

Abstract
Novobiocin, a known DNA gyrase inhibitor, binds to a nucleotide-binding site located on the C-terminus of Hsp90 and induces degradation of Hsp90-dependent client proteins at ~700 μM in breast cancer cells (SkBr3). Although many analogues of novobiocin have been synthesized, it was only recently demonstrated that monomeric species can exhibit anti-proliferative activity against various cancer cell lines. To further refine the essential elements of the coumarin core, a series of modified coumarin derivatives was synthesized and evaluated for elucidation of structure–activity relationships for novobiocin as an anti-cancer agent. Results obtained from these studies have produced novobiocin analogues that manifest low micromolar activity against several cancer cell lines.
Introduction
Increasing resistance to chemotherapeutics and knowledge that cancers employ multiple abnormalities have resulted in an increased interest to target the Hsp90 molecular chaperone for the treatment of cancer.1 Hsp90 is a molecular chaperone that is responsible for the folding and conformational maintenance of more than 100 Hsp90-dependent client proteins.2,3 In vivo, Hsp90 substrates have been implicated in cellular signaling networks such as those mediated by steroid hormone receptors, transcription factors and protein kinases, many of which represent individually sought-after anti-tumor targets.4–9 Because Hsp90 plays a unique role in regulating the function and stability of a growing number of proteins upon which cancer cells depend for survival, as well as many mutated and overexpressed proteins that contribute to cancer cell proliferation, Hsp90 has evolved into a promising drug target.10
Hsp90 inhibition results in the destabilization of a substrate-bound heteroprotein complex, which in turn, leads to degradation of Hsp90-dependent clients via ubiquitination of the unfolded client followed by proteasome-mediated hydrolysis.11–16 As such, small molecule inhibitors of Hsp90 transform the protein folding machinery into a catalyst for protein degradation.17 Through Hsp90 inhibition, depletion of multiple oncogenic proteins readily occurs and the outcome is similar to a combinatorial attack on multiple signaling nodes that contribute to oncogenic signaling and modulation of the malignant phenotype.1,18,19 Hsp90 has been shown to be overexpressed in a wide variety of human malignancies and provides an opportunity for high differential selectivity.1,18,20 The ability to simultaneously disrupt multiple signaling events through a single target makes Hsp90 an attractive protein for the development of chemotherapeutic agents.1,19,21
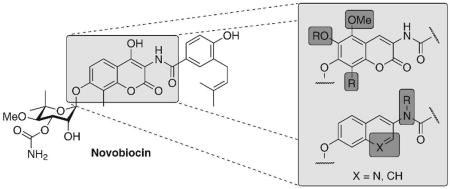
Novobiocin (Figure 1), a member of the coumermycin family of antibiotics, binds to the ATP-binding pocket of DNA gyrase and elicits antimicrobial activity through inhibition of ATP-hydrolysis.22–25 Co-crystal structures of GyrB, the B subunit of DNA gyrase, bound to novobiocin and ADP revealed both molecules bind in a bent conformation,26–28 similar to the manner in which Hsp90 binds ADP.29 With prior knowledge that novobiocin manifests cytotoxicity and binds to a similarly shaped pocket in DNA Gyrase,25,30–33 Neckers and co-workers demonstrated that novobiocin also binds Hsp90 and exhibits anti-tumor activity (~700 μM) against human breast cancer cells. In testing their hypothesis, Neckers and coworkers showed via Western blot analyses of SKBr3 cell lysates that novobiocin induces degradation of Hsp90-dependent clients in a concentration-dependent manner. Related studies in which truncated variants of Hsp90 were eluted from an immobilized novobiocin solid-support revealed that only the C-terminus of Hsp90 was capable of binding novobiocin, which is in contrast to other Hsp90 inhibitors that bind solely to the well-established N-terminal ATP-binding site.34
Figure 1.

Structure of novobiocin.
In addition, it was found that inhibitors bound to the Hsp90 N-terminus were readily displaced by occupation of the C-terminus by novobiocin, which is not recipricol.34,35 It has been proposed that novobiocin may antagonize Hsp90 function by inducing a conformational change that results in separation of the homodimeric C-terminal domains and subsequent release of substrate36, however studies with improved analogues are needed to confirm this hypothesis. It is proposed that the synthesis of improved analogues will not only allow elucidation of the Hsp90 C-terminal nucleotide-binding pocket, but will also provide insight into the unique mechanism exhibited by Hsp90 during the complex protein folding process.36
Several groups have attempted to develop improved analogues of novobiocin in order to improve its comparatively poor Hsp90 inhibitory activity.34 A library of novobiocin analogues disclosed in 2005 demonstrated that A4 (Figure 2) induced degradation of Hsp90-dependent client proteins at ~70-fold lower concentration than novobiocin.11 The structure of A4 included a shortened N-acyl side chain, removal of the 4-hydroxy substituent and an absent carbamoyl group on the noviose appendage. More importantly, this study highlighted that attachment of the noviose moiety to the 7-position and an amide linker at the 3-position of the coumarin ring are critical for anti-Hsp90 activity.11 To confirm the observed SAR trends elucidated from this library, two natural product analogues were prepared and evaluated, DHN1 and DHN2 (Figure 2). Upon evaluation of these molecules in several assays, it was confirmed that the 4-hydroxyl and the 3′-carbamate are detrimental to Hsp90 inhibitory activity, but critical for DNA gyrase inhibition.14
Figure 2.

Structures of A4, DHN1, DHN2, and A4 dimer.
Compound A4 was found to exhibit unique, previously-undocumented activities. A4 induced Hsp90 at concentrations 1000–10000-fold lower than that required for client protein degradation and was thus evaluated for neuroprotective activity. A4 was found to produce an EC50 at 6 nM and exhibited no toxicity at any concentration tested in a model for Alzheimer’s disease.11 In contrast to the monomeric species, the A4-dimer, based on the structure of coumermycin A1, was found to manifest anti-proliferative activity. These results suggested that modification of the amide side chain resulted in conversion of a nontoxic molecule into an anti-proliferative agent.37 Consequently, a series of monomeric species based on A4 was synthesized and evaluated for antitumor activity.38 This later study described the synthesis and evaluation of biaryl and heterocyclic amide-derivatives that explore hydrogen-bonding interactions with the putative novobiocin binding pocket that typically binds the prenylated benzamide of the natural product. Eventually these studies led to the first set of SAR for the amide side chain.
Although the Hsp90 C-terminus does not exhibit ATPase activity, it does play a critical role in conformational rearrangement upon ATP binding.36 To further explore SAR, derivatives of A4 with variations to the coumarin scaffold were designed to probe the importance of interactions typically manifested by the purine ring. These coumarin-derived motifs possess hydrogen bonding capabilities similar to the nucleotide bases, adenine and guanine, and contain strategically placed hydrogen bond acceptors and donors and alkyl groups of variable size to probe the size and nature of the complementary binding pocket. The design, synthesis and evaluation of such compounds are described in this article.
Results and Discussion
Design of new novobiocin analogues
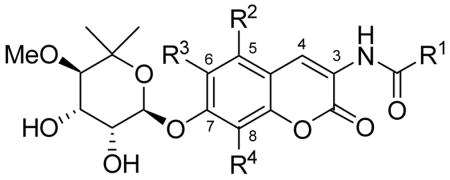
To elucidate structure-activity relationships for the coumarin ring system of novobiocin, we envisioned construction of novobiocin analogues with modified coumarin cores. As shown in Scheme 1, the derivatives were assembled in a modular fashion allowing sequential coupling of noviose and a series of benzoic acids with the modified coumarin cores. We previously demonstrated that the trichloroacetimidate of noviose carbonate couples readily to coumarin phenols in good yield, to afford the corresponding α-anomer.39 The benzoic acids selected were based upon previously obtained SAR for the amide side chain as described by Burlison and co-workers.38
Scheme 1.

Retrosynthesis of novobiocin analogues.
The coumarin scaffolds were designed to complement interactions present on the purine nucleus, via probing the importance of hydrogen bond donor and acceptors in positions surrounding the aromatic ring system. Rationale for these analogues is based on the identification of additional interactions with the nucleotide-binding domain that typically binds the corresponding purine substrate and may lead to enhanced inhibitory affinity for these compounds. Minor perturbations were made on each analogue; however, the addition of one hydrogen bond can produce 1–2 kcal/mol of binding energy and thus increase binding by 10 fold.40 Therefore, these complementary interactions can exhibit a substantial impact on binding and subsequent inhibition. While it has been demonstrated that the N-terminal site is fairly specific for adenine nucleotides, the C-terminal site has been shown to be more promiscuous, and binds both purines and pyrimidines. While adenine specifically binds to the N-terminus, GTP and UTP are specific C-terminal substrates.41 Based on these previous studies, mimics of the guanosine nucleus were chosen to take advantage of this differential. Hydrogen-bond acceptors were placed at the 5-, 6- and 8-positions of the coumarin ring to mimic those at the 6-, 7- and 3-positions of guanine, respectively (Figure 3). Additionally, analogues bearing modification to the coumarin lactone were constructed to probe the importance of hydrogen bond donors/acceptors as well as to potentially improve upon the solubility of the novobiocin scaffold. The activity of such compounds is likely to provide insight into the interactions that are essential or those that can be further optimized.
Figure 3.

Complementarity of GTP and coumarin analogues.
There is limited knowledge regarding the shape and dimension of the pocket since the discovery of the C-terminal binding site is a recent achievement and no Hsp90 co-crystal structure bound to C-terminal inhibitors exists. Therefore, several analogues were designed to probe the breadth of the pocket at positions that potentially project into hydrophobic regions. Alkyl and aryl groups of variable size were attached at the 5-, 6- and 8-positions of the coumarin ring to maximize putative hydrophobic interactions and to optimize affinity. A methoxy group was attached at the 5-poistion of the coumarin ring, while methoxy, propoxy, and isopropoxy ethers were installed at the 6-position. Methyl, methoxy, benzyl and phenyl substituents were placed at the 8-position, offering a variety of possible interactions to fill the pocket that is occupied by the chlorine of chlorobiocin and the methyl substituent of novobiocin. The culmination of structure–activity relationships elucidated by such compounds is likely to provide a platform upon which improved analogues can be sought.
Syntheses of 5-, 6-, and 8-alkyl(oxy) novobiocin analogues
To generate the resorcinol precursors with substitutions at the 4-position, which result in coumarin ring systems with appendages at the 6-position, the phenols of benzaldehyde 1 were protected as the corresponding ethers (Scheme 2). The resulting benzaldehydes (2a–b)42 were converted to their formate esters via Baeyer-Villiger oxidation, and then hydrolyzed to afford phenols 3a–b.43,44 O-Alkylation with the requisite alkyl iodide proceeded in good yield and generated a series of protected 4-substituted resorcinolic ethers (4a–c). Ortho-lithiation of 4a–c, followed by alkylation with methyl iodide provided the 2-methyl protected resorcinols, 5a–c.45 Deprotection46 of the alkoxy ethers by exposure to acidic conditions gave resorcinols 6a–c.
Scheme 2.

Syntheses of 4-substituted resorcinols. OEOM = OCH2OEt.
To generate resorcinol precursors with substitutions at the 5-position, the phenols of 5-methoxy resorcinol 7 were once again protected as the corresponding alkoxy ethers, 8 (Scheme 3). Ortho-lithiation of 8, followed by treatment with methyl iodide, led to installation of a methyl group at the 2-position of 9.45 Acidic deprotection46 was employed to afford resorcinol 10.
Scheme 3.

Synthesis of 5-substituted resorcinol.
To generate the resorcinol precursors with aryl substituents at the 2-position, the phenols of resorcinol 11 were protected as the corresponding alkoxy ethers, 12 (Scheme 4). Subsequent ortho-lithiation of 12, followed by the addition of benzyl bromide provided the benzyl derivative, 13.45 Removal of the ether protecting groups46 gave diphenol 14.45 The anion of resorcinol 12 was also employed to construct the corresponding 2-iodide via reaction with iodine to yield 15.47 A Suzuki coupling in the presence of biaryl ligand S-Phos,48 was used to generate biaryl 16, which underwent deprotection46 to provide 17.
Scheme 4.

Syntheses of 2-substituted resorcinols.
To generate resorcinol precursors with alkyl substitutions at the 2-position, pyragallol (18) was O-alkylated with methyl iodide to generate 2-methoxy resorcinol amongst an inseparable mixture of regioisomers (Scheme 5). The mixture was subsequently subjected to coumarin formation and the corresponding products isolated. Preparation of 2-ethyl resorcinol (21) from 2,6-dihydroxyacetophenone (20) was accomplished according to published procedures.49
Scheme 5.

Synthesis of 2-methoxy resorcinol and 2-ethyl resorcinol.
Once resorcinols 6a–c, 10, 14, 17, 19, and 21 were obtained, the corresponding coumarins 23a–h were synthesized through a modified Pechmann condensation with eneamine 22 as previously described.50,51 The resulting coumarin phenols were noviosylated with the trichloroacetimidate of noviose cyclic carbonate (24) in the presence of catalytic boron trifluoride etherate to generate scaffolds 25a–h in good yield.39 The benzyl carbonate was removed via hydrogenolysis to produce the aminocoumarin, which was readily coupled with preselected benzoic acids in the presence of N-(3-dimethylamino-propyl)-N′-ethylcarbodiimide hydrochloride (EDCI) and pyridine. Benzoic acids were chosen based on previously determined SAR trends reported by Burlison and co-workers.38 The cyclic carbonates were treated with triethylamine in methanol to give the solvolyzed products, 26a–p in moderate to good yield over three steps.
Syntheses of quinoline- and naphthalene-containing novobiocin analogues
Novobiocin analogues containing a quinoline or naphthalene ring in lieu of the 8-methylcoumarin of novobiocin were synthesized to probe the importance of the coumarin lactone moiety in binding the Hsp90 C-terminus, as well as to potentially circumvent the limited solubility of coumarin-containing analogues. Protection of the phenol in 27 as the t-butyl-carbonate52 served two purposes (Scheme 7). Not only did phenol protection remove the quinolone-like properties of 27, introduction of the sterically-hindered tbutyl-carbonate also decreased the relative amount of 6- and 8-bromo regioisomers normally produced upon bromination of 28. Thus, the isolable percentage of desired 3-bromo regioisomer was enriched to 46% yield.53 One-pot t-butyl-carbonate deprotection, followed by immediate reprotection with benzyl bromide afforded intermediate 30. N-arylation of 30 was accomplished with p-methoxybenzylamine under Ullman-like conditions employing CuI and L-(−)-proline as a catalyst to provide 31.54 Acylation of the secondary aniline with the desired benzoyl chloride,55 generated in situ from the appropriate benzoic acid,56 afforded PMB-protected amide 32. Interestingly, subjection of 32 to aluminum trichloride in anisole57 resulted solely in the formation of 7-hydroxy 33; the PMB-protected amide remained intact. Global removal of the PMB and benzyl groups was ultimately accomplished with trifluroacetic acid, to provide phenol 34.58
Scheme 7.

Syntheses of 7-hydroxyquinolines.
Construction of the corresponding naphthalene-containing analogues began by benzyl protection of phenol 35, to provide 36 in high yield (Scheme 8).59 N-arylation of 36 with p-methoxybenzylamine provided 37,54 which was acylated with the desired benzoyl chloride to afford 38.55,56 6-Benzyloxy deprotection of 38 to 39 was performed with aluminum trichloride in anisole,57 while concurrent benzyl- and PMB-deprotection to intermediate 40 was accomplished with trifluoroacetic acid.58
Scheme 8.

