

Abstract
Potent and selective inhibitors of inducible nitric oxide synthase (iNOS) (EC 1.14.13.39) were identified in an encoded combinatorial chemical library that blocked human iNOS dimerization, and thereby NO production. In a cell-based iNOS assay (A-172 astrocytoma cells) the inhibitors had low-nanomolar IC50 values and thus were >1,000-fold more potent than the substrate-based direct iNOS inhibitors 1400W and N-methyl-l-arginine. Biochemical studies confirmed that inhibitors caused accumulation of iNOS monomers in mouse macrophage RAW 264.7 cells. High affinity (Kd ≈ 3 nM) of inhibitors for isolated iNOS monomers was confirmed by using a radioligand binding assay. Inhibitors were >1,000-fold selective for iNOS versus endothelial NOS dimerization in a cell-based assay. The crystal structure of inhibitor bound to the monomeric iNOS oxygenase domain revealed inhibitor–heme coordination and substantial perturbation of the substrate binding site and the dimerization interface, indicating that this small molecule acts by allosterically disrupting protein–protein interactions at the dimer interface. These results provide a mechanism-based approach to highly selective iNOS inhibition. Inhibitors were active in vivo, with ED50 values of <2 mg/kg in a rat model of endotoxin-induced systemic iNOS induction. Thus, this class of dimerization inhibitors has broad therapeutic potential in iNOS-mediated pathologies.
The mammalian nitric oxide synthase (NOS) (EC 1.14.13.39) enzyme family comprises three isoforms: inducible (iNOS), neuronal, and endothelial NOS. NOS isoforms are homodimers that catalyze NADPH-dependent oxidation of l-arginine to NO⋅ and citrulline (1–3). NOS monomers consist of an oxidoreductase domain and an oxygenase domain. The reductase domain is homologous to cytochrome P450 reductase and contains binding sites for NADPH, FAD, and FMN (4, 5). The oxygenase domain has binding sites for l-arginine, the heme prosthetic group, and tetrahydrobiopterin (H4B). Formation of stable NOS homodimers requires structural elements in the oxygenase domain and is an H4B-, substrate-, and heme-dependent process (6–8). Dimerization of NOS is required for fully coupled enzyme activity because the flow of electrons during catalysis occurs in trans from the reductase domain of one monomer subunit to the oxygenase domain of the other monomer (9). The crystal structures of oxygenase domains of murine iNOS monomer (10), murine and human iNOS dimer (11–13), and human and bovine endothelial NOS dimer (13, 14) indicate a high degree of structural similarity within the critical catalytic center and dimer interface regions between NOS isoforms.
NO⋅ plays a pivotal role in the physiology and pathophysiology of the central nervous, cardiovascular, and immune systems (15–17). The reactivity of NO⋅ toward molecular oxygen, thiols, transition metal centers, and other biological targets enables NO⋅ to function both as a rapidly reversible, specific, and local signal transduction molecule as well as a nonspecific mediator of tissue damage (18–20). iNOS is implicated in the pathogenesis of inflammatory and autoimmune diseases including septic shock, hemorrhagic shock, rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, and multiple sclerosis (21–24), and neuronal NOS is implicated in the pathogenesis of cerebral stroke and Parkinson's disease (25–26). Inhibition of endothelial NOS, however, is undesirable because of its role in the maintenance of vascular homeostasis (15–16). In this report, we describe the discovery of a class of potent and selective inhibitors of iNOS activity that function by inhibiting the dimerization of iNOS monomers. A crystal structure of inhibitor bound to murine iNOS oxygenase monomer provides insights into the process of iNOS dimerization.
Materials and Methods
Chemistry.
An encoded chemical library (ECLiPS method) based on a pyrimidineimidazole core was prepared on polyethylene glycol-grafted polystyrene support in three combinatorial steps according to the scheme shown in Fig. 1A (27). Compounds were attached to resin by a photolabile o-nitrobenzyl amide linker. The first synthetic step introduced 31 primary amines. After a pool and split step, the amines were acylated with 31 fluorenylmethoxycarbonyl (Fmoc)-protected amino acids. A second pool and split step was performed, followed by Fmoc deprotection and subsequent heteroarylation of the resulting free amines by electrophilic substitution with a set of nine substituted pyrimidines. The completed library comprised 8,649 compounds, representing the product of the number of synthetic reagents used at each step (31 × 31 × 9). The identity of the library compound synthesized on each bead of the polymeric support was encoded by using polyhalogenated phenyl ethers attached via oxidatively cleavable linkers. By keeping the pools of beads separate after the final synthetic step, screening could focus on compounds from the active sublibraries identified in the initial survey screening. Approximately 200 pmol of compound could be photoeluted from each bead as determined by HPLC analysis and comparison to analytical compound standards.
Figure 1.

