

Abstract
Tumor necrosis factor-α (TNFα) is a proinflammatory cytokine that is elevated in Alzheimer's disease (AD) brains. Because TNFα is released from cell membranes by the TNFα-converting enzyme (TACE), inhibition of TACE has the potential to mitigate TNFα effects in AD brain. TACE also cleaves amyloid precursor protein (APP) and generates sAPPα, precluding the formation of potentially harmful amyloid β (Aβ) peptides by β-site APP cleaving enzymes (BACE). Hence, the anti-inflammatory benefits of TACE inhibition might be offset by an increase in Aβ. We have examined the effects of the highly selective TACE inhibitor, BMS-561392, on APP processing in vitro and in vivo. In Chinese hamster ovary cells expressing APP, BMS-561392 significantly reduced secretion of sAPPα without a corresponding increase in Aβ production. Conversely, a BACE inhibitor decreased sAPPβ and Aβ peptides with no change in the secretion of sAPPα. These data indicate an absence of TACE and BACE competition for the APP substrate. Despite this, we observed competition for APP when TACE activity was enhanced via phorbol ester treatment or if APP was modified such that it was retained within the trans-Golgi network (TGN). These results suggest that BACE and TACE share a common TGN localization, but under normal conditions do not compete for APP. To confirm this finding in vivo, BMS-561392 was infused into the brains of Tg2576 and wild-type mice. Although decreased brain sAPPα levels were observed, steady-state Aβ levels were not significantly changed. Accordingly, it is possible that TACE inhibitors could reduce TNFα levels without increasing Aβ levels within the AD brain.
Keywords: Alzheimer's disease, amyloid-β, inflammation, neuroinflammation, tumor necrosis factor, TNFα
Introduction
Signs of inflammation are evident in Alzheimer's disease (AD) brain (Akiyama et al., 2000), and studies have suggested that chronic inflammation in the brain can exacerbate the disease progression (Sastre et al., 2006). Accordingly, anti-inflammatory drugs, including nonsteroidal anti-inflammatory drugs (NSAIDs), may reduce the severity of the disease (Heneka et al., 2005).
Tumor necrosis factor-α (TNFα) is a proinflammatory cytokine that is upregulated in AD and other CNS diseases (Mogi et al., 1994). In cell-based models, it can increase the production of amyloid β (Aβ), a hallmark peptide found in AD brains (Blasko et al., 1999), and transgenic mice over-expressing TNFα or its receptors in the brain develop chronic inflammation and neurodegeneration (Stalder et al., 1998). Finally, a single nucleotide polymorphism in the TNF gene has been linked to late-onset AD (McGeer and McGeer, 2001; Ramos et al., 2006).
TNFα is proteolytically released from its membrane-bound form (proTNFα) by the ADAM (a disintegrin and metalloproteinase) family proteinases, TNFα converting enzyme (TACE) (Itai et al., 2001). Although pharmacological inhibition of TACE may have therapeutic potential in AD brain by suppressing TNFα-mediated inflammation, it should be noted that TACE is also responsible for the α-secretase cleavage of amyloid precursor protein (APP) (Buxbaum et al., 1998; Lammich et al., 1999). TACE cleaves APP within the Aβ peptide domain, precluding the generation of this amyloidogenic peptide (De Strooper and Annaert, 2000). Therefore, TACE inhibition could potentially increase the availability of APP for cleavage by the β-secretase enzymes (the β-site APP cleaving enzymes BACE 1 and 2) that are involved in Aβ production (Dominguez et al., 2005). This could lead to an unwanted increase in Aβ production that might offset any benefit resulting from a decrease of TNFα in AD.
It is unclear whether α- and β-secretases compete for APP cleavage because conflicting data have been reported. Although some studies showed that α- and β-secretase cleavage of APP are coupled (Buxbaum et al., 1998; Gabuzda et al., 1993; Hung et al., 1993; Jacobsen et al., 1994; Skovronsky et al., 2000; Wolf et al., 1995), others demonstrated that modifying one secretase activity does not affect APP cleavage by the other (Dyrks et al., 1994; LeBlanc et al., 1998; Blacker et al., 2002; Gandhi et al., 2004). However, these previous studies failed to distinguish the proteolytic activity of TACE from other candidate α-secretases, such as ADAM-10 and ADAM-9 (Lammich et al., 1999; Koike et al., 1999), because of the lack of a specific inhibitor.
BMS-561392 is a novel, potent and highly selective TACE inhibitor (supplemental Fig. 1, Table 1, available at www.jneurosci.org as supplemental material) that effectively suppresses excessive TNFα secretion. In this study, we have characterized the effects of BMS-561392 on TACE and BACE cleavage of APP in vitro. We also infused the compound into the brain of human APP-expressing Tg2576 and wild-type mice to examine its effects on APP processing in vivo. Finally, we have addressed the question of whether TACE and BACE compete for APP substrate. Our data support the notion that TACE and BACE share the same pool of APP, but do not normally compete for APP substrate.
Materials and Methods
Cell culture and drug treatment.
Chinese hamster ovary (CHO) cells stably transfected with the wild-type 695- amino acid isoform of APP (APPwt) and a familial AD-associated APP Swedish mutant (APPswe) (Forman et al., 1997) as well as APP-trans-Golgi network (APP-TGN) (Chyung et al., 1997; Skovronsky et al., 2000; Huse et al., 2002) were grown in α-MEM (Invitrogen) containing 5% fetal bovine serum, penicillin/streptomycin, and 50 μg/ml G418 according to standard protocols. Cortices from neonatal Tg2576 mouse brains were isolated and incubated in 0.1% trypsin/HBSS/0.5 mm EDTA without Ca2+ or Mg2+ (Invitrogen), and cells were mechanically dissociated using a fire-polished pipette. Cells were plated in Dulbecco's minimum essential medium (DMEM) plus 10% FBS in poly-d-lysine-coated six-well plates at a density of 0.5 × 106 cells/well. Three hours after plating, medium was replaced with DMEM plus B27 supplements (Invitrogen) to promote neuronal survival and inhibit growth of non-neuronal cells. Neurons were used for experiments after 7 d in vitro. CHO-pro-TNFα cells were generated by transfection of pro-TNFα vector with alkaline phosphatase tagged at its C-terminal (a gift of Dr. Carl Blobel, Hospital for Special Surgery, New York, NY) (Chesneau et al., 2003) into CHO-Pro5, using Nucleofector (Amaxa). After subcloning using Zeocin as a selection agent, they were grown under the same conditions as other CHO cells. CHO cells were plated in six-well plates at a density of 6.75 × 105 cells/well for drug treatment. 24 h after plating, medium was replaced with BMS-561392 (Bristol-Myers Squibb), TAPI-I [(N-R-(2-hydroxyaminocarbonyl)methyl]-4-methylpentanoyl-l-naphthylanyl-l-alanine 2 aminoethyl amide-I; Peptide International), or the BACE inhibitor III (Merck; the molecular structure is proprietary information of Merck) in media containing 0.1% DMSO with or without 10 μm PMA (Phorbol 12-myristate 13-acetate; Sigma). Control wells received media with 0.1% DMSO only.
Osmotic pump implantation and drug treatment in vivo.
The protocol of mouse brain hippocampal microinfusion have been carefully reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at University of Pennsylvania. Briefly, Alzet pumps (type 2004, 100 μl, 0.25 μl/hr; Alzet Osmotic Pumps ) were filled with BMS-561392 at concentration of 8 mg/ml in the citric acid, pH 3.5. A broad-range matrix metalloprotease (MMP) inhibitor TAPI-I was prepared at the same molar concentration. The osmotic pumps then were kept in 0.9% saline at 37°C before implantation. Three groups of six to eight month old Tg2576 and wild-type mice (n = 5 per group) were used in this study. Mice were anesthetized by an intraperitoneal injection of ketamine hydrochloride (1 mg/10 g) and xylazine (0.1 mg/10 g), and canula was stereotactically inserted into the right lateral ventricle using sterotaxic coordinates (anterior–posterior, 0.5 mm; medial–lateral, 1.0 mm; dorsal–ventral, 2.7–3.0 mm; David Kopf Instrument). Instant dental cement was used to secure canula to skull. Osmotic pump was placed subcutaneously on the back of mice. After surgery, mice were housed individually and infusion lasted for 14 d.