Syntheses of 6-hydroxynaphthalenes.
Phenols 33, 34, 39, and 40 were noviosylated with the trichloroacetimide of noviose carbonate (24) in the presence of boron trifluoride etherate to provide noviose carbonate analogues 41a–d.39 In particular, analogues 33 and 34 containing the quinoline nitrogen were both slow to react and low yielding, even when greater than stoichiometric boron trifluoride etherate was employed, suggesting chelation of the quinoline nitrogen to boron was problematic. Solvolysis of carbonates 41a–d with triethylamine in methanol/dichloromethane afforded diols 42a–d in moderate yields.
Biological evaluation of novobiocin analogues
Upon construction of the library of novobiocin analogues, the compounds were evaluated for anti-proliferative activity against SkBr3 (estrogen receptor negative, Her2 over-expressing breast cancer cells), MCF-7 (estrogen receptor positive breast cancer cells), LnCaP (androgen receptor sensitive prostate cancer cells) and PC-3 (androgen receptor insensitive prostate cancer cells) cell lines. As shown in Table 1, the 6-substituted analogues containing the biaryl sidechain (26a–26c) were 3- to 7-fold less active against the two breast cancer cells than analogues containing hydrogen at this position.38 These analogues were more active against prostate cancer cells than breast cancer cells versus the corresponding 6-H derivative. For reasons that remain unclear, the putative binding pocket for biaryl-containing analogues does not appear to tolerate incorporation of steric bulk at the 6-position. Analogues containing the 2-indole sidechain (26i–26k) were consistently more active than their corresponding biaryl derivatives, in-line with previously-observed trends.38 Analogue 26j, containing a 6-propoxy-coumarin, was consistently the most potent derivative in this library exhibiting 2-fold enhanced potency relative to its 6-H analogue against LnCaP cells.
Table 1.
Anti-proliferation activities of coumarin-derived novobiocin analogues.
| Compound (IC50, μM) | R1 | R2 | R3 | R4 | MCF-7 | SKBr3 | PC-3 | LnCaP |
|---|---|---|---|---|---|---|---|---|
| 26a | biaryl | H | OMe | Me | > 100a | 58.8 ± 1.3 | 35.4 | 6.6 |
| 26b | biaryl | H | OPr | Me | > 100 | > 100 | 5.6 ± 5.7 | 3.0 ± 0.6 |
| 26c | biaryl | H | OiPr | Me | 66.9 ± 3.1 | 58.6 ± 5.4 | 60.7 ± 9.1 | 14.4 ± 4.2 |
| 26d | biaryl | OMe | H | Me | 82.8 | 55.7 ± 6.9 | 11.3 ± 2.0 | 2.0 ± 0.8 |
| 26e | biaryl | H | H | Bn | > 100 | > 100 | > 100 | 49.7 ± 25.0 |
| 26f | biaryl | H | H | Ph | > 100 | 17.3 ± 3.4 | > 100 | 1.0 ± 0.1 |
| 26g | biaryl | H | H | OMe | 9.0 ± 5.4 | 13.9 ± 1.2 | 2.3 ± 2.9 | 1.1 ± 0.1 |
| 26h | biaryl | H | H | Et | 41.7 ± 14.0 | 28.6 ± 1.1 | 1.8 ± 0.6 | 1.6 ± 0.3 |
| 26i | 2-indole | H | OMe | Me | 24.4 ± 1.2 | 25.1 ± 7.7 | 20.2 ± 9.8 | 10.5 ± 0.3 |
| 26j | 2-indole | H | OPr | Me | 2.1 ± 0.1 | 2.1 ± 0.8 | 6.2 ± 1.8 | 1.8 ± 0.7 |
| 26k | 2-indole | H | OiPr | Me | 20.0 ± 1.0 | 20.7 ± 0.4 | 11.9 | 11.4 |
| 26l | 2-indole | OMe | H | Me | 6.1 ± 1.7 | 9.0 ± 0.8 | 11.8 ± 1.3 | 12.9 ± 4.4 |
| 26m | 2-indole | H | H | Bn | 13.2 ± 0.6 | 38.0 ± 3.0 | 73.3 ± 3.7 | 67.6 ± 6.3 |
| 26n | 2-indole | H | H | Ph | 22.9 ± 2.1 | 38.8 ± 8.3 | 28.0 ± 12.1 | 27.6 ± 10.8 |
| 26o | 2-indole | H | H | OMe | > 100 | 9.7 ± 1.0 | > 100 | > 100 |
| 26p | 2-indole | H | H | Et | 4.3 ± 2.5 | 4.3 ± 3.4 | > 100 | > 100 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
By comparison, incorporation of a hydrogen-bond acceptor at the 5-position (26d, 26l) resulted in equivalent or decreased activity versus corresponding 6-H analogues, especially against both breast cancer cell lines.38 In general, 5-methoxy functionalized coumarins do not appear beneficial for anti-proliferative activity.
It was previously demonstrated that 8-methyl analogues were ~10-fold more active than the corresponding 8-hydrogen derivatives.38 To further elaborate upon this trend, a larger selection of 8-fuctionalized coumarins were evaluated. Incorporation of an 8-methoxy (26g) led to 2-fold improved activity over its 8-methyl counterpart, and 5-fold increased activity over the similarly-sized 8-ethyl derivative 26h. Introduction of steric bulk (26e, 26f) generally decreased anti-proliferative activity, especially against MCF-7 cells. It appears that while short alkoxy side chains take advantage of putative interactions, steric bulk appears detrimental to inhibitory activity at this location. A similar trend was observed against prostate cancer cells, with 26g and 26h exhibiting 10-fold increased activity versus their 8-methyl counterparts.38 Surprisingly, 8-benzyl 26e was more than twice as active as the 8-methyl derivative against LnCaP cells. In contrast, 8-position analogues containing the 2-indole sidechain (26m–26p) did not exhibit similar, consistent trends against prostate and breast cancer cells. Against breast cancer cells, compounds 26m, 26n, and 26p exhibited significantly reduced activity versus the 8-methyl derivative. The 8-methoxy 26o was inactive against MCF-7 cells, while both 26o and 26p were inactive against both PC-3 and LnCaP cells. The selectivity of 26o and 26p for breast cancer cells versus prostate cancer cells is intriguing and requires further investigation.
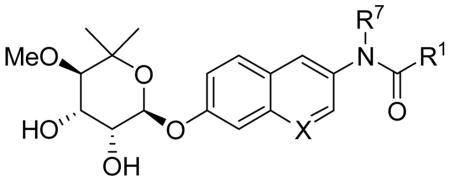
As shown in Table 2, compounds 42a and 42c containing the p-MeOBn-alkylated amides did not exhibit anti-proliferative activity against the cell lines tested. This is in contrast to analogues 42b and 42d lacking the p-MeOBn functionality, which manifested modest antiproliferative activity. This stark difference suggests one of two scenarios regarding the role of the p-MeOBn functionality; either the p-MeOBn group of tertiary amides 42a and 42c is unable to occupy the same pocket as the 4-aryloxy substituted novobiocin analogues60,61 or more simply, the secondary amide is required for benzamide-containing novobiocin analogues to manifest anti-proliferative activity, an observation consistent with prior structure–activity trends.38 It is plausible that the steric congestion of amides 42a and 42c forces adoption of a more static conformation that disallows cis/trans isomerization of the amide, a feature that has been hypothesized to be essential for anti-proliferative activity of novobiocin analogues against bacteria. Further evidence was gathered upon realization that the lack of reactivity for tertiary amides 42a and 42c to all but the harshest conditions for p-MeOBn removal58,62,63 suggest these compounds may adopt a highly-organized and stable conformation.
Table 2.
Anti-proliferation activities of quinoline- and naphthalene-containing novobiocin analogues.
| Compound | R1 | R7 | X | MCF-7 | SkBr3 | PC-3 | LnCaP |
|---|---|---|---|---|---|---|---|
| 42a | biaryl | 4-OMe-Bn | N | > 100a | > 100 | > 100 | > 100 |
| 42b | biaryl | H | N | 13.1 ± 4.1 | 16.5 ± 6.2 | 17.6 ± 4.6 | 14.2 ± 0.4 |
| 42c | biaryl | 4-OMe-Bn | CH | > 100 | > 100 | > 100 | > 100 |
| 42d | biaryl | H | CH | 46.4 ± 5.3 | 38.9 ± 2.4 | 10.9 ± 0.7 | 19.6 ± 1.6 |
Values represent mean ± standard deviation for at least two separate experiments performed in triplicate.
Against breast cancer cells, analogue 42b exhibited similar anti-proliferative activities as the corresponding 8-methylcoumarin analogue, while 42d was between 2- and 5-fold less active.38 In contrast, both 42b and 42d were significantly more active against PC-3 cells than the corresponding 8-methylcoumarin; 42b and 42d exhibited between 7- to 9-fold reduced activity against LnCap cells. Given that both 42b and 42d lack the 8-methyl feature that yields an increased activity of ~10-fold, it is reasonable to hypothesize that the quinoline- and naphthalene-derived analogues that include an 8-methyl substituent could exhibit anti-proliferative activities between 1–5 μM against breast cancer cells and 1–2 μM against prostate cancer cells, approximately an order of magnitude less than the novobiocin analogue containing a coumarin. These results suggest that, while the lactone moiety may provide beneficial hydrogen-bonding interactions with the novobiocin binding pocket, these interactions may not be required to manifest anti-proliferative activity. More importantly, these results implicate that continued optimization of the coumarin scaffold connecting the sugar and benzamide motifs is likely to produce compounds with enhanced anti-proliferative activity. A summary of the observed trends for anti-proliferative activities of coumarin-derived novobiocin analogues is depicted in Figure 4.
Figure 4.

Structure–activity relationships observed for the coumarin scaffold of novobiocin analogues exhibiting anti-proliferative activities.
To provide additional support that the anti-proliferative activities exhibited by coumarin-derived novobiocin analogues results from Hsp90 inhibition, analogues 26g, 26j, and 42d were evaluated for their abilities to induce degradation of Hsp90-dependent client proteins. As shown in Figure 5, the Hsp90 client proteins Her2 and Raf were degraded in MCF-7 cells in a concentration-dependent manner upon treatment with coumarin-derived novobiocin analogs. Moreover, Hsp90 client protein degradation correlates well with observed anti-proliferative IC50s; 26g (IC50 = 9.0 μM) and 26j (IC50 = 2.1 μM) induced client degradation at ~10 μM, while 42d, with its more modest anti-proliferative activity (IC50 = 46.4 μM), induced client degradation at ~50 μM. Since actin, a non-Hsp90-dependent protein, is not affected by these analogues, anti-proliferative activities of these analogues correlate directly with Hsp90-client protein degradation.
Figure 5.

