

Abstract
Typical 2-Cys peroxiredoxins (Prxs) have an important role in regulating hydrogen peroxide-mediated cell signalling1. In this process, Prxs can become inactivated through the hyperoxidation of an active site Cys residue to Cys sulphinic acid. The unique repair of this moiety by sulphiredoxin (Srx) restores peroxidase activity and terminates the signal2. The hyperoxidized form of Prx exists as a stable decameric structure with each active site buried. Therefore, it is unclear how Srx can access the sulphinic acid moiety. Here we present the 2.6 Å crystal structure of the human Srx–PrxI complex. This complex reveals the complete unfolding of the carboxy terminus of Prx, and its unexpected packing onto the backside of Srx away from the Srx active site. Binding studies and activity analyses of site-directed mutants at this interface show that the interaction is required for repair to occur. Moreover, rearrangements in the Prx active site lead to a juxtaposition of the Prx Gly-Gly-Leu-Gly and Srx ATP-binding motifs, providing a structural basis for the first step of the catalytic mechanism. The results also suggest that the observed interactions may represent a common mode for other proteins to bind to Prxs.
Reactive oxygen species, such as hydrogen peroxide (H2O2) and peroxynitrite, have been recognized as compounds capable of modifying protein, DNA and lipids, especially when present at elevated levels3. In contrast, low levels of H2O2 can function as a second messenger signal in cell proliferation, differentiation and migration1,4,5. The dysregulation of these signalling processes are hallmarks of oxidative stress and disease states, including diabetes, cancer and ageing3,6. In this context, the ubiquitous thiol peroxidases, 2-Cys Prxs, function as critical peroxide sensors that can be inactivated through hyperoxidation. The hyperoxidation phenomenon is a fundamental element of the flood-gate hypothesis7. Once the Prx molecules are inactivated, through the formation of a Cys sulphinic acid ( , Fig. 1a) during the catalytic cycle, H2O2 can ‘breach the gate’ to initiate signalling events. Two additional scenarios for Prx-mediated signalling include the sulphinic acid form of 2-Cys Prxs acting as a signal itself and the fostering of disulphide bond formation in other proteins8,9. Thus, the unprecedented repair or retroreduction of 2-Cys Prxs by Srx is essential to restore peroxidase activity and the regulation of signalling events.
Figure 1. Peroxiredoxin hyperoxidation and repair by sulphiredoxin.
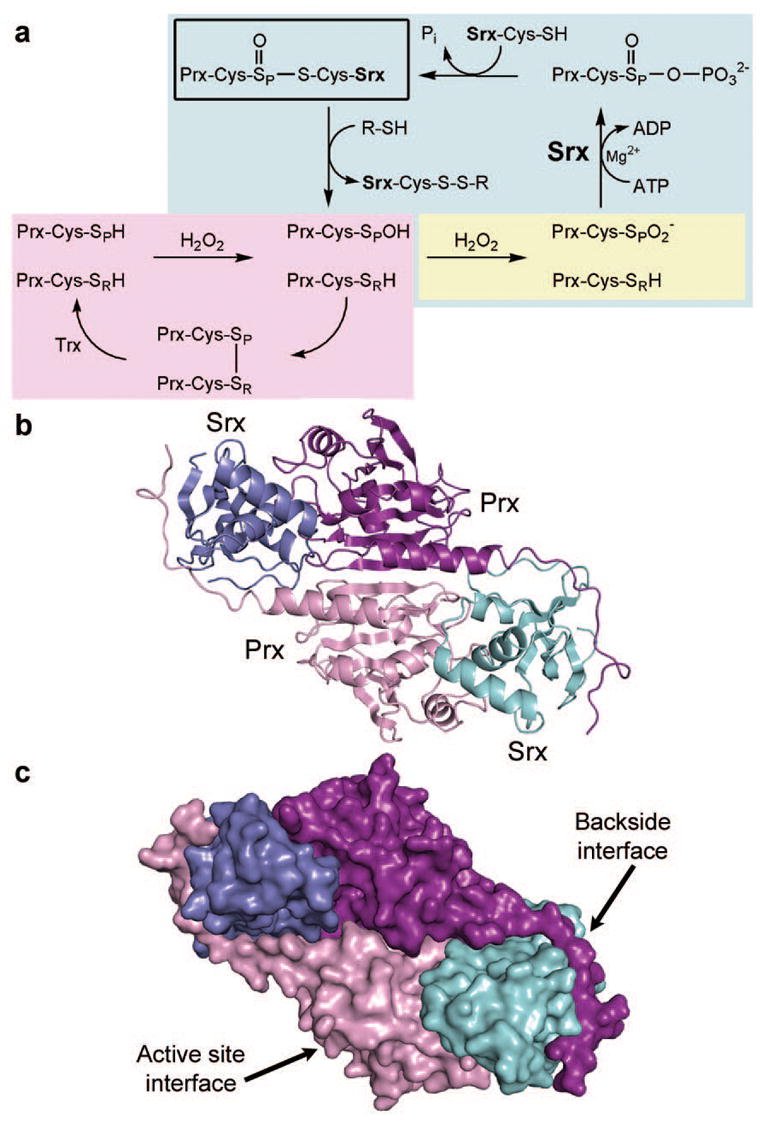
a, In the typical 2-Cys Prx catalytic cycle (violet), the peroxidatic Cys is depicted as a thiol (SPH) or sulphenic acid (SPOH). The disulphide bond between Cys-SP and the resolving Cys, Cys-SRH, from the adjacent monomer is reduced by Trx10. Reaction with a second molecule of H2O2 results in hyperoxidation and sulphinic acid ( ) formation (yellow). The reaction mediated by Srx (blue) is specific for 2-Cys Prxs and dependent upon ATP, Mg2+ and a Cys thiol2,13,14,28. The sulphinic acid moiety is thought to be phosphorylated to form a sulphinic phosphoryl ester ( ) through either a direct attack on the γ-phosphate of ATP or transfer from Cys 99 of human Srx. The phosphoryl ester is subsequently converted to a thiosulphinate bond (for example, Prx-S(O)-S-Srx, boxed in black) which includes the sulphur atom of Cys 99 of human Srx or possibly glutathione (GSH), and inorganic phosphate (Pi) is released. Thioredoxin (Trx) or GSH reduce this complex to release free Srx and Prx-Cys-SPOH. b, Overall structure of the Srx–PrxI complex. Cartoon representation is shown, with secondary structural elements along the two-fold axis. The two monomers of PrxI and Srx are shown in violet/purple and cyan/blue, respectively. c, Surface representation of the Srx–PrxI complex, illustrating active site and backside interfaces.