The mice behaved normally during treatment. Cerebrospinal fluid (CSF) was collected while mice were under anesthesia, and Aβ40/42 levels were measured by sandwich ELISA. Mice were subsequently killed and perfused with PBS, and the cortex and hippocampus from the drug/vehicle-infused right hemisphere were dissected, immediately frozen in dry ice, and kept at −80°C for further processing.
Tg2576 mice were maintained on a C57B6/SJL F2 background by successive backcrossing to wild-type C57B6/SJL F1 females. All mice were generated and handled according to protocols approved by the University of Pennsylvania Institutional Animal Care and Use Committee guidelines.
Sample processing and Western blot analysis.
Cell lysates were collected in RIPA buffer (0.5% sodium deoxycholate, 0.1% SDS, 1% Nonidet P-40, 5 mm EDTA in TBS, pH 8.0) in the presence of protease inhibitors (1 μg/ml each of pepstatin A, leupeptin, l-1-tosylamido-2-phenylethyl chloromethyl ketone, 1-chloro-3-tosylamido-7-amino-2-heptanone, soybean trypsin inhibitor, and 0.5 mm phenylmethylsulfonyl fluoride) and briefly sonicated. Protease inhibitors were also added to conditioned media samples.
Frozen brains of Tg2576 mice were directly lysed in RIPA buffer (150 mg/ml) containing protease inhibitors, sonicated, and centrifuged at 100,000 × g for 20 min at 4°C. The supernatants were saved and used for Aβ sandwich ELISA analysis and Western blotting. To selectively extract Aβ, sAPPα, and sAPPβ, wild-type mouse brains were lysed in 0.2% DEA-containing 50 mm NaCl, homogenized using a pestle, and centrifuged at 100,000 × g for 1 h at 4°C (Miller et al., 2003). The supernatants were saved and neutralized by adding 1/10th volume of 0.5 m Tris-HCl pH 6.8 buffer and used for ELISA analysis and Western blotting of soluble APP cleavage products. The pellets were sonicated in RIPA buffer and centrifuged at 100,000 g for 1 h at 4°C. The supernatants were saved and used for Western blotting of flAPP and α-tubulin.
Cell and tissue lysates were electrophoresed on 7.5% Tris-glycine acrylamide gels for flAPP, sAPPα/β, and α-tubulin, and transferred to nitrocellulose. For CTFs (C-terminal fragments) immunoblotting, samples were immunoprecipitated with 5685, a rabbit polyclonal antibody raised against a C-terminal peptide fraction of APP (Lee et al., 2005), before electrophoresis on 16.5% Tris-tricine gels (Bio-Rad). Full-length APP and CTFs were probed with 5685. sAPPα was probed with 2B3, an end-specific sAPPα monoclonal antibody (IBL America). sAPPβ was specifically probed with C5A4/2, a rabbit polyclonal antibody raised against a synthetic peptide (CSEVKM) corresponding to the C terminus of sAPPβ (Lee et al., 2005). α-Tubulin was used as a loading control for general protein contents. Immunobands were detected with species-specific horseradish peroxidase-conjugated anti-IgG antibodies (Santa Cruz Biotechnology), and developed with enhanced chemiluminescence (ECL; PerkinElmer Life Sciences). ECL signal was visualized and quantified with a LAS-3000 imager and MultiGauge version 2.3 software (Fujifilm Life Science). Signal linearity was verified in a separate experiment (data not shown).
Sandwich ELISA analysis and alkaline phosphatase assay.
To measure Aβ1–40 and Aβ1–42 levels, both conditioned media and RIPA lysates were assayed with Aβ sandwich ELISAs as previously described (Lee et al., 2003). Briefly, BNT-77 (anti-Aβ11–28) was used to capture endogenous mouse Aβ1–40 and Aβ1–42 from wild-type mouse brains. Ban50 (anti-Aβ1–10) was used as a capturing antibody for measuring both Aβ1–40 and Aβ1–42 peptides for all other application. Horseradish peroxidase (HRP)-conjugated BA-27 and BC-05 were used to report Aβ species ending at position 40 and 42. For quantification of Aβ levels, synthetic Aβ1–40 and Aβ1–42 (Bachem Bioscience) were serially diluted in corresponding buffer or media to generate standard curves. LN27, a monoclonal antibody that binds to the N-terminal 200 amino acid residues of APP (Wertkin et al., 1993), was used as capturing antibody for measuring sAPPα in conditioned media. HRP-conjugated Ban50 was used to report sAPPα. C-terminal tagged alkaline phosphatase activity in conditioned media was used as a surrogate marker for TNFα secretion, using the p-Nitrophenyl phosphate liquid substrate system (Sigma).
Results
BMS-561392 is a potent inhibitor of α-cleavage of pro-TNFα and APP
To evaluate the efficacy of BMS-561392 in cell culture, different concentrations of the compound were applied to CHO cells expressing the precursor, membrane-bound form of TNFα (proTNFα). TAPI-I, a broad-range MMP/ADAM inhibitor, was also included for comparison. After 24 h of treatment, we measured the levels of soluble TNFα, the product of TACE cleavage, in culture media. Both inhibitors caused a dose-dependent inhibition of TNFα secretion (Fig. 1A), with BMS-561392 being more potent (50% inhibition with respect to DMSO controls was achieved with 0.15 μm BMS-561392 and 0.90 μm TAPI-I).
Figure 1.

BMS-561392 inhibits secretion of sAPPα and TNFα in a dose-dependent manner. CHO cells expressing human APP695wt and pro-TNFα were treated with BMS-561392 and TAPI-I for 24 h. A, Inhibition of TNFα secretion by TAPI-I and BMS-561392. Inhibition of 50% with respect to DMSO controls was achieved with 0.90 μm TAPI-I and 0.15 μm BMS-561392. B, Inhibition of sAPPα secretion by TAPI-I and BMS-561392 from CHO cells expressing APPwt and APPswe. Inhibition of 50% with respect to DMSO controls was achieved with 59.1 μm TAPI-I and 4.47 μm BMS-561392 on CHO-APPwt cells, and 0.23 μm BMS-561392 on CHO-APPswe cells. Data are expressed as the mean of three independent experiments ±SE measurements. *p < 0.01, two-tailed Student's t test; **, ***p < 0.0001, two-tailed Student's t test.
To test the effect of BMS-561392 on the processing of APP, another TACE substrate, CHO cells expressing either human wild-type APP (APPwt) or human APP with the previously described Swedish mutation (APPswe) (Citron et al., 1992) were treated with different concentrations of this compound for 24 h. Secretion of sAPPα was reduced in a dose-dependent manner by BMS-561392 as detected by a sAPPα sandwich ELISA (Fig. 1B). TAPI-I also caused a decrease in sAPPα secretion from CHO-APPwt cells, although with less potency relative to BMS-561392. No cell toxicity was observed with any of the treatment conditions. Interestingly, BMS-561392 inhibits sAPPα secretion more strongly in the CHO-APPswe cells than in CHO-APPwt cells, and although we are unsure why there is this differential effect, these data confirm that BMS-561392 is a potent inhibitor of TACE in cell culture.
