

Abstract
Histone Deacetylase (HDAC) inhibitors represent a budding class of targeted anti-cancer agents. This structurally diverse group of molecules can induce growth arrest, differentiation, apoptosis, and autophagocytic cell death of cancer cells. Of the different classes of HDAC the class I and Class II are considered the main targets for cancer. For the two classes of HDAC, only a few compounds have emerged as preferential inhibitors and even fewer are able to discriminate efficiently among HDACs in the same class. This limitation has diminutive relevance to the use of HDAC inhibitors as potential anti-tumor drugs. Hence, the four HDACs of class I was modeled and about twelve known inhibitors which are currently under the phase I/II trials were docked using an efficient shape-based search algorithm and the AScore scoring function, to each of the class I HDAC members in order to identify the inhibitor or group with better pharmacological action. The molecular descriptors study and the drug score, drug likeness prediction helped in the identification of potential compounds targeting specific enzymes of HDAC family. The ranking of various groups of ligands helped in the identification of potential groups and better compound that can better target class I HDAC in an effective way.
Keywords: Cancer, HDAC, HDAC-inhibitors, molecular descriptors, modeling, docking, drug, ligands, toxicity
Abbreviations
HDAC - histone deacetylase, HDACi - histone deacetylase inhibitor , TPSA - total polar surface area , SCR - structurally conserved region.
Background
Cancer is a generic term for a group of over 100 chronic diseases, which can affect any part of the body. A defining feature of cancer is the rapid creation of abnormal cells, which grow beyond their usual boundary and can invade adjoining parts of the body. The cells may also spread to other organs, a process referred to as metastasis. It is expected that the Cancer incidence would have a steady increase to 15 million new cases in the year 2020 [1]. Thus, the challenge of developing a novel method to face the impending problem is becoming more significant.
HDAC, Histone Deacetylase, is especially known to play an important role in carcinogenesis. The enzyme has been considered a target molecule for cancer therapy [2]. The members of the classical HDAC family fall into two different phylogenetic classes, namely class I and class II [3, 4]. Deacetylation is a process that removes acetyl group from the histone tails, causing the histones to wrap more tightly around the DNA and interfering with the transcription of genes by blocking access by transcription factors. The overall result of deacetylation is a global (non‐specific) reduction in the gene expression. Therefore, the inhibition of HDAC activity by a specific inhibitor induces growth arrest, differentiation, and apoptosis of transformed or several cancer cells. It is this versatility that makes the discovery and development of specific HDAC inhibitors such a tempting prospect in cancer research.
The aim of this work is to compare the HDAC‐I enzyme inhibitors that are already in the Phase I/II trials based on their pharmacological and ADME (Absorption, Distribution, Metabolism and Excretion) and rank them accordingly to identify the inhibitor(s) group specific for the different enzymes of HDAC‐I with better binding affinity and better pharmacological properties. Since, no theoretical works have been carried out in identifying the properties and specificity we intend to identify the group that could act as potential binding inhibitors.
Methodology
Molecular modeling
Homology modeling of the three dimensional structure of class I Histone Deacetylase (HDAC 1,2,3&8) was carried out using the Modeller9v2 [5]. The template used for the comparative modeling was PDB ID‐’1T64‘ (Crystal Structure of human HDAC8 complexed with Trichostatin A).The class I HDAC shows close similarity in their sequence and hence the structure of HDAC8 was used as template for the modeling of the class I Histone deacetylase. The SCRs (Structurally Conserved Regions), structurally variable regions, N‐terminal and C‐termini of reference structure was assigned to the target sequence based on the satisfactory spatial restraints. Thus, the initial models of the Class I HDAC enzymes were generated. The energy minimization was carried out using the SPDBV [6] and the refined structure for all four HDAC‐I enzymes were obtained.
The Histone Deacetylase Inhibitor (HDACi) structures were drawn using the ACD Chemsketch11.0 [7] which offers functionalities like structure cleaning, optimization etc.,. About 12 known HDACi (Figure 1) from different groups such as hydroxamic acid derivatives, benzamide derivatives, cyclic tetrapeptides and short chain fatty acids [2, 8,9] were taken into account for the study.
Figure 1.