Structural studies on 2-Cys Prxs have revealed that the active site region can exist in fully folded and locally unfolded states7,10. The hyperoxidized form of human PrxII exists in the fully folded state. In this form, the peroxidatic Cys residue, , is located at the amino terminus of an α-helix stabilized by a salt bridge to Arg 127 (Supplementary Fig. 1, residue numbering is one less than in human PrxI) and the resolving Cys-SRH residue is ~14 Å away11. Access to the sulphinic acid moiety is further restricted by the YF and GGLG motifs. The active site helix, however, must locally unfold to allow the formation of a disulphide bond between the Cys-SPH and Cys-SRH residues during the Prx catalytic cycle (Fig. 1a). Given these observations, it is clear that large structural rearrangements must occur in order for the Srx molecule to access the Prx sulphinic acid moiety. This notion is supported by the inability to model the catalytic Cys residues of each enzyme in close proximity to each other12. Therefore, the complex between the two enzymes is not readily predictable.
Using X-ray crystallography, we determined the structure of human Srx in complex with PrxI to 2.6 Å resolution after screening many engineered constructs. This complex contained one PrxI dimer (Fig. 1b) and two Srx monomers. The electron density across the disulphide bond that bridges between the active sites was unambiguous (Supplementary Fig. 2). The Srx molecules were sandwiched between the active site surface of one PrxI monomer and the C-terminal tail from the adjacent PrxI monomer. Complex formation resulted in the burial of ~690 Å2 at each Srx–Prx active site interface and ~960 Å2 between the C-terminal tail and the ‘backside’ of Srx (Fig. 1c). Phe 50 of PrxI packs within an Srx pocket (Fig. 2a) constituted by Leu 52, Leu 82, Phe 96, Val 118, Val 127 and Tyr 128 (Supplementary Fig. 3). A comparison to the structure of hyperoxidized human PrxII (Fig. 2b, c) shows that the moiety (Csd 51) is distant from Srx11. We propose that the hydrophobic surface of Srx triggers the local unfolding of the Prx active site helix to place Phe 50 in the Srx pocket. As a result, of human PrxI moves ~10 Å away from Arg 128 to approach Cys 99 of Srx.
Figure 2. Srx–PrxI active site interactions and structural plasticity.
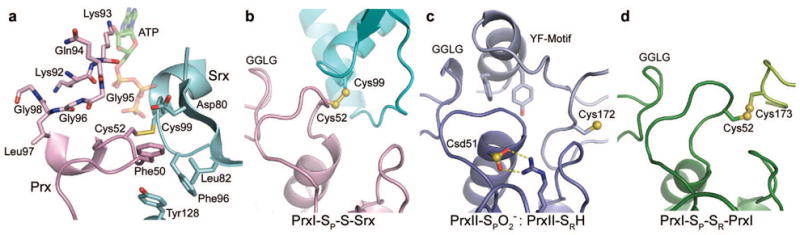
a, ATP modelled (translucent) into the active site of the Srx–PrxI complex containing the disulphide bond between Cys 99 of Srx (cyan) and Cys 52 of PrxI (violet). b, Ribbon diagram of the locally unfolded human PrxI active site in complex with Srx. c, Human active site in the fully folded state (dark purple) with the YF motif from the adjacent monomer (light purple) overlaying the active site. The active site Cys-SPH residue is in the sulphinic acid form (Csd 51). PDB code 1QMV11. Human PrxI and PrxII exhibit 77.8% sequence identity. d, Rat PrxI active site (dark green) present in the oxidized, disulphide form involving the resolving Cys residue, Cys 173 (light green). PDB code 1QQ227. Panels b–d are presented in the same orientation using a superposition of the core β-sheet structure of each Prx dimer.