BMS-561392 does not change the level of β-cleavage products of APP
Because APP is also cleaved by BACE, inhibition of TACE may increase the availability of the APP substrate for BACE cleavage, thereby enhancing the production of the β-secretase cleavage products, including sAPPβ, CTFβ and Aβ40/42. Unexpectedly, BMS-561392 did not change the level of APP β-cleavage products in CHO-APPwt cells, including CTFβ and Aβ40/42, whereas it significantly reduced sAPPα secretion and CTFα levels (Fig. 2A–C). This de-coupling between α- and β-cleavage of APP was also observed in CHO-APPswe cells (data not shown). BMS-561392 also inhibited sAPPα secretion from primary cortical neurons derived from Tg2576 mice, and similar to what was observed with CHO cells, Aβ40/42 secretion levels remained unchanged (Fig. 2D). These results suggest that TACE inhibition does not lead to greater APP processing by BACE.
Figure 2.

Inhibition of sAPPα by BMS-561392 does not coincide with an increase in β-cleavage products of APP. A, Immunoblot of full-length APP and α-tubulin in cell lysates, and sAPPα and sAPPβ in conditioned media, from CHO-APPwt cell. RIPA lysates (20 μg) and 20 μl of conditioned media were resolved in 7.5% Tris-glycine SDS-gels, blotted and probed with 5685 (fl-APP), Ban50 (sAPPα), and α-tubulin antibodies. Whereas the level of sAPPα was reduced by BMS-561392 in a dose-dependent manner, the sAPPβ level in the media remained unchanged. Consistent levels of fl-APP and tubulin confirmed a lack of BMS-561392 toxicity. B, Immunoblot and quantitation of APP C-terminal fragments (CTFs). RIPA lysates (500 μg) were immunoprecipitated with 5685 antibody, resolved in a 16.5% Tris-tricine SDS-gel, blotted and probed with the same antibody. Note that the levels of CTFβ, a pan-precursor protein of Aβ, remain unchanged after BMS-561392 treatment. Quantification was based on the means of three independent experiments ±SE measurements. +p < 0.01, two-tailed Student's t test; n.s.*, not significant, two-tailed Student's t test, p = 0.150; n.s.**, not significant, one-way ANOVA, p = 0.598. #Results are shown in arbitrary unit (DMSO treatment as 1.0). C, Aβ40 and 42 in conditioned media after BMS-561392 treatment. Quantification was based on the mean of six independent experiments ±SE measurements. n.s.*, Not significant, one-way ANOVA, p = 0.902; n.s.**, not significant, one-way ANOVA, p = 0.874. #Results are normalized to fl-APP levels. D, Reduction of sAPPα secretion with no concomitant change in Aβ secretion in primary neurons derived from Tg2576 mice cultured for 7 d with or without BMS-561392 for 24 h. RIPA lysates (20 μg) were in 7.5% Tris-glycine SDS-gels, blotted, and probed with 5685 (fl-APP), Ban50 (sAPPα), C5A4/2 (sAPPβ), and α-tubulin antibodies. Quantification was based on the means of three independent experiments ±SE measurements. n.s.*, Not significant, one-way ANOVA, p = 0.943; n.s.**, not significant, one-way ANOVA, p = 0.996. #Results are normalized to fl-APP levels.
BACE inhibition does not increase sAPPα secretion in APP-expressing CHO cells
Because TACE inhibition did not appear to affect BACE processing of APP, we asked whether conversely BACE inhibition might alter TACE cleavage of APP. To test this, we treated CHO695wt cells with increasing concentrations of the selective Merck BACE inhibitor III. As shown in Figure 3, the secretion of sAPPβ and Aβ40/42 were drastically reduced by this BACE inhibitor, whereas sAPPα secretion was unchanged. This result demonstrates that these enzymes do not compete with each other for the processing of APPwt.
Figure 3.
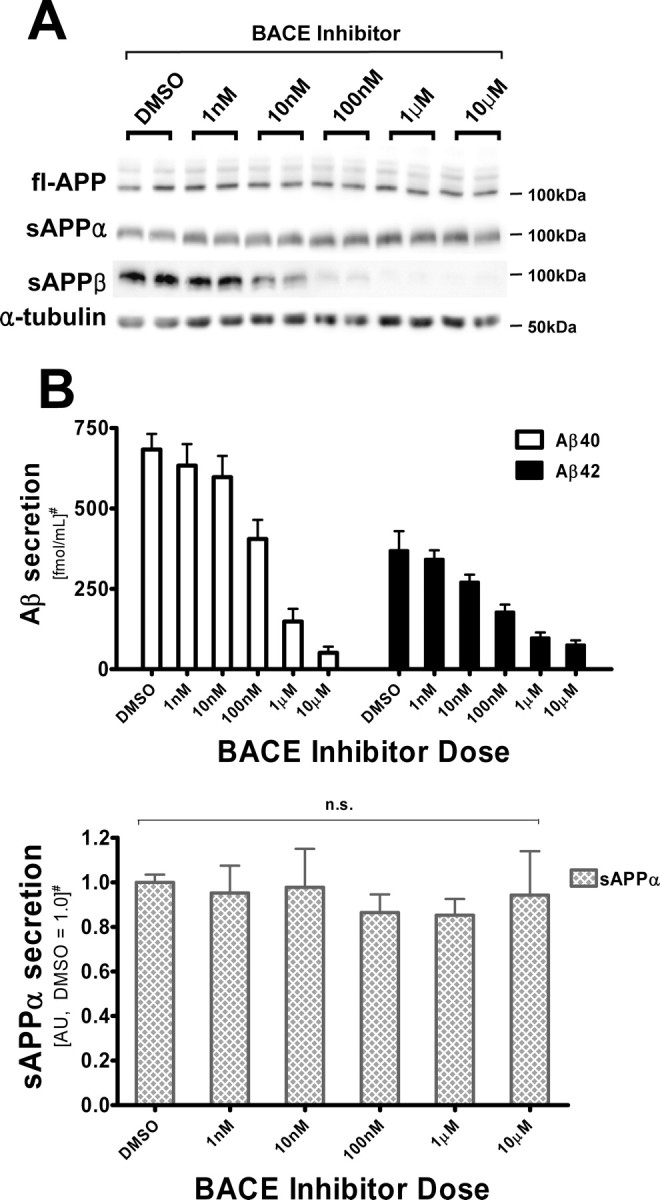
BACE inhibitor does not increase sAPPα secretion from CHO695wt cells. A, Immunoblot of full-length APP and α-tubulin in cell lysates, and sAPPα and sAPPβ in conditioned media, from CHO-APPwt cells. RIPA lysates (20 μg) and 20 μl of conditioned media were resolved in 7.5% Tris-glycine SDS-gels, blotted and probed with 5685 (fl-APP), Ban50 (sAPPα), and α-tubulin antibodies. B, Quantification of sAPPα, Aβ40, and Aβ42 levels in conditioned media by sandwich ELISAs. BACE inhibitor potently decreases the secretion of Aβ, but does not induce higher sAPPα secretion. Quantification was based on the means of three independent experiments ±SE measurements. n.s., Not significant, one-way ANOVA, p = 0.945. #Results are normalized to fl-APP levels. sAPPα levels are shown in arbitrary units (average of DMSO treatments as 1.0).
PMA-activated α-secretase activity results in decreased Aβ secretion that is normalized after BMS-561392 treatment
One explanation for the lack of coupling between TACE and BACE cleavage of APP is that the enzymes “see” two separate pools of substrate; therefore, manipulation of one pathway would not affect the other. However, this hypothesis is inconsistent with a previously published observation that TACE and BACE compete for APP substrate when TACE activity is potentiated by phorbol 12-myristate 13-acetate (PMA)-treatment (Skovronsky et al., 2000). As shown in Figure 4, we also observed a PMA-induced increase of α-secretase activity that resulted in enhanced sAPPα secretion with a concomitant decrease in Aβ40/42 secretion. BMS-561392 treatment in the PMA-activated condition effectively inhibited α-secretase and normalized Aβ40 and Aβ42 secretion to that seen in control cells.
Figure 4.