(A) The synthetic steps used to prepare the encoded combinatorial chemical library on a polymeric support. (B) The distribution frequency of synthons in active compounds from the combinatorial library. (C) Compound 1 (N-[(1,3-benzodioxol-5-yl)methyl]-1-[6-chloro-2-(1H-imidazol-1-yl)pyrimidin-4-yl]piperazine-2-acetamide) is one of the compounds that was found by screening the library. Compound 2 is (N-[(1,3-benzodioxol-5-yl)methyl]-1-[2-(1H-imidazol-1-yl)pyrimidin-4-yl]-4-(methoxycarbonyl)-piperazine-2-acetamide. Compound 3, tritiated on the propionamide moiety, is the analog used in the competitive binding assay (N-[(1,3-benzodioxol-5-yl)methyl]-4-(ethylcarbonyl)-1-[2-(1H-imidazol-1-yl)pyrimidin-4-yl]piperazine-2-acetamide).
Compounds were resynthesized in solution phase by using methods similar to those used for library synthesis for biological evaluation of potency and selectivity. Reaction of the appropriate 4-chloro-2-(1H-imidazol-1-yl)pyrimidine with N-[(1,3-benzodioxan-5-yl)methyl]-4-[(dimethylethoxy)carbonyl]piperazine-2-acetamide in DMSO at 70°C gave the tert-butoxycarbonyl (BOC)-protected compound. Removal of the BOC under standard conditions gave 1. Acylation of the deprotected material with methyl chloroformate or N-succinimidyl-[2,3-3H]propionate gave 2 and 3, respectively (Fig. 1C). The identity of resynthesized compounds was confirmed by MS and NMR spectroscopy, and the compound purity was determined to be >98% by HPLC.
NOS Assays.
The cell-based iNOS assay was done with A-172 human glioblastoma (American Type Culture Collection) cells, which were cultured in DMEM supplemented with 10% FBS. Cells were plated into 96-well tissue culture plates at 100,000 cells per well. After 24 hr, iNOS was induced by the addition of 400 units/ml human IFN-γ, 4 ng/ml of human IL-1β, and 40 ng/ml of human tumor necrosis factor-α (cytokines from Boehringer Mannheim). Inhibitors were added at the time of cytokine induction; reference inhibitors were N-methyl-l-arginine (Sigma) and 1400-W (Alexis Biochemicals). NO⋅ production was measured 22 hr after induction by mixing 0.125 ml of culture medium with an equal volume of Griess reagent as described (28). No detectable NO⋅ was produced by cells in the absence of cytokines.
For NOS isoform selectivity assays, BSC-1 African green monkey kidney epithelial cells (American Type Culture Collection) were maintained in DMEM containing 10% FBS. Cells were plated into 96-well tissue culture plates at 30,000 cells per well. After 6 hr, recombinant vaccinia virus strains encoding human inducible, endothelial, and neuronal NOS isoforms were added, along with 2 or vehicle. After 17 hr, medium was aspirated, and 20 μl of 40 mM Tris, pH 7.5 containing 0.1% NP-40, 5 mg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, and 24 μg/ml of Pefabloc SC was added; the plates then were frozen at −80°C. NOS activity in cell lysates was determined by measuring the conversion of arginine into citrulline (29). The assay mixture contained 40 mM Tris (pH 7.5), 3 mM DTT, 4 μM each H4B, FAD, and FMN, 0.5 μM calmodulin, 15 mM calcium chloride, 1 mM NADPH, and 3 μM [14C]arginine (300 Ci/mol; 1 Ci = 37 GBq). NOS activity in Superdex 200 fractions (Fig. 2) was measured by using the same protocol, except with 4 μM [14C]arginine and 16 μM unlabeled arginine in the assay mixture.
Figure 2.

Inhibition of dimer formation in RAW264.7 cells. The effects of inhibitor treatment on the iNOS monomer/dimer ratio in cells was determined by size-exclusion chromatography of lysates from cells grown in the presence (○) or absence (●) of compound 2. (A) Cytochrome c reductase activity (a property of both iNOS monomers and dimers). (B) iNOS activity, indicating the presence of active dimer. This figure shows one of two experiments, which gave similar results.
Analysis of Cytoplasmic Monomers and Dimers.
The intracellular ratio of iNOS monomers to dimers was determined with the following modifications to a previously published method (30). Cell lysates were prepared from 10 liters of RAW264.7 cells after cytokine induction. Ten milliliters of lysate was chromatographed on a 26 mm × 60 cm Superdex 200 column (Amersham Pharmacia) in 40 mM Bistris propane (pH 7.8), 10% glycerol, 4 μM FAD, 4 μM H4B, and 1 mM DTT. Fractions (3 ml) were collected and analyzed both for iNOS activity as described above, and for cytochrome c reductase activity as described (31) except that the reaction mixture also contained 6 units/ml superoxide dismutase but not EDTA or albumin.
