

Abstract
The effect of the guanidinium cation on the hydrogen bonding strength of water was analyzed using temperature-excursion Fourier transform infrared spectra of the OH stretching vibration in 5% H2O / 95% D2O solutions containing a range of different guanidine-HCl and guanidine-HBr concentrations. Our findings indicate that the guanidinium cation causes the water H-bonds in solution to become more linear than those found in bulk water, and that it also inhibits the response of the H-bond network to increased temperature. Quantum chemical calculations also reveal that guanidinium affects both the charge distribution on water molecules directly H-bonded to it as well as the OH stretch frequency of H-bonds in which that water molecule is the donor. The implications of our findings to hydrophobic solvation and protein denaturation are discussed.
Keywords: chemical denaturation, guanidine, infrared spectroscopy, temperature dependence, hydrogen bonding
Introduction
Guanidine salts, such as guanidine-HCl, have been used for over 70 years to destabilize or completely denature proteins in solution.1-3 Guanidine salts have proven to be particularly useful in the biochemical and biophysical study of protein folding, since a great number of proteins can be successfully refolded by dialyzing out the salt.4 Despite many avenues of investigation over the course of many years, the exact mechanism by which the guanidinium cation (Gdm+, C(NH2)3+) destabilizes the folded structure of proteins is still a matter of debate and speculation.5-7 The primary reason that has been proposed for Gdm+ denaturation of proteins is that Gdm+ directly interacts with the protein or some part of it,2,8-13 while alteration of the solution water structure has been studied much less than for the other common chemical denaturant, urea, and has been ignored or minimized as a possible denaturation mechanism.8,14-18
While there is certainly excellent evidence for the direct interaction model of guanidinium's protein denaturation action, this particular model certainly does not rule out the possibility for other causes. Here, we offer support for an additional mechanism for the destabilization and unfolding of proteins whereby Gdm+ restructures the solution water. We hypothesize, as have others, 7,18-23 that the Gdm+ cation might, in fact, destabilize folded proteins by altering the H-bonding network of water, such that the native, folded state of a given protein is no longer energetically favorable and net equilibrium is shifted towards the unfolded state.
To test this hypothesis, infrared spectroscopy was used to examine the OH stretching vibration of dilute HOD in D2O solutions containing different concentrations of dissolved guanidine salts. Infrared spectroscopy is sensitive to changes in the environment and interactions local to vibrating molecules, and therefore the infrared absorption lineshapes of those vibrating molecules can be used as a probe of their environment. Also, for dilute solutions of HOD in D2O an individual OH oscillator is almost completely decoupled from other OH oscillators, making the OH a selective and independent local probe. In contrast to other experimental methods previously used to probe the structure of water in a guanidinium-containing solution, such as neutron diffraction, calorimetry, and NMR, infrared spectroscopy is a uniquely sensitive probe of H-bond strength.
Since nearly all biological phenomena occur in an aqueous environment, solution FTIR spectroscopy can be a particularly useful technique for investigating biologically relevant interactions. Of particular interest in biological solution spectroscopy is how and to what degree biological solutes affect the structure of water. Because of its ability to form strong H-bonds with both itself and with solutes, water has the ability to solvate a tremendous variety of biologically relevant molecules. In addition, the ability to both donate and accept H-bonds allows water to adopt a flexible, three dimensional H-bond network. This H-bond network is responsive to both external and internal perturbations, such as those caused by the addition or removal of thermal energy or by the addition of solutes.
Temperature excursion IR (TEIR) is very useful for studying H-bonded systems such as aqueous solutions.24-28 The structure of the H-bond network of water is sensitive to changes in temperature, and these temperature-dependent structural rearrangements of the H-bond network are reflected in the spectra.25-27,29 Since TEIR involves the collection of data at many different temperatures, the thermodynamic response of the OH stretch of water containing a solute of interest can be compared to the spectra of pure water to often yield both qualitative and quantitative structural details of the H-bond network.
In this paper, we examine the temperature dependence of water in the presence of guanidine-HCl and guanidine-HBr salts (Gdm+/Cl- and Gdm+/Br- in solution) using TEIR. We analyze the OH stretch spectrum of dilute HOD in D2O samples using a straightforward ratio of absorbances on either side of the OH stretch peak maximum. Our results show that these guanidine salts drive the preferential formation of strong, linear H-bonds over weak, bent H-bonds. Quantum chemical calculations also indicate that a distinct portion of the OH stretch spectrum, a shoulder at ∼3300 cm-1, arises from water molecules directly accepting an amino H-bond from a guanidinium cation. We conclude that the H-bond network of water is substantially perturbed by guanidinium, and we discuss the relevance of this restructuring to Gdm+ denaturation of proteins and contrast it with previous findings for the other commonly used chemical denaturant, urea.
Materials and Methods
Materials
In our experiments, H2O was deionized and then glass distilled. D2O was purchased from Acros Organics (Geel, Belgium). H2O and D2O were degassed by aspiration before sample preparation. C13/3N15 labeled guanidine-HBr was purchased from Cambridge Isotope Laboratories Inc. (Andover, MA) and C13/3N15 labeled guanidine-HCl was purchased from Sigma-Aldrich (St. Louis, MO). Both guanidine salts were completely deuterated by successive rounds of dissolving in excess D2O followed by lyophilization. C13/3N15 salts were used in order to shift the NH stretching vibration to a lower frequency such that it did not overlap with the OH stretching frequency.
Solutions at 7.27 molal (equivalent to ∼5.2 M, or 6.9 water-to-Gdm+/anion mole ratio) for both of the guanidine salts were obtained by dissolving them in a 5% H2O / 95% D2O (v/v) mixture, and then all lower concentrations were obtained by dilution of the 7.27 m stock solutions with this same H2O / D2O mixture.
Spectroscopy
Sample solution was placed between two 2 mm thick calcium fluoride windows, and spectra were collected using a dry nitrogen purged Bruker IFS 66 infrared spectrophotometer (Bruker Optics, Brookline, MA) fitted with an MCT detector. The spectra were collected in transmission mode with a 2 cm-1 resolution.
Using a circulating water bath, temperature at the sample was held to +/-0.1 °C over the course of a spectrum acquisition. Spectra were collected from low temperature to high at 5 °C intervals. Spectra were corrected for H2O atmospheric contribution, baseline corrected, and converted to absorbance using the OPUS software package (Bruker Optics, Brookline, MA) and imported into MATLAB 7.6 (Mathworks, Natick, MA) for further processing. MATLAB was also used to produce Figures 1, 2, and 3. Cubic spline interpolation was used to increase the number of data points from 536 to 51501 and yield a frequency span of 3165 cm-1 to 3680 cm-1. Second degree Savitsky-Golay smoothing was applied to the interpolated data. Plot overlays of interpolated and smoothed data and data with just atmospheric and baseline correction confirmed that smoothing only removed noise and did not perturb the underlying lineshape.