BMS-561392 inhibits PMA-activated α-secretase activity, enhancing Aβ secretion compared with PMA-only treatment. A, Immunoblot of full-length APP and α-tubulin in cell lysates, and sAPPα and sAPPβ in conditioned media, from CHO-APPwt cells. B, Quantitation of sAPPα level in conditioned media, measured by densitometry of a 2B3 Western blot. C, Aβ40 and Aβ42 levels in conditioned media. PMA activates α-secretase activity and reduces Aβ secretion, and BMS-561392 reverses the effect of PMA on CHO695wt cells. Quantification based on the means of three independent experiments ±SE measurements. 1p < 0.05 ∼ 6p < 0.05; two-tailed Student's t test. #Results are normalized to fl-APP levels. sAPPα levels are shown in arbitrary units (average of DMSO only treatments as 1.0).
The results obtained with PMA treatment appear to contradict our previous data which suggested that TACE and BACE do not compete for APP. One way to reconcile both observations is to hypothesize that the two enzymes reside in the same subcellular compartment, as has previously been suggested (Skovronsky et al., 2000), but that the APP levels in the compartment are normally not substrate limiting for TACE and BACE. However, the combined TACE and BACE catalytic activity could exceed the amount of available APP when TACE activity is significantly upregulated by phorbol ester, thereby resulting in enzyme competition for substrate.
BACE inhibition increases sAPPα secretion from trans-Golgi network-targeted APP
To further examine the hypothesis that TACE and BACE share a common compartment, studies were conducted in which the normal intracellular trafficking of APP was altered. CHO cells were stably transfected with APP-TGN, a fusion construct in which the entire cytoplasmic domain of APP is replaced with the rat furin domain that enhances retention in the TGN (Chyung et al., 1997; Skovronsky et al., 2000; Huse et al., 2002). Because the TGN has been proposed to be a site of α- and β-secretase action, increasing APP retention time in the TGN should result in greater accessibility by these enzymes. As shown in Figure 5, addition of the Merck BACE inhibitor III to APP-TGN cells resulted in an increased production of sAPPα relative to the vehicle-treated APP-TGN cells, a result that is consistent with the shared compartment hypothesis. However, this data would appear to conflict with the results obtained with APPwt cells (Fig. 3), where BACE inhibition did not change sAPPα levels. One important difference between the APP-TGN and APPwt cells is that the targeting sequence in the former does not allow for normal APP progression from the TGN outward to the plasma membrane. Thus, whereas APP concentration is unlikely to increase significantly within the TGN as a result of BACE inhibition in APPwt cells because of continued APP transit to the plasma membrane, APP would accumulate within the TGN of APP-TGN cells. Accordingly, in the absence of APP outflow, BACE inhibition would result in an even greater increase of TGN APP concentration that would allow for increased sAPPα production by TACE.
Figure 5.
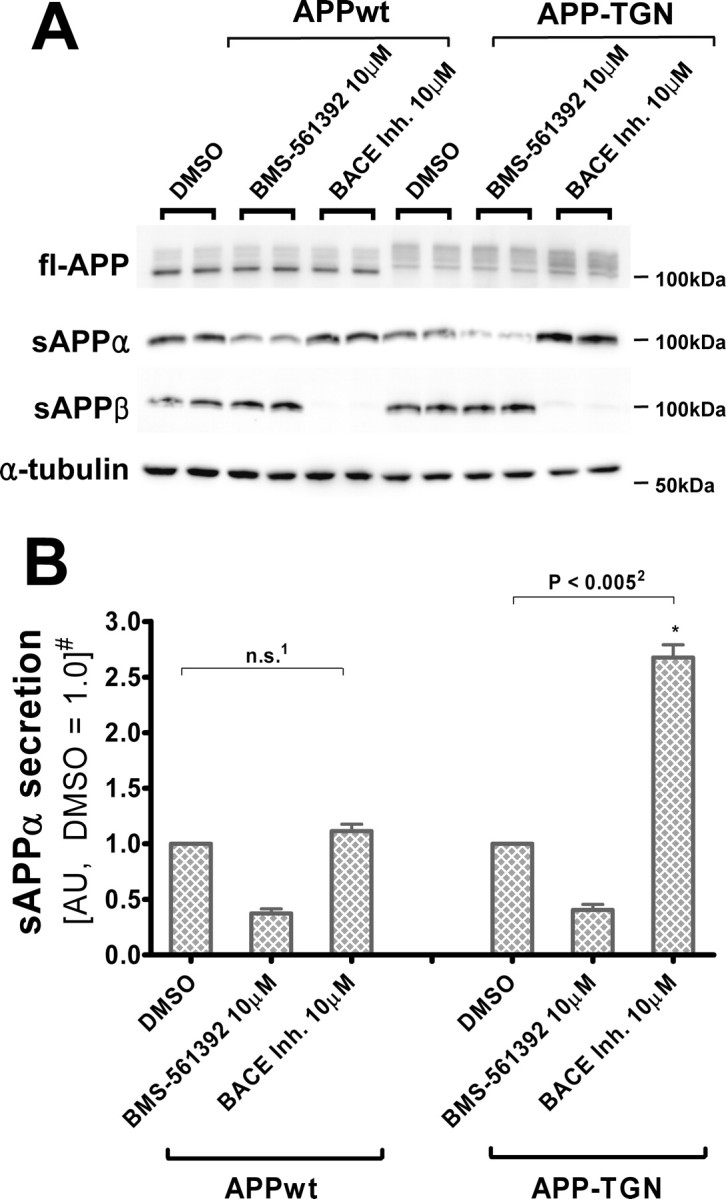
BACE inhibitor increases sAPPα secretion from CHO cells expressing TGN-targeting APP. A, Immunoblot of full-length APP and α-tubulin in cell lysates, and sAPPα and sAPPβ in conditioned media, from CHO-APPwt and CHO-APP-TGN cells. APP-TGN migrated slower because of the addition of the furin sequence (Chyung et al., 1997; Skovronsky et al., 2000; Huse et al., 2002). RIPA lysates (20 μg) and 20 μl of conditioned media were resolved in 7.5% Tris-glycine SDS-gels, blotted and probed with 5685 (fl-APP), Ban50 (sAPPα), and α-tubulin antibodies. B, Quantification of sAPPα in conditioned media. Unlike CHO-APPwt cells, BACE inhibitor increases sAPPα secretion from CHO-APP-TGN cells. Quantification are means of three independent experiments ±SE measurements. n.s., Not significant, two-tailed Student's t test, p = 0.210; p < 0.005, paired two-tailed Student's t test. #Results are normalized to fl-APP levels in each cell line. sAPPα levels are shown in arbitrary units (DMSO treatment as 1.0).
sAPPα levels were significantly reduced in BMS-561392 infused Tg2576 mouse hippocampi without an increase in Aβ levels
To test whether BMS-561392 acts as a potent TACE inhibitor in vivo, we directly infused this compound or vehicle with osmotic pumps into the right lateral ventricle of Tg2576 mice for 14 d. Direct infusion was necessary because BMS-561392 does not penetrate the blood–brain barrier [J. M. Trzaskos (Bristol-Myers Squibb) personal communication]. As shown in Figure 6, A and B, sAPPα levels in BMS-561392-infused mice were significantly reduced in the hippocampus, but not in the cortex. This may be because of the proximity of the hippocampus to the site of infusion, resulting in a greater concentration of BMS-561392 in interstitial fluid in the hippocampus than in the cortex, which is much larger and farther away from the lateral ventricle. These data show that BMS-561392 is an inhibitor of α-cleavage of APP in vivo. Unlike BMS-561392, TAPI-I failed to lower sAPPα in the hippocampus. This may reflect the higher potency of BMS-561392 than TAPI-1 on TACE (Fig. 1). Although the inhibitory effect of BMS-561392 on brain sAPPα levels appears to be somewhat less profound than what was observed in vitro (Fig. 1B), it is difficult to directly compare the cell-based studies with the more complex in vivo model. Nonetheless, we note that the ventricular infusion of BMS-561392 resulted in a highly significant 40% reduction in hippocampal sAPPα, and it is possible that higher doses of compound might have resulted in nearly complete α-secretase inhibition.