Binding of Inhibitors to iNOS Monomers.
iNOS monomers were purified from 6 × 108 HeLa cells after infection with a vaccinia virus expression vector encoding full-length human iNOS (with an N-terminal six-histidine tag). Cells were grown in the absence of hemin chloride for 17 hr after infection with the virus (the multiplicity of infection was 5). The cells were resuspended in 200 ml of 40 mM Tris⋅HCl pH 8.0, 0.2 M NaCl, 10% glycerol, 4 μM FAD, 0.2% NP-40, 1 μg/ml pepstatin, 1 μg/ml leupeptin, 5 μg/ml aprotinin, 24 μg/ml Pefabloc SC. After incubation on ice for 30 min, the sample was centrifuged to remove insoluble material. The extract was diluted 1:2 in column buffer (40 mM BisTris propane, pH 7.8/10% glycerol) and added to 14 ml of Ni-nitrilotriacetic acid resin (Qiagen, Chatsworth, CA), which had been preequilibrated in column buffer. After 1.5 hr at 4°C, the resin was packed into a 1.5 × 100 cm column, washed with 0.3 M NaCl and 20 mM imidazole in column buffer, and eluted with 0.3 M NaCl and 300 mM imidazole in column buffer. The A280 peak was pooled, concentrated to 10 ml, and immediately chromatographed at 3 ml/min on a 2.6 × 60 cm Superdex 200 column in column buffer containing 4 μM FAD, 4 μM H4B, and 1 mM DTT. The monomeric fraction was recovered and stored at −80°C. Reanalysis of this protein by gel filtration indicated that 80% of the protein ran as a monomeric species.
To assess binding of compounds to iNOS monomers, compound 3 was tritium-labeled (104 Ci/mol). Binding reactions contained 0.2 nM 3, various amounts of unlabeled 3 or 2, and 4.6 nM iNOS monomer in 1 ml of binding buffer (40 mM Bistris propane, pH 7.8/4 μM FAD/4 μM FMN/4 μM H4B/5 mM reduced glutathione). After 60 min at 37°C the protein was collected by vacuum filtration onto poly(vinylidene difluoride) membranes in a 96-well filter plate. The membranes were washed three times with 300 μl of binding buffer, and radioactivity was measured by using a Wallac Microbeta scintillation counter after addition of 50 μl of scintillant (Microscint 20, Packard). Further experiments showed that binding reached equilibrium by 60 min and was completely reversible. The Kd for 3 (2.5 nM) was determined from Scatchard analysis of binding data. The Ki for 2 was determined from competitive binding data (IC50 values) and correction using the Cheng–Prusoff equation: Ki = IC50/(1+ [radioligand]/(Kd radioligand).
Rat Endotoxemia Studies.
Endotoxemia was induced in male Lewis rats (n = 8 per dose) by i.p. injection of Escherichia coli lipopolysaccharide (LPS) Serotype 055:B5 (Sigma) at a dose of 5 mg/kg (32). Six hours after the LPS challenge, plasma samples were collected and analyzed for nitrate/nitrite (NOx) levels using the Griess assay (28, 32). One hour after LPS challenge, rats received either vehicle (HCl-acidified water) or compound 2; animals were cared for in accordance with institutional guidelines.
Crystallography.
Murine iNOS Δ114 monomers were expressed from an E. coli expression vector encoding the murine iNOS oxygenase domain lacking the N-terminal 114 residues and with the addition of a six-histidine tag and stop codon after residue 498 and purified as described (10). Orthorhombic crystals of monomeric Δ114 murine iNOS were prepared in the presence of imidazole following the procedures of Crane et al. (10). These crystals were soaked for 50 hr in 5 μM compound 2, 14% PEG-3350, 1 μM NaN3, 50 μM Mes (pH 6.5), and 50 μM Na2SO3. The crystals then were rinsed in 33% ethylene glycol, 9.3% PEG-3350, 0.7 μM NaN3, 33 μM Mes (pH 6.5), and 33 μM Na2SO3, and flash-frozen in liquid nitrogen. The x-ray diffraction data was collected to 2.25 Å at Stanford Synchrotron Radiation Source beamline 7-1, and refined to an R factor of 0.197 and an Rfree of 0.284 (Protein Data Bank Deposit 1DD7). The model contains residues 114–265, 270–325, 335–368, 413–445, 478–497, heme, compound 2, a sulfite ion. and 214 water molecules.
Results and Discussion
An encoded combinatorial chemical library expected to contain NOS inhibitors was designed based on a pyrimidineimidazole core as described in Fig. 1A (27). This template was selected because a structurally related series of compounds, phenylimidazoles, are known to bind heme and inhibit NOS activity, with selectivity toward iNOS (33, 34). In addition, substituted pyrimidineimidazoles are readily accessible via the stepwise nucleophilic displacement of halogen from activated halopyrimidines.
In a screen against partially purified recombinant human iNOS, none of the library compounds exhibited any appreciable direct inhibition of iNOS enzyme activity at <2 μM. However, inhibitors of NO⋅ production by cytokine-stimulated human A-172 cells were identified by screening 20 compounds per well at a predicted concentration of 1 μM for each compound. Potent inhibition (>90%) was observed in the two sublibraries with a 2-(1-imidazolyl)pyrimidine moiety and weaker inhibition in a sublibrary with a 4-(1-imidazolyl)pyrimidine moiety. The three active sublibraries were extensively (≈98% coverage) screened at 200 nM of each compound. Fifty-three different active inhibitors (>60% inhibition) were identified, constituting 0.6% of the library.