A superposition of the model of human Srx with ATP bound to it13, based on the ADP complex determined experimentally, onto the Srx–PrxI complex also suggests that the unfolding of the PrxI active site helix would place near the γ-phosphate of ATP (Fig. 2a and Supplementary Fig. 4). In the ternary complex, the γ-phosphate atom is located 3.0 Å from the Sγ atom of PrxI-Cys52 and 3.5 Å from the Sγ atom of Srx-Cys99. The oxygen atom of is positioned correctly to perform an inline attack on the γ-phosphate, as originally proposed2. In contrast, Cys 99 of Srx points away from the γ-phosphate. This observation implies that this residue does not transfer the phosphate moiety from ATP to the Prx sulphinic acid (Fig. 1a); this is consistent with the weak phosphorylation of the inactive, C99S human Srx variant14. The ternary complex also suggests that the GGLG motif and the preceding residues, Lys 92, Lys 93 and Gln 94, are in a position to generate the second half of the ATP binding site.
A comparison of the Srx–PrxI structure to Prx molecules present in two different oxidation states further supports the necessary flexibility of the GGLG motif, the active site helix containing the Cys-SPH residue, the YF motif, and Cys-SRH movements. The Srx and the YF motif of the adjacent Prx monomer cannot occupy the same space at the same time (Fig. 2b, c). The active site helix containing the sulphinic acid moiety must break its interaction with a conserved Arg residue (Fig. 2c) in order to attack the ATP molecule in the Srx active site, as described above. This locally unfolded state is consistent with the formation of the disulphide bond between Cys-SPH and Cys-SRH (Fig. 2d) during the peroxidase catalytic cycle. The current Srx–PrxI complex also suggests that the ADP molecule would need to be released and that additional structural changes in either of the protein molecules or both would be necessary for Trx or GSH to break down the proposed thiosulphinate intermediate.
The binding of the PrxI C terminus onto the backside of Srx was surprising (Fig. 1). Residues 172–186 of PrxI (Fig. 3a) pack onto a conserved (Supplementary Fig. 5), predominantly hydrophobic groove of Srx. This region of 2-Cys Prxs is also conserved and contains Cys173-SRH (mutated to Ser in this variant), three or more Pro residues, and Trp 177 (Supplementary Fig. 6). This interface suggests that the active site interactions are insufficient for binding, and that the C terminus functions to hold Srx in the correct orientation for catalysis. In order to test this hypothesis and the relevance of the dimeric complex structure to the Prx hyperoxidized decamer, Ile 50, Tyr 92, Phe 93 and Leu 117 (Fig. 3b) of Srx were mutated to Arg to repel the binding of the Prx C terminus. A corresponding alteration or truncation of the PrxI C terminus was not performed, because Prx variants of these types from a variety of organisms become resistant to hyperoxidation and Cys sulphininc acid cannot be formed15–17.
Figure 3. PrxI backside interaction with Srx.