Figure 6.

BMS-561392 reduces the level of sAPPα in Tg2576 transgenic mouse hippocampi without changes in Aβ level. A, BMS-561392 reduces sAPPα level in Tg2576 mouse hippocampi, but TAPI-I fails to do so. RIPA lysates (20 μg) of drug-treated mouse cortices and hippocampi were resolved in 7.5% Tris-glycine SDS-gels, blotted and probed with 5685 (fl-APP), 2B3 (sAPPα), and α-tubulin antibodies. B, Quantification based on the average of n = 5 for each group in six independent immunoblots ±SE measurements. n.s. a, Not significant, one-way ANOVA, p = 0.734; n.s.b, not significant, unpaired two-tailed Student's t test, p = 0.236; +p < 0.0001; unpaired two-tailed Student's t test. #Results are normalized to fl-APP levels and shown in arbitrary units (control as 1.0). C, Aβ40 and Aβ42 levels in brain lysates and CSF showed no statistically significant change. n.s.1 ∼ n.s.6, Not significant, one-way ANOVA, p = 0.735, 0.590, 0.647, 0.480, 0.550, and 0.606, respectively.
We also measured Aβ40/42 levels in brain extracts and CSF of Tg2576 mice. Similar to cell-based experiments, Aβ40/42 levels were not statistically different in vehicle- versus BMS-561392-infused mice (Fig. 6C). Thus, the in vivo result agrees with our observation in cultured cells that α- and β-secretase do not compete for APP under normal conditions. Finally, an analysis of full-length APP did not reveal any significant differences among the treatment groups (data not shown).
sAPPα levels were significantly reduced in BMS-561392- infused wild-type mice without an increase in Aβ levels
Although Aβ levels remained unchanged in BMS-561392-infused Tg2576 mice, the experimental system may not reflect the effect of TACE inhibition on Aβ generation in human brains where APP is not artificially over-expressed. We therefore determined whether BMS-561392 affects Aβ secretion in wild-type mice expressing only endogenous APP. To detect the low level of Aβ and other APP cleavage products, we sequentially extracted wild-type mouse brains with DEA-NaCl and RIPA buffers (Miller et al., 2003). DEA extraction effectively separated sAPPα and sAPPβ from fl-APP (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). Figure 7, A and B, show that sAPPα levels were reduced in the brains of BMS-561392 infused non-transgenic wild type mice. Unlike Tg2576 mice, sAPPα was significantly lower in the cortices as well as the hippocampi of the drug-infused brains compared with vehicle-treated brains. One possible explanation is that the lower level of APP in the cortices of wild-type mice relative to Tg2576 mice enabled more effective inhibition of TACE cleavage of APP by BMS-561392. Whereas BMS-561392 inhibited sAPPα generation in wild-type mouse brains, Aβ levels remained unchanged in both brain lysates and cerebrospinal fluids (Fig. 7C). Similarly, full-length APP levels were not statistically different between control and drug-treated mouse brains (data not shown). These data demonstrate that BMS-561392 does not enhance Aβ secretion either in wild-type mice or in transgenic mice that over-express APP. Thus, anti-inflammatory drugs targeting TACE may not necessarily promote an increase in Aβ production.
Figure 7.

BMS-561392 reduces the level of sAPPα in B6/SJL F1 wild-type mouse brains without changes in Aβ level. A, BMS-561392 reduces sAPPα level in drug-treated SJL/B6 F1 mouse cortices and hippocampi, whereas Aβ40 and Aβ42 levels in brain lysates and CSF were unchanged. Fifty μg of DEA/NaCl lysates (for sAPPα) and RIPA lysates (for fl-APP and α-tubulin) of drug-treated mouse cortices and hippocampi were resolved in 7.5% Tris-glycine SDS-gels, blotted and probed with 5685 (fl-APP), 2B3 (sAPPα), and α-tubulin antibodies. B, Quantification based on the average of n = 6 for each group ±SE measurements. +p < 0.05; 2+p < 0.05, unpaired two-tailed Student's t test. #Results are normalized to fl-APP levels and shown in arbitrary units (control as 1.0). C, Aβ40 and Aβ42 levels in brain lysates and CSF were unchanged. n.s.1 ∼ n.s.6, Not significant, unpaired two-tailed Student's t test, p = 0.877, 0.482, 0.633, 0.574, 0.928, and 0.785, respectively.
Discussion
Although previous studies suggested that suppression of glial activation and a reduction in neuroinflammation might prove beneficial in AD (McGeer et al., 2006), the effects of selective TNFα reduction remain unknown because of a lack of agents that can specifically suppress the biosynthesis or release of this inflammatory cytokine in the brain. In this study, we demonstrated that a highly selective TACE inhibitor, BMS-561392, reduces TNFα secretion in cultured cells and also inhibits the cleavage of another TACE substrate, APP, both in cell culture and in mouse models. It should be noted that TACE cleavage of APP precludes the generation of potentially harmful Aβ peptides, and thus, inhibition of TACE might be predicted to lead to elevated Aβ levels that could offset some or all of the possible therapeutic benefits of TNFα suppression (Buxbaum et al., 1998; Lammich et al., 1999). Our results show that BMS-561392 does not elevate Aβ secretion in cell culture at a dose that causes >50% reduction in TNFα secretion. The level of CTFβ, the precursor that is cleaved by γ-secretase to yield Aβ, also remained unchanged in BMS-561392-treated cells, confirming that TACE inhibition did not affect BACE processing of APP. We observed a similar lack of effect of TACE inhibition on BACE cleavage of APP in an APP-over-expression mouse model as well as in wild-type mice that express only endogenous APP. Whereas sAPPα levels in the hippocampus were significantly reduced in BMS-561392-infused mice, steady-state Aβ levels remained the same. Similarly, CSF Aβ levels also showed no change after TACE inhibition. The lack of correlation between α- and β-cleavage of APP, both in vitro and in vivo, suggests that there is little competition for APP between TACE and BACE. Thus, although BMS-561392 itself is not suitable for the treatment of neuroinflammatory conditions because it does not readily cross the blood–brain barrier, these data suggest that a brain-penetrant TACE inhibitor would be unlikely to induce an undesirable elevation of brain Aβ and therefore may have potential as an AD therapeutic.
Although it appears that TACE and BACE do not normally compete for APP, conditions can be created in which inhibition of one of these enzymes can affect the amount of APP processed by the other. For example, TACE inhibition by BMS-561392 caused an increased production of Aβ peptides relative to untreated cells when the catalytic activity of TACE was significantly enhanced by phorbol ester. Moreover, BACE inhibition in cells that expressed a TGN-retained form of APP resulted in an elevation of sAPPα. These data suggest that TACE and BACE are colocalized within the TGN and that manipulation of the TGN milieu can yield conditions whereby modulation of one secretase can affect APP cleavage by the other. The steady-state level of APP that transiently resides in the TGN during its trafficking to the plasma membrane might normally be in excess of the combined TACE and BACE catalytic activities, but the elevation of TACE activity by phorbol ester may cause enough additional APP cleavage to make the TGN pool of APP limiting. On the other hand, artificially retaining APP within the TGN via a targeting sequence disrupts its normal outward flux such that BACE inhibition would result in an elevation of TGN APP concentration, with a consequent increase of sAPPα production by TACE.
Our observation of limited competition between TACE and BACE for APP only under certain conditions, such as PKC activation, agrees with certain previous studies (Gabuzda et al., 1993; Hung et al., 1993; Jacobsen et al., 1994; Blacker et al., 2002; Gandhi et al., 2004). However, there are other reports that would appear to be in disagreement with our data in regard to whether and under what conditions these two enzymes compete for APP. (Dyrks et al., 1994; Buxbaum et al., 1998; LeBlanc et al., 1998). We suggest that the differences may be attributable to our use of a highly specific TACE inhibitor instead of a pleiotropic inhibitor of the MMP/ADAM family and perhaps to cell-type specific behavior, especially with respect to the response toward PMA treatment. It should be noted, however, that we consistently demonstrated an absence of Aβ enhancement by BMS-561392 in cultured CHO cells and primary neurons, as well as in human APP-expressing transgenic and wild-type mice.
