals; when we compared the additive model to a general model in each study, there was no consistent evidence for departure from an additive model. Because there was no evidence of heterogeneity (I2 = 0%) across the adult studies (14), we combined them using the inverse variance method to pool continuous data (Z scores) and the Mantel-Haenszel method for binary data. Each additional copy of the rs9939609 A allele was associated with a BMI increase of a mean of 0.10 Z-score units (95% CI = 0.08 to 0.12; P = 2 × 10−20), equivalent to ∼0.4 kg/m2. When these data were combined with those from the case-control samples (a total of 30,081 participants), the statistical confidence of the association was further increased (P = 3 × 10−35) (Table 1 and fig. S1). When we applied a Bonferroni correction for the number of tests performed in the initial genome-wide scan (∼400,000), the association remained significant (P = 1.2 × 10−29). This association was present in adults of all ages (Table 1) and of both sexes (fig. S1B and S1C), with no difference between males and females (P = 0.13).
Although BMI is a continuous trait, standard cut-offs are used to assess the burden of increased body weight on health. Hence, we assessed whether the inheritance of the FTO SNP rs9939609 altered the risk of being either overweight or obese compared with being normal weight (<25 kg/m2). In all the studies, the A allele was associated with increased odds of being overweight (Fig. 2A) and also of being obese (Fig. 2B and table S2). In a meta-analysis of the population-based studies, the per-A allele odds ratio (OR) for obesity in the adult general population was 1.31 (95% CI = 1.23 to 1.39; P = 6 × 10−16); for overweight, it was 1.18 (95% CI = 1.13 to 1.24; P = 1 × 10−12). When participants from the type 2 diabetes case and control studies were included, the magnitude of the association was unchanged, although the statistical confidence increased (obesity: OR = 1.32; 95% CI 1.26 to 1.39; P = 3 × 10−26; overweight: OR = 1.18; 95% CI = 1.14 to 1.23], P = 2 × 10−17). Individuals homozygous for the A allele at rs9939609 (16% of the population) are at substantially increased risk of being overweight (OR = 1.38; 95% CI = 1.26 to 1.52]; P = 4 × 10−11) or obese (OR = 1.67; 95% CI = 1.47 to 1.89; P = 1 × 10−14) compared with those homozygous for the low-risk T allele (37% of the population). The extent of the variance in BMI explained by rs9939609 was ∼1%, and the population attributable risk was 20.4% for obesity and 12.7% for overweight.
Fig. 2.