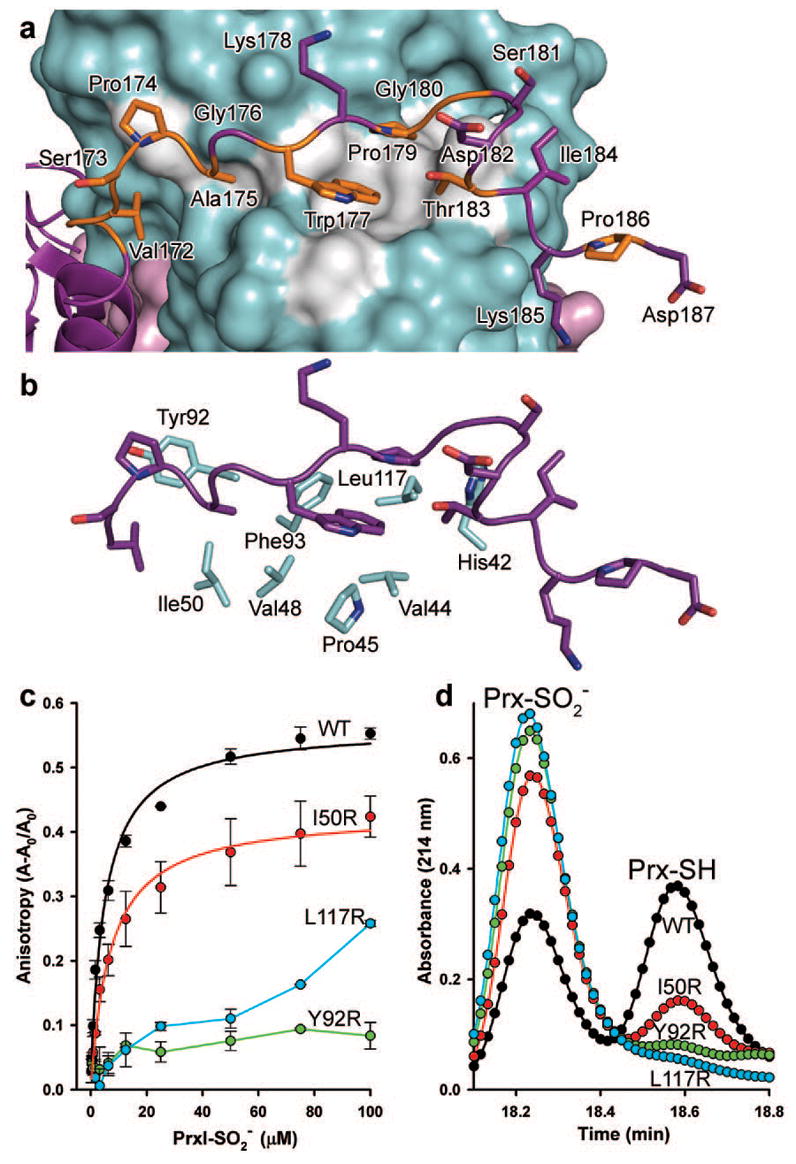
a, The C-terminal tail of PrxI binds to Srx. Conserved residues of PrxI are coloured orange, and the conserved surface of Srx is coloured grey. b, Surface residues of Srx that interact with the C terminus of PrxI. c, interactions measured in solution by changes in fluorescence anisotropy with Oregon Green 514-labelled Srx variants. Data obtained from representative, duplicate titrations are expressed as the fractional change in anisotropy, (A–A0)/A0, versus the concentration of decameric added, with the error bars indicating s.d. The data for WT Srx and the I50R mutant were fitted to a single-site, saturable binding model. d, Representative HPLC traces from the activity analysis of Srx variants.
The Srx variants were analysed by circular dichroic spectroscopy (Supplementary Fig. 7) to verify that their global structure had not been significantly compromised. Despite the careful selection of the sites of mutation, the F93R mutant exhibited a loss in structure. The ability of the Srx variants, including the partially unfolded F93R variant, to bind wild-type (WT), decameric was tested using fluorescence anisotropy (Fig. 3c). WT and I50R Srx bound with similar affinities, 5.1 ± 0.9 μM and 7.2 ± 1.3 μM (mean ± s.d.), respectively. This finding agrees with Ile 50 being the residue farthest from the interface. In contrast, the Y92R and L117R variants of Srx had significantly reduced or no binding, a result similar to that of F93R (data not shown). The catalytic activity of the Srx mutants was also monitored using reverse-phase high-performance liquid chromatography (HPLC; Fig. 3d). WT Srx was able to repair decameric at a rate of 0.23 min−1 (Supplementary Fig. 8), a value similar to the rate previously reported18. The I50R and Y92R variants exhibited 60% and 15% of WT activity, respectively. The L117R mutant and the structurally comprised F93R mutant both exhibited no activity. These observations indicate that decreased binding of Srx to Prx is sufficient to reduce or abolish Srx activity.
The necessity for the C terminus of 2-Cys Prxs to bind and embrace Srx highlights its expanding cellular roles. For example, the interaction of the human PrxI C terminus with the PDZ domain of Omi/HtrA2 is necessary to promote protease activity19. The interactions with c-Abl, c-Myc, MIF, phospholipase D1 and the PDGF receptor also raise the possibility that the binding of the Pro-rich C terminus of Prx to Srx represents a general mechanism for 2-Cys Prxs to associate with key regulatory or signalling proteins5,20–22. Moreover, these latter interactions may modulate the repair process or vice versa.
The importance of Srx is likely to extend beyond the repair of the decameric form of 2-Cys Prxs. The association of Prx decamers into stacks of toroids has been observed via electron microscopy and within the crystal structure of human (refs 11, 23). Confocal microscopy studies also suggest that human can form filamentous structures in cell culture, thereby alerting a perturbation in peroxide homeostasis24. Sphere-like Prx aggregates have also been shown to switch from a peroxidase activity to a protein chaperone function25. In an effort to understand how Srx may interact with the higher-order forms of Prxs, a model of the decameric Srx–PrxI complex was generated. The PrxI dimer of the Srx–Prx complex was superimposed onto each of the five Prx dimers of the structure (Supplementary Fig. 9). No significant steric clashes were observed, suggesting that the Srx–Prx interaction is not influenced by the oligomeric state. The addition of ten Srx molecules, however, did expand the toroid diameter (~110 to 125 Å) and thickness (~45 to 55 Å). These substantial changes suggest that the binding of one or two Srx molecules would be sufficient to destabilize Prx–Prx interactions in higher-order oligomers.