Figure 1.
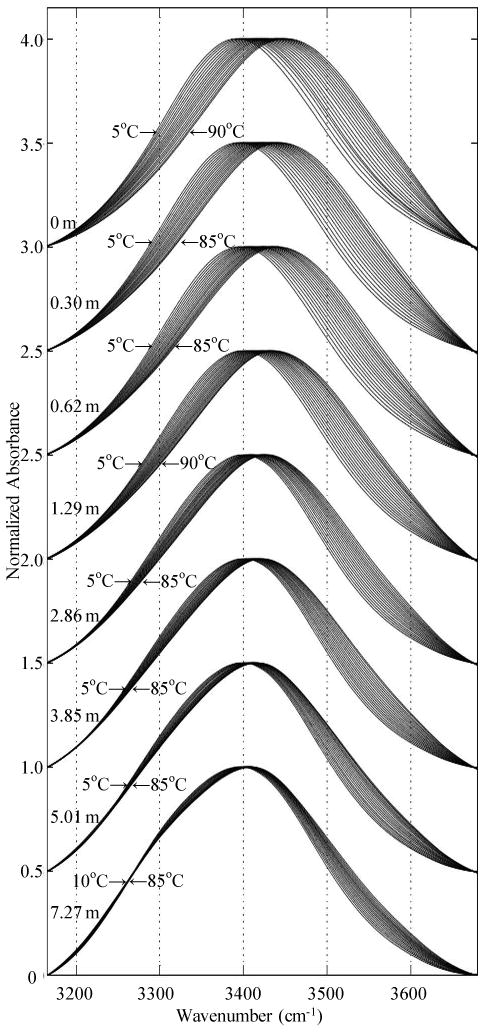
OH stretch TEIR spectra for 5% H2O / 95% D2O solutions containing increasing concentrations of guanidinium chloride.
Figure 2.
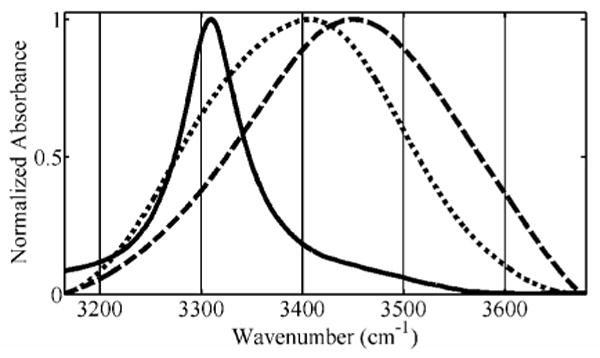
OH stretch spectra of 7.27 m Gdm+/Br- (dot) and 5% H2O / 95% D2O water (dash) at 90°C shown, along with 5% H2O / 95% D2O ice at -10°C (solid).
Figure 3.
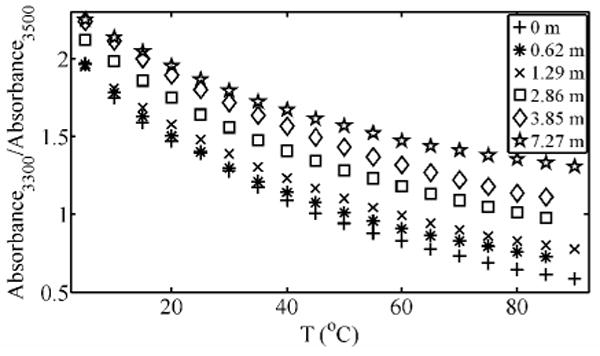
Ratio of spectral intensities at 3300 cm-1 and 3500 cm-1 for selected Gdm+/Cl- OH stretch spectra shown in Figure 1.
Quantum Chemistry
All quantum chemical calculations were carried out using Gaussian 03, Revision D.01,30 and the B3LYP model chemistry31,32 and 6-311++G(d,p) basis set.33,34 We note that such a basis set is only suitable for the sort of qualitative comparison presented here and not for quantitative determinations of H-bond properties.35-39 In the context of the calculations performed, all H-bonds described as “linear” were actually set at 0.1° in order to avoid collinear atoms and the more complicated Z-matrices needed to specify truly linear H-bond angles in the water trimer and guanidinium/dimer cases. The standalone dimer case was tested and showed miniscule and negligible changes to atomic charges, vibrational frequencies, and ground state energy when a truly linear H-bond geometry was used in place of the 0.1° HOO H-bond angle.
A linearly H-bonded HOD/D2O water dimer was constructed as a Z-matrix. This water dimer was partially geometry optimized along the O-O distance, and the infrared frequencies and natural population charges were calculated. Natural Population Analysis, a part of the Natural Bond Orbital formalism,40 was used to calculate atomic charges.
Another D2O water molecule was added to the starting dimer Z-matrix to build a chain of three linearly H-bonded water molecules. Two trimer geometries were used, a hydrogen “up” configuration and a hydrogen “down” configuration, differentiated by a 180° difference in the terminal hydrogen dihedral angle. The trimer systems were each partially geometry optimized along the O-O distances, and the infrared frequencies and natural population charges were calculated.
Finally, a deuterated guanidinium cation was added to the water dimer Z-matrix such that it donated a single linear amino H-bond to the oxygen of the HOD molecule. The internal geometry of the cation was fixed, based on a separate previous geometry optimization, and partial geometry optimization was carried out along the O-O distance of the two water molecules and the guanidinium deuterium/HOD oxygen distance. Infrared frequencies and natural population charges were calculated for the optimized system.
All OH/OD lengths were fixed at 0.991 Å and the HOD/DOD angles were fixed at 105.5°, as per the findings of Silvestrelli and Parrinello concerning condensed phase water.41 Complete optimized Z-matrices used to obtain results are given in Supplementary Materials. Atomic models of the systems studied in the linear HOO angle case are shown in Figure 4, and were visualized using Gaussview 3.07. Electrostatic potential maps (Figures 5 and 6) were generated using Molekel 5.3.0.6. The surfaces shown are the solvent accessible surfaces, probed with a 1.4 Å solvent radius and colored by molecular electrostatic potential. Electron density maps (Figures 7 and 8) were generated using VMD 1.8.642 and rendered using Tachyon 0.97.43
Figure 4.
