

Abstract
A library of ∼51,000 compounds was interrogated by high throughput screening (HTS) using a heparin-induced tau fibrillization assay. HTS was conducted with bacterially expressed recombinant tau fragment K18 and the reaction was monitored by thioflavine T fluorescence. Hits meeting criteria set for selection in HTS were further evaluated in a panel of assays designed (a) to confirm the initial results and (b) to identify possible false positives arising from non-specific mechanisms or assay dependent artifacts. Two 2,3-di(furan-2-yl)-quinoxalines were confirmed as inhibitors of tau fibrillization with IC50s in the low micromolar range (l–3 μM). Among false positives hits, members of the pyrimidotriazines, benzofurans, porphyrins and anthraquinones, inhibited tau fibrillization by generating peroxides via catalytic redox cycles due to the reducing agent dithiothreitol (DTT) in the assay. This study delineates focused strategies for HTS of tau fibrillization inhibitors that are relevant to drug discovery for Alzheimer's disease and related tauopathies.
Introduction
Despite the fact that the precise mechanisms underlying neurodegenerative diseases remain to be fully elucidated, a growing body of evidence clearly indicates that protein misfolding, fibrillization, and aggregation can produce detrimental effects through (a) toxic effects directly mediated by the aggregates; and/or through (b) the loss of the normal function of the sequestered proteins. [1] As such, protein misfolding, fibrillization, and aggregation have emerged as new potential targets for therapeutic intervention. [2] One important example of pathologically relevant aggregates are the neurofibrillary tangles (NFTs) made of paired helical filaments (PHFs), which are aggregated forms of hyperphosphorylated protein tau. NFTs comprise, together with A-β amyloid plaques, the defining lesions of Alzheimer's disease (AD), and NFTs also are pathological hallmarks of other neurodegenerative diseases known as tauopathies. Importantly, the aggregation of tau fibrils to form tangles correlates with the degree of cognitive impairment in AD.
The tau protein is a microtubule-associated protein (MAP) particularly abundant in axons of neurons. The normal tau function is to stabilize the microtubules (MTs) thereby modulating the plasticity of the cytoskeleton. Since the MT-network is a key component of axonal transport, changes in the MT-dynamics caused by a loss of tau function can have profound effects on the transport of protein and other cargo to and from the cell body of neurons. [3] In AD, upon hyperphosphorylation tau becomes sequestered into NFTs thereby losing its MT-stabilizing function. The consequence of this process is the disruption of axonal transport, which ultimately leads to neurodegeneration. For this reason, tau is now recognized as an important therapeutic target for potential new treatments of AD and related tauopathies. Indeed efforts have been made to identify agents that could prevent tau aggregation and/or promote the dissolution of aggregates. [4-7] One of the major obstacles encountered in the study of tau fibrillization was the difficulty in effectively generating tau fibrils in vitro. However, the discovery that anionic cofactors (i.e. heparin, arachidonic acid) efficiently induce the fibrillization process, and the observation that different truncated forms of tau are more prone to fibrillization compared to the full-length protein, enabled the development of in vitro assays amenable to high-throughput screening (HTS) of large compound libraries. This resulted in the identification of some structural classes of compounds including antraquinones, [6] polyphenols [7] and phenothiazines [4,7] that were found to inhibit tau fibrillization. However, despite these findings, clinical candidates have not yet been identified, and further screening as well as additional assay development, are clearly needed. In fact, the tau fibrillization assays reported thus far differ in important ways, such as the tau isoforms or anionic cofactors employed, or concentrations of reducing agents in the reaction mixture etc. The significance of such differences are not yet known and it is unclear which of the reported tau fibrillization assays best reproduces the in vivo situation. However, these differences presumably account for the fact that some compounds (e.g. daunorubicin), generated opposite results in different assays. In the present study, we report HTS of a library of ∼51,000 compounds in a heparin-induced tau fibrillization assay.