The distribution frequency of active synthons at substituents R1, R2, and R3 is shown in Fig. 1B. In addition to preference for 2-(1-imidazolyl)pyrimidine at R3, preference for specific synthons at R1 and R2 was evident. R1 synthons were limited to 4- and 3,4-substituted arylalkyl groups, particularly 3,4-dimethoxyphenethyl (synthon-5) and 1,3-benzodioxolane-5-methyl (synthon-16). R2 synthons were more restricted, with piperazine-2-acetic acid (synthon-31) found on 46% of active beads. Other active R2 synthons included d-thienylalanine (synthon-8), d-methionine (synthon-10), and sarcosine (synthon-24). Compound 1 (Fig. 1C) was composed of the synthons most commonly found in library actives and was one of the most potent (IC50 = 1.1 nM) of 23 inhibitors resynthesized for IC50 determinations.
Because 1 did not inhibit purified iNOS enzyme activity, its mechanism of action was examined further in cell culture systems. Reverse transcriptase–PCR and Western blot analyses indicated 1 had minimal effects on iNOS mRNA and protein expression in A-172 cells (data not shown), suggesting a posttranslational mode of action. This was supported by the observation that the IC50 of 1 increased dramatically with time of addition after cytokine stimulation with an estimated t1/2 of ≈8 hr. We thus hypothesized that 1 was an inhibitor of iNOS dimerization. Further characterization of these inhibitors focused on 2, which lacked the potentially reactive chlorine substituent found in 1.
To substantiate the hypothesis that these compounds inhibited dimerization, the effect of 2 on iNOS dimerization in RAW 264.7 cells was examined. This murine macrophage cell line was chosen because LPS and IFN-γ treatment results in expression of a mixture of iNOS monomers and dimers (30). Preliminary experiments using low-temperature SDS/PAGE to separate monomers and dimers (35) indicated that a concentration-dependent increase in the amount of iNOS monomer occurred when the cells were treated with inhibitors (data not shown). Fig. 2 A and B shows size exclusion chromatographs of extracts from RAW 264.7 cells induced with LPS and IFN-γ. As expected, when cells were grown in the absence of inhibitor, the iNOS cytochrome c reductase activity was in two peaks corresponding to iNOS monomer and dimer, whereas iNOS activity, measured by citrulline formation, was associated only with the dimer peak. Treatment of cells with 2 (10 μM) during cytokine induction ablated iNOS dimer-dependent generation of citrulline, and the shift of cytochrome c reductase activity to a single monomer peak confirmed intracellular iNOS monomer accumulation.
The interaction of 2 with iNOS was characterized in both cell-based and cell-free assays. Compound 2 inhibited NO⋅ production by cytokine-stimulated human glioblastoma A-172 cells with an IC50 of 0.6 nM (SD = 0.5, n = 17). In contrast, >1,000-fold higher concentrations of N-methyl-l-arginine and the inhibitor 1400-W (36) were required to cause similar inhibition of the iNOS activity in these cells. To directly measure the affinities of these inhibitors for human iNOS monomers, the Kd of the radiolabeled compound 3 was determined and found to be 2.5 nM. Scatchard plots were linear, indicating a single class of binding sites and no evidence of homotropic cooperativity (data not shown). Binding of 3 to iNOS monomer also was blocked by imidazole (Ki = 39 μM), consistent with an interaction with heme. Indeed, binding of 3 was substoichiometric with iNOS monomer, but well correlated to the amount of heme-containing monomer in the preparation. As shown in Fig. 3, binding of 3 could be blocked with 2, which had a Ki of 2.2 nM.
Figure 3.

Binding of 2 to purified iNOS monomers. The amount of bound radioligand 3 (cpm) is plotted versus the concentration of 2 (Ki = 2.2 nM). Error bars indicate the SD for each value.
The selectivity of 2 for inhibiting iNOS versus inhibition of neuronal NOS and endothelial NOS dimerization was evaluated by measuring the formation of active dimeric enzymes in BSC-1 cells. The cells were treated with 2 and infected by recombinant vaccinia virus vectors that expressed the three human NOS isoforms. Compound 2 was added at the time of viral infection. Aliquots of the crude cell lysate were assayed for NOS activity by measuring the conversion of arginine into citrulline. Compound 2 potently inhibited human iNOS (IC50 = 28 nM, SD = 7, n = 14), but was about 5-fold less potent against neuronal NOS (IC50 = 140 nM, SD = 55, n = 17) and 1,000-fold less potent against endothelial NOS (IC50 = 32 μM, SD = 15, n = 13). The synthesis of analogs of compounds contained in the combinatorial library resulted in an improvement in the selectivity of the inhibitor series versus neuronal NOS to >100-fold. The whole series of inhibitors was essentially inactive against dimeric iNOS, neuronal NOS, and endothelial NOS (IC50 values >50 μM) in enzyme activity assays.