Western blot analyses of Hsp90 client protein degradation assays against MCF-7 breast cancer cells. Concentrations (in μM) of 26g (top), 26j (middle), and 42d (bottom) are indicated above each lane. GDA (geldanamycin, 500 nM) and DMSO were respectively employed as positive and negative controls.
Conclusions
Compound 26g and 26j demonstrated the most potent anti-proliferative activity against the cancer cell lines tested and represent scaffolds that will be further probed to improve activity. Derivatives 26f and 26o appear to represent compounds that exhibit differential selectivity for one cancer cell lines versus another, for reasons that remain unclear. Since these compounds demonstrated low micromolar activity against one cell line and are inactive against others, they may provide a tool for further exploration and perhaps unraveling of the complicated processes affected. The activities of analogues 42b and 42d, the first documented novobiocin analogues lacking the coumarin functionality, implicate that, while the coumarin ring may participate in hydrogen bonding interactions with Hsp90 that abrogate activity, these interactions are not essential for anti-proliferation activity through inhibition of Hsp90. These analogues provide sufficient evidence to continue the search for optimal ring systems that bridge the benzamide and noviose functionalities. Studies to construct analogues based upon these structure–activity relationships are currently underway and will be reported in due course.
Experimental Section
2,4-Bis(ethoxymethoxy)benzaldehyde (2a)42
N,N-diisopropylethylamine (25.3 mL, 145 mmol) was slowly added to 2,4-dihydroxybenzaldehyde (5.00 g, 36.2 mmol) in anhydrous N,N-dimethylformamide (100 mL) over 5 min at rt. After 30 min, the solution was cooled to 0° C and chloromethyl ethyl ether (14.2 mL, 145 mmol) was added and the mixture warmed to rt over 12 h. The reaction was quenched by the addition of saturated aqueous NH4Cl solution and extracted with EtOAc (3 × 50 mL). The combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 5:1 → 1:1 Hexane:EtOAc) to give 2a as a brown amorphous solid (9.10 g, 99%): 1H NMR (CDCl3, 400 MHz) δ 10.34 (d, J = 2.4 Hz, 1H), 7.81 (dd, J = 8.7, 2.8 Hz, 1H), 6.89 (t, J = 2.5 Hz, 1H), 6.74 (m, 1H), 5.34 (d, J = 2.8, 2H), 5.28 (d, J = 2.8, 2H), 3.81-3.71 (m, 4H), 1.28-1.22 (m, 6H).
2,4-Bis(ethoxymethoxy)phenol (3a)
A solution of 2a (3.78 g, 12.0 mmol) in anhydrous CH2Cl2 (4.0 mL) was slowly added to mCPBA (70%) (3.26 g, 13.2 mmol) in anhydrous CH2Cl2 (16.3 mL) at 0° C. The resulting solution was warmed to rt, then refluxed for 12 h. After cooling to rt, the resulting solution was washed with saturated aqueous NaHCO3 solution (3 × 20 mL) and 10% aqueous Na2S2O3 (30 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was re-dissolved in MeOH (5 mL) and stirred with excess 10% aqueous NaOH for 3 h at rt. The pH was adjusted to 2 with 6M HCl and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated to give 3a as an orange oil (8.21 g, 94%): 1H NMR (CDCl3, 500 MHz) δ 6.89-6.85 (m, 2H), 6.67 (dd, J = 8.8, 2.7 Hz, 1H), 5.81 (d, J = 6.6 Hz, 1H), 5.23 (s, 2H), 5.15 (s, 2H), 3.80-3.73 (m, 4H), 1.29-1.24 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 151.0, 145.0, 141.5, 115.2, 110.6, 106.0, 94.9, 94.2, 64.8, 64.1, 15.1, 15.1; IR (film) νmax 3362, 2887, 1460, 1286, 1162, 735 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C12H18O5, 265.1052; found, 265.1045.
2,4-Bis(methoxymethoxy)phenol (3b)44
Benzaldehyde 2b (700 mg, 3.11 mmol) in CHCl3 (1.80 mL) at 0° C was treated with mCPBA (70% w/w, 1.61 g, 9.33 mmol). After 10 min, the solution was warmed to rt, then refluxed for 12 h. Upon cooling to rt, the solution was washed with saturated aqueous NaHCO3 (3 × 10 mL), saturated aqueous Na2SO3 (20 mL), saturated aqueous NaCl, was dried (Na2SO4), filtered, and concentrated. The residue was dissolved in MeOH (5 mL) and stirred with excess triethylamine for 3 h at rt. The solvent was concentrated and the residue purified by column chromatography (SiO2, 4:1 → 3:1 Hexane:EtOAc) to afford 3b as a yellow oil (320 mg, 50%): 1H NMR (CDCl3, 400 MHz) δ 6.87 (d, J = 8.9 Hz, 1H), 6.86 (s, 1H), 6.67 (dd, J = 11.5, 2.8 Hz, 1H), 5.21 (s, 2H), 5.11 (s, 2H), 3.54 (s, 3H), 3.50 (s, 3H).
2,4-Bis(ethoxymethoxy)-1-methoxybenzene (4a)
Potassium carbonate (14.3 g, 103 mmol) was added to 3a (2.50 g, 10.3 mmol) in N,N-dimethylformamide (103 mL). After 10 min, methyl iodide (6.43 mL, 103 mmol) was added and the solution was heated to reflux for 12 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 50 mL); combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), and concentrated. The residue was purified by column chromatography (SiO2, 4:1 Hexane:EtOAc) to afford 4a as a yellow oil (2.40 g, 91%): 1H NMR (CDCl3, 500 MHz) δ 6.87 (d, J = 2.8 Hz, 1H), 6.72 (d, J = 8.9 Hz, 1H), 6.60 (dd, J = 13.3, 1.7 Hz, 1H), 5.18 (s, 2H), 5.07 (s, 2H), 3.76 (s, 3H), 3.72-3.69 (m, 2H), 3.68-3.63 (m, 2H), 1.17-1.13 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 150.7, 146.2, 143.9, 111.2, 107.9, 105.8, 93.2, 93.0, 63.3, 63.0, 55.4, 14.1, 14.0; IR (film) νmax 2976, 2932, 2899, 2835, 1595, 1508, 1393, 1227, 1153, 1103, 1080, 1009, 847 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C13H20O5, 279.1208; found, 279.1181.
2,4-Bis(methoxymethoxy)-1-propoxybenzene (4b)
Potassium carbonate (322 mg, 2.33 mmol) was added to 3b (50 mg, 0.233 mmol) in N,N-dimethylformamide (2.33 mL) at rt. After 10 min, iodopropane (226 μL, 2.33 mmol) was added and the solution was heated to reflux for 12 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 10 mL); combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified by column chromatography (SiO2, 5:1 Hexane:EtOAc) to afford 4b as a yellow oil (36.4 mg, 61%): 1H NMR (CD2Cl2, 400 MHz) δ 6.87 (s, 1H), 6.84 (d, J = 2.9 Hz, 1H), 6.68 (dd, J = 11.7, 2.8 Hz, 1H), 5.19 (s, 2H), 5.12 (s, 2H), 3.93 (t, J = 6.6 Hz, 2H), 3.53 (s, 3H), 3.49 (s, 3H), 1.86-1.78 (m, 2H), 1.06 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 150.6, 146.5, 143.8, 113.7, 108.5, 106.6, 94.7, 94.2, 70.3, 55.2, 54.9, 21.6, 9.5; IR (film) νmax 2961, 2826, 1595, 1506, 1400, 1261, 1154, 1013, 1076, 924, 800 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C13H20O5, 257.1389; found, 257.1410; [M + Na]+ calcd for C13H20O5, 279.1208; found, 279.1165.
2,4-Bis(ethoxymethoxy)-1-isopropoxybenzene (4c)
Potassium carbonate (2.85 g, 20.7 mmol) was added to 3a (500 mg, 2.07 mmol) in N,N-dimethylformamide (4.10 mL) at rt. After 10 min, 2-iodopropane (2.06 mL, 20.7 mmol) was added and the solution was heated to reflux for 12 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 20 mL); combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 5:1 → 1:1 Hexane:EtOAc) to afford 4c as a yellow oil (0.32 g, 55%): 1H NMR (CD2Cl2, 400 MHz) δ 6.87 (s, 1H), 6.86 (d, J = 4.9 Hz, 1H), 6.66 (dd, J = 11.6, 3.4 Hz, 1H), 5.23 (s, 2H), 5.17 (s, 2H), 4.44-4.38 (m, 1H), 3.83-3.72 (m, 4H), 1.33 (s, 3H), 1.31 (s, 3H), 1.27-1.23 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 152.4, 149.1, 143.2, 118.6, 109.5, 107.5, 94.4, 93.9, 72.8, 64.3, 64.1, 22.2 (2C), 15.1, 15.1; IR (film) νmax 2976, 1591, 1504, 1528, 1391, 1258, 1217, 1107, 1011, 847 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C15H24O5, 285.1702; found, 285.1746; [M + Na]+ calcd for C15H24O5, 307.1522; found, 307.1310.
1,3-Bis(ethoxymethoxy)-4-methoxy-2-methylbenzene (5a)
A solution of 4a (632 mg, 2.27 mmol) in anhydrous THF (1.94 mL) was added dropwise to a solution of nBuLi (2.5 M in hexanes, 1.48 mL, 3.70 mmol) in anhydrous THF (1.62 mL) at rt. After 1 h, the solution was cooled to −78° C and methyl iodide (620 μL, 9.87 mmol) was added. The resulting solution was warmed to rt over 12 h, and the reaction was quenched by the addition of saturated aqueous NH4Cl. Water (5 mL) was added and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 8:1 → 5:1 Hexane:EtOAc) to afford 5a as a yellow oil (353 mg, 53%): 1HNMR (CDCl3, 500 MHz) δ 6.74 (d, J = 9.0 Hz, 1H), 6.60 (d, J = 9.0 Hz, 1H) 5.10 (s, 2H), 5.05 (s, 2H), 3.78 (q, J = 7.1 Hz, 2H), 3.72 (s, 3H), 3.67 (q, J = 7.1 Hz, 2H), 2.14 (s, 3H), 1.18-1.15 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 149.1, 146.5, 121.7, 109.0, 108.4, 96.2, 93.0, 64.3, 63.1, 55.1, 28.7, 14.2, 14.1, 8.8; IR (film) νmax 2918, 2359, 1487, 1260, 1248, 1082, 1055, 945, 798 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C14H22O5, 293.1365; found, 293.1357.
1,3-Bis(methoxymethoxy)-2-methyl-4-propoxybenzene (5b)
A solution of 4b (165 mg, 0.64 mmol) in anhydrous THF (520 μL) was added dropwise to a solution of nBuLi (2.5 M in hexanes, 390 μL, 0.97 mmol) in anhydrous THF (420 μL) at rt. After 1 h, the solution was cooled to −78° C and methyl iodide (160 μL, 2.58 mmol) was added. The resulting solution was warmed to rt over 12 h, and the reaction was quenched by the addition of saturated aqueous NH4Cl. Water (5 mL) was added and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 6:1 Hexane:EtOAc) to afford 5b as a yellow oil (166 mg, 95%): 1H NMR (CDCl3, 500 MHz) δ 6.66 (d, J = 9.0 Hz, 1H), 6.60 (d, J = 9.0 Hz, 1H), 5.02 (s, 2H), 5.00 (s, 2H), 3.80-3.77 (m, 2H), 3.49 (s, 3H), 3.47 (s, 3H), 2.14 (d, J = 7.1 Hz, 3H), 1.73-1.69 (m, 2H), 0.94 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 148.5, 148.5, 147.3, 145.6, 126.7, 123.0, 112.8, 110.8, 110.4, 99.2, 57.7, 57.6, 21.2, 10.9, 10.0; IR (film) νmax 2957, 2924, 2853, 1738, 1597, 1487, 1468, 1391, 1335, 1231, 1157, 974, 798 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C14H22O5, 271.1545; found, 271.1558.
1,3-Bis(ethoxymethoxy)-4-isopropoxy-2-methylbenzene (5c)
A solution of 4c (190 mg, 0.67 mmol) in anhydrous THF (530 μL) was added dropwise to a solution of nBuLi (2.5 M in hexanes, 410 μL, 1.00 mmol) in anhydrous THF (440 μL) at rt. After 1 h, the solution was cooled to −78° C and methyl iodide (170 μL, 2.67 mmol) was added. The resulting solution was warmed to rt over 12 h, and the reaction was quenched by the addition of saturated aqueous NH4Cl. Water (5 mL) was added and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 6:1 Hexane:EtOAc) to afford 5c as a yellow oil (157 mg, 79%): 1H NMR (CDCl3, 500 MHz) δ 6.70 (d, J = 9.0 Hz, 1H), 6.61 (d, J = 9.0 Hz, 1H), 5.10 (s, 2H), 5.08 (s, 2H),4.34 (quintet, J = 6.1 Hz, 1H), 3.78 (q, J = 7.1 Hz, 2H), 3.67 (q, J = 7.1 Hz, 2H), 2.13 (s, 3H), 1.23 (d, J = 6.1 Hz, 6H), 1.24-1.15 (m, 6H); 13C NMR (CDCl3, 125 MHz) δ 150.3, 146.6, 145.3, 122.7, 113.7, 110.1, 97.3, 94.0, 71.5, 65.4, 64.2, 29.4, 22.2, 15.2, 15.2, 9.9; IR (film) νmax 2924, 2853, 2359, 2339, 1591, 1483, 1113, 1057, 974 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C16H26O5, 299.1858; found, 299.1909.
4-Methoxy-2-methylbenzene-1,3-diol (6a)
A solution of 5a (910 mg, 3.37 mmol) in MeOH (28.0 mL) at rt was treated dropwise with 3M HCl (9.00 mL, 26.9 mmol), then heated to reflux for 1 h. Water (30 mL) was added and the solution was extracted with EtOAc (3 × 30 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 6:1 Hexane:EtOAc) to afford 6a as a red amorphous solid (509 mg, 98%): 1H NMR (Acetone-d6, 500 MHz) δ 7.68 (s, 1H), 7.24 (s, 1H), 6.60 (d, J = 11 Hz, 1H), 6.29 (d, J = 11 Hz, 1H), 3.74 (s, 3H), 2.09 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 144.7, 142.1, 139.7, 115.6, 110.2, 108.6, 55.6, 7.6; IR (film) νmax 3583, 2920, 2359, 1616, 1259, 1090, 1020, 798 cm−1.
2-Methyl-4-propoxybenzene-1,3-diol (6b)
A solution of 5b (580 mg, 2.15 mmol) in MeOH (17.9 mL) was treated dropwise with 3M HCl (630 μL, 17.2 mmol), then heated to reflux for 1 h. Water (20 mL) was added and the solution was extracted with EtOAc (3 × 20 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), and concentrated to afford 6b as a red amorphous solid (387 mg, 99%). 1H NMR (CDCl3, 500 MHz) δ 6.51 (d, J = 8.7 Hz, 1H), 6.21 (d, J = 8.6 Hz, 1H), 5.74 (s, 1H), 4.36 (s, 1H), 3.87-3.85 (m, 2H), 2.09 (s, 3H), 1.75-1.71 (m, 2H), 0.96 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 147.5, 143.8, 139.0, 109.8, 108.4, 103.8, 70.2, 21.6, 9.5, 7.3; IR (film) νmax 3520, 3360, 2966, 2880, 2359, 2341, 1636, 1236, 1068, 785, 750 cm−1.
4-Methoxybenzene-1,3-diol (6c)
A solution of 5c (157 mg, 0.53 mmol) in MeOH (4.40 mL) at rt was treated dropwise with 3M HCl (1.40 mL, 4.21 mmol), then heated to reflux for 1 h. Water (5 mL) was added and the solution was extracted with EtOAc (3 × 10 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated to afford 6c as a red amorphous solid (95 mg, 99%): 1H NMR (CDCl3, 500 MHz) δ 6.54 (d, J = 8.7 Hz, 1H), 6.21 (d, J = 8.7 Hz, 1H), 5.78 (s, 1H), 4.37-4.32 (m, 1H), 2.09 (s, 3H), 1.25 (d, J = 6.1 Hz, 6H); 13C NMR (CDCl3, 125 MHz) δ 147.7, 144.9, 137.5, 110.8, 109.8, 104.0, 71.7, 21.3 (2C); IR (film) νmax 3526, 2974, 2924, 2853, 1717, 1607, 1475, 1238, 1113, 1067, 928, 887, 791 cm−1.
1-Methoxy-3,5-bis(methoxymethoxy)benzene (8)
N,N-diisopropylethylamine (3.15 mL, 18.1 mmol) was added to 5-methoxybenzene-1,3-diol (634 mg, 4.52 mmol) in anhydrous N,N-dimethylformamide (12.6 mL) over 5 min at rt. After 30 min, the solution was cooled to 0° C, methoxy methylchoride (3.02 mL, 18.1 mmol) was added, and the solution was warmed to rt over 12 h. The reaction was quenched by the addition of saturated aqueous NaHCO3 at 0°C and extracted with EtOAc (3 × 10 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 6:1→ 4:1 Hexane:EtOAc) to afford 8 as a yellow amorphous solid (441 mg, 43%): 1H NMR (CDCl3, 500 MHz) δ 6.29 (t, J = 2.2 Hz, 1H), 6.21 (d, J = 2.2 Hz, 2H), 5.07 (s, 4H), 3.69 (s, 3H), 3.40 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 161.394, 159.0 (2C), 97.2, 96.2 (2C), 94.5 (2C), 56.1, 55.4 (2C); IR (film) νmax 2997, 2955, 2903, 2827, 1601, 1475, 1400, 1215, 1194, 1146, 1032, 991, 924, 829, 685 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C11H16O5, 251.0895; found, 251.0910.
5-methoxy-1,3-bis(methoxymethoxy)-2-methylbenzene (9)
A solution of 8 (441 mg, 1.93 mmol) in anhydrous THF (1.55 mL) was added dropwise to a solution of nBuLi (2.5 M in hexanes, 1.16 mL, 2.90 mmol) in anhydrous THF (1.26 mL) at rt. After 1 h, the solution was cooled to −78° C and methyl iodide (480 μL, 7.73 mmol) was added. The resulting solution was warmed to rt over 12 h, and the reaction was quenched by the addition of saturated aqueous NH4Cl. Water (5 mL) was added and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 6:1 → 4:1; Hexane:EtOAc) to afford 9 as a yellow oil (314 mg, 67%): 1H NMR (CDCl3, 500 MHz) δ 6.38 (d, J = 2.2 Hz, 1H), 6.24 (d, J = 2.1 Hz, 1H), 5.08 (d, J = 3.6 Hz, 2H), 5.06 (d, J = 2.6 Hz, 2H), 3.72 (s, 3H), 3.40 (s, 6H), 1.97 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 160.3, 157.8, 155.4, 108.2, 93.9, 93.8, 93.7, 92.9, 55.0, 55.0, 54.6, 7.0; IR (film) νmax 2953, 2934, 2905, 1597, 1497, 1396, 1215, 1144, 1126, 1074, 1059, 1028, 922, 822 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C12H18O5, 243.1233; found, 243.1223.
5-Methoxy-2-methylbenzene-1,3-diol (10)
A solution of 9 (314 mg, 1.30 mmol) in MeOH (10.8 mL) at rt was treated dropwise with 3M HCl (3.46 mL, 10.3 mmol), then heated to reflux for 1 h. Water (11 mL) was added and the solution was extracted with EtOAc (3 × 15 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated to afford 10 as a red amorphous solid (177 mg, 99%): 1H NMR (CDCl3, 500 MHz) δ 8.17 (s, 1H), 6.09 (d, J = 1.6 Hz, 1H), 6.04 (s, 1H), 3.67 (d, J = 9.9 Hz, 3H), 2.08 (d, J = 4.1 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 160.3, 157.8, 155.4, 108.4, 93.9, 93.8, 55.0, 7.0; IR (film) νmax 3445, 2924, 2853, 2359, 2332, 1653, 1636, 1456, 1080, 1022, 798, 669 cm−1; HRMS (ESI+) m/z: [2M + H]+ calcd for C8H10O3, 309.1338 found, 309.1332.
1,3-Bis(methoxymethoxy)benzene (12)64
Sodium hydride (872 mg, 36.3 mmol) was added to resorcinol (1.00 g, 9.08 mmol) in anhydrous N,N-dimethylformamide (25.4 mL) at 0° C. After 30 min, methoxy methylchloride (2.76 mL, 36.3 mmol) was added and the resulting solution was warmed to rt over 12 h. The reaction was cooled to 0° C, quenched by the addition of saturated aqueous NaHCO3, and extracted with EtOAc (3 × 30 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 4:1 Hexane:EtOAc) to afford 12 as a yellow oil (1.75 g, 97%): 1H NMR (CDCl3, 400 MHz) δ 7.25-7.20 (m, 1H), 6.80 (d, J = 2.3 Hz, 1H), 6.75 (dd, J = 8.2, 2.4 Hz, 2H), 5.20 (s, 4H), 3.51 (s, 6H).
2-Benzyl-1,3-bis(methoxymethoxy)benzene (13)
A solution of 12 (500 mg, 2.52 mmol) in anhydrous THF (2.02 mL) was added dropwise to a solution of nBuLi (2.5 M in hexanes, 1.51 mL, 3.78 mmol) in anhydrous THF (1.65 mL) at rt. After 1 h, the solution was cooled to −40° C and benzyl bromide (1.22 mL, 10.10 mmol) was added. The resulting solution was warmed to rt over 12 h, and the reaction was quenched by the addition of saturated aqueous NH4Cl. Water (5 mL) was added and the solution was extracted with CH2Cl2 (3 × 10 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 4:1; Hexane:EtOAc) to afford 13 as a yellow oil (214 mg, 30%): 1H NMR (CDCl3, 500 MHz) δ 7.17 (d, J = 7.9 Hz, 2H), 7.17-7.12 (m, 2H), 7.06-7.02 (m, 2H), 6.71 (d, J = 8.3 Hz, 2H), 5.09 (s, 4H), 4.00 (s, 2H), 3.29 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 155.9 (2C), 141.6, 128.5 (2C), 128.0 (2C), 127.5, 125.4, 119.4, 107.7 (2C), 94.3 (2C), 56.0 (2C), 29.1; IR (film) νmax 2953, 2930, 1595, 1470, 1452, 1254, 1153, 1097, 1043, 941, 922, 727, 698 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C17H20O4, 311.1259; found, 311.1201.
2-Benzylbenzene-1,3-diol (14)65
A solution of 13 (214 mg, 0.74 mmol) in MeOH (6.20 mL) was treated dropwise with 3M HCl (0.22 mL, 5.92 mmol), then heated to reflux for 1 h. Water (10 mL) was added and the solution was extracted with EtOAc (3 × 15 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), and concentrated to afford 14 as a red amorphous solid (149 mg, 99%). 1H NMR (CDCl3, 400 MHz) δ 7.31 (d, J = 6.6 Hz, 4H), 7.25-7.19 (m, 1H), 7.01 (t, J = 8.1 Hz, 1H), 6.44 (d, J = 8.1 Hz, 2H), 4.82 (s, 2H), 4.09 (s, 2H).