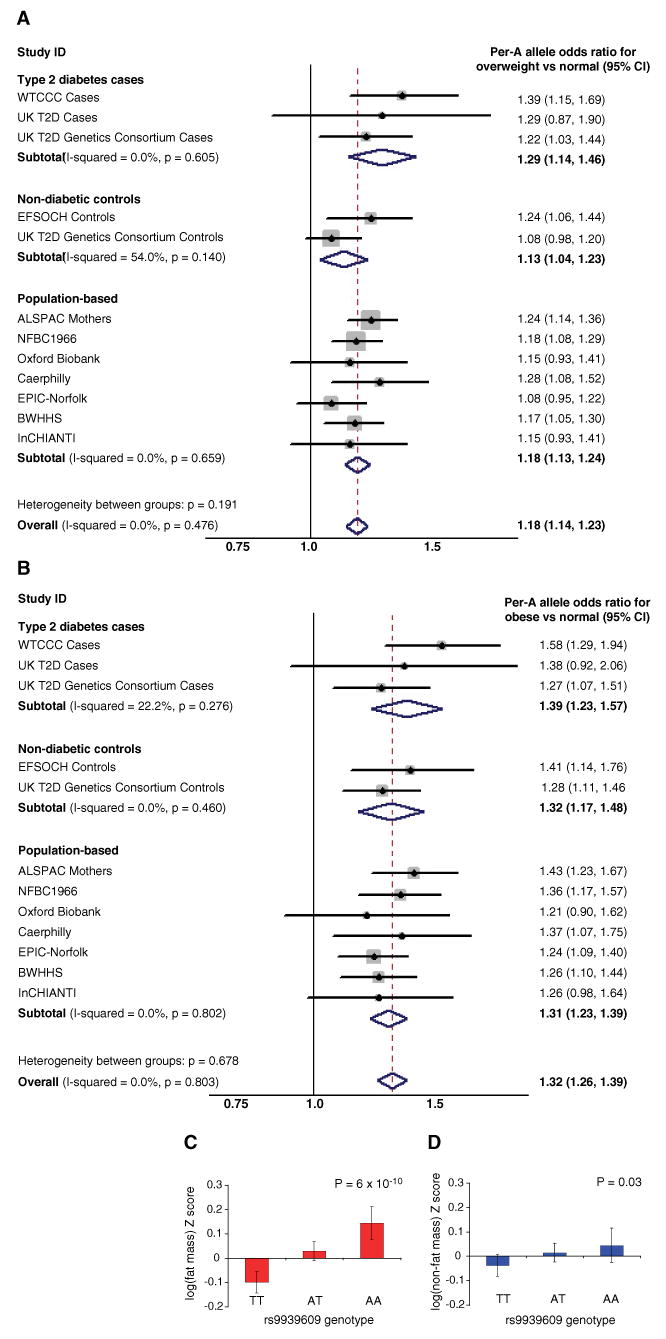
(A and B) Meta-analysis plots for odds of (A) overweight and (B) obesity, compared with normal weight in adults for each copy of the A allele of rs9939609 carried. (C and D) Bar charts showing (C) DEXA-measured fat mass in 9-year-old children and (D) DEXA-measured lean mass in 9-year-old children, both from the ALSPAC study. Error bars represent 95% confidence intervals
Childhood obesity is also increasing rapidly worldwide and is a cause of considerable concern (15). To determine the age at which the association of FTO SNP rs9939609 with BMI first becomes evident, we analyzed two large birth cohorts for which suitable measures were available from birth to early adolescence. These included 7477 UK children from the Avon Longitudinal Study of Parents and Children (ALSPAC) cohort who had anthropometric measures at birth and at 7, 8, 9, 10, and 11 years of age and 4320 children from the Northern Finland 1966 birth cohort (NFBC1966) with birth measures as well as height and weight available at 14 years. rs9939609 was not associated with birth weight (Table 2) or ponderal index at birth (table S3A) in either cohort. In children from the ALSPAC study, each copy of the rs9939609 A allele was associated with an increase in BMI by 0.08 Z-score units (95% CI = 0.04 to 0.12; P = 3 × 10−5; ∼0.2 kg/m2) at age 7, an association maintained up to the most recent assessment at age 11, when the per-allele increase was 0.12 Z-score units (95% CI = 0.08 to 0.16; P = 7 × 10−9; ∼0.4 kg/m2) (Table 2). At all ages, the A allele was associated with an increased risk of childhood obesity (e.g., OR per-A allele at age 11 years = 1.35; CI = 1.14, 1.61; P = 6 × 10−4) and of being overweight (e.g., OR per-A allele at age 11 years = 1.27; CI = 1.16 to 1.39; P= 2 × 10−7), as defined by age-specific BMI (table S2). In the Finnish cohort, each copy of the rs9939609 A allele was associated with an increase in BMI by 0.05 Z-score units (95% CI = 0.003 to 0.09; P = 0.04; ∼0.1 kg/m2) at the age of 14 years (Table 2). We conclude therefore that FTO SNP rs9939609 is not associated with changes in fetal growth but is associated with changes in BMI and obesity in children by the age of 7, changes that persist into the prepubertal period and beyond.
Table 2.
Association of BMI (corrected for sex) and birth weight (corrected for sex and gestational age) with rs9939609 genotypes in children. P values represent the change in log BMI per A allele. BMI presented as geometric means and back-transformed 95% confidence intervals.