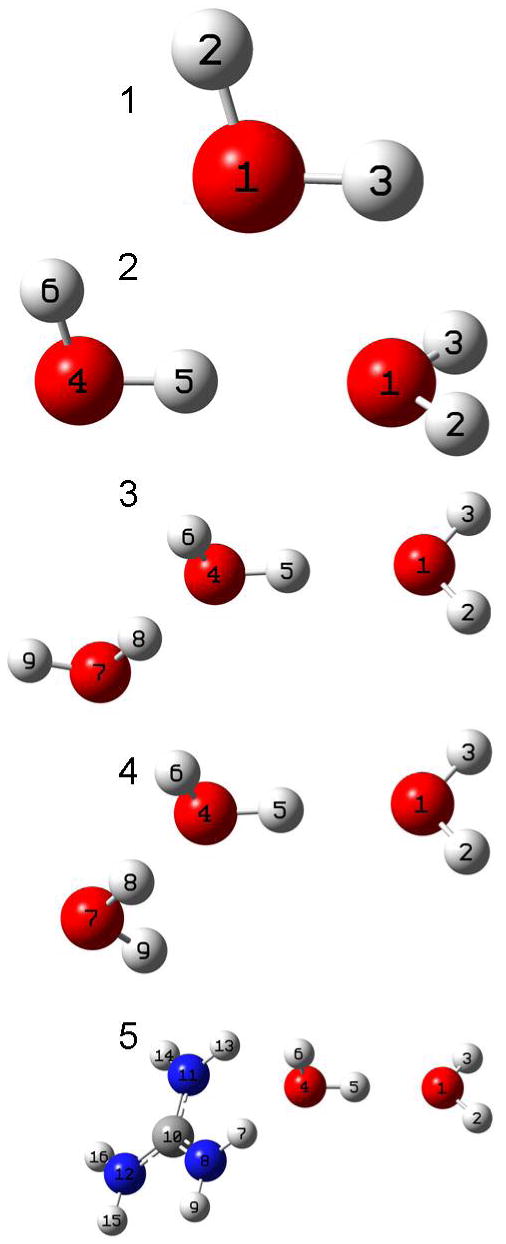
Shown here are graphical representations of the systems for which the OH stretch frequency and natural population charges were calculated. Calculations were also performed for a variation on these systems in which the HOO angle (H5-O4-O1) was set at 45°. From top to bottom, the monomer, dimer, trimer (H up orientation), trimer (H down orientation), and guandinium – water dimer geometries are shown, along with a numerical key to referencing the tables 1 – 4.
Figure 5.
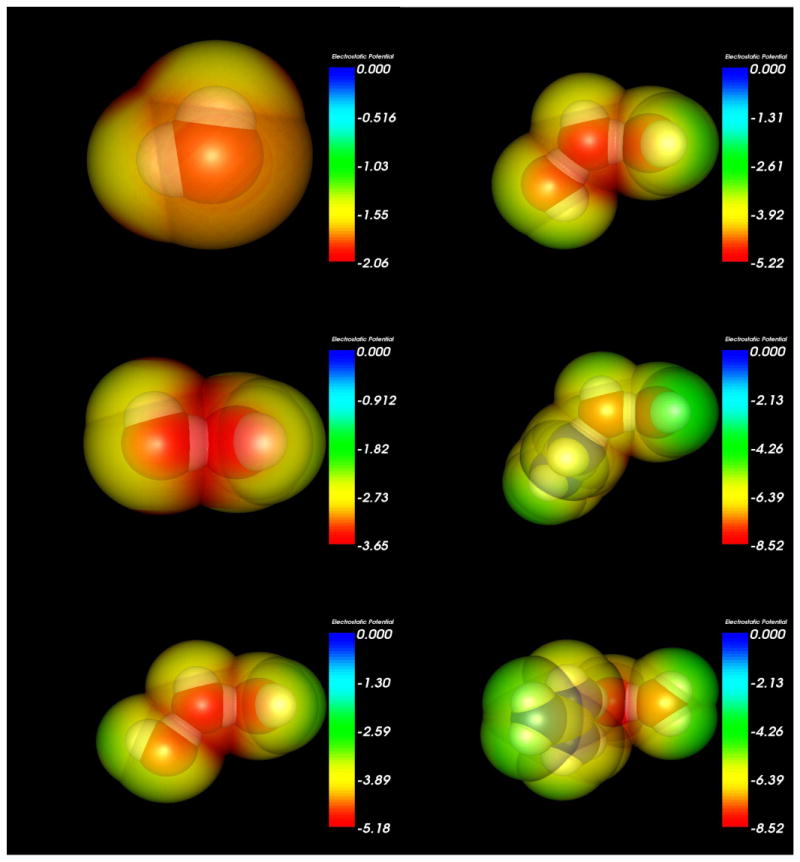
Electrostatic potential maps at the solvent accessible surface (1.4 Å solvent radius probe) generated for the water monomer and linear molecular configurations using Molekel 5.3.0.6 and colored by molecular electrostatic potential.
Figure 6.
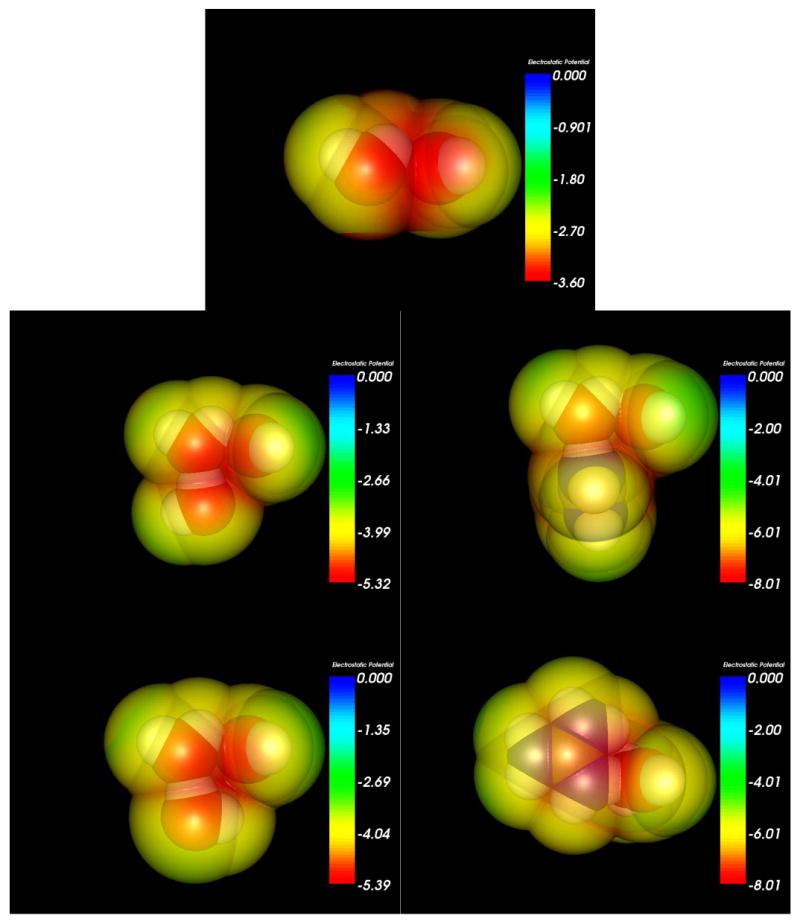
Electrostatic potential maps at the solvent accessible surface (1.4 Å solvent radius probe) generated for the 45° HOO (H5-O4-O1) H-bond angle molecular configurations using Molekel 5.3.0.6 and colored by molecular electrostatic potential.