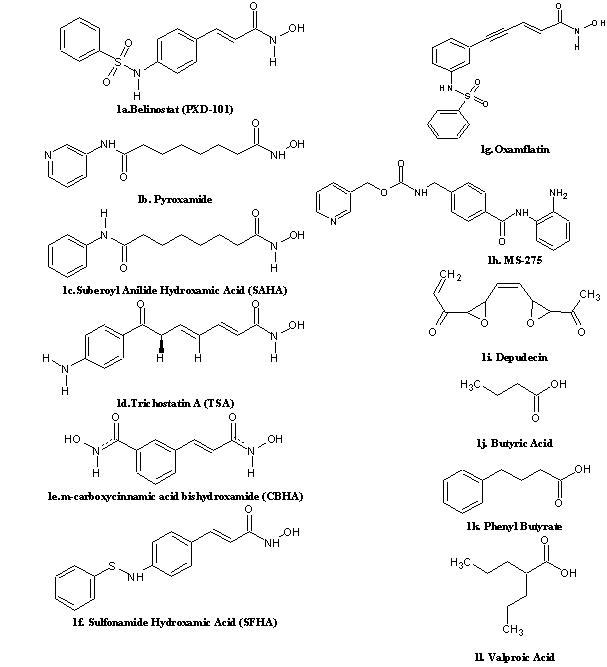
2D structure of different class of compounds with HDACi activity that is currently in Phase I/II clinical trials. 1a‐1g. Hydroxamic acid derivatives; 1h. Benzamide; 1i. Cyclic Peptide; 1j‐1l. Short chain fatty acid derivatives.
Active site prediction
The active sites of the four class I HDAC were identified using the Q‐SiteFinder [10]. The Q‐SiteFinder works by binding hydrophobic (CH3) probes to the protein, and finding clusters of probes with the most favorable binding energy. These clusters are placed in rank order of the likelihood of being a binding site according to the sum total binding energies for each cluster. The higher cavity cluster considered and the residues around the cluster were identified as the binding residues using the SwissPDBViewer.
Docking
Molecular docking and virtual screening based on molecular docking have become an integral part of many modern structure‐based drug discovery efforts and the results are biologically significant. The constructed HDACi's were docked with all the four class I Histone deacetylase viz., HDAC1, HDAC2, HDAC3 & HDAC8 using the Arguslab 4.0.1 [11] which uses a shape‐based search algorithm . The Docking calculations attempt to place Ligands into binding sites.
Molecular descriptors calculation
Quantitative structure‐activity relationships (QSARs) correlate the response with molecular properties of compounds under interest. Any compound to be considered as a lead must possess acceptable scores for all of the descriptors. Molinspiration [12] and Osiris Property explorer [13] were used to calculate twelve descriptors‐logP, solubility, drug likeliness, polar surface area, molecular weight, number of atoms, number of rotatable bonds, volume, drug score and number of violations to Lipinski's rule [14] for all the inhibitors taken for the analysis (Table in supplementary material).
Toxicity prediction
Toxicity is defined as a dose‐linked unsafe effect of a chemical compound on a target life form. Safety issues and ADME (Absorption, Distribution, Metabolism and Excretion) are major factors in drug failure and they are crucial to identify early in the discovery process to reduce late‐stage attrition. ADME‐Tox Box of PharmaAlgorithms [15] was used to predict the various toxicity effects such as rat LD50, mouse LD50, oral bioavailability, Ames test, pKa and ion fraction values.
Discussion
The qualities of the homology modeled proteins were evaluated using the procheck tool [16]. It estimates the stereo‐chemical quality of the modeled structures. On analysis of the Ramachandran plot, it was observed that in the HDAC 1, 2, 3 and 8 around 96.7% of the residues were present in the favored regions. The Q‐SiteFinder analysis produced the ten top most ranked binding sites. The higher cavity site was taken as the most favorable site to dock the ligands. Each ligand showed different affinities with the Class I HDAC family for example SFHA compound showed the best affinity with the HDAC 8 (–10.16 kcal/mol). Whereas the same compound was found to be rank 2 with HDAC1 and HDAC3 (viz. -8.08 kcal/mol and -9.70 kcal/mol) and rank 3 (-9.87 kcal/mol) with HDAC2. Hence, all the ligand compounds were ranked individually based on their binding energies with the four HDAC‐I enzymes in order to identify the more suitable and better binding inhibitor group of the HDAC‐I. The first five ranking inhibitors in each HDAC‐I enzyme were considered for the analysis and eliminating the common ones from the four groups about nine different inhibitors were identified from the first five ranks for each HDAC‐I enzyme. Based on their binding energy ranking TSA and SFHA were found to possess better binding affinity to the four HDAC‐I enzymes because they rank below five in each HDAC‐I enzyme.
The further descriptor analysis and the toxicity prediction helped in the identification of the more suitable inhibitor. From Table 1 (supplementary material), each inhibitor were lacking (red color) in at least one or more properties. TSA was the only compound that had the acceptable range (green color) for drug likeness [17] and also found to possess better binding affinity score (Table 1 in supplementary material). Drug Score and the Drug‐Likeness are the two properties that are important for considering a compound to become a successful drug. Though valproic acid had a drug score of 0.61 and drug likeness property score of 0.36 it was not appropriate as the molecular weight of valproic acid was 116.16 g/mol, a value lower than the preferable range of molecular weight for drug likeness property (160‐480 g/mol) [17]. These values were also taken into account to decide the best inhibitor (Table 1 in supplementary material).
