

Abstract
Pulmonary vascular endothelial injury resulting from lipopolysaccharide (LPS) and oxygen toxicity contributes to vascular simplification seen in the lungs of premature infants with bronchopulmonary dysplasia. Whether the severity of endotoxin-induced endothelial injury is modulated by ambient oxygen tension (hypoxic intra-uterine environment vs. hyperoxic postnatal environment) remains unknown. We posited that ovine fetal pulmonary artery endothelial cells (FPAEC) will be more resistant to LPS toxicity in hypoxic conditions (20-25torr) mimicking the fetal milieu. LPS (10μg/ml) inhibited FPAEC proliferation and induced apoptosis under normoxic conditions (21% O2) in-vitro. LPS-induced FPAEC apoptosis was attenuated in hypoxia (5% O2) and exacerbated by hyperoxia (55% O2). LPS increased intracellular superoxide formation, as measured by 2-hydroxyethidium (2-HE) formation, in FPAEC in normoxia and hypoxia. 2-HE formation in LPS-treated FPAEC increased in parallel with the severity of LPS-induced apoptosis in FPAEC, increasing from hypoxia to normoxia to hyperoxia. Differences in LPS-induced apoptosis between hypoxia and normoxia were abolished when LPS-treated FPAEC incubated in hypoxia were pre-treated with menadione to increase superoxide production. Apocynin decreased 2-HE formation, and attenuated LPS-induced FPAEC apoptosis under normoxic conditions. We conclude that ambient oxygen concentration modulates the severity of LPS-mediated injury in FPAEC by regulating superoxide levels produced in response to LPS.
Keywords: Hypoxia, lipopolysaccharide, superoxide, apoptosis, pulmonary endothelial cells
Introduction
Bronchopulmonary dysplasia (BPD) develops in about 25% of very low birth weight infants, and remains a major cause of pulmonary morbidity and mortality in infancy [1]. Vascular injury characterized by arrested vascular growth with decreased arborization and dysmorphic capillaries is a hallmark of the “new” BPD and may precede alveolar simplification [2, 3]. While the effects of oxygen toxicity on the pulmonary vasculature of the immature lung are well established [2-4], lipopolysaccharide (LPS) mediated endothelial injury can contribute to the vascular remodeling seen in BPD [5, 6] by inhibiting endothelial cell proliferation, migration and angiogenesis. In premature infants, exposure to bacteria is common, can occur in-utero or postnatally, and is associated with subsequent development of BPD [4, 7-9]. However, it is unknown whether the toxic effects of bacteria on vascular development are modulated by the oxygen tension of the environment the immature lung is exposed to (i.e., hypoxic environment of the fetus vs. the normoxic or hyperoxic postnatal environment).
Based on antioxidant inhibitor studies and the use of fluorescence probes, LPS mediated endothelial cell activation/injury is thought to be dependent on the generation of reactive oxygen species (ROS) [10, 11]. LPS-induced ROS production in endothelial cells activates the inflammatory response, promotes cytotoxicity and paradoxically, also activates signaling pathways that inhibit pro-apoptotic signaling [12-15]. Whether LPS mediated ROS generation and its downstream effects on endothelial toxicity are altered by ambient oxygen concentrations remains unknown. Hypoxia has been reported to both increase and decrease ROS production [16, 17]. Therefore, hypoxic conditions might attenuate or exacerbate LPS-induced ROS production and subsequent cytotoxicity in endothelial cells. In the context of BPD, the immature lung of the fetus that develops in moderate hypoxia (20-25 torr) in-utero, might be more vulnerable to LPS toxicity in the relatively hyperoxic extra-uterine environment. Since LPS presents an oxidant stress to endothelial cells we hypothesized that ambient oxygen concentrations will alter the severity of LPS-mediated toxicity in fetal pulmonary artery endothelial cells (FPAEC) with hypoxia (3-5% O2) mimicking the fetal milieu attenuating FPAEC injury while hyperoxia will exacerbate FPAEC injury. Furthermore, we posited that oxygen tension mediated modulation of LPS toxicity in FPAEC occurs via alteration of LPS-induced superoxide (O2▪−) generation in FPAEC.
In this study, we demonstrate that LPS-induced FPAEC apoptosis is attenuated by hypoxia (5%) and exacerbated by hyperoxia (55%). LPS-induced O2▪− levels in FPAEC increased in parallel with increasing oxygen tension and severity of apoptosis. Finally, the severity of LPS-induced apoptosis in FPAEC could be altered by manipulating the intracellular levels of O2▪− generated in response to LPS treatment.
MATERIALS AND METHODS
Isolation and culture of endothelial cells
Pulmonary arteries (until the third generation) dissected from the lung parenchyma of 130-134 day old lambs were stripped of endothelial cells using 0.1% collagenase type A (Roche Molecular Biochemicals, Indianopolis, IN) as previously described [18]. Harvested FPAEC were cultured in 100mm dishes using Dulbecco’s Modified Eagle Medium (DMEM) containing 20% fetal bovine serum (FBS) supplemented with 1% L-Glutamine and 1% Antibiotic/Antimycotic (Invitrogen, Carlsbad, CA) in humidified incubators at 37°C in room air with 5% CO2. Endothelial cell identity was confirmed by acetylated LDL uptake and positive staining for factor VIII-related antigen [18]. FPAEC between passages 5-9 were used for subsequent experiments. For hypoxia and hyperoxia experiments, cells were grown in specialized, humidified incubators fed with a mixture of gas containing 5% O2, 5% CO2 and balance N2 for hypoxia and 55% O2, 5% CO2 and balance N2 for hyperoxia. To ensure quick equilibration in hypoxia or hyperoxia, the lids of the cell culture dishes were removed and the dishes twirled before the lids were replaced.
Cell Proliferation Studies
FPAEC plated at a density of 2.5×103 cells/well in gelatin coated 96-well microplates were allowed to adhere overnight. Cells were growth arrested for 24hrs in DMEM with 2% FBS. FPAEC were then grown in DMEM with 20% FBS under normoxic (21% O2, 5% CO2, balance N2) or hypoxic (5% O2, 5% CO2, balance N2) conditions for 24 or 48 hours with and without E. coli-derived LPS (Sigma, St. Louis, MO). LPS was used at concentrations ranging from 10-1 ng/mL to 10μg/mL. Cell Proliferation was assessed by measuring cellular DNA content via fluorescent dye binding using the CyQuant NF Cell Proliferation Assay Kit (Invitrogen) following the manufacturer’s standard protocol. Briefly, media was gently aspirated and 75μL of 1x DNA dye binding solution was added to each well in the dark. Following 30-minute incubation, the fluorescence intensity of each sample was measured using a LJL BioSystems Analyst HT fluorescence microplate reader with excitation at 485nm and emission detection at 530nm. The noted fluorescence for each experimental condition was averaged and cell numbers determined by regression analysis using a standard curve.