| Cohort | Age (years) | Males (%) |
N | Mean trait value (95% CI) by genotype | P | ||
|---|---|---|---|---|---|---|---|
| TT | AT | AA | |||||
| Children* | |||||||
| ALSPAC | 7 | 51 | 5969 | 16.00 (15.92, 16.07) | 16.11 (16.04, 16.18) | 16.31 (16.19, 16.43) | 3 × 10−5 |
| 8 | 50 | 4871 | 16.80 (16.70, 16.90) | 17.01 (16.92, 17.09) | 17.29 (17.14, 17.45) | 1 × 10−7 | |
| 9 | 50 | 5459 | 17.20 (17.08, 17.31) | 17.53 (17.43, 17.63) | 17.86 (17.69, 18.04) | 5 × 10−11 | |
| 10 | 50 | 5273 | 17.66 (17.54, 17.79) | 18.05 (17.94, 18.17) | 18.37 (18.18, 18.57) | 1 × 10−10 | |
| 11 | 49 | 5010 | 18.46 (18.32, 18.61) | 18.82 (18.70, 18.94) | 19.20 (18.98, 19.42) | 7 × 10−9 | |
| NFBC1966 (age 14) | 14 | 47 | 4203 | 19.14 (19.02, 19.26) | 19.25 (19.14, 19.36) | 19.38 (19.19, 19.57) | 0.04 |
| Birth† | |||||||
| ALSPAC | 0 | 51 | 7477 | 3438 (3422, 3455) | 3452 (3437, 3466) | 3454 (3429, 3480) | 0.21 |
| NFBC1966 | 0 | 47 | 4320 | 3523 (3501, 3546) | 3538 (3518, 3558) | 3536 (3501, 3571) | 0.42 |
ALSPAC children are offspring of the participants included in the adult study (Table 1), and data are shown at five available ages. NFBC1966 children are the same participants as those in the adult study (Table 1).
ALSPAC birth data are for the same participants as those in the children study. NFBC1966 birth data are for the same participants as those in the children and adult studies. Non-singleton births and individuals born at gestation <36 weeks were excluded from the birth-weight analysis.
BMI is a convenient surrogate measure for obesity, but it may be influenced by changes in height, bone mass, and lean mass, as well as adiposity. We used additional anthropometric measurements available in the study samples to address this issue. In all population-based cohorts, the rs9939609 A allele was associated with higher weight (overall per-A allele increase = 0.09 Z-score units; 95% CI = 0.07 to 0.11); P = 4 × 10−17; ∼1.2 kg in adults) (tables S3B and S3C), but there was no difference in height (tables S3C and S3D). Consistent with this observation, we found evidence for higher waist circumference (overall per-A allele = 0.08 Z-score units; 95% CI = 0.05 to 0.11; P = 4 × 10−9; ∼1 cm) (table S3E) and higher subcutaneous mass assessed by skinfold measures (per-A allele difference = 0.11 Z-score units; 95% CI = 0.06 to 0.16; P = 2 × 10−5) (table S3F). In the children from the ALSPAC study, dual-energy x-ray absorptiometry (DEXA)–derived measures of fat mass and lean mass were available at age 9. The association of rs9939609 A allele with weight was almost exclusively attributable to changes in fat mass, with a per-allele difference of 0.12 Z-score units (95% CI = 0.08 to 0.16; P = 6 × 10−10), equivalent to a 14% difference across the three genotype groups) (Fig. 2C). Genotype-related differences in lean mass were, in contrast, a modest 0.04 Z-score units (95% CI = 0.005 to 0.08; P = 0.03), which is equivalent to a 1% increase across the three genotype groups) (Fig. 2D). Therefore, the association of genetic variation at FTO with BMI results from longitudinal changes in fat mass that, on the basis of anthropometric measures, reflect both increased waist circumference and subcutaneous fat.
One important potential source of false-positive associations in genetic studies is population stratification. We do not believe this is likely to be important in the association of the FTO SNP with BMI or type 2 diabetes. In all study cohorts, any individuals who were not European whites were excluded. In addition the cohorts were all recruited from single countries, with the majority coming from specific small geographically defined regions, and the analysis for association was done only within individual cohorts. Analysis of the FTO signal does not support this association resulting from population stratification. In the original genome-wide association study, the principal component analysis (16) implemented in EIGENSTRAT (17) made no difference to the evidence for association for type 2 diabetes (P = 5.3 × 10−8 with EIGENSTRAT adjustment and 5.2 × 10−8 without). Similarly, adjusting for the 11 geographic regions did not alter the significance of the FTO association for BMI (P = 9 × 10−6 adjusted; 8 × 10−6 unadjusted). The minor allele frequency of rs9939609 differs very little across our studies from Finland, Italy, and many different regions in the UK ranging from 0.38 to 0.40 in all except the second-smallest study, where it was 0.44. We found no significant regional variation in allele frequency in UK type 2 diabetic patients, whether testing 4 (P = 0.41) or 11 (P = 0.22) geographical regions of residence. For all these reasons, we do not believe that stratification/structure effects provide a realistic interpretation of our findings.