Compound 2 also effectively suppressed NO⋅ production in vivo. Elevations in plasma NOx levels have been used as an index of systemic iNOS induction (32, 37). Fig. 4 shows that i.p. injection of rats with 2 1 hr after the systemic induction of iNOS by LPS led to dose-dependent inhibition of elevated plasma NOx levels (ED50 = 1.2 mg/kg) measured 6 hr after LPS administration.
Figure 4.

Inhibition of LPS-induced elevations of plasma NOx levels in rats. Plasma levels of NOx were 24.3 ± 1.5 μM in naive rats. Error bars show the SEM; this experiment has been repeated in rats and mice with similar results.
The crystal structure of an inhibitor–iNOS complex was determined to understand the binding of these inhibitors to iNOS and gain insight into the mechanism of iNOS dimerization. Compound 2 was soaked into crystals of the monomeric oxygenase domain of murine iNOS (iNOS Δ114), and the crystal structure of the resulting complex was solved. This revealed that the free imidazole nitrogen of 2 coordinates directly to the heme in the sixth axial position, and that the imidazole, pyrimidine, and part of the piperazine ring are nearly coplanar (Fig. 5). The piperazine ring is in a chair conformation, and the chiral center at the 2-position has an S configuration. Both rotamers of the methyl carbamate in 2 were present in the structure (one is shown). The benzodioxolane moiety fits snugly between residues in the iNOS monomer active site and the inhibitor pyrimidine ring making numerous Van der Waals contacts, which result in the inhibitor adopting a U-shaped conformation. The amide NH of 2 is hydrogen-bonded to one of the heme propionates, and its carbonyl is hydrogen-bonded to two water molecules, which are hydrogen-bonded to Gln-257 and Tyr-367. The methyl carbamate, the carbonyl of the amide bond, and the unsubstituted side of the piperazine are solvent exposed.
Figure 5.
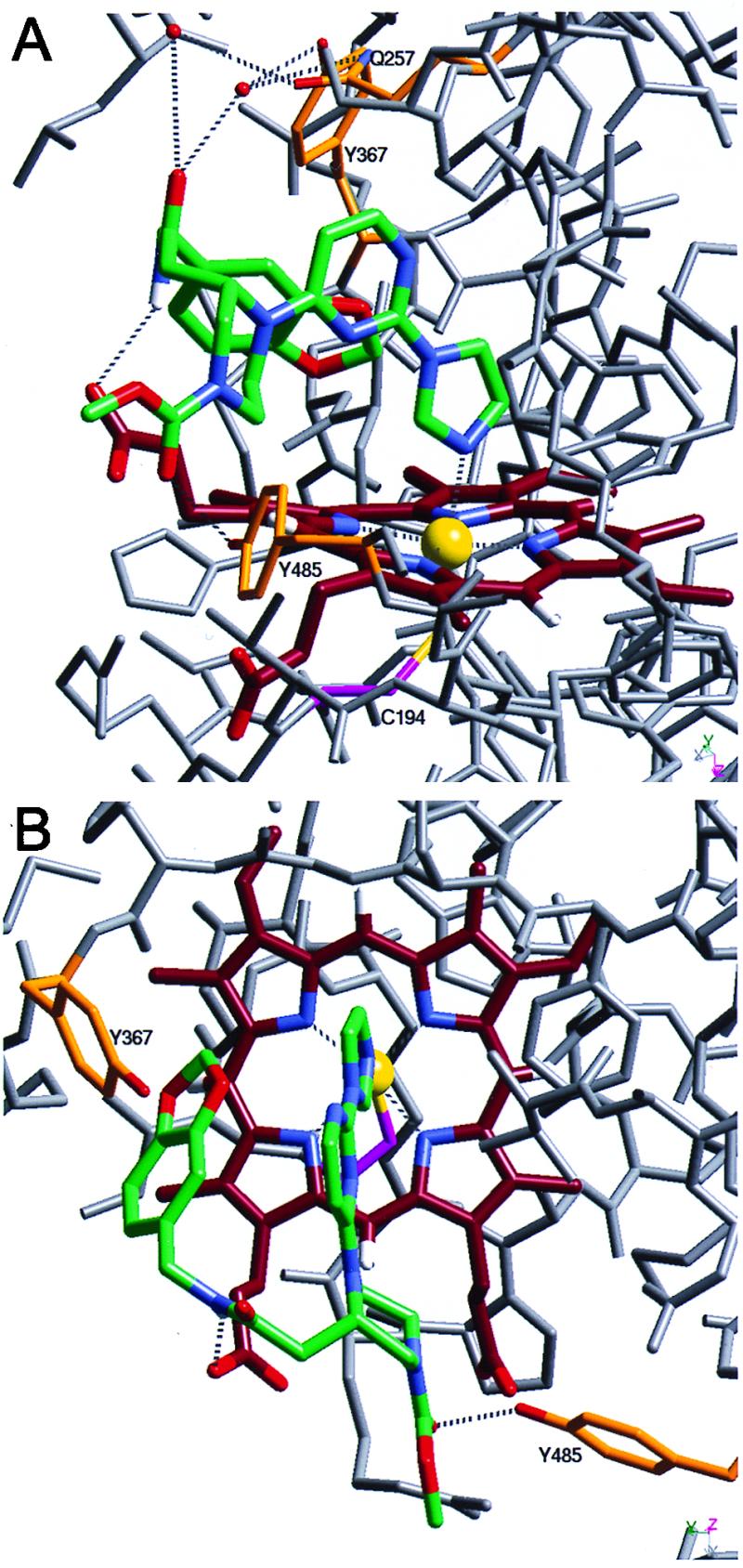
The 2.25-Å resolution crystal structure of the 2–murine iNOS Δ114 complex and comparison to the murine iNOS dimer structure. Two views of the refined model of the 2–murine iNOS Δ114 complex are shown. Compound 2 (green) and heme (dark red) both are shown with blue nitrogens and red oxygens. The sulfur (yellow) of Cys-194 (magenta) coordinates the heme iron (yellow sphere). The water molecules described in the text are shown as red spheres, and in Tyr-367, Tyr-485, and Gln-257 side chains are shown in orange with red oxygens and blue nitrogens. Both iron coordination and hydrogen bonds are shown as dashed lines; other iNOS Δ114 atoms are shown in gray. Numerous residues are not shown for clarity, including residues 486–496 and the side chains of Val-346, Asn-348, and Met-349 in A and Gln-257 and surrounding residues (241–345) in B.