Figure 7.
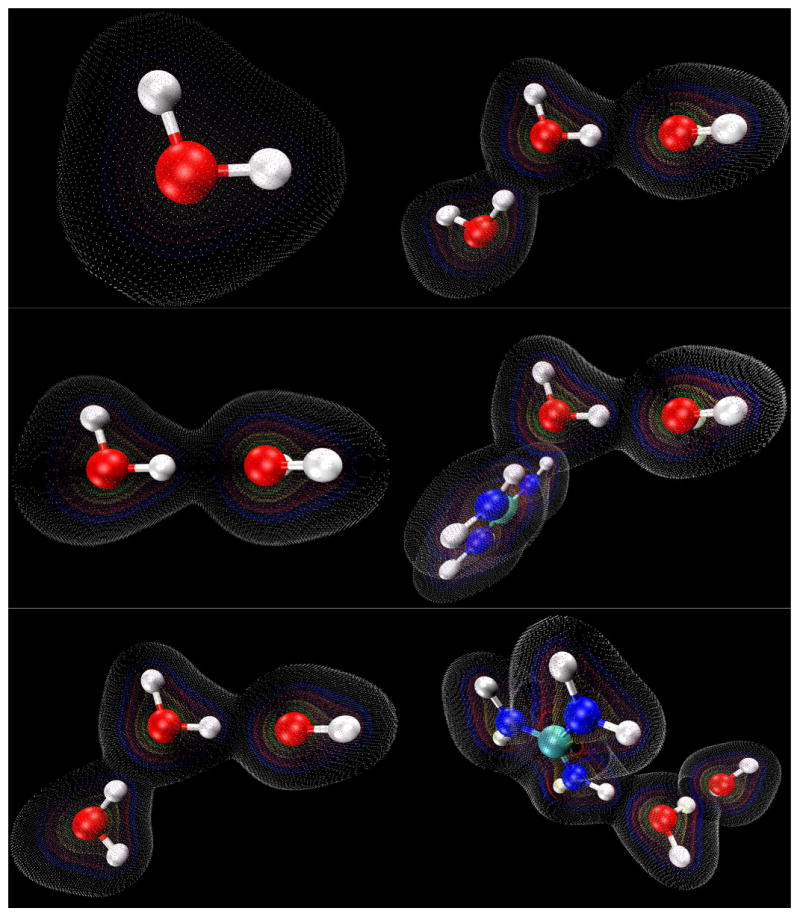
Electron density maps generated for the water monomer and linear molecular configurations using VMD 1.8.6. Contours are shown at values of 0.01 (white), 0.05 (blue), 0.10 (red), 0.25 (yellow), 0.50 (green).
Figure 8.
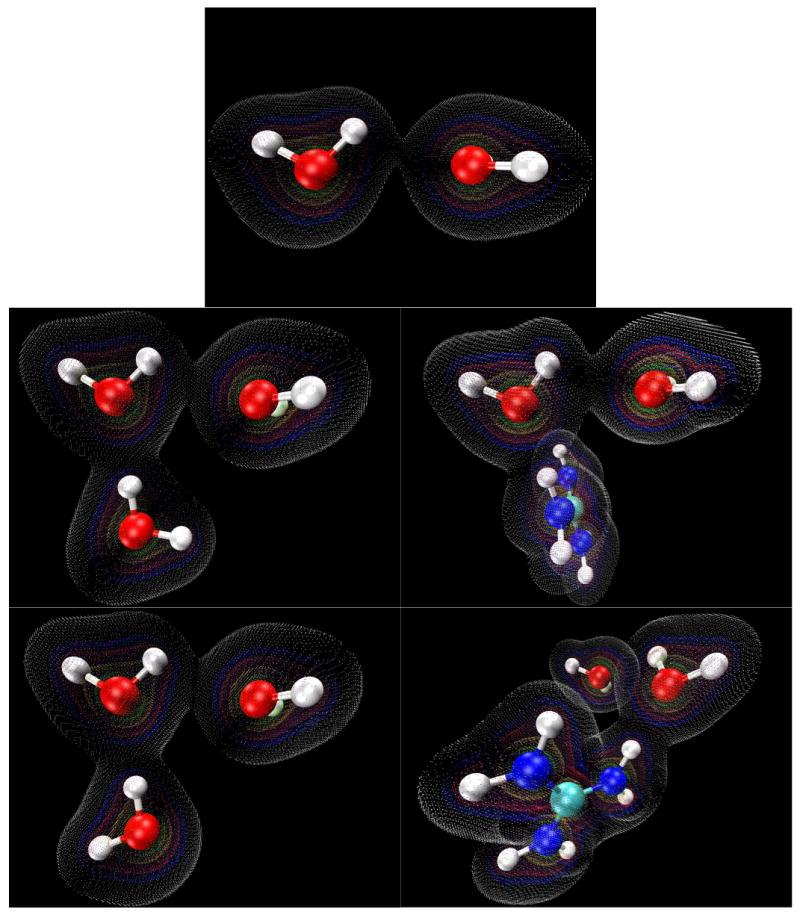
Electron density maps generated for the 45° HOO (H5-O4-O1) H-bond angle molecular configurations using VMD 1.8.6. Contours are shown at values of 0.01 (white), 0.05 (blue), 0.10 (red), 0.25 (yellow), 0.50 (green).
Results
A. IR Spectroscopy. OH stretch spectra for all Gdm+/Cl- concentrations measured are shown in Figure 1, along with a solute free sample containing only 5% H2O / 95% D2O. Similar spectra were obtained at several of the same concentrations for Gdm+/Br- solutions (not shown). Two features of note are immediately visible in the Gdm+/Cl- OH stretch spectra that are not seen in the 5% H2O / 95% D2O spectra. The first is a strong decrease in temperature dependence. The characteristic shifts of both the low and high frequency parts of the OH stretch spectrum are diminished as the concentration of Gdm+/Cl- is increased. This effect is most pronounced at low frequency (∼3200-3400 cm-1), where for the more concentrated samples there is little change in lineshape with increasing temperature, culminating in nearly complete temperature-independence below 3450 cm-1 for the highest concentration sample studied.
The second feature of interest is an increase in absorbance at ∼3300 cm-1. Like the change in lineshape temperature dependence, this localized increase in absorbance can be seen even at low Gdm+/Cl- concentrations, but it is most apparent at the highest concentrations, where a shoulder is easily visible. Though both the infrared and Raman OH stretch spectra of pure water have long been known to possess a pronounced asymmetry, as well as a characteristic temperature dependence,44,45 this data shows a profound change in the lower frequency water OH stretch absorption with the addition of guanidinium.