We have shown that common variation in the FTO gene is reproducibly associated with BMI and obesity from childhood into old age. SNP rs9939609 lies within the first intron of the FTO gene and, based on information from HapMap, is highly correlated (r2 > 0.5), with 45 additional SNPs within a 47-kb region that encompasses parts of the first two introns as well as exon 2 of FTO. There are no features to suggest that any of these SNPs represents the functional variant. Linkage disequilibrium between the BMI-associated SNPs and other variants falls rapidly outside the 47-kb region, such that there are no SNPs correlated at r2 > 0.2 outside a 90-kb interval (Fig. 1 and fig. S2). Sequencing of 47 individuals selected for BMI > 40 kg/m2 has revealed no clear candidate functional variants in the FTO coding region and minimal splice sites or 3′ UTR to explain the association (table S4). FTO is closely adjacent to a gene of unknown function KIAA1005 (Fig. 1 and fig. S2), which is transcribed in the opposite direction. This opens up the possibility that genetic variation affects a regulatory element for KIAA1005; however, there is no obvious such variant within the 47-kb associated region. We conclude that the 47-kb intron within the FTO gene is most likely to contain the predisposing variant(s), but there is, at present, no clear genetic mechanism to explain how this alters the function or expression of FTO, KIAA1005, or more distant genes.
FTO is a gene of unknown function in an unknown pathway that was originally cloned as a result of the identification of a fused-toe (Ft) mutant mouse that results from a 1.6-Mb deletion of mouse chromosome 8 (18). Three genes of unknown function (Fts, Ftm and Fto), along with three members of the Iroquois gene family (Irx3, Irx5, and Irx6 from the IrxB gene cluster), are deleted in Ft mice (18). The homozygous Ft mouse is embryonically lethal and shows abnormal development, including left/right asymmetry (19). Heterozygous animals survive and are characterized by fused toes on the forelimbs and thymic hyperplasia but have not been reported to have altered body weight or adiposity (19). The fused-toe mutant is a poor model for studying the role of altered Fto activity, because multiple genes are deleted. Neither isolated inactivation nor overexpression of Fto has been described.
We used reverse transcription PCR to assess the expression of FTO and KIAA1005 in a human tissue panel (18). FTO was found to be widely expressed in fetal and adult tissues, with expression highest in the brain (fig. S3A). The transcription start site of KIAA1005 lies only 200 base pairs from the 5′ end of FTO and ∼61 kb from the 47-kb interval containing the BMI associations. KIAA1005 is also ubiquitously expressed with relatively high levels in hypothalamus and islet (fig. S3B). The similarity of expression profile between these two transcripts may indicate joint transcriptional regulation but does not provide insights into which of the two genes is more likely to be involved. Further work with both knockout and overexpression models of FTO and KIAA1005 are likely to provide the most fruitful approach to understanding the mechanism and pathways whereby these variants influence the risk of obesity.
Footnotes
Accession numbers for deposited sequence variants from dbSNP are Exon3_A 69374768, 3_UTR_A 69374769, 3_UTR_B 69374770, and 3_UTR_G 69374771.
Supporting Online Material
www.sciencemag.org/cgi/content/full/1141634/DC1
Materials and Methods
SOM Text
Figs. S1 to S3
Tables S1 to S4
References
References and Notes
- 1.Ogden CL, et al. JAMA. 2006;295:1549. doi: 10.1001/jama.295.13.1549. [DOI] [PubMed] [Google Scholar]
- 2.Maes HHM, Neale MC, Eaves LJ. Behav Genet. 1997;27:325. doi: 10.1023/a:1025635913927. [DOI] [PubMed] [Google Scholar]
- 3.Farooqi IS, O'Rahilly S. Endocr Rev. 2006 doi: 10.1210/er.2006-0040. [DOI] [PubMed] [Google Scholar]
- 4.Boutin P, et al. PLoS Biol. 2003;1:361. doi: 10.1371/journal.pbio.0000068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meyre D, et al. Nat Genet. 2005;37:863. doi: 10.1038/ng1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Swarbrick MM, et al. PLoS Biol. 2005;3:1662. [Google Scholar]
- 7.Groves CJ, et al. Diabetes. 2006;55:1884. doi: 10.2337/db05-1674. [DOI] [PubMed] [Google Scholar]
- 8.Weedon MN, et al. Diabetes. 2006;55:3175. doi: 10.2337/db06-0410. [DOI] [PubMed] [Google Scholar]
- 9.Herbert A, et al. Science. 2006;312:279. doi: 10.1126/science.1124779. [DOI] [PubMed] [Google Scholar]
- 10.Loos RJF, Barroso I, O'Rahilly S, Wareham NJ. Science. 2007;315:179c. doi: 10.1126/science.1130012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rosskopf D, et al. Science. 2007;315:179d. [Google Scholar]
- 12.Dina C, et al. Science. 2007;315:179b. doi: 10.1126/science.1129402. [DOI] [PubMed] [Google Scholar]
- 13.Materials and methods are available as supporting material on Science Online.