2-Iodo-1,3-bis(methoxymethoxy)benzene (15)
n-Butyllithium (2.5M in hexanes, 0.22 mL, 0.56 mmol) was added to a solution of 12 (100 mg, 0.50 mmol) in anhydrous THF (790 μL) at 0° C. After 5 min, iodine (141 mg, 0.56 mmol) in anhydrous THF (320 μL) was added. After 2 h at rt, the reaction was quenched via dropwise addition of MeOH and the solvent was concentrated. Water (5 mL) was added and the solution was extracted with EtOAc (3 × 10 mL). Combined organics were washed with saturated aqueous Na2S2O3, saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated to afford 15 as a brown oil (129 mg, 79%): 1H NMR (CDCl3, 100 MHz) δ 7.25-7.18 (m, 1H), 6.79-6.71 (m, 2H), 5.27 (s, 2H), 5.18 (s, 2H), 3.54 (s, 3H), 3.50 (s, 3H); IR (film) νmax 2953, 2924, 2853, 1458, 1377 cm−1.
2,6-Bis(methoxymethoxy)biphenyl (16)
Anhydrous toluene (2.0 mL) was added to a flask charged with Pd2(dba)3 (56.3 mg, 0.062 mmol), dicyclohexyl(2′,6′-dimethoxybiphenyl-2-yl)phosphine (50.5 mg, 0.12 mmol), phenylboronic acid (281 mg, 2.31 mmol), and potassium phosphate (979 mg, 4.61 mmol) at rt. After 15 min, a solution of 15 (500 mg, 1.54 mmol) in anhydrous toluene (1.0 mL) was added and the resulting solution was heated to reflux for 12 h. Upon cooling to rt, ether was added, the solution was filtered through SiO2 and concentrated to give 16 as a colorless amorphous solid (418 mg, 99%): 1H NMR (CDCl3, 500 MHz) δ 7.35-7.28 (m, 2H), 7.28-7.25 (m, 2H), 7.18-7.15 (m, 2H), 6.83 (d, J = 8.3 Hz, 2H), 4.96 (s, 4H), 3.24 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 155.3, 155.0, 134.3, 130.8, 129.5, 128.7, 128.0, 127.6, 126.8, 122.6, 109.4 (2C), 94.9 (2C), 56.0 (2C); IR (film) νmax 2955, 2928, 2901, 2359, 2341, 1587, 1466, 1439, 1400, 1244, 1153, 1099, 1080, 1041, 922, 764, 733, 700 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C16H18O4, 297.1103; found, 297.1052.
Biphenyl-2,6-diol (17)66
A solution of 16 (400 mg, 1.46 mmol) in MeOH (12.0 mL) at rt was treated dropwise with 3M HCl (430 μL, 11.7 mmol), then heated to reflux for 1 h. Water (15 mL) was added and the solution was extracted with EtOAc (3 × 20 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated to afford 17 as an orange amorphous solid (269 mg, 99%): 1H NMR (CDCl3, 400 MHz) δ 7.60 (d, J = 7.6 Hz, 2H), 7.53-7.49 (m, 1H), 7.46-7.44 (m, 2H), 7.18 (t, J = 8.2 Hz, 1H), 6.62 (d, J = 8.2 Hz, 2H), 4.84 (s, 1H), 4.83 (s, 1H).
2-Methoxybenzene-1,3-diol (19)67
Lithium carbonate (281 mg, 1.98 mmol) was added to pyrogallol (100 mg, 0.79 mmol) in N,N-dimethylformamide (3.0 mL) at rt. After 5 min, methyl iodide (130 μL, 1.98 mmol) was added and the resulting solution was heated to 50° C for 48 h. Upon cooling to rt, water (20 mL) was added and the solution was extracted with EtOAc (3 × 20 mL). Combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 5:1 → 1:1 Hexane:EtOAc) to afford 19 as a colorless amorphous solid (44.2 mg, 34%): 1H NMR (CDCl3, 400 MHz) δ 6.89 (td, J = 8.0, 0.9 Hz, H), 6.53 (dd, J = 8.2, 0.8 Hz, 2H), 5.83 (bs, 2H), 3.90 (s, 3H).
Benzyl 7-hydroxy-6-methoxy-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (23a)
A solution of 6a (183 mg, 1.19 mmol) and enamine 22 (331 mg, 1.19 mmol) in glacial acetic acid (7.40 mL) was heated to reflux for 40 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 20 mL); combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 100:1 CH2Cl2:Acetone) to afford 23a as a yellow amorphous solid (195 mg, 46%): 1H NMR (CDCl3, 400 MHz) δ 8.27 (s, 1H), 7.54 (s, 1H), 7.43-7.37 (m, 4H), 6.77 (s, 1H), 6.07 (s, 1H), 5.25 (s, 2H), 3.96 (s, 3H), 2.37 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.0, 153.3 (2C), 145.7, 144.1, 144.0, 135.7, 128.7, 128.5, 128.2 (2C), 122.5, 121.6, 112.1, 111.6, 104.5, 67.4, 56.3, 8.2; IR (film) νmax 2910, 2359, 2339, 1693, 1537, 1354, 1209, 1078, 1024 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C19H17NO6, 378.0954; found, 378.0936.
Benzyl 7-hydroxy-8-methyl-2-oxo-6-propoxy-2H-chromen-3-ylcarbamate (23b)
A solution of 6b (390 mg, 2.14 mmol) and enamine 22 (596 mg, 2.14 mmol) in glacial acetic acid (13.4 mL) was heated to reflux for 36 h. Upon cooling to rt, the precipitated yellow solid was collected by filtration, washed with water, recrystallized from MeOH/water, and extracted with EtOAc (3 × 20 mL). Combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 100:1 CH2Cl2:Acetone) and recrystallized from MeOH/water to afford 23b as a yellow amorphous solid (278 mg, 34%): 1H NMR (CD2Cl2, 400 MHz) δ 8.26 (s, 1H), 7.56 (s, 1H), 7.47-7.38 (m, 5H), 6.84 (s, 1H), 6.28 (s, 1H), 5.25 (s, 2H), 4.09 (t, J = 6.6 Hz, 2H), 2.36 (s, 3H), 1.93-1.88 (m, 2H), 1.10 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.0, 152.2, 144.9, 142.9, 142.3, 134.6, 129.0, 127.6, 127.5 (2C), 127.2 (2C), 121.5, 120.5, 110.9, 110.5, 104.4, 69.8, 66.3, 21.4, 9.4, 7.1; IR (film) νmax 2957, 2920, 2851, 2359, 2341, 1693, 1537, 1358, 1277, 1080, 1024, 910 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C21H21NO6, 384.1447; found, 384.1447.
Benzyl 7-hydroxy-6-isopropoxy-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (23c)
A solution of 6c (142 mg, 0.78 mmol) and enamine 22 (217 mg, 0.78 mmol) in glacial acetic acid (4.90 mL) was heated to reflux for 40 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 10 mL); combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 23c as a yellow amorphous solid (159 mg, 53%): 1H NMR (CD2Cl2, 400 MHz) δ 8.26 (s, 1H), 7.56 (s, 1H), 7.44-7.38 (m, 5H), 6.85 (s, 1H), 6.31 (s, 1H), 5.25 (s, 2H), 4.66 (quintet, J = 6.1 Hz, 1H), 2.35 (s, 3H), 1.42 (d, J = 6.0 Hz, 6H); 13C NMR (CDCl3, 125 MHz) δ 159.1, 154.9, 146.7, 143.9 (2C), 142.0, 135.7, 128.7 (2C), 128.5 128.2 (2C), 122.6 (2C), 111.6, 107.0, 72.3, 67.4, 22.1 (2C), 8.2; IR (film) νmax 3400, 2924, 2853, 2359, 1817, 1699, 1524, 1412, 1354, 1300, 1221, 1204, 1113, 1076, 1022, 824 cm−1 ; HRMS (ESI+) m/z: [M + H]+ calcd for C21H21NO6, 384.1447; found, 384.1452.
Benzyl 7-hydroxy-5-methoxy-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (23d)
A solution of 10 (251 mg, 1.63 mmol) and enamine 22 (680 mg, 2.44 mmol) in glacial acetic acid (10.2 mL) was heated to reflux for 40 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 15 mL); combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 40:1 → 20:1; CH2Cl2:Acetone) to afford 23d as a yellow amorphous solid (204 mg, 35%): 1H NMR (CD2Cl2, 400 MHz) δ 8.48 (s, 1H), 7.46-7.38 (m, 6H), 6.38 (s, 1H), 5.25 (s, 2H), 5.15 (s, 1H), 3.87 (s, 3H), 2.23 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.1, 155.8, 154.3, 153.2, 149.8, 137.0, 128.7, 128.6, 128.6, 128.2, 128.2, 109.3, 109.0, 108.5, 105.6, 96.9, 70.8, 60.2, 7.3; IR (film) νmax 3406, 2935, 2837, 1713, 1670, 1607, 1529, 1501, 1364, 1242, 1101, 1051, 991, 966, 735 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C19H17NO6, 378.0954; found, 378.0974.
Benzyl 8-benzyl-7-hydroxy-2-oxo-2H-chromen-3-ylcarbamate (23e)
A solution of 14 (115 mg, 0.57 mmol) and enamine 22 (160 mg, 0.57 mmol) in glacial acetic acid (4.00 mL) was heated to reflux for 40 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 10 mL); combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 100:1; CH2Cl2:Acetone), followed by recrystallization from MeOH to afford 23e as an orange amorphous solid (296 mg, 48%): 1H NMR (CD2Cl2, 400 MHz) δ 8.29 (s, 1H), 7.53 (s, 1H), 7.46-7.38 (m, 4H), 7.37-7.27 (m, 4H), 7.23-7.19 (m, 2H), 7.01 (t, J = 8.1 Hz, 1H), 6.86 (d, J = 8.4 Hz, 1H), 6.46 (d, J = 8.1 Hz, 1H), 5.25 (s, 2H), 4.25 (s, 2H), 4.06 (s, 1H); 13C NMR (CDCl3, 125 MHz) δ 157.7, 154.3, 153.9, 152.2, 148.0, 137.9, 134.5, 127.6, 127.6, 127.6, 127.6, 127.5, 127.3, 127.2, 127.4, 126.6, 125.4, 125.3, 121.4, 120.4, 114.0, 112.6, 66.5, 27.5; IR (film) νmax 3381, 2957, 2928, 2359, 2341, 1693, 1607, 1526, 1466, 1454, 1383, 1366, 1219, 1204, 1076, 1045, 764, 737, 700 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C24H19NO5, 402.1341; found, 402.1341.
Benzyl 7-hydroxy-2-oxo-8-phenyl-2H-chromen-3-ylcarbamate (23f)
A solution of 17 (400 mg, 2.15 mmol) and enamine 22 (598 mg, 2.15 mmol) in glacial acetic acid (14.3 mL) was heated to reflux for 40 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 30 mL); combined organic fractions were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 100:1; CH2Cl2:Acetone), then recrystallized from MeOH to afford 23f as an orange amorphous solid (264 mg, 27%): 1H NMR (CDCl3, 500 MHz) δ 8.25 (s, 1H), 7.51-7.48 (m, 2H), 7.43-7.40 (m, 2H), 7.35-7.29 (m, 8H), 6.94 (d, J = 8.6 Hz, 1H), 5.16 (s, 2H); 13C NMR (CDCl3,125 MHz) δ 158.5 (2C), 154.3, 153.2, 147.7, 135.6, 130.9, 130.6, 130.5, 129.8, 129.4, 129.2 (2C), 128.7 (2C), 128.6, 128.3, 127.8, 122.2, 121.6, 113.3, 113.5, 67.5; IR (film) νmax 3398, 2957, 2926, 2854, 1815, 1699, 1601, 1524, 1383, 1366, 1308, 1215, 1045, 1009, 764, 750, 698 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C23H17NO5, 388.1185; found, 388.1214.
Benzyl 7-hydroxy-8-methoxy-2-oxo-2H-chromen-3-ylcarbamate (23g)
A solution of 19 (1.10 g, 7.86 mmol) and enamine 22 (2.18 g, 7.86 mmol) in glacial acetic acid (60.0 mL) was heated to reflux for 90 h. Upon cooling to rt, the solution was extracted with EtOAc (3 × 50 mL); combined organic fractions were washed with saturated aqueous NaCl, dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 11:1; Hexane:EtOAc → EtOAc) then recrystallized from MeOH/water to afford 23g as a colorless amorphous solid (207 mg, 7.7%): 1H NMR (CDCl3, 400 MHz) δ 8.30 (s, 1H), 7.50 (s, 1H), 7.43-7.36 (m, 5H), 7.13 (d, J = 8.6 Hz, 1H), 6.97 (d, J = 7.9 Hz, 1H), 6.04 (s, 1H), 5.21 (s, 2H), 4.13 (s, 3H); 13C NMR (Acetone-d6, 100 MHz) δ 157.3, 153.3, 151.5 (2C), 144.1, 136.5, 134.4, 128.4 (2C), 128.1, 128.0 (2C), 122.7, 121.6, 113.6, 113.2, 66.7, 60.7; IR (film) νmax 2920, 2851, 2405, 2357, 1707, 1605, 1522, 1458, 1385, 1364, 1275, 1259, 1213, 1088, 1047, 750 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C18H15NO6, 364.0797; found, 364.0776.
Benzyl 8-ethyl-7-hydroxy-2-oxo-2H-chromen-3-ylcarbamate (23h)
A solution of 21 (1.40 g, 10.1 mmol) and enamine 22 (2.80 g, 10.1 mmol) in glacial acetic acid (50.0 mL) was heated to reflux for 12 h. Upon cooling to rt, the solvent was concentrated. The residue was purified via column chromatography (SiO2, 4:1 → 2:1; Hexane:EtOAc), then recrystallized from acetone/hexanes to afford 23h as a colorless amorphous solid (600 mg, 17%). 1H NMR (DMSO-d6, 400 MHz) δ 9.07 (s, 1H), 8.12 (s, 1H), 7.46-7.32 (m, 6H), 6.86 (d, J = 8.4 Hz, 1H), 5.18 (s, 2H), 2.72 (q, J = 7.6 Hz, 2H), 1.11 (t, J = 7.6 Hz, 3H); 13C NMR (DMSO-d6, 125 MsHz) δ 158.0, 157.1, 153.8 (2C), 149.5, 136.4, 128.4 (2C), 127.9, 127.8, 127.2, 125.8, 120.4, 116.6, 112.8, 111.4, 66.1, 15.7, 13.5; IR (film) νmax 3391, 3339, 2964, 2870, 2357, 1732, 1682, 1620, 1524, 1506, 1454, 1364, 1277, 1188, 1097, 1024, 752, 698 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C19H17NO5, 340.1185; found, 340.1181.
Benzyl 6-methoxy-7-((3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (25a)
Boron trifluoride etherate (5.30 μL, 0.042 mmol) was added to 23a (50.0 mg, 0.14 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (171 mg, 0.47 mmol) in anhydrous CH2Cl2 (3.00 mL). After stirring at rt for 14 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to give 25a as a colorless foam (74.0 mg, 95%): 1H NMR (CD2Cl2, 400 MHz) δ 8.29 (s, 1H), 7.64 (s, 1H), 7.47-7.39 (m, 5H), 6.91 (s, 1H), 5.52 (d, J = 3.4 Hz, 1H), 5.26 (s, 2H), 5.23 (dd, J = 8.4, 3.5 Hz, 1H), 4.95 (t, J = 8.2 Hz, 1H), 3.92 (s, 3H), 3.60 (s, 3H), 3.33 (d, J = 8.0 Hz, 1H), 2.42 (s, 3H), 1.38 (s, 3H), 1.33 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.6, 152.7, 152.1, 148.1, 144.8, 141.8, 134.5, 127.8, 127.7, 127.5, 127.3, 122.3, 120.2, 119.8, 115.1. 109.6, 105.2, 98.3, 82.0, 77.1, 66.5, 65.5, 59.4, 57.4, 55.1, 26.0, 20.9, 8.9; IR (film) νmax 2957, 2928, 2854, 2359, 2341, 1817, 1709, 1522, 1464, 1389, 1371, 1205, 1174, 1111, 1072, 1034, 957, 800 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C28H29NO11, 556.1819; found, 556.1822.
Benzyl 7-((3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-8-methyl-2-oxo-6-propoxy-2H-chromen-3-ylcarbamate (25b)
Boron trifluoride etherate (16.7 μL, 0.13 mmol) was added to 23b (170 mg, 0.44 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (643 mg, 1.77 mmol) in anhydrous CH2Cl2 (11.1 mL). After stirring at rt for 48 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 100:1 → 40:1 CH2Cl2:Acetone) to give 25b as a colorless foam (246 mg, 95%): 1H NMR (CDCl3, 500 MHz) δ 8.17 (s, 1H), 7.35-7.27 (m, 5H), 6.84 (s, 1H), 5.96 (s, 1H), 5.15 (s, 2H), 4.99 (d, J = 7.5 Hz, 1H), 4.59 (d, J = 9.7 Hz, 1H), 4.23 (d, J = 9.6 Hz, 1H), 3.97 (t, J = 6.6 Hz, 1H), 3.82-3.75 (m, 2H), 3.37 (s, 3H), 1.84-1.79 (m, 2H), 1.51 (s, 3H), 1.41 (s, 3H), 1.18 (s, 3H), 1.00 (t, J = 7.5 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.8, 154.3, 152.2, 151.8, 146.7, 144.2, 142.9, 134.6, 127.8, 127.6, 127.5, 127.5, 127.2, 121.0, 120.9, 111.1, 105.3, 101.7, 91.6, 85.7, 82.8, 80.0, 69.8, 58.1, 54.8, 28.3, 28.2, 22.4, 21.3, 9.4; IR (film) νmax 2961, 2939, 2906, 2359, 2341, 1811, 1757, 1726, 1522, 1445, 1371, 1267, 1175, 1113, 1086, 825, 768 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C30H33NO11, 606.1952; found, 606.1950.
Benzyl 6-isopropoxy-7-((3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (25c)
Boron trifluoride etherate (1.30 μL, 0.010 mmol) was added to 23c (13.0 mg, 0.034 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (83.0 mg, 0.23 mmol) in anhydrous CH2Cl2 (1.30 mL). After stirring at rt for 1.5 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to give 25c as a colorless foam (19.0 mg, 95%): 1H NMR (CDCl3, 500 MHz) δ 8.17 (s, 1H), 7.51 (s, 1H), 7.35 (s, 1H), 7.34-7.33 (m, 4H), 6.74 (s, 1H), 5.54 (dd, J = 9.2, 1.2 Hz, 1H), 5.16 (s, 2H), 4.87-4.84 (m, 1H), 4.73 (dd, J = 7.9, 1.9 Hz, 1H), 4.51 (quintet, J = 6.0 Hz, 1H), 3.52 (s, 3H), 3.28 (d, J = 4.8 Hz, 1H), 2.33 (s, 3H), 1.80-1.77 (m, 6H), 1.30 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 161.6, 158.6, 153.2 153.1, 147.1, 146.8, 142.5, 135.5, 128.7, 128.6, 128.3, 123.3, 121.4, 121.1, 116.2, 108.6, 99.4, 83.1, 79.9, 76.1, 74.7, 72.2, 68.0, 60.5, 27.1, 25.6, 21.9, 21.6, 21.0, 10.1; IR (film) νmax 2955, 2922, 2853, 2359, 2339, 1819, 1711, 1520, 1464, 1375, 1171, 1111, 1034, 962, 822, 766 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C30H33NO11, 584.2132; found, 584.2111.
Benzyl 5-methoxy-7-((3aR,4R,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-8-methyl-2-oxo-2H-chromen-3-ylcarbamate (25d)
Boron trifluoride etherate (18.5 μL, 0.15 mmol) was added to 23d (174 mg, 0.49 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (621 mg, 1.71 mmol) in anhydrous CH2Cl2 (11.0 mL). After stirring at rt for 14 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to give 25d as a colorless foam (200 mg, 74%): 1H NMR (CDCl3, 500 MHz) δ 8.49 (s, 1H), 7.34-7.27 (m, 5H), 6.67 (s, 1H), 6.60 (s, 1H), 5.69 (s, 2H), 5.16 (d, J = 5.3 Hz, 1H), 4.89 (t, J = 7.8 Hz, 1H), 4.63 (dd, J = 7.9, 2.4 Hz, 1H), 3.83 (s, 3H), 3.37 (s, 3H), 3.15 (d, J = 8.0 Hz, 1H), 2.16 (s, 3H), 2.16 (s, 3H), 2.12 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.9, 156.0, 155.2, 154.2, 153.1 (2C), 149.4, 135.7, 128.7 (2C), 128.5, 128.2 (2C), 120.8, 117.4, 106.6, 105.4, 94.6, 94.1, 82.9, 67.4, 60.6, 60.6, 56.1, 56.0, 22.2, 22.0, 7.9; IR (film) νmax 2955, 2924, 2853, 1817, 1713, 1526, 1209, 1105, 1072, 1034, 976, 808 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C28H29NO11, 556.1819; found, 556.1826.
Benzyl 8-benzyl-7-((3aR,4R,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-2-oxo-2H-chromen-3-ylcarbamate (25e)
Boron trifluoride etherate (7.80 μL, 0.062 mmol) was added to 23e (80.0 mg, 0.21 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (299 mg, 0.83 mmol) in anhydrous CH2Cl2 (5.20 mL). After stirring at rt for 48 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to give 25e as a colorless foam (47.0 mg, 39%): 1H NMR (CDCl3, 500 MHz) δ 8.22 (s, 1H), 7.46 (s, 1H), 7.35-7.27 (m, 5H), 7.17-7.06 (m, 5H), 6.86 (d, J = 10 Hz, 1H), 6.01 (d, J = 10 Hz, 1H), 5.65 (d, J = 1.6 Hz, 1H), 5.23 (s, 2H), 5.16 (s, 2H), 4.77-4.70 (m, 1H), 4.10 (s, 1H), 3.50 (s, 3H), 3.28 (s, 1H), 3.16 (d, J = 7.4 Hz, 1H), 1.25 (s, 3H), 1.18 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 155.1, 153.2 (2C), 153.1, 148.6 (2C), 139.6 (2C), 128.7, 128.6 (2C), 128.4 (2C), 128.3 (2C), 128.3 (2C), 126.5, 126.2, 123.1, 122.4, 121.7, 117.6, 114.9, 111.5, 94.7, 82.8, 67.6, 60.6 (2C), 29.7, 27.6, 21.9; IR (film) νmax 2926, 2854, 2359, 2341, 1811, 1709, 1607, 1522, 1456, 1381, 1366, 1259, 1209, 1171, 1078, 1049, 968, 766, 700 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C33H31NO10, 602.2026; found, 602.2053.
Benzyl 7-((3aR,4R,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-2-oxo-8-phenyl-2H-chromen-3-ylcarbamate (25f)
Boron trifluoride etherate (14.6 μL, 0.12 mmol) was added to 23f (155 mg, 0.39 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (560 mg, 1.55 mmol) in anhydrous CH2Cl2 (9.70 mL). After stirring at rt for 48 h, triethylamine (150 νL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 100:1 → 40:1 CH2Cl2:Acetone) to give 25f as a colorless foam (225 mg, 99%): 1H NMR (CD2Cl2, 400 MHz) δ 8.37 (s, 1H), 7.75-7.73 (m, 2H), 7.60-7.36 (m, 10H), 7.32 (d, J = 8.8 Hz, 1H), 5.77 (d, J = 1.7 Hz, 1H), 5.26 (s, 2H), 4.76-4.68 (m, 1H), 4.36-4.28 (m, 1H), 3.56 (s, 3H), 3.28 (d, J = 7.2 Hz, 1H), 1.37 (s, 3H), 1.31 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.2, 153.0 (2C), 152.1 (2C), 152.1, 134.4, 129.9, 129.8, 129.4, 127.7 (2C), 127.5, 127.2 (2C), 127.1 (2C), 127.0, 126.3, 121.5 (2C), 120.3, 111.2 (2C), 93.9, 81.9, 66.5, 59.4 (3C), 20.9 (2C); IR (film) νmax 3400, 2959, 2926, 2853, 2359, 2341, 1819, 1715, 1601, 1522, 1381, 1366, 1261, 1215, 1173, 1111, 1059, 970, 800, 700 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C32H29NO10, 588.1870; found, 588.1846.