Apoptosis quantification
1.5×105 − 1.75 ×105 FPAEC were plated in gelatin coated 6-well cell culture plates and allowed to adhere overnight. Following a 24-hour starvation in DMEM containing 2% FBS, media was changed to DMEM with 20% FBS and cells were incubated under normoxic, hypoxic or hyperoxic conditions with or without LPS (10μg/mL) for 24 or 48 hours. Menadione (Sigma, St. Louis, MO) and Apocynin (Calbiochem, San Diego, CA) were diluted according to manufacturer’s instructions and used to pre-treat the cells for 30 minutes before the addition of LPS. Menadione was used in concentrations ranging from 6.25μM to 50μM, while Apocynin was used in concentrations ranging from 100μM to 1mM. Treated and untreated cells were harvested using TrypLE Express (Sigma), combined with supernatant media (to account for cellular detachment), transferred to 5mL round-bottom tubes and centrifuged for 6 minutes at 400 rcf, 4-6°C. After removal of the supernatant, fluorescent detection and differentiation of apoptotic cells, necrotic cells, and viable cells was quantified using an Annexin V-FITC Apoptosis Detection Kit (Sigma) and subsequent FACS analysis using a FACS Calibur (Becton Dickinson, San Jose, CA). Data was analyzed with Cellquest Pro (Becton Dickinson).
Caspase - 3 Activity Assay
4×105 cells were plated in 60 mm dishes and allowed to adhere overnight. After 24-hr starvation, media was changed to DMEM with 20% FBS, and cells were incubated under normoxic or hypoxic conditions with or without LPS (10μg/mL) for 6, 12, 24 or 48 hours. At the end of timed experiments, media was aspirated and cells were washed with ice-cold PBS. Cells were then lysed using 100 μL of RIPA buffer (Sigma, St. Louis, MO) and a small aliquot of cell-lysate used for protein quantification as described below. Caspase-3 activity was measured per 100μg of sample using a Colorimetric Assay Kit (R&D Systems, Minneapolis, MN) following the manufacturer’s instructions.
Protein Quantification
Whole cell-lysates were obtained by using a cell-lysis buffer (RIPA, Sigma) containing protease inhibitor cocktail (Sigma, St. Louis, MO). Protein quantification was done in duplicate using a BCA Protein Assay (Pierce, Rockford, IL) according to the manufacturer’s protocol using bovine serum albumin as a standard.
Detection of Intracellular Superoxide
Superoxide levels were quantified using HPLC analysis of 2-hydroxy ethidium (2-HE) formation as described before [19]. Briefly, 1.75×105 cells were grown in 60mm dishes for 48hours. Following overnight starvation, media was changed to 20% DMEM and cells were incubated under normoxic or hypoxic conditions, with or without LPS (10μg/mL) for 3 or 6 hours. Subsequently, 15μM dihydroethidium (Invitrogen, Carlsbad, CA) was added to the media, and cells were incubated in the dark for thirty minutes. During cell harvest and sample processing the exposure to light was minimized. Media was then aspirated, cells washed and then scrapped into 1mL of ice-cold PBS. Following centrifugation at 12,000g × 10 minutes at 4-6°C, the supernatant was aspirated and the cell pellet frozen at -80°C overnight. On the following day, each cell pellet was resuspended with 300μL of ice-cold 0.1% Triton X-100 (Sigma, St. Louis, MO) in PBS, and lysed by performing 10 syringe strokes with a 1mL 27½-gauge syringe. Samples were centrifuged for 5 minutes at 7500g, 4-6°C. On ice, 200μL of the supernatant was transferred to new 1.5mL microcentrifuge tubes and the residual supernatant was used for protein quantification. Following the addition of 500μL of ice-cold n-Butanol (Sigma, St. Louis, MO) to each 200μL aliquot, samples were vortexed concurrently for 10 minutes at 4-6°C and then centrifuged for 2 minutes at 2500g, 4-6°C. The supernatant was transferred to new 1.5mL microcentrifuge tubes and n-Butanol was evaporated under 100% N2 using an Organomation Multivap Analytical Evaporator for 2-3 hours. Dried sample residues were reconstituted with 100μL of ice-cold 1M phosphate buffer (pH 2.6) and vortexed for 10 minutes at 4-6°C. After centrifuging for 2 minutes at 2500g, 4-6°C, supernatants were transferred to amber-colored HPLC vials. Typically, 50μL of sample was then injected into the HPLC system (HP 1100, Agilent Technologies, Palo Alto, CA) with a C18 column (250 × 4.5 mm) equilibrated with 10% CH3CN in 0.1% trifluoroacetic acid. Hydroethidine, ethidium and 2-dihydroxyethidium were separated by a linear increase in CH3CN concentration from 10% to 70% in 46 min at a flow rate of 0.5 ml/min. The elution was monitored by a variable UV detector at 210 and 350 nm and a fluorescence detector with excitation and emission at 510 and 595 nm, respectively. The area under the 2-HE fluorescent peaks were measured for each sample, compared with known concentrations of the standard and expressed in nmol/mg of protein.
Western Blots
1×106 cells were grown in 100mm dishes for 48hours, starved for 24hrs and then incubated under normoxic or hypoxic conditions, with or without LPS (10μg/mL) for 6, 12, 24 or 48 hours. After aspirating media, cells were washed with PBS and lysed using 200μL of RIPA Buffer containing 2% Protease Inhibitor Cocktail (Sigma, St. Louis, MO). Cell lysate was scraped off dishes, briefly sonicated and centrifuged at 12,000g for 10min at 4°C. Protein content in the supernatant was quantified as described above. Proteins (20 μg per lane) were resolved using SDS-PAGE and transferred onto a nitrocellulose membrane. Blots were blocked 1 hr in 5% nonfat dry milk in Tris-buffered saline with 0.1% Tween 20 and then probed overnight at 4-6°C with rabbit anti-beta actin antibody (1:5000, Novus Biologicals, Littleton, CO), rabbit anti-MnSOD antibody (1:2500, Stressgen, Victoria, BC Canada) or rabbit anti-CuZn SOD antibody (1:1000, Stressgen) diluted in TBS with 0.1% Tween 20. Bands were visualized with the appropriate anti-immunoglobulin horseradish peroxidase conjugate (Bio-Rad Laboratories, Hercules, CA) and a luminol-based ECL substrate (Pierce, Rockford, IL).
NADPH quantification
Intracellular NADPH levels were quantified using a HPLC-based method, as previously described [20]. Briefly, ~5 × 106 cells grown in gelatin-coated cell culture dishes were washed twice with PBS and scraped into 250 μl of PBS followed by centrifugation (2000 × g, 5 min, 4°C). Supernatant was aspirated and 0.3 ml of lysis buffer (0.5 N KOH, HBSS/Hepes, ratio 3:1) was added to the pellet. The samples were mixed for 10 s, incubated on ice for 3 min and mixed further for 1.5 min. Then, 170μL of water was added to the samples followed by centrifugation (10,000 × g, 5 min, 4°C). The pH of the supernatant (0.4 ml) was adjusted to ~ 8 by the addition of 6N HCl (10 μl) and 1 M ammonium acetate, pH 4.7 (10-15 μl). After centrifugation at 10,000 × g for 5 min at 4°C, cell extracts were filtered through a 0.22-μm cellulose acetate syringe filter and 100 μl of the filtrate was injected onto HPLC. HPLC analysis was performed on Kromasil C-18 column (5 μm, 250 mm × 4.6 mm I.D., Alltech) using solvent A (0.1 M potassium phosphate, 4 mM tetrabutylammonium bisulfate, pH 6.0 and water 64:36) and solvent B (0.1 M potassium phosphate, 4 mM tetrabutylammonium bisulfate, pH 6.0 and methanol 64:36) with a flow rate of 1ml/min. The chromatographic conditions were as follows: solvent A 0-2.5 min; a 2.5 min transition to 7:3 solvent A: solvent B; a 5 min transition to 1:1 solvent A: solvent B; a 10 min transition to 3.5:6.5 solvent A: solvent B; a 5 min constant in 3.5:6.5 solvent A: solvent B; a 4 min transition to solvent A, followed by equilibration in solvent A for 11 min. NADPH was detected at 26 min using a fluorescence detector (excitation-330 nm, emission-460 nm). NADPH peak’s were measured for each sample, compared with known concentrations of the standard and expressed in μmol/mg of protein.