In the 2–iNOS Δ114 complex, 2 occupies part of the l-arginine binding site, and residues that normally form part of the site are disordered (Fig. 6). The side chain of Glu-371 in helix 7a hydrogen bonds to l-arginine in the iNOS dimer (11) and imidazole in the monomer (10). However, in the 2–iNOS Δ114 complex the residues in helix 7a are disordered, possibly because 2 displaces Glu-371 from the active site. Alternatively, Glu-371 hydrogen bonding to l-arginine may be required for correct folding of the dimer. The disruption of helix 7a may indirectly disturb the dimer interface because, in the iNOS dimer, helix 7a is packed against helix 8 and residues 460–462. Helix 8 is partially disordered in the 2–iNOS Δ114 structure and is part of the dimer interface. Residues 460–462 make up part of the H4B binding site and part of the dimer interface. The difference between the helix 9 location in the dimer and the 2–iNOS Δ114 structure (Fig. 6) is not thought to be involved in the inhibition of dimerization because the difference also was observed when comparing the imidazole–iNOS monomer and iNOS dimer structures (11). Thus, in occupying the iNOS active site, 2 appears to allosterically prevent appropriate protein–protein interactions between monomers. A similar perturbation of the dimer interface may be the mechanism by which micromolar concentrations of antifungal imidazoles inhibit dimerization (38). The precise mode of binding with iNOS monomer and the NOS isoform selectivity is not known for these antifungal imidazoles.
Figure 6.
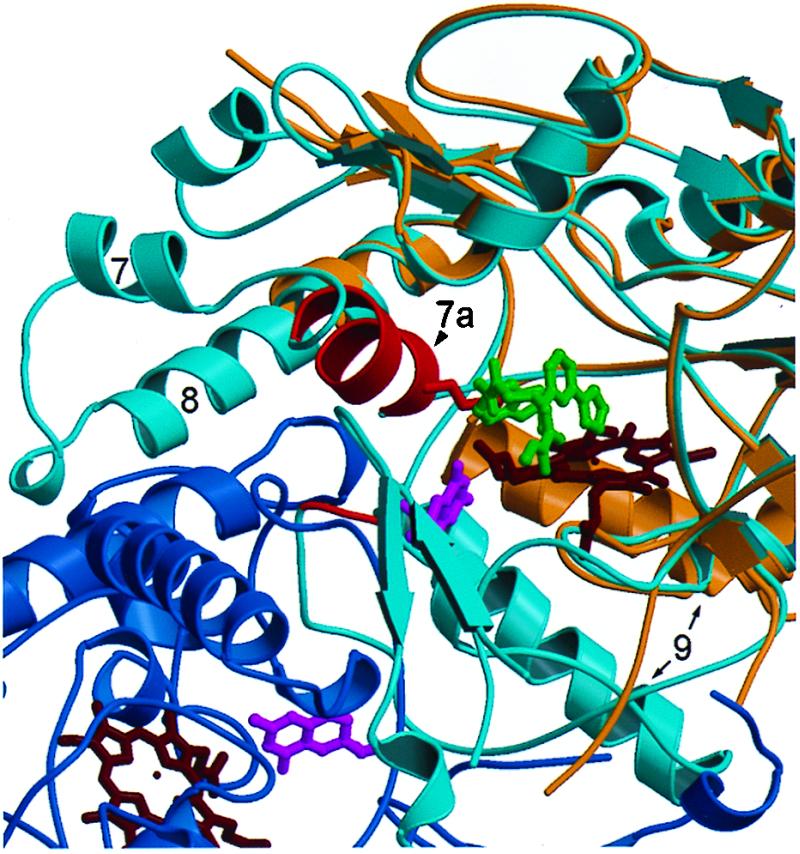
Comparison of the 2–iNOS Δ114 complex with the murine iNOS oxygenase domain dimer. Compound 2 occupies the arginine-binding site. The monomers of the iNOS dimer structure (11) are shown in dark and light blue. The 2–iNOS Δ 114 structure (tan) has been aligned with a monomer (light blue) of the dimer structure; the hemes are shown in dark red. The iNOS Δ 114 heme and the dimer heme align very closely (for clarity only the iNOS Δ 114 heme is shown). Compound 2 (dark gray) displaces helix 7a (red) side chains from the arginine-binding site in the 2–iNOS Δ 114 structure, e.g., Glu-371 (red). The region from the beginning of helix 7a to the middle half of helix 8 is disordered in 2–iNOS Δ 114 structure.