Benzyl 8-methoxy-7-((3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-2-oxo-2H-chromen-3-ylcarbamate (25g)
Boron trifluoride etherate (17.3 μL, 0.14 mmol) was added to 23g (157 mg, 0.46 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (665 mg, 1.83 mmol) in anhydrous CH2Cl2 (11.5 mL). After stirring at rt for 24 h, triethylamine (150 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 → 10:1 CH2Cl2:Acetone) to give 25g as a colorless foam (237 mg, 95%): 1H NMR (CDCl3, 500 MHz) δ 8.20 (s, 1H), 7.48 (s, 1H), 7.33-7.29 (m, 5H), 7.09 (dd, J = 14.2, 8.8 Hz, 2H), 5.72 (d, J = 1.8 Hz, 1H), 5.16 (s, 2H), 5.02 (dd, J = 7.8, 1.8 Hz, 1H), 4.89 (t, J = 7.8 Hz, 1H), 3.88 (s, 3H), 3.52 (s, 3H), 3.21 (d, J = 7.8 Hz, 1H), 1.27 (s, 3H) 1.17 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 158.0, 153.3, 153.2, 153.1, 149.8, 143.8, 137.1, 135.5, 128.7 (2C), 128.6, 128.3 (2C), 122.7, 122.2, 121.4, 116.1, 113.7, 95.3, 74.7, 72.9, 67.6, 61.9, 60.7, 60.6, 29.7, 29.4; IR (film) νmax 3400, 3319, 2984, 2935, 2359, 1815, 1715, 1609, 1526, 1464, 1383, 1364, 1285, 1213, 1175, 1111, 1063, 968, 764, 737, 700 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C27H27NO11, 564.1482; found, 564.1455.
Benzyl 8-ethyl-7-((3aR,4R,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxotetrahydro-3aH-[1,3]dioxolo[4,5-c]pyran-4-yloxy)-2-oxo-2H-chromen-3-ylcarbamate (25h)
Boron trifluoride etherate (19.0 μL, 0.15 mmol) was added to 23h (171 mg, 0.51 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (183 mg, 0.51 mmol) in anhydrous CH2Cl2 (11.0 mL). After stirring at rt for 24 h, triethylamine (150 νL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to give 25h as a colorless foam (138 mg, 51%): 1H NMR (CD2Cl2, 400 MHz) δ 8.29 (s, 1H), 7.62 (s, 1H), 7.47-7.38 (m, 5H), 7.36 (d, J = 8.8 Hz, 1H), 7.19 (d, J = 8.4 Hz, 1H), 5.81 (d, J = 2.4 Hz, 1H), 5.25 (s, 2H), 5.10 (dd, J = 8.0, 2.0 Hz, 1H), 5.02 (t, J = 7.8 Hz, 1H), 3.55 (s, 3H), 3.41 (d, J = 7.2 Hz, 1H), 2.87 (q, J = 7.4 Hz, 2H), 1.42 (s. 3H), 1.28 (s, 3H), 1.21 (t, J = 7.4 Hz, 3H); 13C NMR (CD2Cl2, 100 MHz) δ 158.5, 154.8, 153.2, 153.1, 148.4, 136.0, 128.6 (2C), 128.4, 128.1, 125.6, 122.4, 121.5, 120.7, 114.8, 111.4, 94.8, 82.7, 77.9, 77.2, 76.6, 67.3, 60.3, 27.3, 22.3, 16.4, 13.6; IR (film) νmax 3400, 2980, 2937, 2359, 2339, 1817, 1711, 1607, 1524, 1383, 1366, 1227, 1205, 1175, 1101, 1040, 906, 768, 737, 700 cm−1; [M + Na]+ calcd for C28H29NO10, 562.1689; found, 562.1689.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-6-methoxy-8-methyl-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26a)
Palladium on carbon (10%, 20.0 mg) was added to 25a (100 mg, 0.18 mmol) in anhydrous THF (5.00 mL) and the solution was placed under an atmosphere of H2. After 6.5 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (56.0 mg, 75%).
EDCI (21.4 mg, 0.11 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (23.1 mg, 0.089 mmol) were added to the amine (18.7 mg, 0.045 mmol) in 30% pyridine/CH2Cl2 (0.70 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (10.5 mg, 36%).
Triethylamine (150 μL) was added to the carbonate (10.4 mg, 0.016 mmol) in MeOH (2.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 20:1; CH2Cl2:MeOH) to afford 26a as a colorless amorphous solid (2.00 mg, 20%, 5% over 3 steps): 1H NMR (CDCl3, 500 MHz) δ 8.73 (s, 1H), 8.70 (d, J = 5.4 Hz, 1H), 7.84 (td, J = 6.2, 2.4 Hz, 1H), 7.82 (s, 1H), 7.30 (t, J = 8.0 Hz, 1H), 7.06 (d, J = 7.8 Hz, 1H), 7.03-7.00 (m, 2H), 6.88-6.86 (m, 1H), 6.81 (s, 1H), 4.99 (d, J = 6.6 Hz, 1H), 4.24 (t, J = 4.2 Hz, 1H), 4.00 (dd, J = 6.5, 3.7 Hz, 1H), 3.90 (s, 3H), 3.86 (s, 3H), 3.83 (s, 3H), 3.80 (d, J = 7.4 Hz, 1H), 3.45 (s, 3H), 3.08 (d, J = 4.7 Hz, 1H), 2.67 (s, 1H), 2.42 (s, 3H), 1.28 (d, J = 8.1 Hz, 3H), 1.18 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 164.6, 158.9, 158.3, 158.2, 148.2, 145.6, 142.5, 137.5, 130.1, 129.0, 128.2, 127.2, 124.9, 122.5, 122.2, 121.2, 121.0, 115.1, 144.2, 112.1, 110.0, 105.4, 101.3, 81.7, 76.8, 69.0, 68.0, 59.1, 55.3, 54.9, 54.3, 28.3, 28.2, 9.1; IR (film) νmax 2961, 2928, 1713, 1670, 1601, 1464, 1383, 1261, 1094, 1022, 798, 700 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C34H37NO11, 636.2445; found, 636.2477.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-methyl-2-oxo-6-propoxy-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26b)
Palladium on carbon (10%, 85.0 mg) was added to 25b (425 mg, 0.7283 mmol) in anhydrous THF (4.90 mL) and the solution was placed under an atmosphere of H2. After 6.5 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (325 mg, 99%).
EDCI (116 mg, 0.60 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (125 mg, 0.4821 mmol) were added to the amine (108 mg, 0.2410 mmol) in 30% pyridine/CH2Cl2 (6.70 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 20:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (51.0 mg, 31%).
Triethylamine (150 μL) was added to the carbonate (51.0 mg, 0.074 mmol) in MeOH (2.50 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26b as a colorless amorphous solid (22.8 mg, 47%, 14% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 8.79 (s, 1H), 8.78 (s, 1H), 7.96 (dd, J = 8.6, 2.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.16-7.11 (m, 2H), 6.97-6.94 (m, 2H), 5.97 (s, 1H), 5.14 (d, J = 6.5 Hz, 1H), 4.31 (t, J = 3.5 Hz, 1H), 4.12-4.06 (m, 2H), 4.03 (dd, J = 6.8, 1.8 Hz, 1H), 3.93 (s, 3H), 3.88 (s, 3H), 3.65 (s, 1H), 3.53 (s, 3H), 3.17 (d, J = 4.8 Hz, 1H), 2.80 (s, 1H), 2.48 (s, 3H), 1.95-1.90 (m, 2H), 1.37 (s, 3H), 1.35 (s, 3H), 1.11 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.0, 164.6, 158.8, 158.3, 147.7, 145.7, 142.3, 137.8, 137.5, 131.3, 129.0, 128.2, 127.2, 124.9, 122.3, 121.2, 121.0, 115.111, 114.2, 112.1, 110.0, 106.2, 101.1, 81.7, 70.0, 69.0, 68.0, 64.8, 59.1, 54.9, 54.3, 24.7, 24.0, 21.3, 9.5, 9.1; IR (film) νmax 3398, 3196, 2964, 2935, 2359, 2330, 1705, 1580, 1526, 1504, 1381, 1242, 1124, 1094, 939, 808, 760, 735 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C36H41NO11, 664.2758; found, 664.2754.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-6-isopropoxy-8-methyl-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26c)
Palladium on carbon (10%, 11 mg) was added to 25c (54.5 mg, 0.093 mmol) in anhydrous THF (600 μL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (42.0 mg, 99%).
EDCI (14.9 mg, 0.078 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (16 mg, 0.062 mmol) were added to the amine (14.0 mg, 0.031 mmol) in 30% pyridine/CH2Cl2 (900 μL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (17.5 mg, 82%).
Triethylamine (150 μL) was added to the carbonate (17.5 mg, 0.025 mmol) in MeOH (2.50 mL) and CH2Cl2 (2.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 10:1 CH2Cl2:Acetone) to afford 26c as a colorless amorphous solid (6.0 mg, 35%, 28% over 3 steps): 1H NMR (CD2Cl2, 500 MHz) δ 8.69 (s, 1H), 8.67 (s, 1H), 7.84 (dd, J = 8.6, 2.4 Hz, 1H), 7.80 (d, J = 2.4 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H), 7.25 (t, J = 7.9 Hz, 1H), 7.03-7.01 (m, 2H), 6.87 (s, 1H), 6.87-6.83 (m, 1H), 4.96 (d, J = 6.8 Hz, 1H), 4.61-4.56 (m, 1H), 4.19 (t, J = 4.0 Hz, 1H), 3.89 (dd, J = 6.8, 3.7 Hz, 1H), 3.82 (s, 3H), 3.76 (s, 3H), 3.75 (s, 1H), 3.41 (s, 3H), 3.34 (s, 1H), 3.03 (d, J = 4.5 Hz, 1H), 2.36 (s, 3H), 1.33 (t, J = 6.2 Hz, 6H), 1.25 (s, 3H), 1.23 (s, 3H); 13C NMR (CD2Cl2, 125 MHz) δ 164.6, 159.1, 158.6, 158.3, 146.7, 146.3, 142.5, 138.1, 130.2, 129.1, 128.3, 127.4, 125.2, 122.8, 122.2, 121.2, 121.1, 115.5, 144.5, 112.1, 110.2, 108.4, 101.4, 81.9, 77.0, 71.1, 69.2, 68.3, 59.1, 55.1, 54.5, 28.7, 28.6, 20.8, 20.8, 9.1; IR (film) νmax 2924, 2854, 2359, 2341, 1734, 1684, 1653, 1558, 1541, 1522, 1506, 1458, 1387, 1339, 1286, 1244, 1113, 912, 797 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C36H41NO11, 686.2578; found, 686.2610.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-5-methoxy-8-methyl-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26d)
Palladium on carbon (10%, 40 mg) was added to 25d (200 mg, 0.36 mmol) in anhydrous THF (2.40 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (150 mg, 99%).
EDCI (57.5 mg, 0.30 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (62 mg, 0.24 mmol) were added to the amine (50.6 mg, 0.12 mmol) in 30% pyridine/CH2Cl2 (3.30 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 → 10:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (25.2 mg, 32%).
Triethylamine (150 μL) was added to the carbonate (25.2 mg, 0.038 mmol) in MeOH (2.0 mL) and CH2Cl2 (2.0 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26d as a colorless amorphous solid (17.0 mg, 70%, 22% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 9.02 (s, 1H), 8.97 (s, 1H), 8.66 (s, 1H), 7.96 (dd, J = 8.6, 2.4 Hz, 1H), 7.91-7.90 (m, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.16-7.11 (m, 2H), 6.96 (dd, J = 8.3, 2.6 Hz), 6.85 (d, J = 5.5 Hz, 1H), 5.70 (d, J = 2.1 Hz, 1H), 4.36-4.33 (m, 1H), 4.27 (m, 1H), 3.99 (s, 3H), 3.93 (s, 3H), 3.88 (s, 3H), 3.62 (s, 3H), 3.41-3.38 (m, 1H), 2.24 (s, 3H), 1.41 (s, 3H), 1.19 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 164.2, 158.7, 158.6, 158.3, 155.3, 153.5, 148.6, 137.6, 129.9, 128.9, 128.1, 127.1, 125.1, 121.0, 119.4, 118.9, 114.2, 114.2, 112.1, 110.0, 104.9, 103.7, 96.7, 92.9, 83.2, 70.1, 67.5, 60.9, 60.8, 54.8, 54.3, 21.9, 21.4, 6.8; IR (film) νmax 3405, 2986, 2934, 1713, 1609, 1528, 1383, 1250, 1213, 1053, 999, 914, 878, 737 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C34H37NO11, 636.2445; found, 636.2482.
N-(8-benzyl-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26e)
Palladium on carbon (10%, 46 mg) was added to 25e (230 mg, 0.38 mmol) in anhydrous THF (2.50 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (177 mg, 99%).
EDCI (61.5 mg, 0.32 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (66.3 mg, 0.26 mmol) were added to the amine (60.0 mg, 0.13 mmol) in 30% pyridine/CH2Cl2 (3.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 20:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (12.3 mg, 14%).
Triethylamine (150 μL) was added to the carbonate (12.3 mg, 0.017 mmol) in MeOH (1.5 mL) and CH2Cl2 (1.5 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26e as a colorless amorphous solid (6.00 mg, 51%, 7.1% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 8.84 (s, 1H), 8.72 (s, 1H), 7.96 (dd, J = 10, 2.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H), 7.50 (d, J = 8.8 Hz, 1H), 7.39 (t, J = 7.9 Hz, 1H), 7.31 (d, J = 8.8 Hz, 1H), 7.28-7.25 (m, 5H), 7.21-7.18 (m, 1H), 7.15-7.11 (m, 2H), 6.97-6.94 (m, 1H), 5.54 (d, J = 2.7 Hz, 1H), 4.25 (t, J = 15.1 Hz, 2H), 4.17-4.11 (m, 1H), 4.05 (d, J = 2.6 Hz, 1H), 3.93 (s, 3H), 3.88 (s, 3H), 3.58 (s, 3H), 3.31 (d, J = 8.7 Hz, 1H), 2.64 (s, 1H), 2.04 (s, 1H), 1.40 (s, 3H), 1.03 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.5, 159.8, 159.3, 159.3, 156.4, 148.9, 140.0 (2C), 138.6, 131.1, 130.0, 129.2, 128.5, 128.3, 128.2, 127.0, 126.2 (2C), 126.0 (2C), 124.1, 122.2, 122.0, 117.2, 115.2, 114.4, 113.2, 111.7, 111.0, 98.0, 70.6 (2C), 68.6, 61.6, 55.9, 55.4, 29.3, 28.9, 28.3; IR (film) νmax 3404, 2930, 2359, 2341, 1713, 1670, 1605, 1526, 1502, 1367, 1244, 1180, 1134, 1076, 1026, 960 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C39H39NO10, 682.2652; found, 682.2653.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-2-oxo-8-phenyl-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26f)
Palladium on carbon (10%, 14 mg) was added to 25f (68.0 mg, 0.12 mmol) in anhydrous THF (800 μL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (52.0 mg, 99%).
EDCI (18.5 mg, 0.096 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (19.9 mg, 0.077 mmol) were added to the amine (17.5 mg, 0.039 mmol) in 30% pyridine/CH2Cl2 (1.10 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (14.0 mg, 52%).
Triethylamine (150 μL) was added to the carbonate (14.0 mg, 0.020 mmol) in MeOH (1.5 mL) and CH2Cl2 (1.5 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26f as a colorless amorphous solid (5.20 mg, 39%, 20% over 3 steps): 1H NMR (CD2Cl2, 500 MHz) δ 8.85 (s, 1H), 8.65 (s, 1H), 7.92 (d, J = 2.4 Hz, 1H), 7.90 (d, J = 2.4 Hz, 1H), 7.86 (d, J = 2.4 Hz, 1H), 7.57-7.43 (m, 3H), 7.36-7.33 (m, 4H), 7.11-7.06 (m, 3H), 6.92 (d, J = 0.8 Hz, 1H), 5.52 (d, J = 2.4 Hz, 1H), 4.08 (q, J = 7.2, Hz, 1H), 3.89 (s, 3H), 3.83 (s, 3H), 3.74 (dd, J = 9.0, 3.5 Hz, 1H), 3.50 (s, 3H), 3.23 (d, J = 9.0 Hz, 1H), 2.12 (s, 1H), 2.00 (s, 1H), 1.33 (s, 3H), 1.04 (s, 3H); 13C NMR (CD2Cl2, 125 MHz) δ 164.5, 159.0 (2C), 158.6, 158.1, 154.4, 147.2, 138.0 (2C), 130.7, 130.1, 129.7, 129.1, 128.7, 128.3, 127.4, 127.2, 127.0, 127.0, 125.2, 122.6, 121.6, 121.1, 118.6, 114.5, 113.8, 112.1, 111.3, 110.2, 97.5, 70.1 (2C), 67.4, 60.8, 55.0, 54.5, 21.9, 21.6; IR (film) νmax 3402, 2932, 2359, 2341, 1713, 1603, 1524, 1500, 1367, 1267, 1086, 1040, 964, 750 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C38H37NO10, 668.2496; found, 668.2485.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-methoxy-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26g)
Palladium on carbon (10%, 47 mg) was added to 25g (237 mg, 0.44 mmol) in anhydrous THF (2.93 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (177 mg, 99%).
EDCI (69.4 mg, 0.36 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (74.8 mg, 0.29 mmol) were added to the amine (59.0 mg, 0.14 mmol) in 30% pyridine/CH2Cl2 (4.00 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (26.0 mg, 28%).
Triethylamine (150 μL) was added to the carbonate (26.0 mg, 0.040 mmol) in MeOH (2.0 mL) and CH2Cl2 (2.0 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26g as a colorless amorphous solid (15.7 mg, 63%, 18% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 8.82 (s, 1H), 8.73 (s, 1H), 7.96 (dd, J = 8.6, 2.4 Hz, 1H), 7.91 (d, J = 2.4 Hz, 1H) 7.39 (t, J = 7.9 Hz, 1H), 7.30 (s, 2H), 7.14 (d, J = 8.6 Hz, 2H), 7.12 (d, J = 2.2 Hz, 1H), 6.96 (dd, J = 8.3, 2.5 Hz, 1H), 5.61 (d, J = 2.4 Hz, 1H), 4.29 (t, J = 4.0 Hz, 1H), 4.27-4.25 (m, 1H), 3.98 (s, 3H), 3.93 (s, 3H), 3.88 (s, 3H), 3.62 (s, 3H), 3.47 (s, 1H), 3.37 (d, J = 8.8 Hz, 1H), 2.62 (s, 1H), 1.30 (s, 3H), 1.24 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 164.5, 158.8, 158.3, 157.8, 150.2 (2C), 142.9, 137.5, 135.6, 130.0, 128.9, 128.2, 127.2, 127.2, 124.9, 122.8, 121.6, 121.0, 114.3, 114.2, 112.3, 112.1, 110.0, 97.7, 70.0 (2C), 67.5, 60.8 (2C), 54.9, 54.3, 28.7, 28.3; IR (film) νmax3402, 2961, 2928, 2853, 1713, 1672, 1607, 1526, 1504, 1462, 1367, 1263, 1248, 1086, 1040, 953, 798, 735, 700 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C33H35NO11, 622.2288; found, 622.2307.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-ethyl-2-oxo-2H-chromen-3-yl)-3′,6-dimethoxybiphenyl-3-carboxamide (26h)
Palladium on carbon (10%, 12 mg) was added to 25h (121 mg, 0.22 mmol) in anhydrous THF (5.00 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (90.0 mg, 99%).
EDCI (46.2 mg, 0.24 mmol) and 3′,6-dimethoxybiphenyl-3-carboxylic acid (43.9 mg, 0.19 mmol) were added to the amine (39.0 mg, 0.096 mmol) in 30% pyridine/CH2Cl2 (2.65 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (39.1 mg, 66%).
Triethylamine (150 μL) was added to the carbonate (13.0 mg, 0.020 mmol) in MeOH (1.5 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 1H CH2Cl2:Acetone) to afford 26h as a colorless amorphous solid (4.10 mg, 33%, 22% over 3 steps): 1HNMR (CD2Cl2, 500 MHz) δ 8.70 (s, 1H), 8.61 (s, 1H), 7.83 (dd, J = 8.5, 2.5 Hz, 1H), 7.78 (d, J = 2.5 Hz, 1H), 7.31 (d, J = 9.0 Hz, 1H), 7.27 (t, J = 7.8 Hz, 1H), 7.17 (d, J = 8.5 Hz, 1H), 7.03-6.99 (m, 3H), 6.85-6.82 (m, 1H), 5.49 (d, J = 1.5 Hz, 1H), 4.15 (t, J = 8.5 Hz, 1H), 4.14 (d, J = 8.5 Hz, 1H), 3.90 (s, 2H), 3.87 (s, 3H), 3.81 (s, 3H), 3.28 (s, 3H), 3.27 (d, J = 8.5 Hz, 1H), 2.76 (q, J = 4.5 Hz, 2H), 1.29 (s, 3H), 1.09 (t, J = 7.4 Hz, 3H), 1.08 (s, 3H); 13C NMR (CD2Cl2, 125 MHz) δ 164.5, 159.0, 158.6, 158.5, 155.1, 147.9, 138.1, 130.1, 129.1, 128.3, 127.4, 125.4, 125.2, 123.1, 121.4, 121.2, 119.4, 114.5, 113.5, 112.1, 110.8, 110.2, 97.6, 83.4, 77.7, 70.6, 67.8, 61.0, 55.1, 54.5, 28.2, 21.6, 15.6, 12.9; IR (film) νmax 3404, 2968, 2934, 2359, 2341, 1715, 1605, 1524, 1504, 1367, 1244, 1101, 1024, 995, 960, 800 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C34H37NO10, 620.2496; found, 620.2507.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-6-methoxy-8-methyl-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26i)
Palladium on carbon (10%, 15 mg) was added to 25a (74.0 mg, 0.13 mmol) in anhydrous THF (5.00 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (60.0 mg, 99%).
EDCI (69.0 mg, 0.36 mmol) and 1H-indole-2-carboxylic acid (46.4 mg, 0.29 mmol) were added to the amine (60.0 mg, 0.14 mmol) in 30% pyridine/CH2Cl2 (3.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (68.0 mg, 85%).
Triethylamine (150 μL) was added to the carbonate (68.0 mg, 0.12 mmol) in MeOH (2.5 mL) and CH2Cl2 (2.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26i as a colorless amorphous solid (12.6 mg, 19%, 16% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 8.29 (s, 1H), 7.63 (s, 1H), 7.45-7.39 (m, 3H), 6.92 (s, 1H), 6.85 (s, 1H), 6.19 (s, 1H), 5.09 (d, J = 6.5 Hz, 1H), 4.31-4.28 (m, 1H), 4.01-3.97 (m, 1H), 3.94 (s, 3H), 3.62 (s, 1H), 3.56 (s, 3H), 3.15 (d, J = 4.9 Hz, 1H), 2.46 (s, 3H), 2.36 (s, 1H), 1.38 (d, J = 11.5 Hz, 3H), 1.32 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.6, 152.1, 148.2, 145.4, 142.2, 134.5, 129.9, 127.8, 127.7, 127.5, 127.3, 122.3, 121.2, 120.2, 115.0, 105.3, 105.1, 101.3, 81.7, 69.0, 68.0, 66.5, 59.1, 55.2, 28.7, 24.6, 24.1, 9.1; IR (film) νmax 2926, 1707, 1526, 1464, 1391, 1340, 1296, 1231, 1207, 1086, 1024, 943, 739, 700, 623 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C28H30N2O9, 561.1849; found, 561.1781.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-methyl-2-oxo-6-propoxy-2H-chromen-3-yl)-1H-indole-2-carboxamide (26j)