Statistics
Data are represented as Mean ± Standard error of Mean. Statistical significance for proliferation and apoptosis studies were estimated using the t-test (two-sample assuming equal variances). For 2-HE measurements comparisons between groups were made using one-way ANOVA. P < 0.05 was considered as significant.
RESULTS
Effects of Hypoxia (5%) on LPS-mediated inhibition of FPAEC proliferation
The effect of LPS on FPAEC proliferation was examined under normoxic or hypoxic conditions. FPAEC number increased from 2.5 × 103 cells/well to > 2.6 × 104 cells/well in controls incubated in normoxia or hypoxia for 48 hours. LPS (10μg/mL) treatment decreased FPAEC proliferation in normoxia by 24% (2.6 ± 0.3 × 104 vs. 2.0 ± 0.2 × 104 cells/well, P=0.004, n=4) after 48 hours. FPAEC proliferation in untreated cells was not different under normoxia or hypoxia (2.6 ± 0.3 × 104 vs. 2.7 ± 0.3 × 104 cells/well, P=0.58, n=4) after 48 hours. However, LPS-induced inhibition of FPAEC proliferation was smaller in magnitude in hypoxia vs. normoxia at 48hrs (14% vs. 24%, P=0.04, n=5) and 24hrs (15% vs. 28%, P=0.004, n=4) after LPS treatment (Fig. 1).
Figure 1. Effect of oxygen tension on FPAEC proliferation after LPS treatment.

FPAEC cell growth was assessed after 24- and 48-hrs of LPS (10μg/ml) treatment in normoxia (21% O2) and hypoxia (5% O2). Cell proliferation in LPS treated FPAEC are expressed as a percentage relative to proliferation of untreated cells under normoxic conditions. The data presented above have been pooled from 4 independent experiments at each time-point. Norm C; Normoxia control, Hypox + LPS; LPS treated FPAEC incubated in hypoxia, Norm + LPS; LPS treated FPAEC incubated in normoxia. * - P<0.001, ** -P=0.004, † - P<0.001, ‡ - P=0.04.
Effect of hypoxia on LPS-induced apoptosis and Caspase-3 activity in FPAEC
To examine if the differences in FPAEC proliferation between normoxia and hypoxia after LPS treatment were due to decreased cell death in hypoxia, levels of cellular apoptosis were determined. Treatment with LPS (10μg/mL) induced apoptosis in FPAEC under normoxic conditions at 24hrs (36 ± 2 % vs. 16 ± 3%, P=0.02, n=3) and 48hrs (44 ± 5% vs. 17 ± 3%, P<0.001, n=8). However, LPS-induced FPAEC apoptosis was decreased in hypoxia in comparison with normoxia (Fig. 2A & 2B) at 24 hours (29 ± 1% vs. 36 ± 1%, P<0.015, n=3) and 48 hours (30 ± 3% vs. 44 ± 5%, P=0.001, n=8). There were no differences in apoptosis among untreated FPAEC incubated in hypoxia or normoxia (16 ± 3 % vs. 17 ± 3%, P=0.55, n=4) at 48 hours. These data demonstrate that FPAEC are more resistant to LPS mediated apoptosis at oxygen tensions prevailing in the fetus. While we examined apoptosis at 24 and 48 hours, cell death could have occurred earlier. Although we did not see significant cellular detachment at six and twelve hours after LPS treatment our data could be representative of FPAEC that have survived the initial insult.
Figure 2.
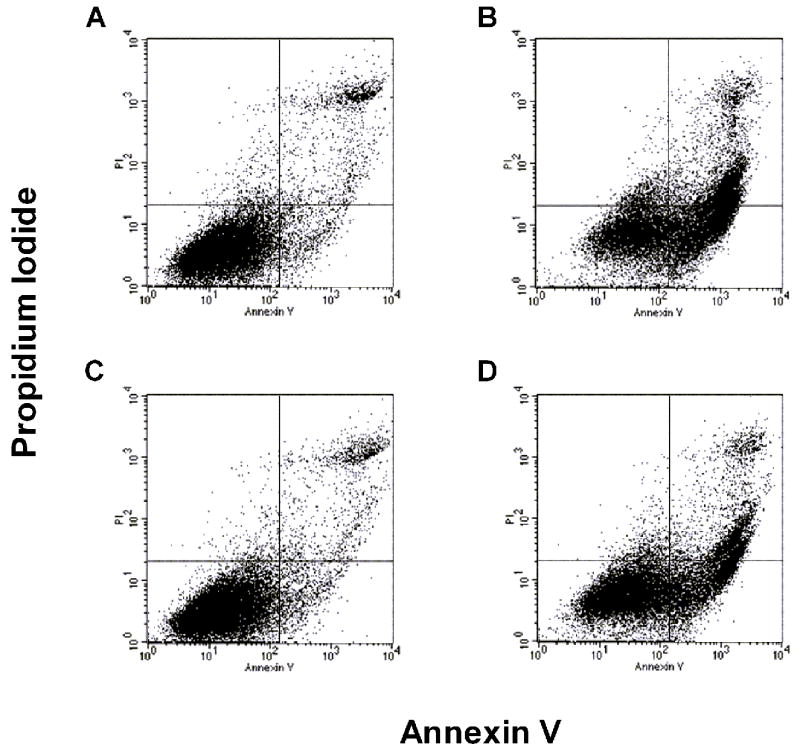

A) Quantification of FPAEC apoptosis by FACS detection of Annexin V binding: Control FPAEC and LPS (10ug/mL) treated FPAEC were incubated in normoxia (21% O2) or hypoxia (5% O2) for 48 hours. A) Untreated FPAEC incubated in normoxia, B) LPS treated FPAEC incubated in normoxia, C) Untreated FPAEC incubated in hypoxia, and D) LPS treated FPAEC incubated in hypoxia.
B) Hypoxia (5%) attenuates LPS-induced apoptosis in FPAEC: LPS-induced apoptosis in FPAEC was reduced by 35% in hypoxia (44% vs. 30%) when compared to normoxia after 48 hours. Cells detected in early and late apoptosis for each condition were pooled and then averaged from eight independent experiments. Norm C; Normoxia control, Norm + LPS; LPS treated FPAEC incubated in normoxia, Hypox C; Hypoxia control, Hypox + LPS; LPS treated FPAEC incubated in hypoxia. * - P<0.001, § -P=0.001, ** - P<0.001.
To elucidate the temporal changes in pro-apoptotic signaling associated with hypoxia-mediated attenuation of LPS injury, we assayed caspase-3 activity at sequential time-points (data not shown). Caspase-3 activity in untreated FPAEC incubated in hypoxia or normoxia were similar. However, caspase-3 activity after 6, 12 and 24 hours of LPS treatment was greater in normoxia in comparison with hypoxia with peak differences observed at 12 hours. Compared to a 3.4 fold increase in caspase-3 activity for LPS treated FPAEC incubated in normoxia, LPS treated FPAEC incubated in hypoxia had only a 2.4-fold increase (3.4 ± 0.6 vs. 2.4 ± 0.5, P<0.05, n=4) at 12 hours. These data, while consistent with our apoptosis data indicate that hypoxia-conferred protection against pro-apoptotic signaling in FPAEC occurs early after LPS exposure.