Materials and Methods
Fibrillization assay
The fibrillization assay adopted here was modified from the one reported by Mandelkow et al [6] with the main difference that recombinant myc tagged truncated tau fragment K18 was employed rather than K19. The myc tag was added in order to evaluate antibody based assays, such as the DELFIA and Alpha Screen, in comparison with the thioflavine T (ThT) fluorescence assay. Since the ThT assay proved to be generally simpler and more reproducible than the antibody based assays, particularly under automated HTS conditions (Z′=0.85; Cv=4.4%), we selected it as the assay of choice for the primary screening. The total volume of the reaction mixture was 25 μl, which included 20 μM myc tagged K18 (1:1: N-terminal myc:C-terminal myc tag), 20 μM heparin, 2 mM DTT in 100 mM sodium acetate pH 7.0. After 18 h of incubation at 37°C, an addition of 25 μl of a 25 μM solution of ThT was made and incubation continued for 1h at room temperature prior to fluorescence reading. (Figure 1). The non-fibrillizing mis-sense K18 mutant obtained by substituting lysine 311 with aspartate (K18-K311D), was used as negative control, while K18 in DMSO provided the positive control. The criteria set for selection in the primary assay was 40% inhibition.
Figure 1.

Time course for tau fibrillization on a 384 well plate format. ThT assay was performed in triplicate and expressed as a percentage of the 18 h time point used in the screen.
Secondary assays
Sedimentation of the ThT reaction was performed at 186,000 g for 30 mins through a 25% sucrose cushion and the supernatant removed from the pellet. Pellets were resuspended in a volume equal to the supernatant and equal amounts of supernatants and pellet were analysed by SDS PAGE on a 12.5% acrylamide gel. N-terminal and C-terminal myc tagged K18 migrated slightly differently and showed up as a double band. Negative staining with uranyl acetate followed by electron microscopy (EM) was performed on the resuspended sedimentation pellets (Figure 2A.).
Figure 2.
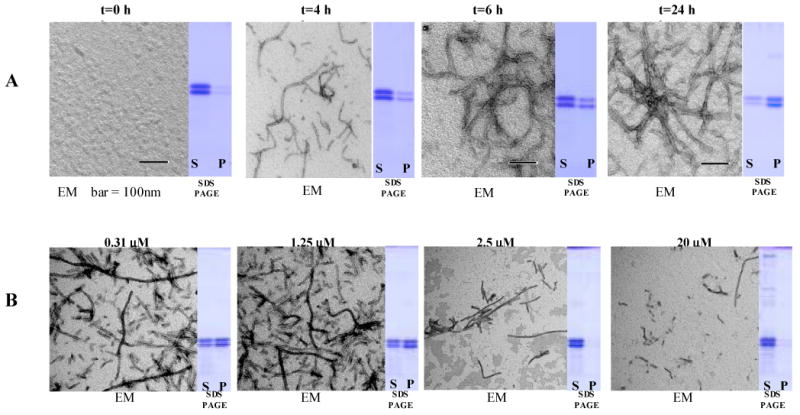
A Sedimentation assay and EM examination of the pellets: (A) positive control (K18 + DMSO) at different time points; (B) after 18 h incubation at different concentrations of 113F08. N-terminal and C-terminal myc tagged K18 migrate slightly differently and give rise to double bands. EM = electron microscopy; S = supernatant; P = pellets.
Compound library
The chemical library screened in this study was designed to maximize structural diversity within chemical space while favoring the representation of “drug-like” structures. The library consisted of 1120 off patent FDA-approved compounds from the Prestwick Chemical Library™ (Prestwick Chemical, France), 240 pure natural product compounds from the Greenpharma Natural Compound Library (Prestwick Chemical, France), a combinatorial library of 30,000 compounds probing 125 different scaffolds from the NOVACore library, (Chembridge, San Diego, California) and 21,000 compounds maximizing sampling of chemical space from the DIVERSet™ library, (Chembridge, San Diego California).
Results and Discussion
Screening
Upon completion of the primary screening, seventy-one compounds (∼1/700) met or exceeded the selection criteria of 40% inhibition by HTS. These compounds were further evaluated through: (a) retest of the primary screening and (b) sedimentation assay followed by examination of the pellets by EM (Figure 2B). Out of the initial set of hits, eleven compounds were confirmed to inhibit the fibrils formation in both primary and secondary assays (see Table 1) representing eight compound classes. Five of the eight classes had been previously published: sulphonated dyes, phenothiazines, anthraquinones, benzofurans and porhyrins were among the classes producing hits in our assay. New compound classes included several quinoxalines and pyrimidotriazines, and a depsidone.