Despite its upregulation in AD and other brain diseases (Fillit et al., 1991; Mogi et al., 1994; Botchkina et al., 1997), the effects of TNFα remain somewhat controversial because proinflammatory cytokines do not usually cause death of neurons. Moreover, it has been suggested that TNFα can act as a neuroprotective agent by suppressing CDK5 hyperactivity (Orellana et al., 2007). However, identification of single nucleotide polymorphisms in the TNFα gene that are linked to AD strongly supports the hypothesis that TNFα is a significant contributing factor to neuronal degeneration (Collins et al., 2000; Lio et al., 2006; Ramos et al., 2006). A G-to-A mutation in the promoter region at position −308 of the TNFα gene has been shown to be associated with higher intrathecal level of this cytokine and earlier onset of Alzheimer's disease. In contrast, an allele at position −863 of the TNFα gene locus results in lower serum TNFα levels and reduced risk of AD. Interestingly, the presence of the APOE ε4 allele, a major risk factor for late-onset AD, is synergetic to the effects of the TNFα position −308 haplotype in lowering the age of onset of the disease.
The prominent brain inflammation and neurodegeneration that is seen in transgenic mice over-expressing TNFα or its receptors indicates that TNFα signaling cascades can lead to significant neuronal loss (Probert et al., 1995; Akassoglou et al., 1997; Stalder et al., 1998). TNFα secretion from activated microglial can be induced by neurotoxic insults, including Aβ40 and 42 peptides (Klegeris et al., 1997; Fiala et al., 1998; Yates et al., 2000), and TNFα can enhance the secretion of Aβ. Thus the interplay between Aβ and TNFα could lead to a vicious and damaging cycle in the AD brain (Blasko et al., 1999). Interestingly, deletion of TNF type I receptor (TNFR1, p55) gene in APP23 transgenic mice diminished amyloid plaque formation and improved cognitive deficits by downregulating BACE1 promoter activity (He et al., 2007). Infliximab and thalidomide, a TNFα antagonist and an inhibitor of TNFα production, were shown to prevent Aβ-mediated LTP inhibition in mice, suggesting cross-interaction between a TNFα signaling cascade and Aβ toxicity (Wang et al., 2005). TNFα can also stimulate other inflammatory pathways, including those mediated by NF-κB and cyclooxygenase (COX) 2 (Meichle et al., 1990; Geng et al., 1995; Perry et al., 2001), of which inhibitors have been suggested to lower the risk of AD (Lim et al., 2000; Sung et al., 2004; Heneka et al., 2005). Thus, TNFα may cause neuronal damage and memory loss through multiple mechanisms (Tarkowski et al., 1999; Sastre et al., 2006).
Although a phase II trial of BMS-561392 for rheumatoid arthritis was halted because of signs of mild hepatotoxicity, several other anti-TNFα agents, such as infliximab, are widely used for the treatment of autoimmune diseases. Interestingly, curcumin, a natural product that reduces amyloid accumulation in mouse models of AD (Yang et al., 2005; Garcia-Alloza et al., 2007), is an anti-inflammatory compound that lowers levels of serum TNFα in a rat model of acute pancreatitis (Gulcubuk et al., 2006). Despite the lack of extensive evidence for the therapeutic effects of TACE-targeted anti-TNFα treatment in AD, this approach may be well worth pursuing. However, it should be recognized that one consequence of TACE inhibition will be a reduction of sAPPα, which has been proposed to have neurotrophic properties in cultured neurons and in a rat traumatic brain injury model (Mattson et al., 1993; Yamamoto et al., 1994; Thornton et al., 2006). Although there is no evidence showing that a reduction of sAPPα by TACE inhibition is detrimental, this possibility deserves further exploration in future studies.
Another consequence of TACE inhibition is that any positive effects of TNFα on neuronal systems will be compromised. For example, it has been suggested that TNFα may protect neurons against cellular stress and is even neurotrophic through TNF receptor II (TNFR2) (Cheng et al., 1994; Yang et al., 2002). However, it is unclear that inhibition of TNFα secretion in adult brain will have negative consequences that will outweigh the likely positive effects of preventing TNFα-mediated inflammation.
In conclusion, we have demonstrated that the potent TACE inhibitor, BMS-561392, does not induce or increase Aβ secretion under normal conditions. Therefore, although TACE and BACE appear to reside in a shared TGN compartment, the use of TACE-targeted anti-inflammatory drugs in AD may not be associated with higher Aβ generation in brain. Although BMS-561392 itself is unlikely to be a candidate drug for AD because of its poor brain penetration, the identification and development of improved TACE inhibitors that gain access to the brain may be of be potential value in reducing the glial-mediated inflammation that has been suggested to contribute to AD neuropathology.
Footnotes
This work was supported by National Institutes of Health Grant AG11542 and the Marian S. Ware Program for Drug Discovery. V.M.-Y.L. is the John H. Ware III Professor for Alzheimer's disease research. We would like to thank Dr. Diana Shineman for her advice and helpful suggestions on this manuscript. We thank Susan Leight and Jennifer Bruce for excellent technical support. We also thank Drs. James Trzaskos and James Duan at Bristol-Myers Squibb for BMS-561392.
References
- Akassoglou K, Probert L, Kontogeorgos G, Kollias G. Astrocyte-specific but not neuron-specific transmembrane TNF triggers inflammation and degeneration in the central nervous system of transgenic mice. J Immunol. 1997;158:438–445. [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, Finch CE, Frautschy S, Griffin WS, Hampel H, Hull M, Landreth G, Lue L, Mrak R, Mackenzie IR, McGeer PL, et al. Inflammation and Alzheimer's disease. Neurobiol Aging. 2000;21:383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker M, Noe MC, Carty TJ, Goodyer CG, LeBlanc AC. Effect of tumor necrosis factor-alpha converting enzyme (TACE) and metalloprotease inhibitor on amyloid precursor protein metabolism in human neurons. J Neurochem. 2002;83:1349–1357. doi: 10.1046/j.1471-4159.2002.01228.x. [DOI] [PubMed] [Google Scholar]
- Blasko I, Marx F, Steiner E, Hartmann T, Grubeck-Loebenstein B. TNFalpha plus IFNgamma induce the production of Alzheimer beta-amyloid peptides and decrease the secretion of APPs. FASEB J. 1999;13:63–68. doi: 10.1096/fasebj.13.1.63. [DOI] [PubMed] [Google Scholar]
- Botchkina GI, Meistrell ME, 3rd, Botchkina IL, Tracey KJ. Expression of TNF and TNF receptors (p55 and p75) in the rat brain after focal cerebral ischemia. Mol Med. 1997;3:765–781. [PMC free article] [PubMed] [Google Scholar]
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- Cheng B, Christakos S, Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron. 1994;12:139–153. doi: 10.1016/0896-6273(94)90159-7. [DOI] [PubMed] [Google Scholar]
- Chesneau V, Becherer JD, Zheng Y, Erdjument-Bromage H, Tempst P, Blobel CP. Catalytic properties of ADAM19. J Biol Chem. 2003;278:22331–22340. doi: 10.1074/jbc.M302781200. [DOI] [PubMed] [Google Scholar]