Effect of hypoxia on LPS-dependent superoxide formation measured by the oxidation of DHE to 2-HE
To test the hypothesis that hypoxia-conferred protection against LPS-induced apoptosis is associated with decreased O2▪− levels, we quantified 2-HE formation from DHE in FPAEC after LPS treatment. In comparison with untreated cells, LPS treatment increased 2-HE levels in FPAEC under normoxic and hypoxic conditions (Fig. 3A). Intracellular 2-HE formation was 40% lower in LPS treated FPAEC incubated in hypoxia when compared to normoxia (0.10 nmol/mg protein vs. 0.19 nmol/mg protein, P=0.001, n=4) at 3 hours post-treatment (Fig. 3B). Similar differences in 2-HE formation between normoxia and hypoxia were observed at 6 hours post-LPS treatment (Fig. 3B). These data demonstrate that increased apoptosis seen in LPS treated FPAEC in normoxia is associated with a parallel increase in intracellular 2-HE formation, strongly indicative of increases in cellular O2▪− formation.
Figure 3.


A) Quantification of intracellular 2-HE levels in LPS treated and untreated FPAEC in hypoxia: 2-HE peaks were quantified in FPAEC by HPLC after n-Butanol extraction. There was a 30% increase in 2-HE levels after LPS treatment in hypoxia-incubated FPAEC at 3 hours. Hypox C; Hypoxia control, Hypox + LPS; LPS treated FPAEC incubated in hypoxia. * - P=0.04.
B) 2-HE levels in LPS treated FPAEC are lower in hypoxia: 2-HE levels were 40% lower in hypoxia vs. normoxia at 3- and 6-hours post LPS treatment. 2-HE levels in LPS treated FPAEC incubated in hypoxia are expressed relative to 2-HE levels in normoxia. Norm + LPS; LPS treated FPAEC incubated in normoxia, Hypox + LPS; LPS treated FPAEC incubated in hypoxia. * - P=0.003 & ** - P<0.001 (n=4 each).
LPS-dependent Expression of CuZnSOD and MnSOD in hypoxia and normoxia
To examine if the difference in O2▪− formation between hypoxia and normoxia was due to increased dismuation, we quantified sequential changes in expression of the two superoxide dismutase (CuZnSOD and MnSOD) proteins at 6, 12 and 24 hours after LPS treatment (Figure 4). LPS treatment strongly induced MnSOD expression in FPAEC by 6 hours with persistent, increased expression seen at 12- and 24-hours after LPS exposure. However, LPS-dependent MnSOD induction was not quantifiably different between normoxia and hypoxia. CuZnSOD expression decreased with LPS treatment in FPAEC after 12hrs to the same extent in hypoxia or normoxia. These data suggest that observed differences in 2-HE levels between LPS treated FPAEC in normoxia and hypoxia were not attributable to changes in expression of CuZnSOD or MnSOD.
Figure 4. Western Blot to quantify sequential changes in MnSOD and CuZnSOD expression.

CuZnSOD and MnSOD protein were quantified at 12- and 24-hours post LPS treatment in FPAEC incubated in normoxia and hypoxia. Protein (20 μg) obtained from cell lysates was used for blotting using standard protocols. β-actin expression was used as loading control. Data are representative of three independent experiments. Norm C; Normoxia control, Norm + LPS; LPS treated FPAEC incubated in normoxia, Hypox C; Hypoxia control, Hypox + LPS; LPS treated FPAEC incubated in hypoxia.
Effect of manipulating superoxide generation on 2-HE formation and apoptosis
LPS-induced, NADPH-oxidase-dependent O2▪− generation has been previously linked to endothelial activation and injury [21, 22]. We therefore investigated whether the observed differences in LPS-induced FPAEC apoptosis between hypoxia and normoxia could be abrogated by exogenous supplementation of O2▪−. For this, menadione, a compound which increases superoxide production via redox cycling [23], was used. Menadione, at identical concentrations used in subsequent apoptosis experiments, increased intracellular formation of 2-HE in LPS-treated FPAEC incubated in hypoxia (Fig. 5A). Pre-treatment with menadione augmented LPS-induced FPAEC apoptosis in hypoxia in a dose-dependent manner (12.5μM to 50μM concentrations) (data not shown). When FPAEC in hypoxia were pre-treated with menadione (12.5μM), LPS-induced apoptosis was similar in normoxia and hypoxia (38 ± 4% vs. 36 ± 4%, P=0.36, n=6), (Fig. 5B). Menadione alone did not increase apoptosis in FPAEC at 12.5μM concentrations. To exclude the possibility that menadione could have augmented FPAEC apoptosis by NADPH depletion we quantified intracellular NADPH levels 3 hours after menadione (12.5μM) treatment. NADPH levels were not different between control FPAEC and menadione-treated FPAEC (0.192 ± 0.01μM vs. 0.193 ± 0.005μM, P=0.86, n=2). This suggests that NADPH depletion may not be a significant contributor to FPAEC apoptosis at 12.5 μM concentrations.
Figure 5.


A) Menadione (12.5μM) increases intracellular levels of 2-HE in LPS-treated FPAEC incubated in hypoxia: 2-HE levels were quantified in FPAEC by HPLC after n-Butanol extraction. 2-HE levels increased by greater than two-fold when LPS treated FPAEC were pre-treated with menadione (0.09 vs. 0.25 nmol 2-HE/mg protein). Hypox + LPS; LPS treated FPAEC incubated in hypoxia, Hypox + LPS + Menadione; LPS and menadione treated FPAEC incubated in hypoxia. * - P<0.001.
B) Pre-treatment of FPAEC incubated in hypoxia with menadione abolishes differences in LPS-induced apoptosis between normoxia and hypoxia: Quantification of apoptosis in FPAEC treated with LPS (10μg/mL) and incubated under normoxic or hypoxic conditions (5% O2) for 48 hours with or without 12.5μM of menadione. Cells detected in early and late apoptosis for each condition were pooled and averaged from six independent experiments. Menadione alone; FPAEC treated with menadione alone, Norm + LPS; LPS treated FPAEC incubated in normoxia, Hypox + LPS; LPS treated FPAEC incubated in hypoxia, Hypox + LPS + Menadione; LPS and menadione treated FPAEC incubated in hypoxia. § - P<0.001, * - P=0.003, ** - P=0.39.
Apocynin (4-acetovanillone), a NADPH oxidase inhibitor [22], was examined as an inhibitor of cellular superoxide generation. Apocynin (400μM) decreased 2-HE formation in FPAEC treated with LPS in normoxia (Fig. 6A). Pre-treatment with apocynin attenuated LPS-induced FPAEC apoptosis in normoxia at concentrations between 200μM (data not shown) and 400μM, with maximum protection observed at 400μM (Fig. 6B). Apocynin decreased the percentage of apoptotic cells after LPS-treatment from 48 ±1 % to 39 ± 1% in normoxia (P=0.002, n=4). These data demonstrate that manipulation of NADPH oxidase dependent O2▪− generation after LPS exposure alters the degree of apoptosis strongly suggesting that hypoxia attenuates LPS-induced FPAEC apoptosis by decreasing O2▪− generated in response to LPS.
Figure 6.