In summary, the embrace observed in the Srx–Prx complex represents an unexpected structural rearrangement fundamentally important for the repair of Prxs in higher organisms. A structural basis is now available for designing future biochemical and cellular studies to dissect additional aspects of the Srx reaction mechanism and the roles of Srx and Prxs in modulating cell signalling.
METHODS SUMMARY
One key to stabilizing the Srx–Prx crystals was to mimic the proposed thiosulphinate intermediate (Fig. 1a) with a disulphide bond. An intermolecular disulphide bond was formed between the active site residues, Cys 99 of human Srx and Cys52-SPH of the C71S, C83E, C173S variant of PrxI. Both proteins were separately overexpressed in Escherichia coli, and disulphide bond formation was facilitated by pre-treatment of the PrxI variant with 5,5′-dithiobis-(2-nitrobenzoic acid). The comparable disulphide-bonded species has also been observed in vivo and in vitro2,14. A dimeric form of PrxI was also necessary, and was generated by introducing charged residues, Cys83Glu on each monomer, juxtaposed at the dimer–dimer interface of the PrxI decamer26. An N-terminal truncation of human Srx, residues 1–37, was required to remove a non-conserved, glycine-rich region13. Crystals were grown by vapour diffusion, and diffraction data collected on beamline X8C at the National Synchrotron Light Source (NSLS). The structure was solved by molecular replacement using the human PrxI dimer and Srx as a search models13,27. The final model has Rwork and Rfree values of 23.9% and 30.8%, respectively. The binding of Srx variants to the hyperoxidized, decameric form of human PrxI was determined by fluorescence anisotropy by labelling Srx with Oregon Green 514. Hyperoxidized PrxI was generated by forcing the enzyme to go through the catalytic cycle many times by the addition of H2O2 and dithiothreitol. The activity of Srx variants was determined by quantifying the conversion of to Prx-SH by reverse-phase HPLC. Detailed procedures are presented in Supplementary Information.
METHODS
Protein purification and generation of PrxI–Srx complex crystals
Several approaches were used to obtain crystals of the human Srx–Prx complex. Both His-tagged and untagged recombinant PrxI and human PrxII constructs were simultaneously manipulated to obtain crystals. Attempts to crystallize hyperoxidized PrxI and PrxII decamers in association with Srx and the presence or absence of ATP/ADP were not successful. In order to facilitate crystallization, a covalent disulphide bond between the active site Cys 99 of Srx and the peroxidatic Cys in Prx (Cys 52 in PrxI and Cys 51 in PrxII) was generated as described below. The dimer–dimer interface of the Prx decamer was manipulated by mutating Cys 83 to Glu/Ser/Val in PrxI or the corresponding Thr 82 in PrxII to Glu or Val. Some of these mutant constructs were also truncated at the C terminus (position 170 or 185 in PrxI and the corresponding positions in PrxII). Three different forms of Srx were used in the crystallization attempts as well: full length (residues 1–137), ET-Srx (32–137) or TT-Srx (38–137)13. More than 15 different disulphide-linked complexes were generated and screened for crystallization. The typical yields for each complex were 2.5–20 mg. The best crystals obtained were from the complex that contained the PrxI variant C71S, C83E, C173S disulphide linked to ET-Srx.
Human Srx was expressed using pET19 (Novagen) or pMALc2 (New England Biolabs) vector derivatives containing a PreScission protease (GE Healthcare) cleavage site between Srx and the N-terminal His-tag or maltose binding protein. The proteins were expressed in C41(DE3) E. coli cells and purified using nickel affinity or amylose resins followed by size-exclusion chromatography13,29. All site-directed mutants were generated using the QuikChange Site-directed Mutagenesis Kit from Stratagene. All Srx variants were analysed by circular dichroic spectroscopy in 20 mM HEPES pH 7.5, 100 mM NaCl at a concentration from 0.3 to 0.45 mg ml−1. Three 20 nm min−1, room temperature scans from a JASCO-720 spectropolarimeter were averaged. The PrxI variant was expressed from the pET17b vector in C41(DE3) cells and purified as previously described with alterations27. Briefly, PrxI for crystallographic studies was purified in the presence of 1 mM DTT and sequential chromatography on phenyl sepharose, S-sepharose and hydroxyapatite columns. The protein was then passed through a 250 ml Superdex 200 size exclusion column to facilitate buffer exchange and removal of DTT. Ellman’s reagent (500 μM), 5,5′-dithiobis-(2-nitrobenzoic acid) (DTNB), was added immediately to all fractions corresponding to the dimeric form of PrxI. Excess DTNB and generated TNB were removed using a 75 ml G25 size exclusion column. ET-Srx was titrated into the PrxI solution until no further release of TNB was observed at 412 nm (<2-fold excess Srx over PrxI). The disulphide-bonded Srx–PrxI complex was passed over the Superdex 200 column to remove excess ET-Srx and TNB. The complex was concentrated, aliquoted, flash frozen with liquid nitrogen, and stored at −80 °C. Crystals of the Srx–PrxI complex were obtained by hanging-drop vapour diffusion. Equal volumes of protein (10–14.5 mg ml−1 in 20 mM HEPES pH 7.5 and 100 mM NaCl) and well solution (14% PEG 8000, 16% ethylene glycol and 100 mM HEPES pH 7.2) were mixed. Crystals were mounted in nylon loops and cryo-cooled at −170 °C for data collection.