- Chyung AS, Greenberg BD, Cook DG, Doms RW, Lee VM-Y. Novel beta-secretase cleavage of beta-amyloid precursor protein in the endoplasmic reticulum/intermediate compartment of NT2N cells. J Cell Biol. 1997;138:671–680. doi: 10.1083/jcb.138.3.671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citron M, Oltersdorf T, Haass C, McConlogue L, Hung AY, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe DJ. Mutation of the beta-amyloid precursor protein in familial Alzheimer's disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- Collins JS, Perry RT, Watson B, Jr, Harrell LE, Acton RT, Blacker D, Albert MS, Tanzi RE, Bassett SS, McInnis MG, Campbell RD, Go RC. Association of a haplotype for tumor necrosis factor in siblings with late-onset Alzheimer disease: the NIMH Alzheimer Disease Genetics Initiative. Am J Med Genet. 2000;96:823–830. doi: 10.1002/1096-8628(20001204)96:6<823::aid-ajmg26>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- De Strooper B, Annaert W. Proteolytic processing and cell biological functions of the amyloid precursor protein. J Cell Sci. 2000;113:1857–1870. doi: 10.1242/jcs.113.11.1857. [DOI] [PubMed] [Google Scholar]
- Dominguez D, Tournoy J, Hartmann D, Huth T, Cryns K, Deforce S, Serneels L, Camacho IE, Marjaux E, Craessaerts K, Roebroek AJ, Schwake M, D'Hooge R, Bach P, Kalinke U, Moechars D, Alzheimer C, Reiss K, Saftig P, De Strooper B. Phenotypic and biochemical analyses of B. J Biol Chem. 2005;280:30797–30806. doi: 10.1074/jbc.M505249200. [DOI] [PubMed] [Google Scholar]
- Dyrks T, Mönning U, Beyreuther K, Turner J. Amyloid precursor protein secretion and beta A4 amyloid generation are not mutually exclusive. FEBS Lett. 1994;349:210–214. doi: 10.1016/0014-5793(94)00671-7. [DOI] [PubMed] [Google Scholar]
- Fiala M, Zhang L, Gan X, Sherry B, Taub D, Graves MC, Hama S, Way D, Weinand M, Witte M, Lorton D, Kuo YM, Roher AE. Amyloid-beta induces chemokine secretion and monocyte migration across a human blood–brain barrier model. Mol Med. 1998;4:480–489. [PMC free article] [PubMed] [Google Scholar]
- Fillit H, Ding WH, Buee L, Kalman J, Altstiel L, Lawlor B, Wolf-Klein G. Elevated circulating tumor necrosis factor levels in Alzheimer's disease. Neurosci Lett. 1991;129:318–320. doi: 10.1016/0304-3940(91)90490-k. [DOI] [PubMed] [Google Scholar]
- Forman MS, Cook DG, Leight S, Doms RW, Lee VM-Y. Differential effects of the Swedish mutant amyloid precursor protein on beta-amyloid accumulation and secretion in neurons and nonneuronal cells. J Biol Chem. 1997;272:32247–32253. doi: 10.1074/jbc.272.51.32247. [DOI] [PubMed] [Google Scholar]
- Gabuzda D, Busciglio J, Yankner BA. Inhibition of beta-amyloid production by activation of protein kinase C. J Neurochem. 1993;61:2326–2329. doi: 10.1111/j.1471-4159.1993.tb07479.x. [DOI] [PubMed] [Google Scholar]
- Gandhi S, Refolo LM, Sambamurti K. Amyloid precursor protein compartmentalization restricts beta-amyloid production: therapeutic targets based on BACE compartmentalization. J Mol Neurosci. 2004;24:137–143. doi: 10.1385/JMN:24:1:137. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Borrelli LA, Rozkalne A, Hyman BT, Bacskai BJ. Curcumin labels amyloid pathology in vivo, disrupts existing plaques, and partially restores distorted neurites in an Alzheimer mouse model. J Neurochem. 2007;102:1095–1104. doi: 10.1111/j.1471-4159.2007.04613.x. [DOI] [PubMed] [Google Scholar]
- Geng Y, Blanco FJ, Cornelisson M, Lotz M. Regulation of cyclooxygenase-2 expression in normal human articular chondrocytes. J Immunol. 1995;155:796–801. [PubMed] [Google Scholar]
- Gulcubuk A, Altunatmaz K, Sonmez K, Haktanir-Yatkin D, Uzun H, Gurel A, Aydin S. Effects of curcumin on tumour necrosis factor-alpha and interleukin-6 in the late phase of experimental acute pancreatitis. J Vet Med A Physiol Pathol Clin Med. 2006;53:49–54. doi: 10.1111/j.1439-0442.2006.00786.x. [DOI] [PubMed] [Google Scholar]
- He P, Zhong Z, Lindholm K, Berning L, Lee W, Lemere C, Staufenbiel M, Li R, Shen Y. Deletion of tumor necrosis factor death receptor inhibits amyloid beta generation and prevents learning and memory deficits in Alzheimer's mice. J Cell Biol. 2007;178:829–841. doi: 10.1083/jcb.200705042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C, O'Banion K, Klockgether T, Van Leuven F, Landreth GE. Acute treatment with the PPARgamma agonist pioglitazone and ibuprofen reduces glial inflammation and Abeta1–42 levels in APPV717I transgenic mice. Brain. 2005;128:1442–1453. doi: 10.1093/brain/awh452. [DOI] [PubMed] [Google Scholar]
- Hung AY, Haass C, Nitsch RM, Qiu WQ, Citron M, Wurtman RJ, Growdon JH, Selkoe DJ. Activation of protein kinase C inhibits cellular production of the amyloid beta-protein. J Biol Chem. 1993;268:22959–22962. [PubMed] [Google Scholar]
- Huse JT, Liu K, Pijak DS, Carlin D, Lee VM-Y, Doms RW. Beta-secretase processing in the trans-Golgi network preferentially generates truncated amyloid species that accumulate in Alzheimer's disease brain. J Biol Chem. 2002;277:16278–16284. doi: 10.1074/jbc.M111141200. [DOI] [PubMed] [Google Scholar]
- Itai T, Tanaka M, Nagata S. Processing of tumor necrosis factor by the membrane-bound TNF-alpha-converting enzyme, but not its truncated soluble form. Eur J Biochem. 2001;268:2074–2082. doi: 10.1046/j.1432-1327.2001.02085.x. [DOI] [PubMed] [Google Scholar]
- Jacobsen JS, Spruyt MA, Brown AM, Sahasrabudhe SR, Blume AJ, Vitek MP, Muenkel HA, Sonnenberg-Reines J. The release of Alzheimer's disease beta amyloid peptide is reduced by phorbol treatment. J Biol Chem. 1994;269:8376–8382. [PubMed] [Google Scholar]
- Klegeris A, Walker DG, McGeer PL. Interaction of Alzheimer beta-amyloid peptide with the human monocytic cell line THP-1 results in a protein kinase C-dependent secretion of tumor necrosis factor-alpha. Brain Res. 1997;747:114–121. doi: 10.1016/s0006-8993(96)01229-2. [DOI] [PubMed] [Google Scholar]
- Koike H, Tomioka S, Sorimachi H, Saido TC, Maruyama K, Okuyama A, Fujisawa-Sehara A, Ohno S, Suzuki K, Ishiura S. Membrane-anchored metalloprotease MDC9 has an alpha-secretase activity responsible for processing the amyloid precursor protein. Biochem J. 1999;343:371–375. [PMC free article] [PubMed] [Google Scholar]
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci U S A. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeBlanc AC, Koutroumanis M, Goodyer CG. Protein kinase C activation increases release of secreted amyloid precursor protein without decreasing Aβ production in human primary neuron cultures. J Neurosci. 1998;18:2907–2913. doi: 10.1523/JNEUROSCI.18-08-02907.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM-Y. Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem. 2003;278:4458–4466. doi: 10.1074/jbc.M210105200. [DOI] [PubMed] [Google Scholar]