In summary, we have described a class of potent iNOS inhibitors, with high iNOS versus endothelial NOS selectivity, that inhibit iNOS dimerization. The compounds have exceptional potency in cell-based assays, indicating superior cell permeability compared with other iNOS inhibitors such as N-methyl-l-arginine and 1400W. The crystal structure of the monomer–inhibitor complex shows that the inhibitors ligate heme and occupy the active site in the iNOS monomer in such a way as to grossly perturb key structural elements in the iNOS monomer that are critical for stable dimer formation. From a pharmaceutical discovery perspective, we have shown that high-throughput encoded combinatorial chemistry methods can be applied to the discovery of potent, selective, cell-permeable iNOS inhibitors that are active in vivo and potentially of broad therapeutic utility.
Acknowledgments
We thank D. J. Stuehr (Cleveland Research Clinic, Cleveland, OH) for advice on NOS enzymology and for the murine iNOS Δ114 E. coli expression system; J. A. Trainer and A. Arvai (The Scripps Research Clinic, La Jolla, CA) for providing iNOS Δ114 crystal coordinates and advice on crystal growth; A. Cohen, P. Kuhn, and M. Soltis at the Stanford Synchrotron Radiation Laboratory (funded by the Department of Energy, Office of Basic Energy Sciences) for help collecting x-ray diffraction data; J. G. Keck, S. Vijay, Y. Zhu, and M. Carnine for developing the BSC-1 selectivity assay; and H. Daniel Perez for his support.
Abbreviations
- H4B
tetrahydrobiopterin
- LPS
lipopolysaccharide
- NOS
nitric-oxide synthase
- iNOS
inducible NOS
- Fmoc
fluorenylmethoxycarbonyl
Footnotes
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID code 1DD7).
References
- 1.Griffith O W, Stuehr D J. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 2.Marletta M A. Cell. 1994;78:927–930. doi: 10.1016/0092-8674(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 3.Masters B S, McMillan K, Sheta E A, Nishima J S, Roman L J, Martasek P. FASEB J. 1996;10:552–558. doi: 10.1096/fasebj.10.5.8621055. [DOI] [PubMed] [Google Scholar]
- 4.Wang M, Roberts D L, Paschke R, Shea T M, Masters B S, Kim J J. Proc Natl Acad Sci USA. 1997;94:8411–8416. doi: 10.1073/pnas.94.16.8411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bredt D S, Hwang P M, Glatt C E, Lowenstein C, Reed R R, Snyder S H. Nature (London) 1991;351:714–718. doi: 10.1038/351714a0. [DOI] [PubMed] [Google Scholar]
- 6.Ghosh D K, Wu C, Pitters E, Moloney M, Werner E R, Mayer B, Stuehr D S. Biochemistry. 1997;36:10609–10619. doi: 10.1021/bi9702290. [DOI] [PubMed] [Google Scholar]
- 7.Klatt P, Pfeiffer S, List B M, Lehner D, Glatter O, Bächingers H P, Werner E R, Schmidt K, Mayer B. J Biol Chem. 1996;271:7336–7342. doi: 10.1074/jbc.271.13.7336. [DOI] [PubMed] [Google Scholar]
- 8.List B M, Klösch B, Völker C, Gorren A C F, Sessa W C, Werner E R, Kukovetz W R, Schmidt K, Mayer B. Biochem J. 1997;323:159–165. doi: 10.1042/bj3230159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siddhanta U, Presta A, Fan B, Wolan D, Rousseau D L, Stuehr D J. J Biol Chem. 1998;273:18950–18958. doi: 10.1074/jbc.273.30.18950. [DOI] [PubMed] [Google Scholar]
- 10.Crane B R, Arvai A S, Gachhui R, Wu C, Ghosh D K, Getzoff E D, Stuehr D J, Tainer J A. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- 11.Crane B R, Arvai A S, Ghosh D K, Wu C, Getzoff E D, Stuehr D J, Tainer J A. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 12.Li H, Raman C S, Glaser C B, Blasko E, Young T A, Parkinson J F, Whitlow M, Poulos T L. J Biol Chem. 1999;274:21276–21284. doi: 10.1074/jbc.274.30.21276. [DOI] [PubMed] [Google Scholar]