The shoulder that grows in at ∼3300 cm-1 shows a minimal decrease in absorbance with temperature, and therefore the solvation environment that produces this feature must be retained over the whole temperature range for high guanidinium concentrations. To emphasize this feature, in Figure 2, the spectra of a solution of 7.27 m Gdm+/Br- and that of 5% H2O / 95% D2O water at 90°C are overlaid. The figure also shows the spectrum of 5% H2O / 95% D2O ice at -10°C. A quantitative measure of this shoulder's appearance with an increase in guanidinium concentration can be seen in Figure 3.
B. Computation. In order to rationalize the experimental results, quantum calculations of water – water interactions and water – guanidinium ion were undertaken. The graphical representations of the systems studied are shown in Figure 4. This figure shows geometries in which the HOO angle (H5-O4-O1) was set at 0.1°, as well as the water monomer. Calculations were also performed for a variation on these H-bonded systems in which the HOO angle (H5-O4-O1) was set at 45°.
The calculated atomic charges are given in Table 1 for the water monomer, in Table 2 for the water dimer, in Table 3 for the water trimers, and in Table 4 for guanidinium plus water dimer. Images of the electrostatic potentials are given in Figures 5 and 6. The Z-matrix geometries of the systems studied are given in the Supporting Information.
Table 1.
Calculated Charges for the Water Monomer Systems
| atom type | atom number | charge |
|---|---|---|
| O | 1 | -0.92048 |
| D | 2 | 0.46024 |
| H | 3 | 0.46024 |
Atom numbers reference Model 1 in Figure 4.
Table 2.
Calculated Charges for the Water Dimer Systems
| atom type | atom number | charge (0.1° H5-O4-O1) |
charge (45° H5-O4-O1) |
|---|---|---|---|
| O | 1 | -0.94514 | -0.92934 |
| D | 2 | 0.47788 | 0.46579 |
| D | 3 | 0.47788 | 0.46579 |
| O | 4 | -0.94971 | -0.92511 |
| H | 5 | 0.48857 | 0.47427 |
| D | 6 | 0.45053 | 0.44859 |
Atom numbers reference Model 2 in Figure 4.
Table 3.
Calculated Charges for the Water Trimer Systems
| atom type | atom number | H-up orientation charge (0.1° H5-O4-O1) | H-up orientation charge (45° H5-O4-O1) | H-down orientation charge (0.1° H5-O4-O1) | H-down orientation charge (45° H5-O4-O1) |
|---|---|---|---|---|---|
| O | 1 | -0.94915 | -0.93924 | -0.94846 | -0.93648 |
| D | 2 | 0.48211 | 0.47072 | 0.48224 | 0.47024 |
| D | 3 | 0.48211 | 0.47072 | 0.48224 | 0.47024 |
| O | 4 | -0.97304 | -0.94904 | -0.97323 | -0.95070 |
| H | 5 | 0.50282 | 0.49129 | 0.50572 | 0.49504 |
| D | 6 | 0.47001 | 0.46823 | 0.46624 | 0.46349 |
| O | 7 | -0.95637 | -0.95792 | -0.95622 | -0.95300 |
| D | 8 | 0.49414 | 0.49484 | 0.49422 | 0.49519 |
| D | 9 | 0.44737 | 0.45039 | 0.44725 | 0.44596 |
Table 4.
Calculated Charges for the Guanidinium/Water Dimer Systems
| atom type | atom number | charge (0.1° H5-O4-O1) |
charge (45° H5-O4-O1) |
|---|---|---|---|
| O | 1 | -0.96130 | -0.95662 |
| D | 2 | 0.49595 | 0.48289 |
| D | 3 | 0.49256 | 0.47736 |
| O | 4 | -0.99675 | -0.97202 |
| H | 5 | 0.52080 | 0.51517 |
| D | 6 | 0.48922 | 0.48770 |
| D | 7 | 0.45315 | 0.45507 |
| N | 8 | -0.75218 | -0.74675 |
| D | 9 | 0.40918 | 0.40890 |
| C | 10 | 0.67050 | 0.67021 |
| N | 11 | -0.75132 | -0.75149 |
| N | 12 | -0.75143 | -0.75185 |
| D | 13 | 0.43031 | 0.43020 |
| D | 14 | 0.41468 | 0.41459 |
| D | 15 | 0.42009 | 0.42003 |
| D | 16 | 0.41653 | 0.41658 |
Atom numbers reference Model 5 in Figure 4.
In solution, water will be H-bonded to other molecules, so for the discussion of charges, the atoms of most interest are O4 and H5, since this OH oscillator is the one that is H-bonded to a neighboring water molecule. The results show that the donation of an H-bond makes the donor water molecule's oxygen take on a more negative charge while the donor hydrogen becomes more positive. When that H-bond donating water molecule accepts an H-bond itself from a third water molecule its oxygen becomes even more negative and its hydrogen becomes more positive. When the third water molecule is replaced by a guanidinium cation, this effect is amplified, making the donor water molecule's oxygen its most negative and the donor hydrogen its most positive.
With the addition of the guanidinium cation the charge difference between the water molecule's hydrogen and oxygen grows to almost 1.52 e, a charge disparity nearly 0.17 e greater than that found in the isolated water monomer. For the case where the H-bond of the water molecule of interest is increased to 45° the O4-H5 charge disparity decreases by approximately 0.03 e. The atomic charges of the water molecule that is outside of the first sphere of interaction with guanidium is likewise affected by interaction with guanidinium, though considerably less so than O4 and H5.
The frequencies of the stretching vibration of O4-H5 for the five model systems are given in Table 5. As is expected, the vibrational frequency drops considerably (3500 cm-1 to 3462 cm-1) when the HOD monomer donates a linear H-bond to another water molecule. The frequency becomes even lower when the H-bond donating water accepts an H-bond from another water molecule (3462 cm-1 in the dimer to 3444 cm-1 or 3447 cm-1 in the trimer). However, the most dramatic change in frequency occurs when the H-bond donating water accepts an amino H-bond from a guanidinium cation. In this case, the O4-H5 vibrational frequency drops to 3395 cm-1. This large shift is of the order of magnitude that is experimentally observed.
Table 5.