A) Apocynin (400μM) decreases 2-HE levels in LPS-treated FPAEC incubated in normoxia: Intracellular levels of 2-HE were quantified in FPAEC by HPLC after n-Butanol extraction. 2-HE levels decreased by almost 60% when LPS treated FPAEC were pre-treated with apocynin (0.15 vs. 0.06 nmol 2-HE/mg protein). Norm + LPS; LPS treated FPAEC incubated in normoxia, Norm + LPS + Apocynin; LPS and apocynin treated FPAEC incubated in normoxia. * - P=0.015.
B) Apocynin attenuates LPS-induced FPAEC apoptosis in normoxia: Quantification of apoptosis in FPAEC treated with LPS (10μg/mL) ± apocynin (400μM) for 48 hours in normoxia. Cells detected in early and late apoptosis for each condition were pooled and averaged from four independent experiments. Norm C; Normoxia control, Norm + Apocynin; FPAEC treated with apocynin, Norm + LPS; LPS treated FPAEC incubated in normoxia, Norm + LPS + Apocynin; LPS and apocynin treated FPAEC incubated in normoxia. * - P=0.002, ** - P<0.001, § - P=0.71.
Effect of hyperoxia (55% oxygen) on LPS-induced apoptosis in FPAEC
To further demonstrate that ambient levels of oxygen modulate the degree of LPS-induced injury in FPAEC, we compared the severity of LPS-induced apoptosis between normoxia and hyperoxia. Compared to LPS-treated cells incubated in normoxia, LPS-treated FPAEC incubated in hyperoxia (55% O2) exhibited increased apoptosis (67% vs. 49%, P<0.001, n=4) after 48 hours (Fig. 7A). There were no significant differences in the degree of apoptosis among untreated cells incubated in hyperoxia or normoxia (26% vs. 19%, P=0.08, n=4). HPLC analysis revealed a two-fold increase in 2-HE formation in LPS-treated FPAEC in hyperoxia (P<0.001, n=2) when compared to normoxia (Fig. 7B). These data demonstrate that ambient levels of oxygen modulate the severity of LPS-induced apoptosis in FPAEC, and that the toxic effects of oxygen and LPS on FPAEC are cumulative.
Figure 7.


A) LPS-mediated apoptosis in FPAEC is exacerbated in hyperoxia (55%O2): Compared to LPS (10μg/mL) treated cells incubated in normoxia, LPS treated FPAEC incubated in hyperoxia had a 35% increase in apoptosis after 48hrs of treatment. Norm C; Normoxia control, Hyperox C; Hyperoxia control, Norm + LPS; LPS treated FPAEC incubated in normoxia, Hyperox + LPS; LPS treated FPAEC incubated in hyperoxia. Data is representative of four independent experiments. * - P<0.001, ** - P< 0.001, § -P=0.001.
B) 2-HE levels in LPS treated FPAEC are increased by 2-fold in hyperoxia: 2-HE levels in FPAEC were two-fold greater in hyperoxia when compared to normoxia three hours after LPS (10μg/mL) treatment. 2-HE levels in LPS treated FPAEC incubated in hyperoxia are expressed relative to 2-HE levels in normoxia. Norm + LPS; LPS treated FPAEC incubated in normoxia, Hyperox + LPS; LPS treated FPAEC incubated in hyperoxia. * - P<0.002.
DISCUSSION
This study demonstrates that LPS mediated apoptosis in FPAEC is modulated by ambient oxygen concentrations. While hypoxic conditions mimicking the fetal milieu attenuate LPS-induced cytotoxicity, hyperoxia exacerbates it, highlighting the cumulative effect of LPS and oxygen on endothelial toxicity. LPS-dependent 2-HE generation in FPAEC increased in parallel with severity of FPAEC apoptosis from 5%O2 to 55% O2 strongly suggesting that LPS-dependent O2▪− generation is a key mediator of endothelial toxicity. Finally, manipulation of O2▪− production after LPS exposure abolished differences in FPAEC apoptosis between normoxia and hypoxia suggesting that oxygen tension modulates LPS-induced apoptosis in FPAEC by regulating intracellular levels of O2▪−. While this study has generated novel data, a limitation of our study is that we have not elucidated downstream signaling events that link LPS exposure to superoxide generation, though a NADPH oxidase -dependent mechanism is probably involved.
While the effects of varying oxygen tension on LPS-induced cytotoxicity have not been explored before, multiple studies have reported contrasting results regarding the effects of hypoxia on cellular injury[24-27]. Hypoxia has been shown to directly induce apoptosis in human umbilical vein endothelial cells, glomerular endothelial cells and it also potentiates nitric oxide-mediated apoptosis in bovine aortic endothelial cells [24, 26, 28]. Paradoxically, hypoxia protects against etoposide induced apoptosis in a human alveolar epithelial cell line, confers protection against cisplatin-induced tubular epithelial apoptosis and preconditioning with hypoxia attenuates ischemia/re-perfusion injury in myocardiocytes [25, 27, 29]. Our experiments did not demonstrate differences in apoptosis between untreated FPAEC incubated in hypoxia or normoxia, although hypoxia attenuated LPS-induced FPAEC apoptosis. In our model, FPAEC had been subcultured in room air for 5-8 passages before comparisons were made between LPS injury in hypoxia and normoxia. Our experiments were designed to study the effects of LPS at different oxygen concentrations (normoxia, “fetal” normoxia and postnatal hyperoxia) in the immature lung of premature infants; with a view towards understanding the development of vascular injury in bronchopulmonary dysplasia. As the endothelial cells had been conditioned in normoxia before they were exposed to fetal oxygen concentrations, our data is representative of LPS-induced FPAEC injury in premature infants who have transitioned to the extra-uterine environment. Another interesting, alternate approach would have been the continuous subculture of freshly harvested FPAEC in 5-10% oxygen before LPS treatment in normoxia or hyperoxia. We used 5% oxygen as our hypoxic threshold to mimic the fetal pulmonary milieu. Our results are similar to the data obtained by Balasubramaniam et al [30] in ovine FPAEC demonstrating no differences in the baseline apoptotic rates in FPAEC incubated in normoxia or hypoxia (3%). Consistent with our data elucidating the protective effects of moderate hypoxia, Yang et al [31] noted that rats maintained in a high-altitude chamber (mimicking hypoxic preconditioning) were more resistant to intraperitoneal LPS exposure compared to rats maintained at sea-level. Changes in intracellular ROS production seen with varying degrees of hypoxia may account for differential effects of hypoxia on apoptosis [16, 17, 32]. Muzaffar et al [32] were able to demonstrate increased O2▪− production with acute anoxia in porcine pulmonary arteries while Fresquet et al [33] reported increased ROS generation in mouse pulmonary arteries with chronic hypoxia. Our experiments demonstrate that moderate hypoxia (5% O2) decreases LPS-induced O2▪− generation in FPAEC when compared to normoxia, and that LPS dependent O2▪− generation increases in parallel with ambient oxygen concentrations. Our results are consistent with Mehta et al [17] who reported decreased ROS production in human pulmonary artery smooth muscle cells and human coronary smooth muscle cells incubated in hypoxia (5%). Based on these studies and the current data, we speculate that anoxic conditions augment apoptosis while more moderate degrees of hypoxia confer protection against apoptogenic stimuli in association with increased or decreased ROS generation, respectively.