Data collection and structure determination
A single wavelength (1 Å) data set was collected on beamline X8C at NSLS. Diffraction intensities were integrated using d*Trek (Rigaku/MSC) and scaled to 2.6 Å resolution (Supplementary Table 1). 5.1% of the reflections were set aside for cross-validation. The space group of the crystal was P212121 with unit cell dimensions a = 54.9 Å, b = 85.0 Å, c = 130.8 Å. The structure of the Srx–PrxI complex was solved by molecular replacement using PHASER30. The search models used were the following: the ET-Srx model (residues 38–137, PDB code 1XW3)13 and only the regions of rat PrxI (PDB entry 1QQ2)27 that were structurally conserved with human PrxII (PDB entry 1QMV)11. Importantly, the entire active site helix of PrxI (residues 46–69), the GGLG motif (residues 89–102) and the C terminus (residues 169–199) were removed from the search model. Two monomers of Srx and one dimeric PrxI were found in the asymmetric unit with rotational Z-scores from 5.4 to 10.6 and translational Z-scores from 21.1 to 22.8. Electron densities for the regions not present in the search model were clearly visible except for the residues 188–199. A combination of simulated-annealing composite omit and sigmaA-weighted 3Fobs – 2Fcalc, 2Fobs – Fcalc and Fobs – Fcalc maps were used for model building in O and COOT31,32. The model was refined with CNS using alternating cycles of simulated annealing, positional and B-factor refinement33. At the final stage of refinement REFMAC5 was used and ten water molecules were added to 3σ positive features within a Fobs – Fcalc map34. Structural figures were generated using PYMOL (DeLano Scientific). Model quality was assessed using MOLPROBITY35. Intermolecular surface accessible areas were calculated using AREAIMOL within the CCP4 package36.
Srx activity assay
In order to generate the hyperoxidized form of PrxI for Srx to repair, WT PrxI was added to a solution containing 50 mM Tris pH 7.5 and 100 mM KCl. Hyperoxidation was achieved by four step-wise additions of 5 mM H2O2 and 10 mM DTT with incubation for 30 min at 37 °C. Excess DTT and H2O2 were removed by extensive buffer exchange (50 mM Tris-HCl pH 7.5 and 100 mM KCl) via ultrafiltration (Vivaspin by Sartorious). Repair of was performed by incubating 50 μM with 10 μM WT Srx or variant in a 30 μl reaction mixture containing 50 mM Tris pH 7.5, 100 mM KCl, 1 mM ATP, 1 mM MgCl2 and 2 mM DTT. The reaction was stopped at various times by the addition of 15 μl 1 M H3PO4 to prevent disulphide shuffling and potential reoxidation. Five microlitres of the sample was injected using an autosampler onto a Waters analytical HPLC system. The and PrxI-SH species were separated from each other on a C4 column (Vydac) using a 60–63% acetonitrile/0.1% TFA gradient over 19 min. A similar reaction containing five-fold excess (250 μM) of Srx over was performed to determine the amount of non-repairable present. The peaks from duplicate samples were integrated and the amount of subtracted. The fraction of available that was repaired is reported as the mean ± s.d.