The ADME‐TOX box results showed that the TSA has an oral bioavailability of more than 70% i.e., good solubility and stability. It acts as a non‐substrate and non‐inhibitor of P‐gp. TSA does not undergo significant first‐pass metabolism [17]. The predicted LD50 values of TSA in mouse and rat was found to be 1700mg/kg and 1800mg/kg respectively when administered orally. TSA acts as a non‐substrate when checked for the P‐glycoprotein substrate specificity.
Conclusion
The few compounds predicted to inhibit the HDAC family are not effective to all forms of HDAC. This analysis involves a comparative investigation by the ranking the inhibitors based on diverse strategies. It is clear that only TSA (a natural compound that can be isolated from the metabolites of strains of Streptomyces hygroscopicus) satisfied almost all the properties i.e., binding affinity scores of TSA in the four HDAC‐I enzymes was -8.99 kcal/mol, -8.58 kcal/mol, -8.90 kcal/mol & -9.34 kcal/mol respectively, the Drug Likeness value (1.24) and drug score (0.37) with 70% oral bioavailability and the hydroxyl group of these compounds fits well into the active site of the target where the zinc is present [18] (Figure 2). Hence, better hydroxamate derivatives with more potency could be developed as potential inhibitors of class‐I HDAC and valuable anti‐cancer‐agents.
Figure 2.
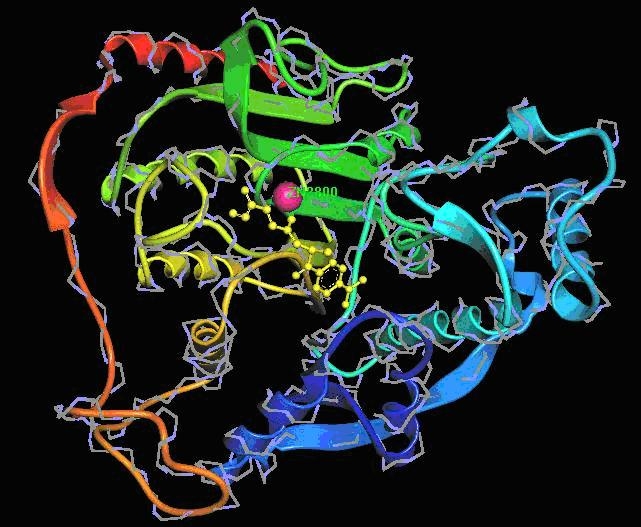
Structure showing a bound Hydroxamic acid derivative, TSA with HDAC 8.
Supplementary material
Acknowledgments
The authors thank Mr. G. Aravindhan, JRF, AU‐KBC Research Centre for assisting in preparing the documentation.
References
- 01. http://www.who.int/mediacentre/news/releases/2003/pr27/en/
- 2.Kim DH, et al. J Biochem Mol Biol. 2003;36:110. doi: 10.5483/bmbrep.2003.36.1.110. [DOI] [PubMed] [Google Scholar]
- 3.Hu E, et al. The J Biol Chem. 2000;275:15254. doi: 10.1074/jbc.M908988199. [DOI] [PubMed] [Google Scholar]
- 4.de Ruijter AJ, et al. J Biochem Soc. 2003;370:737. doi: 10.1042/BJ20021321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eswar N, et al. Current Prot in Bioinform. 2006;15:5.6. [Google Scholar]
- 6.Guex N, Peitsch MC. Electrophoresis. 1997;18:2714. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 7. http://www.acdlabs.com/download/chemsk.html.
- 8.Johnstone RW. Nat rev drug dis. 2002;4:287. doi: 10.1038/nrd772. [DOI] [PubMed] [Google Scholar]
- 9.Jung M. Curr Med Chem. 2001;8:1505. doi: 10.2174/0929867013372058. [DOI] [PubMed] [Google Scholar]
- 10.Laurie AT, Jackson RM. Bioinformatics. 2005;21:1908. doi: 10.1093/bioinformatics/bti315. [DOI] [PubMed] [Google Scholar]
- 11. http://www.arguslab.com.
- 12. http://www.molinspiration.com/
- 13. http://www.organic-chemistry.org/prog /peo/
- 14.Lipinski CA, et al. Adv Drug Del Rev. 1997;23:3. [Google Scholar]
- 15. http://pharma-algorithms.com/physchem.htm.
- 16. http://www.biochem.ucl.ac.uk/~roman/procheck/procheck.html.
- 17. http://pubs.acs.org/cgi-bin/abstract.cgi/jcchff/1999/1/i01/abs/cc9800071.html.
- 18.Dokmanovic M, Marks PA. J Cell Biochem. 2005;96:293. doi: 10.1002/jcb.20532. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.