Frequency of O4-H5 Stretching Vibration for All Quantum Chemical Models
| O1 – H3 stretching vibration | O4 – H5 stretching vibration HOO angle (H5-O4-O1) = 0.1° |
O4 – H5 stretching vibration HOO angle (H5-O4-O1) = 45° |
|
|---|---|---|---|
| Model 1, monomer | 3499.7268 cm-1 | - | - |
| Model 2, dimer | - | 3461.9444 cm-1 | 3500.5098 cm-1 |
| Model 3, trimer (H up) | - | 3446.7225 cm-1 | 3503.1583 cm-1 |
| Model 4, trimer (H down) | - | 3443.8779 cm-1 | 3501.9835 cm-1 |
| Model 5, guanidinium – water dimer | - | 3395.1482 cm-1 | 3502.1586 cm-1 |
We note that the effect of the guanidinium cation's amino H-bond donation on the OH stretch vibrational frequency is canceled out when the HOO (H5-O4-O1) angle is increased to 45° (Table 5). This is true for the trimer cases as well, showing that a simple redistribution of charges cannot fully account for the large changes seen in the O4-H5 vibrational frequency. However, we do observe decreased electrostatic potential (Figures 5 and 6) in the region of the H5-O1 H-bond when the angle of that bond is increased, as well as a very large decrease in electron density overlap (Figures 7 and 8).
Finally, we need to point out caveats for these calculations. In a true liquid solution there is a distribution of H-bond angles, and each water molecule is further H-bonded to others to form the H-bond network, therefore these calculations represent a simplified picture of the chemistry. Although these calculations do not exactly mimic the experimental reality, they do offer strong support for the trends observed in the infrared spectra – that the H-bonding network in guanidinium-containing solutions favors linear H-bonds, which accounts for the overall shift to lower frequency absorption seen in the OH stretch band of guanidinium solutions, and that the acceptance of a guanidinium H-bond by an OH oscillator that is itself donating a linear H-bond causes a large decrease in that OH's vibrational frequency.
Discussion
The results presented in this study demonstrate that the guanidinium cation has a substantial effect on water structure. These results are significant in that the techniques employed experimentally probe the H-bond network directly and correlate well with model quantum chemistry calculations. Changes in the OH stretch spectra are indicative of changes in the local H-bonding environment of OH oscillators,46-50 and as can be seen qualitatively in Figure 1, the OH stretch spectra are highly altered by the addition of guanidinium.
The fact that the OH stretching vibrations go to lower frequency, and therefore lower energy, with the addition of guanidinium is important. Low energy OH vibrations can be attributed to shorter, more linear H-bonds.25-27,50-53 The ultrafast spectroscopic research of Laenen et. al.,52 Wang et. al.50,53, Woutersen et. al.54 and other groups indicate distinct structural subpopulations of the OH stretching vibrational spectrum that give rise to different OH stretch relaxation lifetimes, and the extensive computational studies of Sharp et. al.51,55 and others56 confirm these findings for a number of different water models. Taken together, the experimental and theoretical evidence points to a water network consisting of distinct H-bond populations: strong, linear H-bonds and distorted, weak H-bonds.
Since the solutes studied are free from vibrational contributions that would overlap with the OH stretch of water, the shoulder at ∼3300 cm-1 that grows in with Gdm+/anion concentration can be attributed to a restructuring of the water H-bond network by the ions. The same reasoning holds for the differences observed in temperature dependence of the solutions. The decrease in temperature dependence of the OH stretch spectrum with the addition of guanidinium means that the rearrangement response of the water network to the addition of thermal energy is diminished. Our spectra indicate that the presence of the guanidinium cation in high concentrations structures water into a network that is, on average, resistant to reordering when thermal energy is added.
The response of water to the presence of guanidinium is quite different from urea, which produces very little change in the OH stretch spectrum of water.51 Urea is a net uncharged molecule, and so it is unlikely to have a substantial effect on the natural charge distributions of the water molecules to which it H-bonds. Like guanidinium, there is a great deal of evidence for direct binding of urea to the folded protein or peptide backbone.12,57-59 However, there is also a substantial amount of research, both computational51,60,61 and experimental,51,62,63 that urea is “floppy”64 in solution and fits neatly into the bulk water H-bond network, leaving water structure mostly unperturbed.
Given the complex structure of the guanidinium cation and its potential for multiple types of interactions with water, anions, and other guanidinium cations, it seems most likely that guanidinium, and not the anions, plays the primary role in the network-wide restructuring of water. The OH stretch spectrum of a high concentration KCl solution supports this idea.51 The K+/Cl- solution does not show the visible low frequency structuring of the OH stretch that a similar concentration of Gdm+/Cl- does, indicating that the low frequency OH stretch component that grows in with Gdm+/Cl- concentration is not simply due to an increase in strong OH–Cl interactions. To fully simulate the IR absorption spectra would require a more elaborate calculation than was carried out here, and would also need to include the counter ion, but our quantum chemical calculations do qualitatively indicate a trend that accounts for the experimental spectral features; that both the charge distribution and OH stretch frequency of water are significantly altered by H-bonding to guanidinium, and that the effect on OH oscillator frequency is dependent on that oscillator's donating its own linear H-bond.
Our results show that though the addition of a guandinium amino H-bond to a water molecule does alter that water's charge distribution, the effect on charge distribution is only minimally responsive to the angle at which that water molecule donates an H-bond of its own. In both the water trimer and guanidinium-dimer calculations, the angle of the H5-O4-O1 H-bond had very little effect on distribution of charges. Instead, the H5-O4-O1 H-bond angle had its most significant impact on the O4-H5 vibrational stretch frequency and the degree to which electron density was shared between H-bonded water molecules, along with a decrease in electrostatic potential at the H-bond site. Therefore, these results are consistent with the notion of a covalent component to the H-bond that is dependent primarily on H-bond angle.65
As guanidinium ions increase in concentration it can reasonably be expected that an increasing percentage of solution water is involved in first shell guanidinium solvation. Since it is known that guanidine salts do not partition out of solution at the concentrations used in these experiments, we can reason that it must either a) fit neatly into the bulk water H-bonding network, thereby not disrupting water structure at all, or b) rearrange the H-bonding network of water such that it can solvate the ions. Our data strongly indicates rearrangement of the H-bond network by high concentrations of guanidinium, and an increase, on average, in strong, linear H-bonds relative to weak, bent H-bonds. This relative deficit in weak H-bonds implies that guanidinium is forcing water to be over structured, as compared to pure water. This idea is also strongly supported by the reduction in OH stretch temperature dependence of the concentrated guanidinium solutions.
The results we present here have very interesting implications for the chemical denaturation of proteins by dissolved guanidine salts. Direct interactions between guanidinium cations and proteins have typically been indicated as the mechanism through which guanidine salts destabilize and unfold proteins, and it seems clear that direct binding must play a primary role in protein denaturation. Though our experiments were not designed to test the direct binding mechanism directly, our results do show a substantial restructuring of the water network, and these denaturation mechanisms need not be mutually exclusive. The hydrophobic collapse model of protein folding states that the primary force driving protein folding and stability is the shedding of highly structured, entropically unfavorable, low-density solvation water from hydrophobic residues exposed to solvent.66-69 The collapse of these hydrophobic residues into the protein interior releases the linearly H-bonded, low-density waters back to the bulk solvent, thereby allowing the protein/solvent system to attain a net lower energy state.