- 14.Higgins JP, Thompson SG, Deeks JJ, Altman DG. BMJ. 2003;327:557. doi: 10.1136/bmj.327.7414.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lobstein T, Baur L, Uauy R. Obes Rev. 2004;5:4. doi: 10.1111/j.1467-789X.2004.00133.x. [DOI] [PubMed] [Google Scholar]
- 16.Zhang S, Zhu X, Zhao H. Genet Epidemiol. 2003;24:44. doi: 10.1002/gepi.10196. [DOI] [PubMed] [Google Scholar]
- 17.Price AL, et al. Nat Genet. 2006;38:904. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- 18.Peters T, Ausmeier K, Dildrop R, Rüther U. Mamm Genome. 2002;13:186. doi: 10.1007/s00335-001-2142-7. [DOI] [PubMed] [Google Scholar]
- 19.van der Hoeven F, et al. Development. 1994;120:2601. doi: 10.1242/dev.120.9.2601. [DOI] [PubMed] [Google Scholar]
- 20.Collection of the type 2 diabetes cases was supported by Diabetes UK, British Diabetic Association Research, and the UK Medical Research Council (Biomedical Collections Strategic Grant G0000649). The UK Type 2 Diabetes Genetics Consortium collection was supported by the Wellcome Trust (Biomedical Collections Grant GR072960). The ALSPAC study was supported by the UK Medical Research Council (MRC), the Wellcome Trust, and the University of Bristol. The British Women's Heart and Health Study was funded by the Department of Health and the British Heart Foundation. The Caerphilly study was funded by MRC and the British Heart Foundation. The Caerphilly study was undertaken by the former MRC Epidemiology Unit (South Wales) and was funded by the MRC. The data archive is maintained by the Department of Social Medicine, University of Bristol. The Exeter Family Study of Childhood Health was supported by NHS Research and Development and the Wellcome Trust. The work on the Northern Finland birth cohort study was supported by the Academy of Finland (104781), MRC (G0500539), and the Wellcome Trust (Project Grant GR069224). Oxford Biobank was supported by the British Heart Foundation. The Invecchiare in Chianti (InCHIANTI) study was supported by contract funding from the U.S. National Institute on Aging (NIA), and the research was supported in part by the Intramural Research Program, NIA, NIH. The European Prospective Investigation of Cancer (EPIC)–Norfolk cohort study is supported by program grants from MRC and Cancer Research UK. The EPIC-Norfolk obesity case-cohort study and its genotyping was funded by the Wellcome Trust and MRC. The genome-wide association genotyping was supported by the Wellcome Trust (076113), and replication genotyping was supported by the Wellcome Trust, Diabetes UK, the European Commission (EURODIA LSHG-CT-2004-518153), and the Peninsula Medical School. Personal funding comes from the Wellcome Trust (A.T.H., Research Leave Fellow; I.B., Investigator; E.Z., Research Career Development Fellow; L.R.C., Principal Research Fellow); MRC (J.R.B.P.); Diabetes UK (R.M.F.); and Throne-Holst Foundation (C.M.L.). L.W.H. is a Research Councils UK Diabetes and Metabolism Academic Fellow. M.N.W. is Vandervell Foundation Research Fellow at the Peninsula Medical School. C.N.A.P. and A.D.M. are supported by the Scottish executive as part of the Generation Scotland Initiative. We acknowledge the assistance of many colleagues involved in sample collection, phenotyping, and DNA extraction in all the different studies. We thank K. Parnell, C. Kimber, A. Murray, K. Northstone, and C. Boustred for technical assistance. We thank S. Howell, M. Murphy, and A. Wilson (Diabetes UK) for their long-term support for these studies. We also acknowledge the efforts of J. Collier, P. Robinson, S. Asquith, and others at Kbiosciences.