Fluorescence anisotropy binding assay
The N-terminal amine of ET-Srx (present in 75 mM NaHCO3 adjusted to pH 8.3) was derivatized by incubation with a two-fold molar excess of the fluorophor Oregon Green 514 succinimidyl ethyl ester (Molecular Probes) for 30 min at room temperature in the dark. The reaction was quenched with 200 mM glycine in 75 mM NaHCO3 which had been adjusted to pH 8.3. The labelled protein was separated from excess dye by a G-25 sephadex desalting column (Bio-Rad). The correct labelling of the N terminus was confirmed by limited trypsin digest, which cleaves ET-Srx at residue Arg 37. This truncated version of Srx, TT-Srx, lacked fluorescence as judged by SDS–PAGE separation and imaging. The reconstituted protein concentrations and the degree of labelling, typically 0.5–1 mol Oregon Green 514 per mol protein, were determined by UV-VIS spectroscopy. Fluorescence intensity and anisotropy measurements were carried out on a Safire II plate reader (Tecan Instruments) equipped with dual monochromators and the ability to rapidly collect data from quadruplicate 20 μl samples in the wells of a microtitre plate. Samples of Oregon Green-labelled ET-Srx mutants were excited at 470 nm and emission measured at 530 nm using 5 nm slits. The instrument computed anisotropy data from samples illuminated with vertically polarized light from the vertically and horizontally polarized components (Iv and Ih respectively) of the emitted light: A = Iv − (GIh/Iv) + GIh. The G-factor, which corrects for differential detector responses to the vertically and horizontally polarized emitted light, was calculated optimally over the plate. Oregon Green 514 (1 nM) was used as the reference. Oregon green labelled ET-Srx (50 nM) was mixed with for 10 min at 30 °C at the indicated concentrations in a 20 μl reaction mixture containing 50 mM Tris pH 7.5, 100 mM KCl and 1 mM ATP before measurements were performed at the same temperature. In these experiments Mg2+ was omitted from the reaction to prevent Srx-mediated catalysis. Representative, duplicate samples with their s.d. are shown in Fig. 3c.
Supplementary Material
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
Acknowledgments
We thank M. Murray for contributions to the structure determination, L. B. Poole, P. A. Karplus and N. H. Heintz for discussions, the staff of the NSLS and beamline X8C for their assistance during data collection and the RapiData course, and R. R. Hantgan for help with the circular dichroism and fluorescence anisotropy experiments. This work was supported by an NIH grant (W.T.L.) and an American Heart Association Postdoctoral Fellowship (T.J.J.). NSLS is supported by the US Department of Energy and NIH.
Footnotes
Author Contributions
T.J. and L.J. performed all biochemical and crystallization experiments. T.J. and T.L solved the structure. T.J. and T.L. wrote the paper. All authors discussed the results and commented on the manuscript.
References
- 1.Rhee SG. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312:1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 2.Biteau B, Labarre J, Toledano MB. ATP-dependent reduction of cysteine-sulphinic acid by S. cerevisiae sulphiredoxin. Nature. 2003;425:980–984. doi: 10.1038/nature02075. [DOI] [PubMed] [Google Scholar]
- 3.Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- 4.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 5.Kang SW, Rhee SG, Chang TS, Jeong W, Choi MH. 2-Cys peroxiredoxin function in intracellular signal transduction: Therapeutic implications. Trends Mol Med. 2005;11:571–578. doi: 10.1016/j.molmed.2005.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- 7.Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 8.Bozonet SM, et al. Oxidation of a eukaryotic 2-Cys peroxiredoxin is a molecular switch controlling the transcriptional response to increasing levels of hydrogen peroxide. J Biol Chem. 2005;280:23319–23327. doi: 10.1074/jbc.M502757200. [DOI] [PubMed] [Google Scholar]
- 9.Vivancos AP, et al. A cysteine-sulfinic acid in peroxiredoxin regulates H2O2-sensing by the antioxidant Pap1 pathway. Proc Natl Acad Sci USA. 2005;102:8875–8880. doi: 10.1073/pnas.0503251102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wood ZA, Schröder E, Harris RJ, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 11.Schröder E, et al. Crystal structure of decameric 2-Cys peroxiredoxin from human erythrocytes at 1.7 Å resolution. Structure. 2000;8:605–615. doi: 10.1016/s0969-2126(00)00147-7. [DOI] [PubMed] [Google Scholar]
- 12.Lee DY, et al. Mutagenesis and modeling of the peroxiredoxin (Prx) complex with the NMR structure of ATP-bound human sulfiredoxin implicate aspartate 187 of Prx I as the catalytic residue in ATP hydrolysis. Biochemistry. 2006;45:15301–15309. doi: 10.1021/bi061824h. [DOI] [PubMed] [Google Scholar]