When the results presented here are contemplated in the context of hydrophobic collapse, it can be seen how the restructuring of the water network by guanidinium may play a role in the denaturation of proteins. Guanidinium in high concentrations, in addition to replacing much of the bulk water in solution, causes an overall structuring of the water network, making its H-bonds much more linear than those found in pure water. The consequence of this solution-wide reordering for folded proteins is that their hydrophobic residues would no longer have as great an entropic drive to pack with one another in the interior of the protein as they do in a non-denaturing solution. With a large portion of the H-bonds in the high concentration guanidinium solution already very linear, there would be a reduced entropic penalty for their solvation of a hydrophobic residue. We believe that this reduction of the impetus for hydrophobic packing may be a factor in the denaturing action of guanidine salts. This rationale may also help in explaining the high relative solubility of molecules like benzene,23 hydrophobic amino acids,70 cyclic dipeptides,71 and hydrocarbons20 in Gdm+/Cl- solutions. The data presented here does not provide for an estimate of the relative contribution of denaturation mechanisms, but given that solvation water structure plays such an important role in protein folding, it is difficult for us to envision guanidinium water structuring being a negligible component to the overall denaturation phenomenon.
Conclusions
We have presented here OH stretch spectra of the water present in several different concentrations of Gdm+/Cl- solutions. We showed that the spectra, and therefore the water H-bond network, are altered by Gdm+. The TEIR solution spectra show that the Gdm+ cation causes an increase in strong, linear H-bonds. We performed quantum chemical calculations that indicated a subpopulation of water OH oscillators in the presence of guanidinium, causing the peculiar spectral features we observed. Finally, we have indicated how these results could play a role in destabilization and denaturation of folded proteins by guanidine salts.
Supplementary Material
Full Z-Matrix specification of the quantum chemical systems presented in this article is available free of charge via the Internet at http://pubs.acs.org.
Acknowledgments
This research was supported by National Institute of Health grants P01 48130 and F31NS53399.
References
- 1.Svedberg T. Nature (London, U K) 1937;139:1051–62. [Google Scholar]
- 2.Pace CN. Methods Enzymol. 1986;131:266–80. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 3.Greenstein JP. J Biol Chem. 1938;125:501–13. [Google Scholar]
- 4.Neurath H, Cooper GR, Erickson JO. J Biol Chem. 1942;142:249–63. [Google Scholar]
- 5.Schellman JA. Biophys Chem. 2002;96:91–101. doi: 10.1016/s0301-4622(02)00009-1. [DOI] [PubMed] [Google Scholar]
- 6.Pace CN, Grimsley GR, Scholtz JM. Protein Folding Handbook. Vol. 1. 2005. pp. 45–69. [Google Scholar]
- 7.Tanford C. Adv Protein Chem. 1970;24:1–95. [PubMed] [Google Scholar]
- 8.Mason PE, Neilson GW, Dempsey CE, Barnes AC, Cruickshank JM. Proc Natl Acad Sci U S A. 2003;100:4557–61. doi: 10.1073/pnas.0735920100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson DR, Jencks WP. J Am Chem Soc. 1965;87:2462–70. doi: 10.1021/ja01089a028. [DOI] [PubMed] [Google Scholar]
- 10.Schellman JA. Biopolymers. 1987;26:549–59. doi: 10.1002/bip.360260408. [DOI] [PubMed] [Google Scholar]
- 11.Timasheff SN. Biochemistry. 1992;31:9857–64. doi: 10.1021/bi00156a001. [DOI] [PubMed] [Google Scholar]
- 12.O'Brien EP, Dima RI, Brooks B, Thirumalai D. J Am Chem Soc. 2007;129:7346–53. doi: 10.1021/ja069232+. [DOI] [PubMed] [Google Scholar]
- 13.Roseman M, Jencks WP. J Am Chem Soc. 1975;97:631–40. [Google Scholar]
- 14.Mountain RD, Thirumalai D. J Phys Chem B. 2004;108:19711–6. [Google Scholar]
- 15.Batchelor JD, Olteanu A, Tripathy A, Pielak GJ. J Am Chem Soc. 2004;126:1958–61. doi: 10.1021/ja039335h. [DOI] [PubMed] [Google Scholar]
- 16.Mason PE, Neilson GW, Enderby JE, Saboungi ML, Dempsey CE, MacKerell AD, Jr, Brady JW. J Am Chem Soc. 2004;126:11462–70. doi: 10.1021/ja040034x. [DOI] [PubMed] [Google Scholar]
- 17.Shimizu A, Fumino K, Yukiyasu K, Taniguchi Y. J Mol Liq. 2000;85:269–78. [Google Scholar]
- 18.Vanzi F, Madan B, Sharp K. J Am Chem Soc. 1998;120:10748–53. [Google Scholar]
- 19.Schiffer CA, Dotsch V. Curr Opin Biotechnol. 1996;7:428–32. doi: 10.1016/s0958-1669(96)80119-4. [DOI] [PubMed] [Google Scholar]
- 20.Wetlaufer DB, Malik SK, Stoller L, Coffin RL. J Am Chem Soc. 1964;86:508–14. [Google Scholar]
- 21.Rupley JA. J Phys Chem. 1964;68:2002–3. [Google Scholar]
- 22.Alonso DOV, Dill KA. Biochemistry. 1991;30:5974–85. doi: 10.1021/bi00238a023. [DOI] [PubMed] [Google Scholar]
- 23.Breslow R, Guo T. Proc Natl Acad Sci U S A. 1990;87:167–9. doi: 10.1073/pnas.87.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Imamura K, Sakaura K, Ohyama Ki, Fukushima A, Imanaka H, Sakiyama T, Nakanishi K. J Phys Chem B. 2006;110:15094–9. doi: 10.1021/jp057527o. [DOI] [PubMed] [Google Scholar]
- 25.Dashnau JL, Nucci NV, Sharp KA, Vanderkooi JM. J Phys Chem B. 2006;110:13670–7. doi: 10.1021/jp0618680. [DOI] [PubMed] [Google Scholar]
- 26.Nucci NV, Vanderkooi JM. J Phys Chem B. 2005;109:18301–9. doi: 10.1021/jp051068+. [DOI] [PubMed] [Google Scholar]
- 27.Vanderkooi JM, Dashnau JL, Zelent B. Biochim Biophys Acta, Proteins Proteomics. 2005;1749:214–33. doi: 10.1016/j.bbapap.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Rozenberg M, Shoham G, Reva I, Fausto R. Spectrochim Acta, Part A. 2004;60A:463–70. doi: 10.1016/s1386-1425(03)00251-8. [DOI] [PubMed] [Google Scholar]
- 29.Dashnau JL, Conlin LK, Nelson HCM, Vanderkooi JM. Biochim Biophys Acta. 2007 doi: 10.1016/j.bbagen.2007.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Frisch, M. J. T., G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Bakken, V.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C.; and Pople, J. A. Gaussian 03, Revision D.01; Gaussian, Inc.: Wallingford, CT, 2004.