Table 1.
Hits and false positives obtained from the screening.
| Cpd Class | Cpd Name | Structure | % Inhib. (ThT)a | Sed. Assayb | % Inhib. (ThT) No DTT | Sed. Assay No DTT | IC50 (μM) |
|---|---|---|---|---|---|---|---|
| Antraquinones | 31G03 | 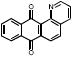 |
76 | ++ | 39 | - | 0.63 |
| Quinoxalines | 113F08 | 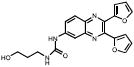 |
69 | ++ | 70 | +++ | 2.4 |
| 330B06 | 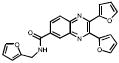 |
27 | +++ | 85 | ++ | 3.1 | |
| Pyrimidotriazines | 300C04 |  |
94 | +++ | 20 | - | 0.36 |
| Sulfonated Dyes | 6C06 | 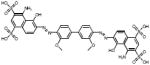 |
102 | +++ | 51 | - | 3.2 |
| Depsidones | 15B02 | 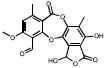 |
98 | +++ | 100 | +++ | 1.6 |
| Porphyrins | 06D04 | 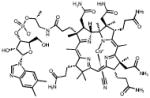 |
75 | +++ | -2.6 | nd | 4.5 |
| Phenothiazines | 30H07 | 100 | +++ | 99 | nd | 0.29 | |
| Benzofuran | 348E10 | 84 | +++ | 9.8 | nd | 0.61 |
Percentage inhibition of K18 fibrillization followed by ThT assay;
Sedimentation assay data: “+++” = 100% inhibition, “-“ = 0% inhibition, IC50 determined in the presence of DTT. Additional hits from the pyrimidotriazines, 17H03 and 93D02, behaved similarly to 300C04 and are not shown.
Among the compounds that successfully passed all assays, a sufonated dye (06C06), a phenothiazine (30H07) and stictic acid (15B02), a natural product extracted from lichens, were both given lower priority either due to concerns over their chemical reactivity and stability (15B02), or because the class of compounds had been already studied (sufonated dyes, and phenothiazines).
Testing for False Positives
Next we turned our attention towards identifying possible false positives. In order to eliminate possible false positives that could inhibit tau fibirillzation through a non specific mechanism involving the generation of peroxides, all hits were evaluated for their ability to inhibit tau fibril formation in the absence of DTT, which is known to participate in catalytic redox cycles with specific classes of compounds, with rapid generation of peroxides. [11] Seven compounds lost activity in the absence of DTT representing 5 compound classes (Table 1). The anthraquinone (31G03), porphyrin (6D04), benzofuran (348E10), and all three pyrimidotriazines (17H03, 93D02, and 300C04) showed dependence on the presence of DTT.
We tested several published representatives of the anthraquinones, and porphyrins for dependence on the presence of DTT (daunorubicin, doxorubicin, hemin, and protoporphyrin IX). As opposed to the compounds in these classes obtained from our screen, none of these representative compounds showed DTT sensitivity, highlighting the need to be mindful of individual chemistries within any compound class.
Since non-specific binding of aggregates of small molecules onto proteins, is often the source of false positives in high-throughput screenings, [9,10] dose response curves were generated at different concentrations of K18 for the remaining 2,3-di(furan-2yl)-quinoxalines (Figure 3). It is expected that IC50s of compounds that exhibit inhibitory activity through the formation of aggregates would be independent from protein concentration. For both of the compounds in this class the IC50 varied with protein concentration, indicating that non-specific inhibition due to a compound aggregation mechanism of inhibition is not occurring.
Figure 3.

Dose dependence of quinoxalines at different concentrations of K18. ThT assay result was expressed as a percent inhibition relative to controls containing DMSO alone. Suffix following compound name indicates concentration of K18 in micromolar.