- 13.Fischmann T O, Hruza A, Niu X D, Fossetta J D, Lunn C A, Dolphin E, Prongay A J, Reichert P, Lundell D J, Narula S K, Weber P C. Nat Struct Biol. 1999;6:233–242. doi: 10.1038/6675. [DOI] [PubMed] [Google Scholar]
- 14.Raman C S, Huiying L, Martasek P, Kral V, Masters B S S, Poulos T L. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 15.Moncada S, Higgs A. N Engl J Med. 1993;329:2002–2012. doi: 10.1056/NEJM199312303292706. [DOI] [PubMed] [Google Scholar]
- 16.Nathan C, Xie Q W. Cell. 1994;78:915–918. doi: 10.1016/0092-8674(94)90266-6. [DOI] [PubMed] [Google Scholar]
- 17.Bredt D S, Snyder S H. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- 18.Kerwin J F, Lancaster J R, Jr, Feldman P L. J Med Chem. 1995;38:4343–4362. doi: 10.1021/jm00022a001. [DOI] [PubMed] [Google Scholar]
- 19.Stamler J S, Singel D J, Loscalzo J. Science. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- 20.Dawson V L, Dawson T M. Adv Pharmacol. 1995;34:323–342. doi: 10.1016/s1054-3589(08)61095-9. [DOI] [PubMed] [Google Scholar]
- 21.Nathan C. J Clin Invest. 1997;100:2417–2423. doi: 10.1172/JCI119782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Amin A R, Abramson S B. Curr Opin Rheumatol. 1998;10:263–268. doi: 10.1097/00002281-199805000-00018. [DOI] [PubMed] [Google Scholar]
- 23.Stichtenoth D O, Frolich J C. Br J Rheumatol. 1998;37:246–257. doi: 10.1093/rheumatology/37.3.246. [DOI] [PubMed] [Google Scholar]
- 24.Parkinson J F, Mitrovic B, Merrill J E. J Mol Med. 1997;75:174–186. doi: 10.1007/s001090050102. [DOI] [PubMed] [Google Scholar]
- 25.Samdani A F, Dawson T M, Dawson V L. Stroke. 1997;28:1283–1288. doi: 10.1161/01.str.28.6.1283. [DOI] [PubMed] [Google Scholar]
- 26.Beal M F. Ann Neurol. 1998;44:S110–S114. doi: 10.1002/ana.410440716. [DOI] [PubMed] [Google Scholar]
- 27.Ohlmeyer M H, Swanson R N, Dillard L W, Reader J C, Asouline G, Kobayashi R, Wigler M, Still W C. Proc Natl Acad Sci USA. 1993;90:10922–10926. doi: 10.1073/pnas.90.23.10922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stuehr D J, Marletta M A. Proc Natl Acad Sci USA. 1985;82:7738–7742. doi: 10.1073/pnas.82.22.7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bredt D S, Snyder S H. Proc Natl Acad Sci USA. 1990;87:682–685. doi: 10.1073/pnas.87.2.682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albakri Q A, Stuehr D J. J Biol Chem. 1996;271:5414–5421. doi: 10.1074/jbc.271.10.5414. [DOI] [PubMed] [Google Scholar]
- 31.Gerber N C, de Montellano P R O. J Biol Chem. 1995;270:17791–17796. doi: 10.1074/jbc.270.30.17791. [DOI] [PubMed] [Google Scholar]
- 32.Tracey W R, Tse J, Carter G. J Pharmacol Exper Ther. 1995;272:1011–1015. [PubMed] [Google Scholar]
- 33.Wolff D J, Lubeskie A, Umansky S. Arch Biochem Biophys. 1994;314:360–366. doi: 10.1006/abbi.1994.1454. [DOI] [PubMed] [Google Scholar]
- 34.Chabin R M, McCauley E, Calaycay J R, Kelly T M, MacNaul K L, Wolfe G C, Hutchinson N I, Madhusudanaraju S, Schmidt J A, Kozarich J W, Wong K K. Biochemistry. 1996;35:9567–9575. doi: 10.1021/bi960476o. [DOI] [PubMed] [Google Scholar]
- 35.Klatt P, Schmidt K, Lehner D, Glatter O, Bächinger H P, Mayer B. EMBO J. 1995;14:3687–3695. doi: 10.1002/j.1460-2075.1995.tb00038.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Garvey E P, Oplinger J A, Furfine E S, Kiff R J, Laszlo F, Whittle B J, Knowles R G. J Biol Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- 37.Kengatharan K M, De Kimpe S J, Thiemermann C. Br J Pharmacol. 1996;119:1411–1421. doi: 10.1111/j.1476-5381.1996.tb16053.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sennequier N, Wolan D, Stuehr D J. J Biol Chem. 1999;274:930–938. doi: 10.1074/jbc.274.2.930. [DOI] [PubMed] [Google Scholar]