- 31.Becke AD. J Chem Phys. 1992;96:2155–60. [Google Scholar]
- 32.Parr RG, Yang W. Density-functional Theory of Atoms and Molecules. 1989. [Google Scholar]
- 33.McLean AD, Chandler GS. J Chem Phys. 1980;72:5639–48. [Google Scholar]
- 34.Krishnan R, Binkley JS, Seeger R, Pople JA. J Chem Phys. 1980;72:650–4. [Google Scholar]
- 35.Boese AD, Martin JML, Klopper W. J Phys Chem A. 2007;111:11122–33. doi: 10.1021/jp072431a. [DOI] [PubMed] [Google Scholar]
- 36.Inada Y, Orita H. J Comput Chem. 2007;29:225–32. doi: 10.1002/jcc.20782. [DOI] [PubMed] [Google Scholar]
- 37.Lee JS. J Chem Phys. 2007;127:085104/1-/5. doi: 10.1063/1.2761881. [DOI] [PubMed] [Google Scholar]
- 38.Riley KE, Hobza P. J Phys Chem A. 2007;111:8257–63. doi: 10.1021/jp073358r. [DOI] [PubMed] [Google Scholar]
- 39.Santra B, Michaelides A, Scheffler M. J Chem Phys. 2007;127:184104/1-/9. doi: 10.1063/1.2790009. [DOI] [PubMed] [Google Scholar]
- 40.Rives AB, Weinhold F. International Journal of Quantum Chemistry, Quantum Chemistry Symposium. 1980;14:201–9. [Google Scholar]
- 41.Silvestrelli PL, Parrinello M. J Chem Phys. 1999;111:3572–80. [Google Scholar]
- 42.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33–8. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 43.Stone J. An Efficient Library for Parallel Ray Tracing and Animation. Computer Science Department, University of Missouri-Rolla; 1998. [Google Scholar]
- 44.Iwata T, Koshoubu J, Jin C, Okubo Y. Appl Spectrosc. 1997;51:1269–75. [Google Scholar]
- 45.Walrafen GE. J Chem Phys. 1967;47:114–26. [Google Scholar]
- 46.Senior WA, Verrall RE. J Phys Chem. 1969;73:4242–9. [Google Scholar]
- 47.Palamarev H, Georgiev G. Vib Spectrosc. 1994;7:255–64. [Google Scholar]
- 48.Falk M, Ford TA. Can J Chem. 1966;44:1699–707. [Google Scholar]
- 49.Murphy WF, Bernstein HJ. J Phys Chem. 1972;76:1147–52. [Google Scholar]
- 50.Wang Z, Pakoulev A, Pang Y, Dlott DD. J Phys Chem A. 2004;108:9054–63. [Google Scholar]
- 51.Sharp KA, Madan B, Manas E, Vanderkooi JM. J Chem Phys. 2001;114:1791–6. [Google Scholar]
- 52.Laenen R, Rauscher C, Laubereau A. Phys Rev Lett. 1998;80:2622–5. [Google Scholar]
- 53.Wang Z, Pakoulev A, Pang Y, Dlott DD. Chem Phys Lett. 2003;378:281–8. [Google Scholar]
- 54.Woutersen S, Emmerichs U, Bakker HJ. Science (Washington, D C) 1997;278:658–60. [Google Scholar]
- 55.Gallagher KR, Sharp KA. J Am Chem Soc. 2003;125:9853–60. doi: 10.1021/ja029796n. [DOI] [PubMed] [Google Scholar]
- 56.Kumar R, Schmidt JR, Skinner JL. J Chem Phys. 2007;126:204107/1-/12. doi: 10.1063/1.2742385. [DOI] [PubMed] [Google Scholar]
- 57.Caballero-Herrera A, Nordstrand K, Berndt KD, Nilsson L. Biophys J. 2005;89:842–57. doi: 10.1529/biophysj.105.061978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Das A, Mukhopadhyay C. J Phys Chem B. 2008;112:7903–8. doi: 10.1021/jp800370e. [DOI] [PubMed] [Google Scholar]
- 59.Stumpe MC, Grubmueller H. J Am Chem Soc. 2007;129:16126–31. doi: 10.1021/ja076216j. [DOI] [PubMed] [Google Scholar]
- 60.Kuharski RA, Rossky PJ. J Am Chem Soc. 1984;106:5786–93. [Google Scholar]
- 61.Kokubo H, Rosgen J, Bolen DW, Pettitt BM. Biophys J. 2007;93:3392–407. doi: 10.1529/biophysj.107.114181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hayashi Y, Katsumoto Y, Omori S, Kishii N, Yasuda A. J Phys Chem B. 2007;111:1076–80. doi: 10.1021/jp065291y. [DOI] [PubMed] [Google Scholar]
- 63.Rezus YLA, Bakker HJ. Proc Natl Acad Sci U S A. 2006;103:18417–20. doi: 10.1073/pnas.0606538103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hermida-Ramon JM, Oehrn A, Karlstroem G. J Phys Chem B. 2007;111:11511–5. doi: 10.1021/jp073579x. [DOI] [PubMed] [Google Scholar]
- 65.Weinhold F, Robert LB, David B. Advances in Protein Chemistry. Vol. 72. Academic Press; 2005. Resonance Character of Hydrogen-bonding Interactions in Water and Other H-bonded Species; pp. 121–55. [DOI] [PubMed] [Google Scholar]
- 66.Wiggins PM. Physica A (Amsterdam) 1997;238:113–28. [Google Scholar]
- 67.Levy Y, Onuchic JN. Annu Rev Biophys Biomol Struct. 2006;35:389–415. doi: 10.1146/annurev.biophys.35.040405.102134. [DOI] [PubMed] [Google Scholar]
- 68.Sorin EJ, Rhee YM, Shirts MR, Pande VS. J Mol Biol. 2006;356:248–56. doi: 10.1016/j.jmb.2005.11.058. [DOI] [PubMed] [Google Scholar]
- 69.Creighton TE. Biopolymers. 1983;22:49–58. doi: 10.1002/bip.360220110. [DOI] [PubMed] [Google Scholar]
- 70.Nozaki Y, Tanford C. J Biol Chem. 1970;245:1648–52. [PubMed] [Google Scholar]
- 71.Venkatesu P, Lee MJ, Lin Hm. J Phys Chem B. 2007;111:9045–56. doi: 10.1021/jp0701901. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full Z-Matrix specification of the quantum chemical systems presented in this article is available free of charge via the Internet at http://pubs.acs.org.