Palladium on carbon (10%, 85 mg) was added to 25b (425 mg, 0.729 mmol) in anhydrous THF (4.90 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (325 mg, 99%).
EDCI (116 mg, 0.6026 mmol) and 1H-indole-2-carboxylic acid (77.7 mg, 0.4821 mmol) were added to the amine (108 mg, 0.2410 mmol) in 30% pyridine/CH2Cl2 (6.70 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (91.0 mg, 64%).
Triethylamine (150 μL) was added to the carbonate (91.0 mg, 0.1536 mmol) in MeOH (2.5 mL) and CH2Cl2 (2.50 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26j as a colorless amorphous solid (17.5 mg, 20%, 13% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 9.32 (s, 1H), 8.80 (s, 1H), 8.76 (s, 1H), 7.77 (d, J = 8.0 Hz, 1H), 7.53 (d, J = 7.5 Hz, 1H), 7.40-7.36 (m, 1H), 7.24-7.20 (m, 1H), 6.98 (s, 1H), 6.01 (s, 1H), 5.15 (d, J = 6.5 Hz, 1H), 4.32-4.25 (m, 1H), 4.11-4.04, (m, 1H), 3.62-3.59 (m, 2H), 3.53 (s, 3H), 3.18-3.12 (m, 1H), 2.64 (s, 1H), 2.49 (s, 3H), 2.18 (s, 1H), 1.95-1.91 (m, 2H), 1.36 (d, J = 9.6 Hz, 3H), 1.29 (d, J = 9.8 Hz, 3H), 1.12 (t, J = 7.4 Hz, 3H); 13C NMR (CDCl3, 125 MHz) δ 160.1, 159.0, 148.8, 146.9, 143.4, 136.9, 129.8, 127.6, 125.5, 123.5, 123.0, 122.6, 122.3, 121.2, 116.0, 112.0, 107.3, 104.3, 102.2, 82.8, 71.0, 70.1, 69.1, 60.2, 59.7, 25.7, 23.1, 23.4, 10.5, 10.2; IR (film) νmax 3630, 3304, 2926, 2854, 2359, 2332, 1713, 1705, 1539, 1387, 1240, 1103, 947, 930, 822, 739 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C30H34N2O9, 567.2342; found, 567.2367.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-6-isopropoxy-8-methyl-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26k)
Palladium on carbon (10%, 4 mg) was added to 25c (19.0 mg, 0.033 mmol) in anhydrous THF (220 μL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (14.5 mg, 99%).
EDCI (15.6 mg, 0.081 mmol) and 1H-indole-2-carboxylic acid (10.5 mg, 0.065 mmol) was added to the amine (14.5 mg, 0.033 mmol) in 30% pyridine/CH2Cl2 (1.00 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (10.0 mg, 50%).
Triethylamine (150 μL) was added to the carbonate (10.0 mg, 0.017 mmol) in MeOH (2.5 mL) and CH2Cl2 (2.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 10:1 CH2Cl2:Acetone) to afford 26k as a colorless amorphous solid (6.00 mg, 46%, 23% over 3 steps): 1H NMR (CDCl3, 500 MHz) δ 8.16 (s, 1H), 7.52 (s, 1H), 7.34-7.30 (m, 5H), 6.75 (s, 1H), 4.93 (d, J = 5.0 Hz, 1H), 4.56-4.51 (m, 1H), 4.23 (t, J = 4.0 Hz, 1H), 3.98-3.96 (m, 1H), 3.76 (s, 1H), 3.43 (s, 3H), 3.06 (d, J = 4.3 Hz, 1H), 2.65 (s, 1H), 2.38 (s, 3H), 1.33 (dd, J = 11.2, 6.1 Hz, 6H), 1.29 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 157.6, 152.1, 146.6, 146.0, 142.1, 134.5, 127.7, 127.5, 127.2, 122.2, 121.3 (2C), 120.2 (2C), 115.0 (2C), 108.1 (2C), 101.2, 81.6, 71.2, 68.9, 68.1, 66.5, 59.0, 24.8, 23.6, 20.8 (2C), 9.1; IR (film) νmax cm−1 3406, 2930, 2375, 1705, 1522, 1394, 1229, 1205, 1111, 1078, 1049, 933, 793, 739, 698; HRMS (ESI+) m/z: [M + Na]+ calcd for C30H34N2O9, 589.2162; found, 589.2111.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-5-methoxy-8-methyl-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26l)
Palladium on carbon (10%, 40 mg) was added to 25d (200 mg, 0.36 mmol) in anhydrous THF (2.40 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, whichwas used without further purification (150 mg, 99%).
EDCI (57.5 mg, 0.30 mmol) and 1H-indole-2-carboxylic acid (38.7 mg, 0.24 mmol) were added to the amine (50.6 mg, 0.12 mmol) in 30% pyridine/CH2Cl2 (3.30 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (26.3 mg, 39%).
Triethylamine (150 μL) was added to the carbonate (26.3 mg, 0.047 mmol) in MeOH (2.00 mL) and CH2Cl2 (2.00 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26l as a colorless amorphous solid (6.60 mg, 26%, 10% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 9.26 (s, 1H), 8.96 (s, 1H), 8.68 (s, 1H), 7.74 (d, J = 8.1 Hz, 1H), 7.52 (d, J = 8.3 Hz, 1H), 7.38-7.34 (m, 1H), 7.22-7.16 (m, 1H), 6.84 (s, 1H), 6.00 (s, 1H), 5.65 (d, J = 1.7 Hz, 1H), 4.26-4.21 (m, 2H), 3.96 (s, 3H), 3.59 (s, 3H), 3.35 (d, J = 8.6 Hz, 1H), 2.25 (s, 3H), 1.53 (s, 3H), 1.20 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.1, 157.4, 156.5, 149.7, 136.7, 134.7, 127.7, 125.3, 122.5, 121.1, 120.0, 111.9, 104.6, 103.8, 97.7, 94.0, 84.3, 84.2, 82.6, 69.6, 69.1, 66.1, 62.2, 62.0, 59.7, 23.1, 22.7, 14.2; IR (film) νmax 3389, 2924, 2853, 1697, 1605, 1535, 1460, 1340, 1211, 1101, 1088, 962, 729 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C28H30N2O9, 539.2030; found, 539.2056.
N-(8-benzyl-7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26m)
Palladium on carbon (10%, 46 mg) was added to 25e (230 mg, 0.38 mmol) in anhydrous THF (2.50 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (177 mg, 99%).
EDCI (61.5 mg, 0.32 mmol) and 1H-indole-2-carboxylic acid (41.4 mg, 0.26 mmol) were added to the amine (60.0 mg, 0.13 mmol) in 30% pyridine/CH2Cl2 (3.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a yellow solid, which was used without further purification (66.2 mg, 85%).
Triethylamine (150 μL) was added to the carbonate (66.2 mg, 0.11 mmol) in MeOH (2.50 mL) and CH2Cl2 (2.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26m as a colorless amorphous solid (6.50 mg, 10%, 8.4% over 3 steps): 1H NMR (CD2Cl2, 500 MHz) δ 8.68 (s, 1H), 8.62 (s, 1H), 7.63-7.61 (m, 1H), 7.47 (dd, J = 5.7, 3.3 Hz, 1H), 7.16-7.10 (m, 4H), 7.10-7.04 (m, 4H), 6.99 (t, J = 8.2 Hz, 1H), 6.74 (d, J = 8.0 Hz, 1H), 6.43 (dd, J = 8.1, 0.7 Hz, 1H), 5.31 (d, J = 2.9 Hz, 1H), 4.17 (t, J = 6.8 Hz, 1H), 4.01 (dd, J = 8.5, 2.9 Hz, 1H), 3.90 (d, J = 13.1 Hz, 2H), 3.45 (s, 3H), 3.16 (d, J = 8.6 Hz, 1H), 2.43 (s, 1H), 2.21 (s, 1H), 1.25 (s, 3H), 0.95 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 166.8, 155.4, 153.8, 140.2, 131.6, 45 130.2, 128.0, 127.6, 127.5 (2C), 127.4 (2C), 126.9, 125.1 (2C), 115.2, 108.3 (2C), 105.8 (2C), 97.1 (2C), 83.3 (2C), 77.2, 70.2, 67.9 (2C), 65.0, 60.6, 21.9, 13.1, 13.0; IR (film) νmax 3333, 2961, 2926, 2854, 1717, 1601, 1466, 1261, 1090, 1076, 1041, 800, 750 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C33H32N2O8, 607.2056; found, 607.2056.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-2-oxo-8-phenyl-2H-chromen-3-yl)-1H-indole-2-carboxamide (26n)
Palladium on carbon (10%, 14 mg) was added to 25f (68.0 mg, 0.12 mmol) in anhydrous THF (800 μL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (52.0 mg, 99%).
EDCI (18.5 mg, 0.096 mmol) and 1H-indole-2-carboxylic acid (12.4 mg, 0.077 mmol) were added to the amine (17.5 mg, 0.039 mmol) in 30% pyridine/CH2Cl2 (1.10 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (8.20 mg, 36%).
Triethylamine (150 μL) was added to the carbonate (8.2 mg, 0.014 mmol) in MeOH (1.00 mL) and CH2Cl2 (1.00 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26n as a colorless amorphous solid (4.00 mg, 51%, 18% over 3 steps): 1H NMR (CD2Cl2, 500 MHz) δ 9.23 (s, 1H), 8.80 (s, 1H), 8.67 (s, 1H), 7.71 (dd, J = 8.0, 0.7 Hz, 1H), 7.57 (d, J = 8.8 Hz, 1H), 7.51-7.48 (m, 3H), 7.46-7.44 (m, 1H), 7.37-7.32 (m, 4H), 7.19-7.17 (m, 2H), 5.53 (d, J = 2.4 Hz, 1H), 3.86 (s, 1H), 3.76-3.73 (m, 2H), 3.51 (s, 3H), 3.23 (d, J = 9.1 Hz, 1H), 2.41 (s, 1H), 1.34 (s, 3H), 1.05 (s, 3H); 13C NMR (CD2Cl2, 125 MHz) δ 159.1, 157.9, 154.5, 147.3, 136.0 (2C), 130.7 (2C), 129.8, 129.2,. 127.3, 127.1, 127.0, 126.9, 124.5, 122.8, 121.6, 121.2, 120.3, 113.7, 111.4, 111.1, 103.1 (2C), 97.5, 83.2, 77.7, 70.1, 67.5, 60.8, 21.9, 21.7; IR (film) νmax 3427, 2961, 2924, 2853, 2062, 1643, 1614, 1537, 1362, 1236, 1094, 1041, 962, 791, 739, 698 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C32H30N2O8, 593.1900; found, 593.1890.
N-(7-((2S,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-methoxy-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26o)
Palladium on carbon (10%, 47 mg) was added to 25g (237 mg, 0.44 mmol) in anhydrous THF (2.93 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (177 mg, 99%).
EDCI (69.4 mg, 0.36 mmol) and 1H-indole-2-carboxylic acid (46.7 mg, 0.29 mmol) were added to the amine (59.0 mg, 0.14 mmol) in 30% pyridine/CH2Cl2 (4.00 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 Hexane:Ether → 40:1 CH2Cl2:Acetone) to afford a colorless solid, which was used without further purification (32.0 mg, 49%).
Triethylamine (150 μL) was added to the carbonate (32.0 mg, 0.071 mmol) in MeOH (2.00 mL) and CH2Cl2 (2.00 mL). After 48 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 3:1 CH2Cl2:Acetone) to afford 26o as a colorless amorphous solid (22.1 mg, 73%, 35% over 3 steps): 1H NMR (CD2Cl2, 400 MHz) δ 9.28 (s, 1H), 8.78 (s, 1H), 7.77 (d, J = 8.1 Hz, 1H), 7.53 (dd, J = 8.3, 0.8 Hz, 1H), 7.38 (m, 1H), 7.31 (s, 2H), 7.24 (d, J = 0.9 Hz, 1H), 7.22-7.20 (m, 1H), 6.02 (s, 1H), 5.62 (d, J = 2.3 Hz, 1H), 4.25 (t, J = 3.5 Hz, 1H), 3.99 (s, 3H), 3.75 (dd, J = 9.0, 3.6 Hz, 1H), 3.62 (s, 3H), 3.13 (d, J = 3.6 Hz, 1H), 1.30 (s, 3H), 1.27 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 163.8, 159.1, 157.7, 150.6, 143.3, 136.0, 135.8, 129.2, 126.9, 124.6, 122.8, 121.8, 121.6, 121.4, 120.3, 114.4, 112.5, 111.2, 103.1, 98.0, 83.2, 77.9, 74.0, 60.9, 58.7, 22.3, 21.8; IR (film) νmax 3420, 2957, 2924, 2854, 2359, 1653, 1558, 1541, 1246, 1001, 798 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C27H28N2O9, 525.1873; found, 525.1875.
N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyltetrahydro-2H-pyran-2-yloxy)-8-ethyl-2-oxo-2H-chromen-3-yl)-1H-indole-2-carboxamide (26p)
Palladium on carbon (10%, 12 mg) was added to 25h (121 mg, 0.22 mmol) in anhydrous THF (5.00 mL) and the solution was placed under an atmosphere of H2. After 12 h, the solution was filtered through SiO2 (1:1 CH2Cl2:Acetone) and the eluent was concentrated to afford a yellow solid, which was used without further purification (90.0 mg, 99%).
EDCI (28.3 mg, 0.15 mmol) and 1H-indole-2-carboxylic acid (19.0 mg, 0.12 mmol) were added to the amine (24.0 mg, 0.059 mmol) in 30% pyridine/CH2Cl2 (1.63 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford an colorless foam, which was used without further purification (23.8 mg, 73%).
Triethylamine (150 μL) was added to the carbonate (14.1 mg, 0.026 mmol) in MeOH (1.50 mL). After 12 h, the solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 CH2Cl2:Acetone) to afford 26p as a yellow amorphous solid (5.00 mg, 37%, 27% over 3 steps): 1H NMR (CD2Cl2, 500 MHz) δ 8.65 (s, 1H), 7.62 (d, J = 8.0 Hz, 1H), 7.41 (d, J = 8.5 Hz, 1H), 7.33 (d, J = 8.5 Hz, 1H), 7.23 (t, J = 7.8 Hz, 1H), 7.20 (d, J = 8.5 Hz, 2H), 7.13 (s, 2H), 7.07 (t, J = 7.5 Hz, 1H), 5.46 (d, J = 2.0 Hz, 1H), 4.07 (dd, J = 9.3, 3.5 Hz, 1H), 4.04 (t, J = 3.5 Hz, 1H), 3.51 (s, 3H), 3.25 (d, J = 7.2 Hz, 1H), 2.78 (q, J = 7.0 Hz, 2H), 1.27 (s, 3H), 1.10 (t, J = 7.5 Hz, 3H), 1.06 (s, 3H); 13C NMR (CD2Cl2, 125 MHz) δ 158.4, 157.3, 154.2, 146.8, 135.3, 128.1, 125.5, 124.0, 123.0, 123.0, 122.8, 120.1, 119.5, 118.7, 118.2, 112.0, 110.1, 109.7, 102.4, 97.0, 82.1, 76.5, 69.3, 66.4, 26.7, 20.3, 14.3, 11.4; IR (film) νmax 3435, 3416, 2974, 2935, 2469, 2359, 2339, 1715, 1651, 1520, 1456, 1435, 1379, 1354, 1259, 1180, 1113, 1088, 1026, 997, 962, 798, 739 cm−1; HRMS (ESI+) m/z: [M + Na]+ calcd for C28H30N2O8, 545.1900; found, 545.1909.
tert-Butyl quinolin-7-yl carbonate (28)
Di-tert-butyl dicarbonate (7.40 g, 33.92 mmol) and 4-(dimethylamino)pyridine (222 mg, 1.81 mmol) were added in sequence to 7-hydroxyquinoline (2.00 g, 13.75 mmol) in anhydrous N, N-dimethylformamide (20.0 mL) at rt. After 18 h, the reaction was diluted with EtOAc (250 mL). The organic layer was washed with 1.0 M NaOH (250 mL), water (3 × 250 mL), saturated aqueous NaCl solution (250 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 1:1 Hexanes:EtOAc) to give 28 as a colorless amorphous solid (3.26 g, 97%): 1H NMR (CDCl3, 500 MHz) δ 8.93 (dd, J = 4.2, 1.7 Hz, 1H), 8.17 (bd, J = 8.4 Hz, 1H), 7.90 (d, J = 2.2 Hz, 1H), 7.84 (d, J = 8.9 Hz, 1H), 7.43 (dd, J = 8.8, 2.3 Hz, 1H), 7.40 (dd, J = 8.3, 4.3 Hz, 1H), 1.60 (s, 9H); 13C NMR (CDCl3, 125 MHz) δ 151.7, 151.7, 148.9, 135.9, 129.0, 126.3, 122.0, 121.0, 120.2, 84.2, 27.8 (3C); IR (film) νmax 1759, 1277, 1240, 1142, 768, 750 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C14H15NO3, 246.1130; found, 246.1113.
N-(4-Methoxybenzyl)-7-(benzyloxy)quinolin-3-amine (31)
Bromine (790 μL, 2.48 g, 15.38 mmol) was added to 28 (3.26 g, 13.30 mmol) in CCl4 (30.0 mL) at rt. This solution was heated to reflux, anhydrous pyridine (1.20 mL,1.17 g, 14.84 mmol) was added over 10 min, and the solution was stirred at reflux for 18 h. The cooled reaction was diluted with EtOAc/MeOH (250 mL) and saturated aqueous NaHCO3 (200 mL), then extracted with EtOAc (4 × 250 mL). Combined organic layers were dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 4:1 Hexanes:EtOAc) to give 29 as a light yellow solid containing >80% 3-bromo isomer (2.00 g, 46%), which was used without further purification.
Hydrogen chloride was bubbled through 29 (2.00 g, 6.16 mmol) in anhydrous MeOH (44.0 mL) for 3 min at rt, then the solution was stirred at 50° C for 5 min. The solvent was concentrated and the residue placed under high vacuum for 6 h to ensure complete removal of MeOH. The yellow residue was dissolved in anhydrous dimethylformamide (44.0 mL) and cooled to 0° C, then NaH (997 mg, 24.93 mmol) was added. After 15 min, BnBr (1.20 mL, 1.73 g, 10.09 mmol) was added and the reaction was warmed to rt over 18 h. Reaction contents were partitioned between saturated aqueous NaHCO3 (500 mL) and EtOAc (500 mL), then extracted with EtOAc (3 × 500 mL). The combined organic layers were washed with water (3 × 1 L), saturated aqueous NaCl solution (1 L), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 9:1 Hexanes:EtOAc) to give 30 as a light orange solid containing >80% 3-bromo isomer (1.70 g, 88%), which was used without further purification.
4-Methoxy-benzylamine (1.77 mL, 1.87 g, 13.64 mmol) and anhydrous DMSO (2.90 mL) were added to a high-pressure flask charged with 30 (1.65 g, 5.26 mmol), K3PO4 (2.36 g, 11.10 mmol), CuII (167 mg, 0.88 mmol), and L-(−)-proline (141 mg, 1.22 mmol); the sealed flask was heated to 80° C for 44 h. After cooling to rt, reaction contents were partitioned between water (50 mL) and EtOAc (100 mL), then were extracted with EtOAc (2 × 100 mL). The combined organic layers were washed with saturated aqueous NaCl solution (200 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 1:1 Hexanes:EtOAc) to give 31 as a light yellow amorphous solid (1.43 g, 73%): 1H NMR (CDCl3, 500 MHz) δ 8.44 (d, J = 2.8 Hz, 1H), 7.52 (d, J = 8.9 Hz, 1H), 7.50 (m, 2H), 7.43-7.38 (m, 3H), 7.36-7.32 (m, 3H), 7.20 (dd, J = 8.9, 2.6 Hz, 1H), 7.06 (d, J = 2.7 Hz, 1H), 6.91 (m, 2H), 5.17 (s, 2H), 4.35 (d, J = 4.8 Hz, 2H), 4.17 (bt, J = 4.8 Hz, 1H), 3.82 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.3, 156.9, 143.4, 143.3, 140.5, 137.0, 130.6, 129.1 (2C), 128.8 (2C), 128.2, 127.9 (2C), 127.3, 124.7, 120.4, 114.4 (2C), 112.0, 109.0, 70.3, 55.5, 48.0; IR (film) νmax 3279, 2953, 2833, 1609, 1510, 1377, 1354, 1302, 1225, 1175, 1124, 1028, 995, 868, 818, 762, 708 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C24H22N2O2, 371.1759; found, 371.1732.
N-(4-Methoxybenzyl)-N-(7-(benzyloxy)quinolin-3-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (32)
Thionyl chloride (395 μL, 644 mg, 5.42 mmol) was added to 4-methoxy-3-(3-methoxyphenyl)-benzoic acid (464 mg, 1.80 mmol) in anhydrous THF (6.10 mL) at rt and the resulting solution was heated at reflux for 4.5 h. The solvent was removed and the resulting acid chloride was used without further purification.
Sodium hydride (93 mg, 2.31 mmol) was added to 31 (508 mg, 1.37 mmol) in anhydrous THF (9.10 mL). After stirring for 2 h at rt, a solution of the freshly-prepared acid chloride (1.80 mmol) in anhydrous THF (3.00 mL) was added and the reaction was heated to reflux for 18 h. The reaction was cooled, partitioned between saturated aqueous NaHCO3 (50 mL) and CH2Cl2 (50 mL), then extracted with CH2Cl2 (3 × 100 mL). Combined organic extracts were washed with saturated aqueous NaCl solution (200 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 1:1 EtOAc:Hexanes to 2:1 EtOAc:Hexanes) to give 32 as a pale yellow amorphous solid (684 mg, 82%): 1H NMR (CDCl3, 500 MHz) δ 8.35 (d, J = 2.3 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.57 (d, J = 9.0 Hz, 1H), 7.48 (m, 2H), 7.44-7.34 (m, 5H), 7.29 (m, 2H), 7.23 (m, 2H), 7.13 (t, J = 7.9 Hz, 1H), 6.81 (m, 2H), 6.78 (dd, J = 2.5, 1.0 Hz, 1H), 6.74 (d, J = 8.6 Hz, 1H), 6.64 (m,1H), 6.61 (m, 1H), 5.19 (s, 2H), 5.15 (s, 2H), 3.77 (s, 3H), 3.72 (s, 3H), 3.66 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.2, 160.2, 159.3, 159.3, 157.9, 150.9, 147.9, 138.9, 136.4, 136.0, 132.6, 132.4, 130.5, 130.2 (2C), 130.1, 129.4, 129.0, 129.0 (2C), 128.5, 127.9 (2C), 127.4, 123.1, 122.1, 121.2, 114.8, 114.2 (2C), 113.3, 110.6, 108.6, 70.5, 55.7, 55.4, 55.3, 53.8; IR (film) νmax 3032, 3001, 2953, 2935, 2835, 1645, 1603, 1512, 1456, 1429, 1385, 1331, 1248, 1209, 1178, 1034, 1026, 818, 735, 698 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C39H34N2O5, 611.2546; found, 611.2574.
N-(4-Methoxybenzyl)-N-(7-(hydroxy)quinolin-3-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (33)