Generation of ROS can lead to imbalances between pro- and antioxidant states, resulting in oxidative stress and cell injury [34]. Our data are consistent with other investigators who have shown that agents that decrease ROS generation protect against LPS-induced cytotoxicity [10, 11]. We attempted to explain decreased O2▪− generation after LPS exposure in hypoxia by quantifying changes in the expression of MnSOD and CuZnSOD. Consistent with published reports [35, 36], we noted a strong induction of MnSOD protein after LPS exposure, which was similar in hypoxia and normoxia. Since LPS-induced O2▪− production has been implicated in endothelial activation and injury [21], we hypothesized that differences in O2▪− generation between hypoxia and normoxia resulted in the observed differences in LPS-induced FPAEC apoptosis. Treatment of FPAEC incubated in hypoxia with menadione abolished the differences in LPS-induced apoptosis between normoxia and hypoxia at concentrations which were not directly toxic to FPAEC. Menadione augmented FPAEC apoptosis in association with increased O2▪− generation, but without a concomitant decrease in NADPH. Our data is consistent with Warren et al [37] who noted that bovine capillary endothelial cells were resistant to menadione at lower concentrations but menadione directly induced apoptosis in a dose-dependent manner at higher concentrations in parallel with increasing O2▪− levels. To test our hypothesis further, we studied the effect of apocynin on LPS-induced O2▪− generation and apoptosis in FPAEC under normoxic conditions. Consistent with other investigators who have shown that apocynin attenuates LPS mediated endothelial activation and injury [21, 22, 38]; apocynin decreased O2▪− levels in LPS-treated FPAEC and attenuated FPAEC apoptosis in normoxia. While apocynin inhibited O2▪− formation by greater than 60%, apoptosis was decreased by only 25% suggesting that LPS-induced FPAEC apoptosis is also propagated by other mechanisms. Apocynin-conferred protection against LPS-induced apoptosis in FPAEC could result from an inhibition of NADPH oxidase activity or due to a direct antioxidant effect [22, 39]. Our data, while confirming that O2▪− generated in response to LPS is a key mediator of endothelial cytotoxicity, also suggests that observed differences in LPS-induced apoptosis under varying oxygen concentration is attributable to differences in intracellular levels of O2▪−.
Since the developing lung of premature infants is frequently exposed to hyperoxia and endotoxin together [7, 40, 41], we explored the effects of moderate hyperoxia (55% O2) and LPS on FPAEC apoptosis. Our data demonstrates that there is a cumulative toxic effect of hyperoxia and LPS on FPAEC apoptosis. Evidence of a cumulative effect of hyperoxia and LPS on the inflammatory response and pulmonary injury has been reported before [42, 43]. O’Brien-Ladner et al [43] reported that while unstimulated alveolar macrophages did not release TNF-α in hyperoxia (95% O2), the addition of LPS resulted in a significant augmentation of the TNF-α response in hyperoxia when compared to normoxia. Kohno M et al [42] demonstrated that hyperoxia (75%) exacerbated the emphysematous changes in the rodent lung during the sub-acute phase of chronic endotoxin injury in rats. However, the above studies are in contrast to the observations made by Frank et al [44, 45]. They reported that adult rats that were incubated in 40% oxygen (or an increasing oxygen gradient 40%-60%-85%) and administered intra-peritoneal LPS were protected against subsequent lethal hyperoxia (95% O2) [44]. Mechanistically, pre-treatment with LPS induced MnSOD and other enzymes that attenuate oxidant stress resulting in protection against subsequent hyperoxia stress. In neonatal rats, LPS did not confer protection against lethal hyperoxia during the two weeks of hyperoxia treatment; however, differences in survival became apparent during the subsequent wean to room air [45]. Our use of sub-lethal concentrations of oxygen to mimic hyperoxia (55% O2) is more relevant to the clinical scenario prevalent in premature infants now, and is consistent with the results obtained by Kohno et al [42] using sub-lethal oxygen concentrations (75%). Some of the paradoxical results observed among the various animal and cell-culture models probably relate to the differences in the concentration of oxygen used, the timing and duration of the LPS exposure with respect to the hyperoxia stress and the maturity of the animal or cells (adult vs. fetal) used. Although our aim for the experiments in hyperoxia was to demonstrate a dose-response relationship between LPS toxicity and ambient oxygen tension in fetal pulmonary endothelial cells, it is unlikely that FPAEC will be exposed to significant hyperoxia in-vivo. The paO2 in the pulmonary artery relates closely to the mixed venous paO2 and fetal hemoglobin acts a buffer against hyperoxia. While the end-points being studied were different, it is interesting to note that LPS-induced downstream effects on inflammation, apoptosis and emphysema varied with ambient oxygen concentrations. This warrants the speculation that NADPH oxidase-dependent O2▪− production might be the critical pathway by which the redox state modulates LPS responses at a cellular level. Pursuing this hypothesis, we could demonstrate that LPS induced O2▪− generation was two-fold higher in FPAEC incubated in hyperoxia when compared to normoxia.
This study enhances our understanding of vascular injury in BPD and has potential implications for premature infants. The protective effects of hypoxia on endotoxin mediated lung injury might explain why intra-amniotic endotoxin exposure in-utero is not associated with severe inflammation and lung injury [46]. We also speculate that cumulative toxicity from LPS and oxygen might explain why premature infants exposed to chorioamnionitis develop “new” BPD despite not being ventilated [47]. Moreover, injudicious use of supplemental oxygen during endotoxemia might exacerbate endothelial injury. In conclusion, our study demonstrates a protective effect of moderate hypoxia against LPS-mediated oxidative stress and endothelial toxicity. Future studies will seek corroboration of the current observations in animal models and will determine the signaling pathways by which oxygen tension modulates LPS-induced O2▪− generation and endothelial toxicity.
Acknowledgments
We would like to thank Dr Jacek Zielonka, PhD and Jeremy T. Leverence, BS with the Departments of Biophysics and Free Radical Research Center (JZ), and Department of Pediatrics (JL), Medical College of Wisconsin, for their help with HPLC quantification and partial pressure estimations. We would also like to thank Dr Ronald N. Hines, PhD, Professor of Pediatrics and Pharmacology/Toxicology, Medical College of Wisconsin for his critical input in the preparation of this manuscript.