The remaining compounds shared an identical 2,3-di(furan-2yl)-quinoxaline structure (113F08 and 330B06), exhibited IC50s in the 1–3 μM range. Interestingly, examination of the compound library revealed ∼200 entries containing the quinoxaline substructure, most of them, however, completely inactive at 10uM. This would suggest the presence of a structure-activity relationship, yet resolving the differences between test compounds that exhibit moderate or low activities, proved to be challenging in the current assay. In the course of our testing we found the activity to be dependent on the presence of the 2,3 di(furan-2yl) moieties on the quinoxoline scaffold. Of the other 2,3 di(furan-2yl) quinoxalines with low activity we found 6 additional compounds which show dose dependent activity. The 2,3-di(furan-2yl)-quinoxalines represent the first examples of a new scaffold exhibiting inhibition of tau fibrils formation in vitro and as such, they may be useful in further defining the structural requirements for inhibition of fibril formation.
In conclusion, this study delineates analytical strategies for the interrogation of compound libraries by HTS to identify tau fibrillization inhibitors that may become candidates for further structure function and in vivo studies in drug discovery efforts to develop better treatments for Alzheimer's disease and related tauopathies.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med. 2004;10(Suppl):S10–7. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- 2.Skovronsky DM, Lee VMY, Trojanowski JQ. NEURODEGENERATIVE DISEASES: New Concepts of Pathogenesis and Their Therapeutic Implications. Annual Review of Pathology: Mechanisms of Disease. 2006;1(1):151–170. doi: 10.1146/annurev.pathol.1.110304.100113. [DOI] [PubMed] [Google Scholar]
- 3.Trojanowski JQ, Smith AB, Huryn D, Lee VMY. Microtubule-stabilizing drugs for therapy of Alzheimer's disease and other neurodegenerative disorders with axonal transport impairments. Expert Opin Pharmacother. 2005;6(5):683–6. doi: 10.1517/14656566.6.5.683. [DOI] [PubMed] [Google Scholar]
- 4.Wischik CM, Edwards PC, Lai RYK, Roth M, Harrington CR. Selective inhibition of Alzheimer disease-like tau aggregation by† phenothiazines. PNAS. 1996;93(20):11213–11218. doi: 10.1073/pnas.93.20.11213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chirita C, Necula M, Kuret J. Ligand-dependent inhibition and reversal of tau filament formation. Biochemistry. 2004;43(10):2879–87. doi: 10.1021/bi036094h. [DOI] [PubMed] [Google Scholar]
- 6.Pickhardt M, Gazova Z, von Bergen M, Khlistunova I, Wang Y, Hascher A, Mandelkow EM, Biernat J, Mandelkow E. Anthraquinones inhibit tau aggregation and dissolve Alzheimer's paired helical filaments in vitro and in cells. J Biol Chem. 2005;280(5):3628–35. doi: 10.1074/jbc.M410984200. [DOI] [PubMed] [Google Scholar]
- 7.Taniguchi S, Suzuki N, Masuda M, Hisanaga S, Iwatsubo T, Goedert M, Hasegawa M. Inhibition of heparin-induced tau filament formation by phenothiazines, polyphenols, and porphyrins. J Biol Chem. 2005;280(9):7614–23. doi: 10.1074/jbc.M408714200. [DOI] [PubMed] [Google Scholar]
- 8.Arkin MR, Wells JA. Small-molecule inhibitors of protein-protein interactions: progressing towards the dream. Nat Rev Drug Discov. 2004;3(4):301–17. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]
- 9.McGovern SL, Caselli E, Grigorieff N, Shoichet BK. A common mechanism underlying promiscuous inhibitors from virtual and high-throughput screening. J Med Chem. 2002;45(8):1712–22. doi: 10.1021/jm010533y. [DOI] [PubMed] [Google Scholar]
- 10.McGovern SL, Helfand BT, Feng B, Shoichet BK. A specific mechanism of nonspecific inhibition. J Med Chem. 2003;46(20):4265–72. doi: 10.1021/jm030266r. [DOI] [PubMed] [Google Scholar]
- 11.Guertin KR, Setti L, Qi L, Dunsdon RM, Dymock BW, Jones PS, Overton H, Taylor M, Williams G, Sergi JA, Wang K, Peng Y, Renzetti M, Boyce R, Falcioni F, Garippa R, Olivier AR. Identification of a novel class of orally active pyrimido[5,4-3][1,2,4]triazine-5,7-diamine-based hypoglycemic agents with protein tyrosine phosphatase inhibitory activity. Bioorg Med Chem Lett. 2003;13(17):2895–8. doi: 10.1016/s0960-894x(03)00623-1. [DOI] [PubMed] [Google Scholar]