A solution of AlCl3 (44 mg, 0.33 mmol) in anhydrous anisole (150 μL) was added to 32 (43 mg, 0.07 mmol) in anhydrous anisole (150 μL) and the resulting solution was stirred at rt for 18 h. The reaction was diluted with MeOH (150 μL) and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 80:20:1 EtOAc:Hexanes:MeOH) to give 33 as a yellow amorphous solid in quantitative yield: 1H NMR (CDCl3, 500 MHz) δ 8.35 (s, 1H), 7.74 (s, 1H), 7.58 (d, J = 8.9 Hz, 1H), 7.52 (bs, 1H), 7.37 (m, 1H), 7.31 (m, 1H), 7.25-7.11 (m, 5H), 6.84-6.77 (m, 3H), 6.74 (d, J = 8.6 Hz, 1H), 6.67 (d, J = 7.6 Hz, 1H), 6.64 (s, 1H), 5.16 (s, 2H), 3.76 (s, 3H), 3.70 (s, 3H), 3.68 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.3, 159.4, 159.3, 158.0, 138.8, 135.5, 132.3, 130.4, 130.3, 130.1 (2C), 129.6, 129.1 (2C), 127.1, 123.0, 122.0, 114.9, 114.3 (2C), 113.3, 110.7, 55.8, 55.4, 53.9; IR (film) νmax 2926, 1601, 1506, 1248, 1177, 1030, 818 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C32H28N2O5, 521.2076; found, 521.2030.
N-(7-Hydroxyquinolin-3-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (34)
Trifluoromethanesulfonic acid (120 μL, 204 mg, 1.36 mmol) was added to 32 (184 mg, 0.30 mmol) in 1:1 CH2Cl2:TFA (1.36 mL) and the solution was stirred at rt for 2 h. The resulting solution was diluted with CH2Cl2 (100 mL), washed with saturated aqueous NaHCO3 (3 × 50 mL), saturated aqueous NaCl solution (2 × 50 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 100:1 EtOAc:MeOH to 4:1 EtOAc:MeOH) to give 34 as a yellow oil (51 mg, 43%): 1H NMR (CD3OD, 500 MHz) δ 9.01 (bs, 1H), 8.61 (bs, 1H), 8.02 (dd, J = 8.5, 1.7 Hz, 1H), 7.97 (d, J = 1.9 Hz, 1H), 7.74 (d, J = 8.9 Hz, 1H), 7.30 (t, J = 8.2 Hz, 1H), 7.27 (d, J = 1.9 Hz, 1H), 7.19 (dd, J = 8.9, 2.1 Hz, 1H), 7.17 (d, J = 8.8 Hz, 1H), 7.11-7.08 (m, 2H), 6.89 (m, 1H), 3.88 (s, 3H), 3.81 (s, 3H); 13C NMR (CD3OD, 125 MHz) δ 168.7, 161.2, 160.9, 160.2, 147.1, 146.2, 140.5, 132.0, 132.0, 131.5, 130.4, 130.2, 130.1, 128.5, 127.6, 124.2, 123.2, 121.5, 116.5, 113.8, 112.4, 109.7, 56.5, 55.9; IR (film) νmax 3281, 3203, 2930, 2835, 1605, 1545, 1499, 1371, 1252, 1036, 764, 735 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C24H20N2O4, 401.1501; found, 401.1481.
2-(Benzyloxy)-6-bromonaphthalene (36)
Sodium hydride (1.16 g, 29.12 mmol) was added to 6-bromo-2-naphthol (5.00 g, 22.41 mmol) in anhydrous N, N-dimethylformamide (162 mL) at 0° C. After 15 min, benzyl bromide (2.40 mL, 3.45 g, 20.18 mmol) was added and the reaction warmed to rt over 18 h. The reaction was diluted with EtOAc (500 mL), saturated aqueous NaHCO3 (200 mL) was added, and the solution was extracted with EtOAc (3 × 500 mL). The combined organic layers were washed with water (3 × 1 L), saturated aqueous NaCl solution (1 L), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 5:1 Hexanes:CH2Cl2) to give 36 as a colorless amorphous solid (5.71 g, 90%): 1H NMR (CDCl3, 500 MHz) δ 7.94 (d, J = 1.9 Hz, 1H), 7.68 (d, J = 9.0 Hz, 1H), 7.61 (d, J = 8.7 Hz, 1H), 7.53-7.47 (m, 3H), 7.46-7.40 (m, 2H), 7.37 (m, 1H), 7.26 (dd, J = 9.0, 2.5 Hz, 1H), 7.20 (d, J = 2.5 Hz, 1H), 5.18 (s, 2H); 13C NMR (CDCl3, 125 MHz) δ 157.2, 136.8, 133.2, 130.3, 129.9, 129.9, 128.9 (2C), 128.8, 128.7, 128.4, 127.8 (2C), 120.3, 117.4, 107.3, 70.3; IR (film) νmax 1585, 1452, 1256, 1219, 1204, 1165, 1065, 997, 924, 852, 820, 800, 733, 698, 476 cm−1.
N-(4-Methoxybenzyl)-6-(benzyloxy)naphthalen-2-amine (37)
4-Methoxy-benzylamine (3.40 mL, 3.59 g, 26.20 mmol) and anhydrous DMSO (5.20 mL) were added to a high-pressure flask charged with 36 (3.00 g, 9.58 mmol), K3PO4 (4.19 g, 19.73 mmol), CuII (290 mg, 1.52 mmol), and L-(−)-proline (255 mg, 2.21 mmol); the sealed flask was heated to 80° C for 44 h. After cooling to rt, reaction contents were partitioned between water (100 mL) and EtOAc (200 mL), then were extracted with EtOAc (2 × 200 mL). The combined organic layers were washed with saturated aqueous NaCl solution (500 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 7:1:1 Hexanes:CH2Cl2:EtOAc) to give 37 as a light orange amorphous solid (952 mg, 27%): 1H NMR (CDCl3, 500 MHz) δ 7.55 (d, J = 8.7 Hz, 2H), 7.49 (m, 2H), 7.41 (m, 2H), 7.37-7.32 (m, 3H), 7.15 (dd, J = 8.8, 2.6 Hz, 1H), 7.13 (m, 1H), 6.92 (dd, J = 8.8, 2.3 Hz, 1H), 6.90 (d, J = 8.7 Hz, 2H), 6.87 (m, 1H), 5.14 (s, 2H), 4.35 (s, 2H), 3.82 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 159.1, 154.6, 137.4, 130.7, 129.3 (2C), 128.8 (2C), 128.1, 128.0, 127.8 (2C), 119.4 (2C), 118.6, 114.3 (2C), 107.9, 70.3, 55.5; IR (film) νmax 3734, 1558, 1456, 1259, 1167, 847, 746, 702, 660 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C25H23NO2, 370.1807; found, 370.1786.
N-(4-Methoxybenzyl)-N-(6-(benzyloxy)naphthalen-2-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (38)
Thionyl chloride (308 μL, 502 mg, 4.22 mmol) was added to 4-methoxy-3-(3-methoxyphenyl)-benzoic acid (365 mg, 1.41 mmol) in anhydrous THF (4.75 mL) at rt, and the resulting solution was heated at reflux for 4.5 h. The solvent was removed and the resulting acid chloride was used without further purification.
Sodium hydride (85 mg, 2.12 mmol) was added to 37 (378 mg, 1.02 mmol) in anhydrous THF (7.50 mL). After stirring for 2 h at rt, a solution of the freshly-prepared acid chloride (1.41 mmol) in anhydrous THF (2.40 mL) was added and the reaction was heated to reflux for 18 h. The reaction was cooled, partitioned between saturated aqueous NaHCO3 (50 mL) and CH2Cl2 (50 mL), then extracted with CH2Cl2 (3 × 100 mL). Combined organic extracts were washed with saturated aqueous NaCl solution (200 mL), dried (Na2SO4), filtered, and concentrated. The residue was purified via column chromatography (SiO2, 4:1:1 Hexanes:EtOAc:CH2Cl2) to give 38 as a brown-yellow amorphous solid (515 mg, 83%): 1H NMR (CDCl3, 500 MHz) δ 7.58 (d, J = 9.0 Hz, 1H), 7.55 (d, J = 8.7 Hz, 1H), 7.50-7.45 (m, 3H), 7.44-7.40 (m, 2H), 7.38-7.34 (m, 2H), 7.29 (d, J = 2.2 Hz, 1H), 7.25 (m, 2H), 7.21 (dd, J = 8.9, 2.6 Hz, 1H), 7.15 (d, J = 2.4 Hz, 1H), 7.09 (m, 1H), 7.01 (dd, J = 8.7, 2.1 Hz, 1H), 6.80 (m, 2H), 6.77 (ddd, J = 8.2, 2.7, 0.9 Hz, 1H), 6.74 (d, J = 8.7 Hz, 1H), 6.59-6.56 (m, 2H), 5.16 (s, 2H), 5.14 (s, 2H), 3.78 (s, 3H), 3.72 (s, 3H), 3.62 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.0, 159.3, 159.0, 157.6, 157.3, 139.9, 139.1, 136.8, 133.0, 132.4, 130.5, 130.1, 130.1 (2C), 129.6, 129.5, 129.1, 128.9, 128.9 (2C), 128.4, 128.2, 127.9, 127.7 (2C), 127.3, 125.8, 122.1, 120.0, 114.7, 114.0 (2C), 113.2, 110.4, 107.1, 70.3, 55.7, 55.4, 55.2, 53.9; IR (film) νmax 3059, 3032, 2999, 2934, 2835, 1636, 1601, 1506, 1456, 1389, 1248, 1209, 1026, 854, 818, 735, 698 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C40H35NO5, 610.2593; found, 610.2567.
N-(4-Methoxybenzyl)-N-(6-hydroxynaphthalen-2-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (39)
A solution of AlCl3 (203 mg, 1.52 mmol) in anhydrous anisole (750 μL) was added to 38 (191 mg, 0.31 mmol) in anhydrous anisole (750 μL) and the resulting solution was stirred at rt for 18 h. The reaction was diluted with MeOH (750 μL) and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 1:1 EtOAc:Hexanes) to give 39 as a light yellow amorphous solid (126 mg, 77%): 1H NMR (CDCl3, 500 MHz) δ 7.56 (m, 1H), 7.50 (d, J = 8.7 Hz, 1H), 7.47 (dd, J = 8.6, 2.3 Hz, 1H), 7.36 (d, J = 2.0 Hz, 1H), 7.29 (d, J = 2.2 Hz, 1H), 7.25 (m, 2H), 7.12-7.06 (m, 3H), 7.00 (dd, J = 8.7, 2.1 Hz, 1H), 6.80 (m, 2H), 6.77 (m, 1H), 6.73 (d, J =8.7 Hz, 1H), 6.59-6.55 (m, 2H), 5.43 (s, 1H), 5.15 (s, 2H), 3.77 (s, 3H), 3.71 (s, 3H), 3.62 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.2, 159.3, 159.0, 157.6, 154.2, 139.7, 139.0, 133.1, 132.4, 130.5, 130.1, 130.1 (2C), 130.0, 129.5, 128.9, 128.9, 128.0, 127.5, 127.3, 125.9, 122.1, 118.7, 114.7, 114.0 (2C), 113.2, 110.4, 109.5, 55.7, 55.4, 55.3, 54.0; IR (film) νmax 3236, 2934, 2835, 1599, 1508, 1456, 1437, 1394, 1248, 1177, 1036 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C33H29NO5, 520.2124; found, 520.2120.
N-(4-Methoxybenzyl)-N-(7-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)quinolin-3-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (42a)
Boron trifluoride etherate (45 μL, 52 mg, 0.36 mmol) was added to 33 (160 mg, 0.31 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (181 mg, 0.50 mmol) in anhydrous CH2Cl2 (5.00 mL). After stirring at rt for 40 h, triethylamine (30 μL) was added and the solvent was concentrated. The residue was partially purified via column chromatography (SiO2, 1:1 EtOAc:Hexanes) to provide 41a, which was used without further purification.
Carbonate 41a was added to MeOH (22.0 mL), CH2Cl2 (1.5 mL), and triethylamine (2.2 mL) and stirred for 18 h at rt. The solvent was concentrated and the residue purified via column chromatography (SiO2, 50:1 EtOAc:MeOH) to give 42a as a near-colorless amorphous solid (33 mg, 16% over two steps): 1H NMR (CDCl3, 500 MHz) δ 8.38 (d, J = 2.4 Hz, 1H), 7.67 (d, J = 2.4 Hz, 1H), 7.65 (d, J = 2.3 Hz, 1H), 7.54 (d, J = 9.1 Hz, 1H), 7.41 (dd, J = 8.6, 2.3 Hz, 1H), 7.26 (d, J = 2.3 Hz, 1H), 7.22 (m, 2H), 7.17 (dd, J = 8.9, 2.4 Hz, 1H), 7.10 (t, J = 7.8 Hz, 1H), 6.80 (m, 2H), 6.75 (dd, J = 8.4, 1.0 Hz, 1H), 6.74 (d, J = 8.7 Hz, 1H), 6.62-6.57 (m, 2H), 5.69 (d, J = 2.3 Hz, 1H), 5.15 (AB, JAB = 14.6 Hz, 1H), 5.15 (AB, JAB = 14.6 Hz, 1H), 4.23 (dd, J = 9.1, 3.4 Hz, 1H), 4.15 (m, 1H), 3.76 (s, 3H), 3.70 (s, 3H), 3.64 (s, 3H), 3.59 (s, 3H), 3.38 (d, J = 9.1 Hz, 1H), 1.39 (s, 3H), 1.17 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.3, 159.3, 159.2, 158.1, 157.9, 150.9, 147.4, 138.8, 136.1, 132.7, 132.4, 130.5, 130.1 (2C), 129.3, 129.0, 128.9, 127.3, 123.4, 122.0, 120.7, 114.8, 114.2 (2C), 113.2, 111.3, 110.7, 98.2, 84.5, 78.7, 71.1, 68.6, 62.0, 55.7, 55.4, 55.3, 53.8, 29.0, 23.0; IR (film) νmax 2937, 2833, 1601, 1246, 1209, 1117, 1033, 989, 964 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C40H42N2O9, 695.2969; found, 695.2891.
N-(7-((2R,3R,4S,5R)-3,4-Dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)quinolin-3-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (42b)
Boron trifluoride etherate (46 μL, 53 mg, 0.37 mmol) was added to 34 (51 mg, 0.13 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (180 mg, 0.50 mmol) in anhydrous CH2Cl2 (5.00 mL). After stirring at rt for 40 h, triethylamine (30 μL) was added and the solvent was concentrated. The residue was purified via column chromatography (SiO2, 1:1 EtOAc:Hexanes) to give 41b (29 mg, 37%) in a 10:1 ratio of anomers (α:β), which was used without further purification.
Carbonate 41b was added to MeOH (7.5 mL), CH2Cl2 (500 μL), and triethylamine (750 μL) and stirred for 18 h at rt. The solvent was concentrated and the residue purified via column chromatography (SiO2, 40:1 EtOAc:MeOH) to give 42b as a near-colorless amorphous solid (11 mg, 15% over two steps): 1H NMR (CDCl3, 500 MHz) δ 8.81 (bs, 2H), 7.98 (dd, J = 8.7, 2.4 Hz, 1H), 7.89 (d, J = 2.3 Hz, 1H), 7.72 (d, J = 9.0 Hz, 1H), 7.70 (d, J = 1.9 Hz, 1H), 7.37 (t, J = 8.0 Hz, 1H), 7.23 (dd, J = 9.0, 2.3 Hz, 1H), 7.13 (dt, J = 7.8, 1.2 Hz, 1H), 7.10 (m, 1H), 7.09 (d, J = 8.6 Hz, 1H), 6.94 (ddd, J = 8.2, 2.5, 0.8 Hz, 1H), 5.73 (d, J = 2.0 Hz, 1H), 4.30-4.22 (m, 2H), 3.91 (s, 2H), 3.86 (s, 3H), 3.61 (s, 3H), 3.39 (d, J = 8.9 Hz, 1H), 1.40 (s, 3H), 1.21 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.8, 159.9, 159.6, 157.1, 146.7, 144.7, 138.9, 131.1, 130.6, 129.9, 129.4, 129.0, 128.7, 126.6, 125.0, 124.0, 122.2, 120.5, 115.6, 113.2, 111.5, 111.4, 98.0, 84.6, 78.6, 71.4, 68.8, 62.1, 56.1, 55.6, 29.2, 23.0; IR (film) νmax 3288, 2976, 2934, 2835, 1373, 1250, 1117, 731 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C32H34N2O8, 575.2393; found, 575.2368.
N-(4-Methoxybenzyl)-N-(6-((2R,3R,4S,5R)-3,4-dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)naphthalen-2-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (42c)
Boron trifluoride etherate (10 μL, 12 mg, 0.08 mmol) was added to 39 (77 mg, 0.15 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (214 mg, 0.59 mmol) in anhydrous CH2Cl2 (5.00 mL). After stirring at rt for 40 h, triethylamine (10 μL) was added and the solvent was concentrated. The residue was partially purified via column chromatography (SiO2, 1:1 EtOAc:Hexanes) to give 41c, which was used without further purification (69 mg, 64%).
Carbonate 41c (42 mg, 0.06 mmol) was added to MeOH (9.1 mL), CH2Cl2 (650 μL), and triethylamine (910 μL) and stirred for 18 h at rt. The solvent was concentrated and the residue purified via column chromatography (SiO2, 2:1 EtOAc:Hexanes to EtOAc) to give 42c as a colorless oil (27 mg, 66%, 42% over two steps): 1H NMR (CDCl3, 500 MHz) δ 7.58 (d, J = 8.8 Hz, 1H), 7.55 (d, J = 9.0 Hz, 1H), 7.48 (dd, J = 8.7, 2.2 Hz, 1H), 7.37 (d, J = 2.3 Hz, 1H), 7.35 (d, J = 2.0 Hz, 1H), 7.28 (d, J = 2.3 Hz, 1H), 7.25 (m, 2H), 7.14 (dd, J = 8.9, 2.4 Hz, 1H), 7.09 (t, J = 7.9 Hz, 1H), 7.04 (dd, J = 8.7, 2.1 Hz, 1H), 6.80 (m, 2H), 6.76 (ddd, J = 8.2, 2.5, 0.9 Hz, 1H), 6.73 (d, J = 8.7 Hz, 1H), 6.58 (m, 1H), 6.54 (brd, J = 7.7 Hz, 1H), 5.66 (d, J = 2.1 Hz, 1H), 5.14 (s, 2H), 4.25 (m, 1H), 4.22 (m, 1H), 3.77 (s, 3H), 3.70 (s, 3H), 3.63 (s, 3H), 3.61 (s, 3H), 3.38 (d, J = 9.2 Hz, 1H), 2.84 (brs, 1H), 2.71 (brs, 1H), 1.39 (s, 3H), 1.20 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 170.2, 159.2, 159.0, 157.6, 155.1, 140.1, 139.0, 132.9, 132.4, 130.5, 130.1 (2C), 129.5, 129.5, 129.4, 128.9, 128.3, 128.1, 127.1, 125.8, 122.1, 119.5, 114.7, 114.0, 113.2 (2C), 110.4, 109.9, 97.9, 94.6, 78.6, 71.5, 68.7, 62.1, 55.7, 55.4, 55.3, 53.9, 29.4, 22.9; IR (film) νmax 3420, 2932, 2835, 1601, 1506, 1394, 1387, 1248, 1178, 1117, 1026, 993, 910, 733 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C41H43NO9, 694.3016; found, 694.3010.
N-(6-((2R,3R,4S,5R)-3,4-Dihydroxy-5-methoxy-6,6-dimethyl-tetrahydro-2H-pyran-2-yloxy)naphthalen-2-yl)-4-methoxy-3-(3-methoxyphenyl)-benzamide (42d)
Trifluoromethanesulfonic acid (130 μL, 221 mg, 1.47 mmol) was added to 38 (198 mg, 0.33 mmol) in 1:1 CH2Cl2:TFA (1.48 mL) and the solution was stirred at rt for 2 h. The resulting solution was diluted with CH2Cl2 (100 mL), washed with saturated aqueous NaHCO3 (3 × 50 mL), saturated aqueous NaCl solution (2 × 50 mL), dried (Na2SO4), filtered, and concentrated. The residue was partially purified via column chromatography (SiO2, 1:1:1 Hexanes:EtOAc:CH2Cl2) to give 40 as a purple solid, which was used without further purification (28 mg, 22%).
Boron trifluoride etherate (4 μL, 5 mg, 0.03 mmol) was added to 40 (28 mg, 0.07 mmol) and (3aR,4S,7R,7aR)-7-methoxy-6,6-dimethyl-2-oxo-tetrahydro-3aH-[1.3]dioxolo[4,5-c]pyran-4-yl 2,2,2-trichloroacetimidate (83 mg, 0.23 mmol) in anhydrous CH2Cl2 (1.85 mL). After stirring at rt for 40 h, triethylamine (10 μL) was added and the solvent was concentrated. The residue was partially purified via column chromatography (SiO2, 3:1:1 Hexanes:EtOAc:CH2Cl2) to provide 41d, which was used without further purification (30 mg).
Carbonate 41d was added to MeOH (10.0 mL), CH2Cl2 (1.0 mL), and triethylamine (1.0 mL) and stirred for 18 h at rt. The solvent was concentrated and the residue purified via column chromatography (SiO2, 2:1 EtOAc:Hexanes, then EtOAc) to give 42d as a colorless oil (12 mg, 6% in three steps): 1H NMR (CDCl3, 500 MHz) δ 8.28 (d, J = 1.9 Hz, 1H), 7.96 (dd, J = 8.5, 2.4 Hz, 1H), 7.90 (brs, 1H), 7.86 (d, J = 2.4 Hz, 1H), 7.75 (brs, 1H), 7.73 (brs, 1H), 7.55 (dd, J = 8.9, 2.2 Hz, 1H), 7.42 (d, J = 2.3 Hz, 1H), 7.38 (t, J = 8.0 Hz, 1H), 7.18 (dd, J = 8.9, 2.5 Hz, 1H), 7.15 (m, 1H), 7.11 (m, 1H), 7.09 (d, J = 8.7 Hz, 1H), 6.94 (ddd, J = 8.2, 2.5, 0.8 Hz, 1H), 5.69 (d, J = 2.1 Hz, 1H), 4.28 (dt, J = 9.2, 3.6 Hz, 1H), 4.25 (m, 1H), 3.91 (s, 3H), 3.87 (s, 3H), 3.62 (s, 3H), 3.39 (d, J = 9.2 Hz, 1H), 2.61 (d, J = 2.5 Hz, 1H), 2.56 (d, J = 3.7 Hz, 1H), 1.41 (s, 3H), 1.22 (s, 3H); 13C NMR (CDCl3, 125 MHz) δ 165.4, 159.6 (2C), 154.2, 139.1, 134.2, 131.8, 131.0, 129.9, 129.7, 129.4, 129.4, 128.6, 128.2, 127.4, 122.2, 120.9, 119.5, 117.3, 115.5, 113.2, 111.3, 110.1, 97.9, 84.7, 78.5, 71.6, 68.8, 62.1, 56.1, 55.6, 29.4, 22.9; IR (film) νmax 3400, 2970, 2930, 2835, 1605, 1535, 1342, 1250, 1178, 1117, 1024, 995, 966, 908, 733 cm−1; HRMS (ESI+) m/z: [M + H]+ calcd for C33H35NO8, 574.2441; found, 574.2461.
Anti-Proliferation Assays
Cells were maintained in a 1:1 mixture of Advanced DMEM/F12 (Gibco) supplemented with non-essential amino acids, L-glutamine (2 mM), streptomycin (500 μg/mL), penicillin (100 units/mL), and 10% FBS. Cells were grown to confluence in a humidified atmosphere (37° C, 5% CO2), seeded (2000/well, 100 μL) in 96-well plates, and allowed to attaché overnight. Compound or GDA at varying concentrations in DMSO (1% DMSO final concentration) was added, and cells were returned to the incubator for 72 h. At 72 h, the number of viable cells was determined using an MTS/PMS cell proliferation kit (Promega) per the manufacturer’s instructions. Cells incubated in 1% DMSO were used at 100% proliferation, and values were adjusted accordingly. IC50 values were calculated from separate experiments performed in triplicate using GraphPad Prism.
Western Blot Analyses
MCF-7 cells were cultured as described above and treated with various concentrations of drug, GDA in DMSO (1% DMSO final concentration), or vehicle (DMSO) for 24 h. Cells were harvested in cold PBS and lysed in RIPA lysis buffer containing 1 mM PMSF, 2 mM sodium orthovanadate, and protease inhibitors on ice for 1 h. Lysates were clarified at 14000g for 10 min at 4° C. Protein concentrations were determined using the Pierce BCA protein assay kit per the manufacturer’s instructions. Equal amounts of protein (20 μg) were electrophoresed under reducing conditions, transferred to a nitrocellulose membrane, and immunoblotted with the corresponding specific antibodies. Membranes were incubated with an appropriate horseradish peroxidase-labeled secondary antibody, developed with a chemiluminescent substrate, and visualized.
Supplementary Material
Full experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Scheme 6.