Abbreviations
- LPS
Lipopolysaccharide
- BPD
Bronchopulmonary dysplasia
- ROS
Reactive oxygen species
- FPAEC
Fetal pulmonary artery endothelial cells
- O2▪−
Superoxide
- DMEM
Dulbecco’s Modified Eagle medium
- FBS
Fetal Bovine serum
- 2-HE
2-Hydroxyethidium
- CuZnSOD
Copper Zinc Superoxide dismutase
- MnSOD
Manganese Superoxide dismutase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fanaroff AA, Stoll BJ, Wright LL, Carlo WA, Ehrenkranz RA, Stark AR, Bauer CR, Donovan EF, Korones SB, Laptook AR, Lemons JA, Oh W, Papile LA, Shankaran S, Stevenson DK, Tyson JE, Poole WK. Trends in neonatal morbidity and mortality for very low birthweight infants. American journal of obstetrics and gynecology. 2007;196(147):e141–148. doi: 10.1016/j.ajog.2006.09.014. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH. Bronchopulmonary dysplasia: “a vascular hypothesis”. American journal of respiratory and critical care medicine. 2001;164:1755–1756. doi: 10.1164/ajrccm.164.10.2109111c. [DOI] [PubMed] [Google Scholar]
- 3.Jobe AJ. The new BPD: an arrest of lung development. Pediatric research. 1999;46:641–643. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Kallapur SG, Jobe AH. Contribution of inflammation to lung injury and development. Archives of disease in childhood. 2006;91:F132–135. doi: 10.1136/adc.2004.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kallapur SG, Bachurski CJ, Le Cras TD, Joshi SN, Ikegami M, Jobe AH. Vascular changes after intra-amniotic endotoxin in preterm lamb lungs. American journal of physiology. 2004;287:L1178–1185. doi: 10.1152/ajplung.00049.2004. [DOI] [PubMed] [Google Scholar]
- 6.Wang W, Wei W, Ning Q, Luo XP. Effect of intra-amniotic endotoxin priming plus hyperoxic exposure on the expression of vascular endothelial growth factor and its receptors in lungs of preterm newborn rats. Zhonghua er ke za zhi. 2007;45:533–538. [PubMed] [Google Scholar]
- 7.Cordero L, Ayers LW, Davis K. Neonatal airway colonization with gram-negative bacilli: association with severity of bronchopulmonary dysplasia. The Pediatric infectious disease journal. 1997;16:18–23. doi: 10.1097/00006454-199701000-00005. [DOI] [PubMed] [Google Scholar]
- 8.Young KC, Del Moral T, Claure N, Vanbuskirk S, Bancalari E. The association between early tracheal colonization and bronchopulmonary dysplasia. J Perinatol. 2005;25:403–407. doi: 10.1038/sj.jp.7211297. [DOI] [PubMed] [Google Scholar]
- 9.Speer CP. Inflammation and bronchopulmonary dysplasia: a continuing story. Seminars in fetal & neonatal medicine. 2006;11:354–362. doi: 10.1016/j.siny.2006.03.004. [DOI] [PubMed] [Google Scholar]
- 10.Brigham KL, Meyrick B, Berry LC, Jr, Repine JE. Antioxidants protect cultured bovine lung endothelial cells from injury by endotoxin. J Appl Physiol. 1987;63:840–850. doi: 10.1152/jappl.1987.63.2.840. [DOI] [PubMed] [Google Scholar]
- 11.Kim KY, Shin HK, Choi JM, Hong KW. Inhibition of lipopolysaccharide-induced apoptosis by cilostazol in human umbilical vein endothelial cells. The Journal of pharmacology and experimental therapeutics. 2002;300:709–715. doi: 10.1124/jpet.300.2.709. [DOI] [PubMed] [Google Scholar]
- 12.Bannerman DD, Goldblum SE. Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. American journal of physiology. 2003;284:L899–914. doi: 10.1152/ajplung.00338.2002. [DOI] [PubMed] [Google Scholar]
- 13.Irani K. Oxidant signaling in vascular cell growth, death, and survival : a review of the roles of reactive oxygen species in smooth muscle and endothelial cell mitogenic and apoptotic signaling. Circulation research. 2000;87:179–183. doi: 10.1161/01.res.87.3.179. [DOI] [PubMed] [Google Scholar]
- 14.Zhuang S, Lynch MC, Kochevar IE. Caspase-8 mediates caspase-3 activation and cytochrome c release during singlet oxygen-induced apoptosis of HL-60 cells. Experimental cell research. 1999;250:203–212. doi: 10.1006/excr.1999.4501. [DOI] [PubMed] [Google Scholar]
- 15.Fleury C, Mignotte B, Vayssiere JL. Mitochondrial reactive oxygen species in cell death signaling. Biochimie. 2002;84:131–141. doi: 10.1016/s0300-9084(02)01369-x. [DOI] [PubMed] [Google Scholar]
- 16.Li AE, Ito H, Rovira II, Kim KS, Takeda K, Yu ZY, Ferrans VJ, Finkel T. A role for reactive oxygen species in endothelial cell anoikis. Circulation research. 1999;85:304–310. doi: 10.1161/01.res.85.4.304. [DOI] [PubMed] [Google Scholar]
- 17.Mehta JP, Li Campian J, Guardiola J, Cabrera JA, Weir KE, Eaton JW. Generation of Oxidants by Hypoxic Human Pulmonary and Coronary Smooth Muscle Cells. Chest. 2008 doi: 10.1378/chest.07-2984. [DOI] [PubMed] [Google Scholar]
- 18.Konduri GG, Bakhutashvili I, Eis A, Pritchard K., Jr Oxidant stress from uncoupled nitric oxide synthase impairs vasodilation in fetal lambs with persistent pulmonary hypertension. Am J Physiol Heart Circ Physiol. 2007;292:H1812–1820. doi: 10.1152/ajpheart.00425.2006. [DOI] [PubMed] [Google Scholar]
- 19.Zhao H, Joseph J, Fales HM, Sokoloski EA, Levine RL, Vasquez-Vivar J, Kalyanaraman B. Detection and characterization of the product of hydroethidine and intracellular superoxide by HPLC and limitations of fluorescence. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:5727–5732. doi: 10.1073/pnas.0501719102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Merker MP, Bongard RD, Kettenhofen NJ, Okamoto Y, Dawson CA. Intracellular redox status affects transplasma membrane electron transport in pulmonary arterial endothelial cells. American journal of physiology. 2002;282:L36–43. doi: 10.1152/ajplung.00283.2001. [DOI] [PubMed] [Google Scholar]
- 21.Al Ghouleh I, Magder S. Nicotinamide adenine dinucleotide phosphate (reduced form) oxidase is important for LPS-induced endothelial cell activation. Shock (Augusta, Ga) 2008;29:553–559. doi: 10.1097/SHK.0b013e318157ebc8. [DOI] [PubMed] [Google Scholar]
- 22.Matthiesen S, Lindemann D, Warnken M, Juergens UR, Racke K. Inhibition of NADPH oxidase by apocynin inhibits lipopolysaccharide (LPS) induced up-regulation of arginase in rat alveolar macrophages. European journal of pharmacology. 2008;579:403–410. doi: 10.1016/j.ejphar.2007.10.043. [DOI] [PubMed] [Google Scholar]
- 23.Criddle DN, Gillies S, Baumgartner-Wilson HK, Jaffar M, Chinje EC, Passmore S, Chvanov M, Barrow S, Gerasimenko OV, Tepikin AV, Sutton R, Petersen OH. Menadione-induced reactive oxygen species generation via redox cycling promotes apoptosis of murine pancreatic acinar cells. The Journal of biological chemistry. 2006;281:40485–40492. doi: 10.1074/jbc.M607704200. [DOI] [PubMed] [Google Scholar]
- 24.Eguchi R, Suzuki A, Miyakaze S, Kaji K, Ohta T. Hypoxia induces apoptosis of HUVECs in an in vitro capillary model by activating proapoptotic signal p38 through suppression of ERK1/2. Cellular signalling. 2007;19:1121–1131. doi: 10.1016/j.cellsig.2006.12.004. [DOI] [PubMed] [Google Scholar]