- Lee EB, Zhang B, Liu K, Greenbaum EA, Doms RW, Trojanowski JQ, Lee VM-Y. BACE overexpression alters the subcellular processing of APP and inhibits Abeta deposition in vivo. J Cell Biol. 2005;168:291–302. doi: 10.1083/jcb.200407070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim GP, Yang F, Chu T, Chen P, Beech W, Teter B, Tran T, Ubeda O, Ashe KH, Frautschy SA, Cole GM. Ibuprofen suppresses plaque pathology and inflammation in a mouse model for Alzheimer's disease. J Neurosci. 2000;20:5709–5714. doi: 10.1523/JNEUROSCI.20-15-05709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lio D, Annoni G, Licastro F, Crivello A, Forte GI, Scola L, Colonna-Romano G, Candore G, Arosio B, Galimberti L, Vergani C, Caruso C. Tumor necrosis factor-alpha -308A/G polymorphism is associated with age at onset of Alzheimer's disease. Mech Ageing Dev. 2006;127:567–571. doi: 10.1016/j.mad.2006.01.015. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Cheng B, Culwell AR, Esch FS, Lieberburg I, Rydel RE. Evidence for excitoprotective and intraneuronal calcium-regulating roles for secreted forms of the beta-amyloid precursor protein. Neuron. 1993;10:243–254. doi: 10.1016/0896-6273(93)90315-i. [DOI] [PubMed] [Google Scholar]
- McGeer PL, McGeer EG. Polymorphisms in inflammatory genes and the risk of Alzheimer disease. Arch Neurol. 2001;58:1790–1792. doi: 10.1001/archneur.58.11.1790. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Rogers J, McGeer EG. Inflammation, anti-inflammatory agents and Alzheimer disease: the last 12 years. J Alzheimers Dis. 2006;9:271–276. doi: 10.3233/jad-2006-9s330. [DOI] [PubMed] [Google Scholar]
- Meichle A, Schütze S, Hensel G, Brunsing D, Krönke M. Protein kinase C-independent activation of nuclear factor kappa B by tumor necrosis factor. J Biol Chem. 1990;265:8339–8343. [PubMed] [Google Scholar]
- Miller BC, Eckman EA, Sambamurti K, Dobbs N, Chow KM, Eckman CB, Hersh LB, Thiele DL. Amyloid-beta peptide levels in brain are inversely correlated with insulysin activity levels in vivo. Proc Natl Acad Sci USA. 2003;100:6221–6226. doi: 10.1073/pnas.1031520100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogi M, Harada M, Riederer P, Narabayashi H, Fujita K, Nagatsu T. Tumor necrosis factor-alpha (TNF-alpha) increases both in the brain and in the cerebrospinal fluid from parkinsonian patients. Neurosci Lett. 1994;165:208–210. doi: 10.1016/0304-3940(94)90746-3. [DOI] [PubMed] [Google Scholar]
- Orellana DI, Quintanilla RA, Maccioni RB. Neuroprotective effect of TNFalpha against the beta-amyloid neurotoxicity mediated by CDK5 kinase. Biochim Biophys Acta. 2007;1773:254–263. doi: 10.1016/j.bbamcr.2006.10.010. [DOI] [PubMed] [Google Scholar]
- Perry RT, Collins JS, Wiener H, Acton R, Go RC. The role of TNF and its receptors in Alzheimer's disease. Neurobiol Aging. 2001;22:873–883. doi: 10.1016/s0197-4580(01)00291-3. [DOI] [PubMed] [Google Scholar]
- Probert L, Akassoglou K, Pasparakis M, Kontogeorgos G, Kollias G. Spontaneous inflammatory demyelinating disease in transgenic mice showing central nervous system-specific expression of tumor necrosis factor alpha. Proc Natl Acad Sci U S A. 1995;92:11294–11298. doi: 10.1073/pnas.92.24.11294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos EM, Lin MT, Larson EB, Maezawa I, Tseng LH, Edwards KL, Schellenberg GD, Hansen JA, Kukull WA, Jin LW. Tumor necrosis factor alpha and interleukin 10 promoter region polymorphisms and risk of late-onset Alzheimer disease. Arch Neurol. 2006;63:1165–1169. doi: 10.1001/archneur.63.8.1165. [DOI] [PubMed] [Google Scholar]
- Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer's disease: molecular mechanisms. Int J Dev Neurosci. 2006;24:167–176. doi: 10.1016/j.ijdevneu.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Skovronsky DM, Moore DB, Milla ME, Doms RW, Lee VM. Protein kinase C-dependent alpha-secretase competes with beta-secretase for cleavage of amyloid-beta precursor protein in the trans-golgi network. J Biol Chem. 2000;275:2568–2575. doi: 10.1074/jbc.275.4.2568. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Carson MJ, Pagenstecher A, Asensio VC, Kincaid C, Benedict M, Powell HC, Masliah E, Campbell IL. Late-onset chronic inflammatory encephalopathy in immune-competent and severe combined immune-deficient (SCID) mice with astrocyte-targeted expression of tumor necrosis factor. Am J Pathol. 1998;153:767–783. doi: 10.1016/S0002-9440(10)65620-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S, Yang H, Uryu K, Lee EB, Zhao L, Shineman D, Trojanowski JQ, Lee VM, Praticò D. Modulation of nuclear factor-kappa B activity by indomethacin influences A beta levels but not A beta precursor protein metabolism in a model of Alzheimer's disease. Am J Pathol. 2004;165:2197–2206. doi: 10.1016/s0002-9440(10)63269-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarkowski E, Blennow K, Wallin A, Tarkowski A. Intracerebral production of tumor necrosis factor-alpha, a local neuroprotective agent, in Alzheimer disease and vascular dementia. J Clin Immunol. 1999;19:223–230. doi: 10.1023/a:1020568013953. [DOI] [PubMed] [Google Scholar]
- Thornton E, Vink R, Blumbergs PC, Van Den Heuvel C. Soluble amyloid precursor protein alpha reduces neuronal injury and improves functional outcome following diffuse traumatic brain injury in rats. Brain Res. 2006;1094:38–46. doi: 10.1016/j.brainres.2006.03.107. [DOI] [PubMed] [Google Scholar]
- Wang Q, Wu J, Rowan MJ, Anwyl R. Beta-amyloid inhibition of long-term potentiation is mediated via tumor necrosis factor. Eur J Neurosci. 2005;22:2827–2832. doi: 10.1111/j.1460-9568.2005.04457.x. [DOI] [PubMed] [Google Scholar]
- Wertkin AM, Turner RS, Pleasure SJ, Golde TE, Younkin SG, Trojanowski JQ, Lee VM-Y. Human neurons derived from a teratocarcinoma cell line express solely the 695-amino acid amyloid precursor protein and produce intracellular beta-amyloid or A4 peptides. Proc Natl Acad Sci U S A. 1993;90:9513–9517. doi: 10.1073/pnas.90.20.9513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf BA, Wertkin AM, Jolly YC, Yasuda RP, Wolfe BB, Konrad RJ, Manning D, Ravi S, Williamson JR, Lee VM. Muscarinic regulation of Alzheimer's disease amyloid precursor protein secretion and amyloid beta-protein production in human neuronal NT2N cells. J Biol Chem. 1995;270:4916–4922. doi: 10.1074/jbc.270.9.4916. [DOI] [PubMed] [Google Scholar]
- Yamamoto K, Miyoshi T, Yae T, Kawashima K, Araki H, Hanada K, Otero DA, Roch JM, Saitoh T. The survival of rat cerebral cortical neurons in the presence of trophic APP peptides. J Neurobiol. 1994;25:585–594. doi: 10.1002/neu.480250510. [DOI] [PubMed] [Google Scholar]
- Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, Chen PP, Kayed R, Glabe CG, Frautschy SA, Cole GM. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280:5892–5901. doi: 10.1074/jbc.M404751200. [DOI] [PubMed] [Google Scholar]
- Yang L, Lindholm K, Konishi Y, Li R, Shen Y. Target depletion of distinct tumor necrosis factor receptor subtypes reveals hippocampal neuron death and survival through different signal transduction pathways. J Neurosci. 2002;22:3025–3032. doi: 10.1523/JNEUROSCI.22-08-03025.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, Brunden KR. Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem. 2000;74:1017–1025. doi: 10.1046/j.1471-4159.2000.0741017.x. [DOI] [PubMed] [Google Scholar]