Preparation of 5-, 6-, and 8-modified novobiocin analogues.
Scheme 9.

Preparation of quinoline- and naphthalene-derived novobiocin analogues.
Acknowledgments
The authors gratefully acknowledge support of this project by the NIH/NCI (CA120458) and the DOD Prostate Cancer Research Program (QH815179). The authors acknowledge Donna Lubbers for her preliminary studies toward the preparation of 14, 17, and 21.
References
- 1.Powers MV, Workman P. In Endocrine-Related Cancer. 2006;13:S125–S135. doi: 10.1677/erc.1.01324. [DOI] [PubMed] [Google Scholar]
- 2.Pratt WB, Toft DO. Exp Biol Med. 2003;228:111–133. doi: 10.1177/153537020322800201. [DOI] [PubMed] [Google Scholar]
- 3.Issacs JS, Xu W, Neckers L. Cancer Cell. 2003;3:213–217. doi: 10.1016/s1535-6108(03)00029-1. [DOI] [PubMed] [Google Scholar]
- 4.Terasawa K, Minami M, Minami YJ. Biochem. 2005;137:443–447. doi: 10.1093/jb/mvi056. [DOI] [PubMed] [Google Scholar]
- 5.Buchner J. Trends Biochem Sci. 1999;24:136–141. doi: 10.1016/s0968-0004(99)01373-0. [DOI] [PubMed] [Google Scholar]
- 6.Picard D. Cell Mol Life Sci. 2002;59:1640–1648. doi: 10.1007/PL00012491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yonehara M, Minami Y, Kawata Y, Nagai J, Yahara I. J Biol Chem. 1996;271:2641–2645. doi: 10.1074/jbc.271.5.2641. [DOI] [PubMed] [Google Scholar]
- 8.Xiao L, Lu X, Ruden DM. Mini-Reviews in Medicinal Chemistry. 2006;6:1137–1143. doi: 10.2174/138955706778560166. [DOI] [PubMed] [Google Scholar]
- 9.Zhao R, Houry WA. Biochem Cell Biol. 2005;83:703–710. doi: 10.1139/o05-158. [DOI] [PubMed] [Google Scholar]
- 10.Neckers L, Ivy SP. Curr Opin Oncol. 2003;15:419–424. doi: 10.1097/00001622-200311000-00003. [DOI] [PubMed] [Google Scholar]
- 11.Yu XM, Shen G, Neckers L, Blake H, Holzbeierlein J, Cronk B, Blagg BSJ. J Am Chem Soc. 2005;127:12778–12779. doi: 10.1021/ja0535864. [DOI] [PubMed] [Google Scholar]
- 12.Zhang H, Burrows F. J Mol Med. 2004;82:488–499. doi: 10.1007/s00109-004-0549-9. [DOI] [PubMed] [Google Scholar]
- 13.Chiosis G, Vilenchik M, Kim J, Solit D. Drug Discuss Today. 2004;9:881–888. doi: 10.1016/S1359-6446(04)03245-3. [DOI] [PubMed] [Google Scholar]
- 14.Burlison JA, Neckers L, Smith AB, Maxwell A, Blagg BSJ. J Am Chem Soc. 2006;128:15529–15536. doi: 10.1021/ja065793p. [DOI] [PubMed] [Google Scholar]
- 15.Toft DO. Trends Endocrin Metab. 1998;9:238–243. doi: 10.1016/s1043-2760(98)00060-5. [DOI] [PubMed] [Google Scholar]
- 16.Walter S, Buchner J. J Agnew Chem, Int Ed. 2002;41:1098–1113. doi: 10.1002/1521-3773(20020402)41:7<1098::aid-anie1098>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 17.Blagg BSJ, Kerr TD. Med Res Rev. 2006;26:310–338. doi: 10.1002/med.20052. [DOI] [PubMed] [Google Scholar]
- 18.Maloney A, Workman P. Expert Opin Biol Ther. 2002;2:3–24. doi: 10.1517/14712598.2.1.3. [DOI] [PubMed] [Google Scholar]
- 19.Workman P. Cancer Letters. 2004;206:149–157. doi: 10.1016/j.canlet.2003.08.032. [DOI] [PubMed] [Google Scholar]
- 20.Sreedhar AS, Kalmar E, Csermely P, Shen YF. FEBS Letters. 2004;562:11–15. doi: 10.1016/s0014-5793(04)00229-7. [DOI] [PubMed] [Google Scholar]
- 21.Workman P. Cold Spring Harbor Symposia on Quantitative Biology. 2005;70:499–515. doi: 10.1101/sqb.2005.70.020. [DOI] [PubMed] [Google Scholar]
- 22.Lewis RJ, Tsai FT, Wigley DB. BioEssays. 1996;18:661–671. doi: 10.1002/bies.950180810. [DOI] [PubMed] [Google Scholar]
- 23.Reece RJ, Maxwell A. Crit Rev Biochem Mol Biol. 1991;26:335–375. doi: 10.3109/10409239109114072. [DOI] [PubMed] [Google Scholar]
- 24.Laurin P, Ferroud D, Schio L, Klich M, Dupuis-Hamelin C, Mauvais P, Lassaigne P, Bonnefoy A, Musicki B. Bioorg Med Chem Lett. 1999;9:2875–2880. doi: 10.1016/s0960-894x(99)00492-8. [DOI] [PubMed] [Google Scholar]
- 25.Ali JA, Jackson AP, Howells AJ, Maxwell A. Biochemistry. 1993;32:2717–2724. doi: 10.1021/bi00061a033. [DOI] [PubMed] [Google Scholar]
- 26.Holdgate GA, Tunnicliffe A, Ward WHJ, Weston SA, Rosenbrock G, Barth PT, Taylor IWF, Paupit RA, Timms D. Biochemistry. 1997;36:9663–9673. doi: 10.1021/bi970294+. [DOI] [PubMed] [Google Scholar]
- 27.Lewis RJ, Singh OMP, Smith CV, Skarzyknski T, Maxwell A, Wonacott AJ, Wigley DB. EMBO J. 1996;15:1412–1420. [PMC free article] [PubMed] [Google Scholar]
- 28.Tsai FTF, Singh OMP, Skarzynski T, Wonacott AJ, Weston S, Tucker A, Pauptit RA, Breeze AL, Poyser JP, O’Brien R, Ladbury JE, Wigley DB. Proteins: Struct, Funct, Genet. 1997;28:41–52. [PubMed] [Google Scholar]
- 29.Roe SM, Prodromou C, O’Brien R, Ladbury JE, Piper PW, Pearl LH. J Med Chem. 1999;42:260–266. doi: 10.1021/jm980403y. [DOI] [PubMed] [Google Scholar]
- 30.Gobernado M, Canton E, Santos M. Eur J Clin Microbiol. 1984;3:371. doi: 10.1007/BF01977500. [DOI] [PubMed] [Google Scholar]
- 31.Schwartz GN, Teicher BA, Eder JP, Jr, Korbut T, Holden SA, Ara G, Herman TS. Cancer Chemother Pharmacol. 1993;32:455–462. doi: 10.1007/BF00685890. [DOI] [PubMed] [Google Scholar]
- 32.Nordenberg J, Albukrek D, Hadar T, Fux A, Wasserman L, Novogrodsky A, Sidid Y. Br J Cancer. 1992;65:183–188. doi: 10.1038/bjc.1992.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hombrouck C, Capmau M, Moreau N. Cell Mol Biol. 1999;45:347–352. [PubMed] [Google Scholar]
- 34.Marcu MG, Schulte TW, Neckers L. J Natl Cancer Inst. 2000;92:242–248. doi: 10.1093/jnci/92.3.242. [DOI] [PubMed] [Google Scholar]
- 35.Marcu MG, Chadli A, Bohouche I, Catelli B, Neckers LM. J Biol Chem. 2001;276:37181–37186. doi: 10.1074/jbc.M003701200. [DOI] [PubMed] [Google Scholar]
- 36.Allan RK, Mok D, Ward BK, Ratajczak T. J Biol Chem. 2006;281:7161–7171. doi: 10.1074/jbc.M512406200. [DOI] [PubMed] [Google Scholar]
- 37.Burlison JA, Blagg BSJ. Org Lett. 2006;8:4855–4858. doi: 10.1021/ol061918j. [DOI] [PubMed] [Google Scholar]
- 38.Burlison JA, Avila C, Vielhauer G, Lubbers DJ, Holzbeierlein J, Blagg BSJ. J Org Chem. 2008;73:2130–2137. doi: 10.1021/jo702191a. [DOI] [PubMed] [Google Scholar]
- 39.Shen G, Yu XM, Blagg BSJ. Bioorg Med Chem Lett. 2004;14:5903–5906. doi: 10.1016/j.bmcl.2004.09.017. [DOI] [PubMed] [Google Scholar]
- 40.Klaholz BP, Moras D. Structure. 2002;10:1197–1204. doi: 10.1016/s0969-2126(02)00828-6. [DOI] [PubMed] [Google Scholar]
- 41.Soti C, Vermes A, Haystead TAJ, Csermely P. Eur J Biochem. 2003;270:2421–2428. doi: 10.1046/j.1432-1033.2003.03610.x. [DOI] [PubMed] [Google Scholar]
- 42.Nabaei-Bidhendi G, Bannerjee NR. J Indian Chem Soc. 1990;67:43–45. [Google Scholar]
- 43.Horvath RF, Chan TH. J Org Chem. 1986;52:4489–4494. [Google Scholar]
- 44.Miyake M, Hanaoka Y, Fujimoto Y, Sato Y, Taketomo N, Yokota I, Yoshiyama Y. Heterocycles. 1996;43:665–674. [Google Scholar]
- 45.Carreno MC, Garcia Ruano JL, Toledo MA, Urbano A. Tetrahedron: Asymmetry. 1997;8:913–921. [Google Scholar]
- 46.Wang Y, Tan W, Li WZ, Li Y. J Nat Prod. 2001;64:196–199. doi: 10.1021/np0001124. [DOI] [PubMed] [Google Scholar]
- 47.Ruenitz PC, Bagley JR, Nanavati NT. J Med Chem. 1988;31:1471–1475. doi: 10.1021/jm00402a037. [DOI] [PubMed] [Google Scholar]
- 48.Milne JE, Buchwald SL. J Am Chem Soc. 2004;126:13028–13032. doi: 10.1021/ja0474493. [DOI] [PubMed] [Google Scholar]
- 49.Elliger CA. Synth Commun. 1985;15:1315–1324. [Google Scholar]
- 50.Robinson AJ, Lim CY, He L, Ma P, Li HY. J Org Chem. 2001;66:4141–4147. doi: 10.1021/jo001151n. [DOI] [PubMed] [Google Scholar]
- 51.Toplak R, Svete J, Stanovnik B, Grdadolnik SG. J Hetero Chem. 1999;36:225–235. [Google Scholar]
- 52.Hansen MM, Riggs JR. Tetrahedron Lett. 1998;39:2705–2706. [Google Scholar]
- 53.Zymalkowski F, Tinapp P. Justus Liebigs Ann Chem. 1966;699:98–106. doi: 10.1002/jlac.19666990110. [DOI] [PubMed] [Google Scholar]
- 54.Zhang H, Cai Q, Ma D. J Org Chem. 2005;70:5164–5173. doi: 10.1021/jo0504464. [DOI] [PubMed] [Google Scholar]
- 55.Ulbrich HK, Luxenburger A, Prech P, Eriksson EE, Soehnlein O, Rotzius P, Lindborn L, Dannhardt G. J Med Chem. 2006;49:5988–5999. doi: 10.1021/jm060468y. [DOI] [PubMed] [Google Scholar]
- 56.Jin Y, Zhou ZY, Tian W, Yu Q, Long YQ. Bioorg Med Chem Lett. 2006;16:5864–5869. doi: 10.1016/j.bmcl.2006.08.058. [DOI] [PubMed] [Google Scholar]
- 57.Akiyama T, Takesue Y, Kumegawa M, Nishimoto H, Ozaki S. Bull Chem Soc Jpn. 1991;64:2266–2269. [Google Scholar]
- 58.Das J, Chen P, Norris D, Padmanabha R, Lin J, Moquin RV, Shen Z, Cook LS, Doweyko AM, Pitt S, Pang S, Shen DR, Fang Q, de Fex HF, McIntyre KW, Shuster DJ, Gillooly KM, Behnia K, Schieven GL, Wityak J, Barrish JC. J Med Chem. 2006;49:6819–6832. doi: 10.1021/jm060727j. [DOI] [PubMed] [Google Scholar]
- 59.Ling KQ, Sayre LM. J Am Chem Soc. 2006;127:4777–4784. doi: 10.1021/ja0455603. [DOI] [PubMed] [Google Scholar]
- 60.Radanyi C, La Bras G, Messaoudi S, Couclier C, Peyrat JF, Brion JD, Marsaud V, Renoir JM, Alami M. Bioorg Med Chem Lett. 2008;18:2495–2498. doi: 10.1016/j.bmcl.2008.01.128. [DOI] [PubMed] [Google Scholar]
- 61.Le Bras G, Radanyi C, Peyrat JF, Brion JD, Alami M, Marsaud V, Stella B, Renoir JM. J Med Chem. 2007;50:6189–6200. doi: 10.1021/jm0707774. [DOI] [PubMed] [Google Scholar]
- 62.Faraoni R, Blanzat M, Kubicek S, Braun C, Schweizer WB, Gramlich V, Diederich F. Org Biomol Chem. 2004;2:1962–1964. doi: 10.1039/b404311a. [DOI] [PubMed] [Google Scholar]
- 63.Nadin A, Lopez JMS, Owens AP, Howells DM, Talbot AC, Harrison T. J Org Chem. 2003;68:2844–2852. doi: 10.1021/jo026860a. [DOI] [PubMed] [Google Scholar]
- 64.Akritopoulou-Zanze I, Patel JR, Hartandi K, Brenneman J, Winn M, Pratt JK, Grynfarb M, Goos-Nisson A, von Geldern TW, Kym PR. Bioorg Med Chem Lett. 2004;14:2079–2082. doi: 10.1016/j.bmcl.2004.02.048. [DOI] [PubMed] [Google Scholar]
- 65.Klarmann E. J Am Chem Soc. 1926;48:791–794. [Google Scholar]
- 66.Greig LM, Slawin AMZ, Smith MH, Philp D. Tetrahedron. 2007;63:2391–2403. [Google Scholar]
- 67.Green K. J Org Chem. 1991;56:4325–4326. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Full experimental procedures and characterization for all compounds. This material is available free of charge via the Internet at http://pubs.acs.org.