- 25.Lee SM, Lee CT, Kim YW, Han SK, Shim YS, Yoo CG. Hypoxia confers protection against apoptosis via PI3K/Akt and ERK pathways in lung cancer cells. Cancer letters. 2006;242:231–238. doi: 10.1016/j.canlet.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 26.Tanaka T, Miyata T, Inagi R, Kurokawa K, Adler S, Fujita T, Nangaku M. Hypoxia-induced apoptosis in cultured glomerular endothelial cells: involvement of mitochondrial pathways. Kidney international. 2003;64:2020–2032. doi: 10.1046/j.1523-1755.2003.00301.x. [DOI] [PubMed] [Google Scholar]
- 27.Wang J, Biju MP, Wang MH, Haase VH, Dong Z. Cytoprotective effects of hypoxia against cisplatin-induced tubular cell apoptosis: involvement of mitochondrial inhibition and p53 suppression. J Am Soc Nephrol. 2006;17:1875–1885. doi: 10.1681/ASN.2005121371. [DOI] [PubMed] [Google Scholar]
- 28.Walford GA, Moussignac RL, Scribner AW, Loscalzo J, Leopold JA. Hypoxia potentiates nitric oxide-mediated apoptosis in endothelial cells via peroxynitrite-induced activation of mitochondria-dependent and -independent pathways. The Journal of biological chemistry. 2004;279:4425–4432. doi: 10.1074/jbc.M310582200. [DOI] [PubMed] [Google Scholar]
- 29.Wu X, Liu X, Zhu X, Tang C. Hypoxic preconditioning induces delayed cardioprotection through p38 MAPK-mediated calreticulin upregulation. Shock (Augusta, Ga) 2007;27:572–577. doi: 10.1097/01.shk.0000246901.58068.a8. [DOI] [PubMed] [Google Scholar]
- 30.Balasubramaniam V, Maxey AM, Fouty BW, Abman SH. Nitric oxide augments fetal pulmonary artery endothelial cell angiogenesis in vitro. American journal of physiology. 2006;290:L1111–1116. doi: 10.1152/ajplung.00431.2005. [DOI] [PubMed] [Google Scholar]
- 31.Yang CC, Ma MC, Chien CT, Wu MS, Sun WK, Chen CF. Hypoxic preconditioning attenuates lipopolysaccharide-induced oxidative stress in rat kidneys. The Journal of physiology. 2007;582:407–419. doi: 10.1113/jphysiol.2006.122747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muzaffar S, Shukla N, Angelini GD, Jeremy JY. Acute hypoxia simultaneously induces the expression of gp91phox and endothelial nitric oxide synthase in the porcine pulmonary artery. Thorax. 2005;60:305–313. doi: 10.1136/thx.2003.018796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fresquet F, Pourageaud F, Leblais V, Brandes RP, Savineau JP, Marthan R, Muller B. Role of reactive oxygen species and gp91phox in endothelial dysfunction of pulmonary arteries induced by chronic hypoxia. British journal of pharmacology. 2006;148:714–723. doi: 10.1038/sj.bjp.0706779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pohlman TH, Harlan JM. Adaptive responses of the endothelium to stress. The Journal of surgical research. 2000;89:85–119. doi: 10.1006/jsre.1999.5801. [DOI] [PubMed] [Google Scholar]
- 35.Visner GA, Block ER, Burr IM, Nick HS. Regulation of manganese superoxide dismutase in porcine pulmonary artery endothelial cells. The American journal of physiology. 1991;260:L444–449. doi: 10.1152/ajplung.1991.260.6.L444. [DOI] [PubMed] [Google Scholar]
- 36.Visner GA, Dougall WC, Wilson JM, Burr IA, Nick HS. Regulation of manganese superoxide dismutase by lipopolysaccharide, interleukin-1, and tumor necrosis factor. Role in the acute inflammatory response. The Journal of biological chemistry. 1990;265:2856–2864. [PubMed] [Google Scholar]
- 37.Warren MC, Bump EA, Medeiros D, Braunhut SJ. Oxidative stress-induced apoptosis of endothelial cells. Free radical biology & medicine. 2000;29:537–547. doi: 10.1016/s0891-5849(00)00353-1. [DOI] [PubMed] [Google Scholar]
- 38.Ben-Shaul V, Lomnitski L, Nyska A, Carbonatto M, Peano S, Zurovsky Y, Bergman M, Eldridge SR, Grossman S. Effect of natural antioxidants and apocynin on LPS-induced endotoxemia in rabbit. Human & experimental toxicology. 2000;19:604–614. doi: 10.1191/096032700666138364. [DOI] [PubMed] [Google Scholar]
- 39.Heumuller S, Wind S, Barbosa-Sicard E, Schmidt HH, Busse R, Schroder K, Brandes RP. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension. 2008;51:211–217. doi: 10.1161/HYPERTENSIONAHA.107.100214. [DOI] [PubMed] [Google Scholar]
- 40.Stoll BJ, Hansen N, Fanaroff AA, Wright LL, Carlo WA, Ehrenkranz RA, Lemons JA, Donovan EF, Stark AR, Tyson JE, Oh W, Bauer CR, Korones SB, Shankaran S, Laptook AR, Stevenson DK, Papile LA, Poole WK. Late-onset sepsis in very low birth weight neonates: the experience of the NICHD Neonatal Research Network. Pediatrics. 2002;110:285–291. doi: 10.1542/peds.110.2.285. [DOI] [PubMed] [Google Scholar]
- 41.Stoll BJ, Hansen NI, Higgins RD, Fanaroff AA, Duara S, Goldberg R, Laptook A, Walsh M, Oh W, Hale E. Very low birth weight preterm infants with early onset neonatal sepsis: the predominance of gram-negative infections continues in the National Institute of Child Health and Human Development Neonatal Research Network, 2002-2003. The Pediatric infectious disease journal. 2005;24:635–639. doi: 10.1097/01.inf.0000168749.82105.64. [DOI] [PubMed] [Google Scholar]
- 42.Kohno M, Ishizaka A, Sawafuji M, Koh H, Hirayama Y, Ikeda E, Shiomi T, Ohashi A, Okada Y, Kobayashi K. Hyperoxia-induced emphysematous changes in subacute phase of endotoxin-induced lung injury in rats. American journal of physiology. 2004;287:L184–190. doi: 10.1152/ajplung.00324.2003. [DOI] [PubMed] [Google Scholar]
- 43.O’Brien-Ladner AR, Nelson ME, Cowley BD, Jr, Bailey K, Wesselius LJ. Hyperoxia amplifies TNF-alpha production in LPS-stimulated human alveolar macrophages. American journal of respiratory cell and molecular biology. 1995;12:275–279. doi: 10.1165/ajrcmb.12.3.7873193. [DOI] [PubMed] [Google Scholar]
- 44.Frank L. Endotoxin reverses the decreased tolerance of rats to greater than 95% O2 after preexposure to lower O2. J Appl Physiol. 1981;51:577–583. doi: 10.1152/jappl.1981.51.3.577. [DOI] [PubMed] [Google Scholar]
- 45.Frank L. Oxygen toxicity in neonatal rats: the effect of endotoxin treatment on survival during and post-O2 exposure. Pediatric research. 1987;21:109–115. doi: 10.1203/00006450-198702000-00001. [DOI] [PubMed] [Google Scholar]
- 46.Kallapur SG, Nitsos I, Moss TJ, Kramer BW, Newnham JP, Ikegami M, Jobe AH. Chronic endotoxin exposure does not cause sustained structural abnormalities in the fetal sheep lungs. American journal of physiology. 2005;288:L966–974. doi: 10.1152/ajplung.00389.2004. [DOI] [PubMed] [Google Scholar]
- 47.Ikegami M, Jobe AH. Postnatal lung inflammation increased by ventilation of preterm lambs exposed antenatally to Escherichia coli endotoxin. Pediatric research. 2002;52:356–362. doi: 10.1203/00006450-200209000-00008. [DOI] [PubMed] [Google Scholar]