- 13.Jönsson TJ, Murray MS, Johnson LC, Poole LB, Lowther WT. Structural basis for the retroreduction of inactivated peroxiredoxins by human sulfiredoxin. Biochemistry. 2005;44:8634–8642. doi: 10.1021/bi050131i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jeong W, Park SJ, Chang TS, Lee DY, Rhee SG. Molecular mechanism of the reduction of cysteine sulfinic acid of peroxiredoxin to cysteine by mammalian sulfiredoxin. J Biol Chem. 2006;281:14400–14407. doi: 10.1074/jbc.M511082200. [DOI] [PubMed] [Google Scholar]
- 15.Koo KH, et al. Regulation of thioredoxin peroxidase activity by C-terminal truncation. Arch Biochem Biophys. 2002;397:312–318. doi: 10.1006/abbi.2001.2700. [DOI] [PubMed] [Google Scholar]
- 16.Moon JC, et al. Oxidative stress-dependent structural and functional switching of a human 2-Cys peroxiredoxin isotype II that enhances HeLa cell resistance to H2O2-induced cell death. J Biol Chem. 2005;280:28775–28784. doi: 10.1074/jbc.M505362200. [DOI] [PubMed] [Google Scholar]
- 17.Sayed AA, Williams DL. Biochemical characterization of 2-Cys peroxiredoxins from Schistosoma mansoni. J Biol Chem. 2004;279:26159–26166. doi: 10.1074/jbc.M401748200. [DOI] [PubMed] [Google Scholar]
- 18.Chang TS, et al. Characterization of mammalian sulfiredoxin and its reactivation of hyperoxidized peroxiredoxin through reduction of cysteine sulfinic acid in the active site to cysteine. J Biol Chem. 2004;279:50994–51001. doi: 10.1074/jbc.M409482200. [DOI] [PubMed] [Google Scholar]
- 19.Hong SK, Cha MK, Kim IH. Specific protein interaction of human Pag with Omi/HtrA2 and the activation of the protease activity of Omi/HtrA2. Free Radic Biol Med. 2006;40:275–284. doi: 10.1016/j.freeradbiomed.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 20.Choi MH, et al. Regulation of PDGF signalling and vascular remodelling by peroxiredoxin II. Nature. 2005;435:347–353. doi: 10.1038/nature03587. [DOI] [PubMed] [Google Scholar]
- 21.Jung H, Kim T, Chae HZ, Kim KT, Ha H. Regulation of macrophage migration inhibitory factor and thiol-specific antioxidant protein PAG by direct interaction. J Biol Chem. 2001;276:15504–15510. doi: 10.1074/jbc.M009620200. [DOI] [PubMed] [Google Scholar]
- 22.Xiao N, Du G, Frohman MA. Peroxiredoxin II functions as a signal terminator for H2O2-activated phospholipase D1. FEBS J. 2005;272:3929–3937. doi: 10.1111/j.1742-4658.2005.04809.x. [DOI] [PubMed] [Google Scholar]
- 23.Harris JR, et al. Comparison of the decameric structure of peroxiredoxin-II by transmission electron microscopy and X-ray crystallography. Biochim Biophys Acta. 2001;1547:221–234. doi: 10.1016/s0167-4838(01)00184-4. [DOI] [PubMed] [Google Scholar]
- 24.Phalen TJ, et al. Oxidation state governs structural transitions in peroxiredoxin II that correlate with cell cycle arrest and recovery. J Cell Biol. 2006;175:779–789. doi: 10.1083/jcb.200606005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jang HH, et al. Two enzymes in one: Two yeast peroxiredoxins display oxidative stress-dependent switching from a peroxidase to a molecular chaperone function. Cell. 2004;117:625–635. doi: 10.1016/j.cell.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 26.Parsonage D, et al. Analysis of the link between enzymatic activity and oligomeric state in AhpC, a bacterial peroxiredoxin. Biochemistry. 2005;44:10583–10592. doi: 10.1021/bi050448i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hirotsu S, et al. Crystal structure of a multifunctional 2-Cys peroxiredoxin heme-binding protein 23 kDa/proliferation-associated gene product. Proc Natl Acad Sci USA. 1999;96:12333–12338. doi: 10.1073/pnas.96.22.12333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Woo HA, et al. Reduction of cysteine sulfinic acid by sulfiredoxin is specific to 2-Cys peroxiredoxins. J Biol Chem. 2005;280:3125–3128. doi: 10.1074/jbc.C400496200. [DOI] [PubMed] [Google Scholar]
- 29.Mazur DJ, Perrino FW. Excision of 3′ termini by the TREX1 and TREX2 3′→ 5′ exonucleases. Characterization of the recombinant proteins. J Biol Chem. 2001;276:17022–17029. doi: 10.1074/jbc.M100623200. [DOI] [PubMed] [Google Scholar]
- 30.McCoy AJ. Solving structures of protein complexes by molecular replacement with Phaser. Acta Crystallogr D. 2007;63:32–41. doi: 10.1107/S0907444906045975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones TA, Kjeldgaard M. Electron-density map interpretation. Methods Enzymol. 1997;277:173–208. doi: 10.1016/s0076-6879(97)77012-5. [DOI] [PubMed] [Google Scholar]
- 32.Emsley P, Cowtan K. Coot: Model-building tools for molecular graphics. Acta Crystallogr D. 2004;60:2126–2132. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- 33.Brunger AT, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 34.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 35.Davis IW, Murray LW, Richardson JS, Richardson DC. MOLPROBITY: Structure validation and all-atom contact analysis for nucleic acids and their complexes. Nucleic Acids Res. 2004;32:W615–W619. doi: 10.1093/nar/gkh398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Collaborative Computational Project Number 4. The CCP4 suite: Programs for protein crystallography. Acta Crystallogr D. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.